

Abstract
The invasive signal amplification reaction has been previously developed for quantitative detection of nucleic acids and discrimination of single-nucleotide polymorphisms. Here we describe a method that couples two invasive reactions into a serial isothermal homogeneous assay using fluorescence resonance energy transfer detection. The serial version of the assay generates more than 107 reporter molecules for each molecule of target DNA in a 4-h reaction; this sensitivity, coupled with the exquisite specificity of the reaction, is sufficient for direct detection of less than 1,000 target molecules with no prior target amplification. Here we present a kinetic analysis of the parameters affecting signal and background generation in the serial invasive signal amplification reaction and describe a simple kinetic model of the assay. We demonstrate the ability of the assay to detect as few as 600 copies of the methylene tetrahydrofolate reductase gene in samples of human genomic DNA. We also demonstrate the ability of the assay to discriminate single base differences in this gene by using 20 ng of human genomic DNA.
Previously, we described the invasive signal amplification assay for direct genetic analysis of nucleic acids by using invasive cleavage with structure-specific 5′ nucleases (1). The method requires annealing of two oligonucleotides, called the upstream oligonucleotide and the probe, to a target sequence, which results in the formation of a unique substrate for the 5′ nuclease (Fig. 1A). The probe contains two regions, an analyte-specific region that forms a duplex with the target and a noncomplementary 5′ arm region, which is not required for enzyme activity but serves as a reporter molecule precursor. Cleavage of the probe occurs only when the probe and upstream oligonucleotide overlap (2, 3); therefore, two target DNAs differing only by a single nucleotide that affects formation of the cleavage structure can be differentiated. This extraordinary specificity of substrate structure recognition by the 5′ nuclease enables detection of single point mutations with a discrimination level required for single-nucleotide polymorphism analysis (1, 4).
Figure 1.

Schematic representations of the serial invasive signal amplification reaction (SISAR). (A) Proposed secondary structure of the overlapping substrate of the primary reaction. The upstream oligonucleotide and the primary probe are bound with the target strand so that the 3′ terminal nucleotide (T) of the upstream oligonucleotide overlaps with the terminal A-T base pair of the duplex formed between the probe and the target. The arrow indicates the site of cleavage, which generates a cleaved 5′ arm (shown in bold) that contains one nucleotide of the analyte-specific region (A) bearing a 3′-OH. (B) Proposed secondary structure of the overlapping substrate of the secondary reaction. The cleaved 5′ arm, produced in the primary reaction, forms an invasive substrate with target and probe strands linked into a hairpin structure called the secondary probe. Fl and Cy3 dyes, forming a FRET pair, are denoted by Fl and Cy3, respectively. Bt denotes a biotin modification. The arrow indicates the cleavage site. (C) The X-structure formed by the uncut primary probe and the secondary probe, which contributes to the background of the reaction (see text).
Performing the invasive reaction at elevated temperatures allows for rapid turnover of the probe, thus enabling the 5′ nuclease to produce multiple cleaved probes per target molecule, thereby amplifying the signal. The cleaved 5′ arm of the probe serves as a reporter molecule whose presence in a sample provides a means for qualitative and quantitative analysis of target sequences. The previously reported version of the invasive assay achieved a typical signal amplification level of approximately 3,000 cleavages per target molecule in 90 min and thus required an additional signal amplification step for detection of small amounts of target molecules (1). Also the second step of the assay, which involved the capture and enzymatic labeling of the cleaved 5′ arm with fluorescein (Fl) or digoxigenin followed by chemiluminescence detection, was not compatible with a homogeneous format nor did it result in the detection of subattomol levels of a target.
Here we describe improvements in sensitivity of the invasive signal amplification assay that permit a limit of detection (LOD) of zeptomol (10−21 mol) levels of a target DNA in a homogeneous format. This was accomplished by combining two invasive signal amplification reactions in series in a single-tube format. The cleaved 5′ arm from the target-specific primary reaction is used to drive a secondary invasive reaction, resulting in a total signal amplification of more than seven orders of magnitude in 4 h. To simplify detection, the secondary probe is labeled with a fluorescence resonance energy transfer (FRET) dye pair, in which a donor dye is quenched by an acceptor dye. After cleavage, the dyes are separated, and the increased fluorescence of the donor dye is directly detected by a conventional fluorescence plate reader. We call this simple more sensitive assay the serial invasive signal amplification reaction (SISAR).
In this work, we determine the kinetic parameters of both the primary and secondary reactions and show that the overall SISAR can be described by a simple kinetic model. The analysis identifies a single nonspecific structure as the major source of background generation that limits the LOD of the reaction. An application of the assay is demonstrated in which ≈6,000 copies of the human methylene tetrahydrofolate reductase (MTHFR) gene in a human genomic DNA sample (20 ng) are assayed for a C to T polymorphism at position 667.
Materials and Methods
Materials.
Chemicals and buffers were from Fisher Scientific unless otherwise noted. Structure-specific 5′ nuclease from Archaeoglobus fulgidus was expressed, purified, and quantitated as described (1). The enzyme was dialyzed and stored in 50% glycerol/20 mM Tris⋅HCl, pH 8/50 mM KCl/0.5% Tween 20/0.5% Nonidet P-40/100 μg/ml BSA. Unless otherwise noted, A, G, C, and T refer to deoxyribonucleotides.
Oligonucleotide Synthesis.
All oligonucleotides were synthesized on a PerSeptive Biosystems (Framingham, MA) instrument by using standard phosphoramidite chemistries including Fl, Cy3 dye, and biotin (Bt) modifications (Glen Research, Sterling, VA). The secondary probes were purified by ion exchange HPLC by using a Resource Q column (Amersham Pharmacia Biotech). All other oligonucleotides were purified by separating the primary synthesis products on a 20% denaturing polyacrylamide gel followed by excision and elution of the major band. Oligonucleotide concentrations were determined by measuring absorption at 260 nm and using specific extinction coefficients for A, T, G, and C (5).
Preparation of the Hepatitis B Virus (HBV) Target PCR Product for the Model System.
A 608-bp region corresponding to bases 220–827 of the HBV genome (6) was PCR amplified by using the HotStarTaq master mix kit (Qiagen, Chatsworth, CA) and HBV DNA extracted from a serum sample. The PCR product was cloned by using the TOPO TA cloning kit (Invitrogen), and the sequence of the cloned fragment was determined with an Applied Biosystems Prism 377 DNA sequencer. The plasmid containing the HBV fragment was used as a template to PCR amplify the 608-bp target DNA used in the model invasive reaction in this study. The target DNA was purified with a High-pure PCR product purification kit (Boehringer Mannheim) and quantitated by absorption measurement at 260 nm. Dilutions of the amplicon were made in a solution containing 5 μg/ml human genomic DNA (Novagen).
Determination of the Cycling Cleavage Rate of the Primary Reaction.
The 25-μl reactions were carried out with 0.4 μM primary probe labeled with Fl at the 5′ end/40 nM upstream oligonucleotide/0.1 nM target PCR product/100 ng AfuFEN enzyme in a reaction buffer containing 10 mM 4-morpholinepropanesulfonic acid, pH 7.5, 7.5 mM MgCl2, 3.2% polyethylene glycol, 0.05% Tween 20, 0.05% Nonidet P-40, and 2 μg/ml of human genomic DNA as a carrier. The target PCR product was denatured before assembling the reactions by heating at 95°C for 3 min in 10 mM Tris⋅HCl, pH 8/0.1 mM EDTA. The assays were assembled on ice, and reactions were initiated by transferring the samples to a Mastercycler (Eppendorf) heating block. The reactions were stopped by cooling the samples in an ice bath followed by addition of 15 μl of 95% formamide containing 10 mM EDTA and 0.02% methyl violet (Sigma). The samples were analyzed by electrophoresis through a 20% denaturing polyacrylamide gel, and the gels were then scanned on an FMBIO-100 fluorescence gel scanner (Hitachi, Alameda, CA) by using a 532-nm laser and 585-nm filter, as described previously (3). The fraction of cleaved product was determined from the intensities of bands corresponding to uncut and cut substrate with fmbio analysis software (Ver. 6.0, Hitachi). The fraction of cut product did not exceed 20%, ensuring that measurements approximated initial cleavage rates. The cycling cleavage rate was defined as the concentration of cut product divided by the target concentration and the time of the reaction (in minutes), as described previously (7).
Determination of the Cycling Cleavage Rate of the Secondary Reaction.
Unless otherwise noted, reaction conditions for determining the secondary reaction rate constants were the same as the reaction conditions for determining the primary reaction rates. Reactions were performed with 0.2 μM secondary probe and different concentrations of the 5′ arm (0, 0.4, 1, 2, 4, 6, 10, 15, or 20 pM) in either the absence or presence of 0.4 μM primary probe. To allow for signal normalization, a passive reference (PE Biosystems, Foster City, CA) was added to a final concentration of 0.12 μM. The reagents were mixed in an ice bath and transferred to a 96-well reaction plate in quadruplicate for each target level. Twenty-five microliters of Chill-out liquid wax (MJ Research, Cambridge, MA) was added to each well to prevent evaporation and optical caps (PE Biosystems) were placed on top of the wells. The plate was centrifuged for 2 min at 500 × g to remove any bubbles. Reactions were initiated by placing the plate into an Applied Biosystems Prism 7700 sequence detector (PE Biosystems) and raising the temperature to 63°C (≈1.5°C/sec). Emission spectra were measured in a range from 500 to 655 nm every minute for 90 min. The Fl, Cy3, and passive reference signals were analyzed by using software provided by the manufacturer. A relative fluorescence signal was defined as the Fl signal normalized to the passive reference signal to account for well-to-well variation.
To determine the relationship between the relative fluorescence units and the concentration of cleaved secondary probe, the relative fluorescence signal was measured for standard solutions with different ratios of the secondary probe and Fl-CC. The fluorescence signal of those standard mixtures was collected on the same plate during the invasive reaction in quadruplicate. A plot of the relative fluorescence signal as a function of Fl-CC concentration followed a linear relationship and was used as a standard curve to determine the concentration of cleaved secondary probe from the raw data.
Kinetics of SISAR.
The SISAR kinetics were determined with 0, 0.01, 0.03, 0.1, 0.3, 1, or 3 pM HBV target under the reaction conditions described above. The target PCR product was denatured before reagent assembly by incubating at 95°C for 5 min in the presence of 5 μg/ml human genomic DNA as a carrier. The reactions were run for 4 h at 63°C in quadruplicate. The relative fluorescence signal was collected at time intervals from 20 sec to 5 min. The relative fluorescence signal was converted into concentration of the cleaved secondary probe using the standard curve as described above. The average SISAR signals were fit to either linear or quadratic equations by using the sigmaplot program (SPSS, Chicago)
Detection and Polymorphism Analysis of the Human MTHFR Gene.
Assays were performed by using the MTHFR polymorphism (C677T) detection kit (Third Wave Technologies). The upstream oligonucleotide was 5′-CAAAGAAAAGCTGCGTGATGATGAAATCGC. The C667 and T667 primary probes blocked at the 3′ end with an amino group (Glen Research) were 5′-AACGAGGCGCACGCTCCCGCAGACAC-NH3 and 5′-AACGAGGCGCACACTCCCGCAGACACC-NH3, respectively (where boldface indicates the nucleotide that is complementary to one or the other nucleotide at the polymorphic position in the target DNAs). The secondary probe was 5′-Fl-CCTC-Cy3-GTCTCGGTTTTCCGAGACGAGGGTGCGCCTCGTTT, where boldface denotes 2′-O-methyl modified nucleotides. The reaction conditions were the same as those described for SISAR kinetic measurements except that the reaction volume was reduced to 20 μl, and 100 ng tRNA (Sigma) was used as a carrier. The purified human genomic samples were obtained either from an in-house sample for the C/C alleles or from a commercial source for the T/T and C/T alleles (Coriell Cell Repositories, Camden, NJ). The sample genotypes were verified by in-house restriction fragment length polymorphism analysis (8), and DNA concentrations were determined by using the PicoGreen assay (Molecular Probes). The DNA samples were denatured before reaction assembly by incubating at 95°C for 5 min in the presence of 100 ng tRNA. The serial invasive reactions were carried out for 4 h at 63°C by using an Applied Biosystems Prism 7700 as described above. All reactions were performed in quadruplicate. The net relative fluorescence signal was obtained by subtracting the average signal generated by the secondary probe in the absence of target from the average signal generated from each target level assayed with the identical probe at the same time point.
Results
Principle of the SISAR.
A model system used in this work for the analysis of SISAR is shown in Fig. 1. In the primary reaction, developed for detection of HBV DNA, binding of the target strand with the upstream oligonucleotide, and the primary probe aligns them so that the 3′ terminal nucleotide of the upstream oligonucleotide overlaps the first base paired nucleotide of the primary probe (Fig. 1A). Although this overlap, or “invasion,” is required for cleavage, the 3′ terminal nucleotide does not need to base pair with the target and can be any of the four common nucleotides (2, 3). The structure-specific 5′ nuclease AfuFEN used in this work (1, 3) cleaves the primary probe on the 3′ side of the first base-paired nucleotide at the position dictated by the 3′ end of the upstream oligonucleotide. This cleavage event releases the noncomplementary 5′ arm and one nucleotide (Fig. 1A) of the analyte-specific region of the primary probe (shown in bold in Fig. 1A). The 5′ arm is not required for enzymatic activity and can have variable length and almost any sequence; however, very long arms may inhibit cleavage activity (9).
Because the target strand is not cleaved, it can be used to direct cleavage of multiple probes, being limited only by the rate at which the 3′ portion of the probe bound to the target is replaced by an intact probe after cleavage. Optimal replacement of cleaved probe with an intact one is accomplished by incubation near the melting temperature of the probe/target duplex (7). Previously, we demonstrated (7) that the kinetics of the invasive signal amplification reaction can be described by the following equation when the probe is in excess over the target and only a small fraction of the probes has been cleaved during the reaction:
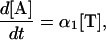 |
1 |
where [A] and [T] are the concentrations of the cleaved 5′ arm and the target strand, respectively, and α1 is the cycling cleavage rate of the primary reaction, defined as the number of primary probes cleaved per target molecule per minute (7).
In the secondary reaction, the target strand and probe are linked in a hairpin structure, called the secondary probe (Fig. 1B). The secondary probe is labeled at the 5′ end with Fl and internally with a Cy3 dye. These dyes function as a FRET donor-acceptor pair in which the fluorescence of the Fl is quenched by the Cy3. Annealing of this secondary probe with the 5′ arm that was released by cleavage in the primary reaction provides another substrate for the 5′ nuclease. In contrast to the primary reaction, signal amplification in the secondary reaction results from the cycling of the limiting cleaved 5′ arm, which is provided by the primary reaction. Increased fluorescence from the released Fl in the secondary reaction is a quantitative measure of the amount of target.
Assuming that the secondary probe is in excess over the 5′ arm and only a small fraction of the probes has been cleaved during the reaction, the cleavage rate of the secondary probe can be described by the following equation:
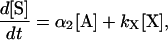 |
2 |
where [S] is the concentration of product generated in the secondary reaction, α2 is the cycling cleavage rate of the secondary reaction, and [X] is the concentration of the X-structure formed by annealing of the secondary probe with the uncut primary probe (Fig. 1C). The term kx[X] describes background generation caused by nonspecific cleavage of the X-structure with the background generation constant kx. Earlier, we showed that the X-structure exhibits the highest cleavage rate among all nonspecific structures that are likely to form during SISAR (7). Thus, the background component of Eq. 2 becomes an issue only during the secondary reaction.
Under conditions where only a small fraction of the secondary probe has been cleaved during the reaction, concentration of the X-structure can be assumed to be constant, and the entire term kx [X] can be condensed to kb, which does not change during the time of incubation. Thus, Eq. 2 can be written as:
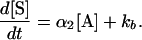 |
3 |
Cycling Cleavage Rates of the Primary and Secondary Reactions.
The SISAR conditions used in this work were previously optimized for the best performance by varying reaction parameters such as the concentrations of the enzyme and each probe, temperature, the buffer content, the sequences of the 5′ arm, and the secondary probe (data not shown). These conditions were then used to determine the kinetic parameters of both the primary and the secondary reactions.
The cycling cleavage rate α1 of the primary reaction shown in Fig. 1A was determined by using the Fl-labeled primary probe (0.4 μM) as described in Materials and Methods. The kinetics of the primary probe cleavage were measured by gel electrophoresis analysis of the cleaved products, and the cycling cleavage rate α1 = 15 min−1 was determined as described (7) (data not shown). The optimal temperature of the primary reaction was 62–63°C.
The cycling cleavage rate α2 of the secondary reaction shown in Fig. 1B was determined from the kinetics of the secondary probe (0.2 μM) cleavage under the SISAR conditions. Kinetic data corresponding to the initial 10% of secondary probe cleavage were used to determine the initial rates of the reaction, d[S]/dt, at different 5′ arm concentrations, [A], by adding an oligonucleotide identical to the authentic cleaved 5′ arm that would be generated in a primary reaction. These rates were measured in the absence and in the presence of 0.4 μM primary probe to determine how this probe would contribute to the background of the reaction. As shown in Fig. 2, the initial rates can be approximated by linear functions of [A]. According to Eq. 3, defining the initial rate as α2[A] + kb under the conditions of a constant concentration of the limiting 5′ arm, the cycling cleavage rate α2 and the background constant kb can be determined from the slopes and the intercepts of the linear functions, respectively.
Figure 2.

Dependence of the initial cleavage rate, d[S]/dt, of the secondary reaction on the concentration of added 5′ arms, [A], such as would be generated by cleavage in the primary reaction. The initial cleavage rates, measured as the concentration of the secondary probe cleaved by the enzyme in one minute (nM⋅min−1), were determined at different concentrations of 5′ arm. The reactions were run with 0.2 μM secondary probe, in the absence (●) or presence (○) of 0.4 μM primary probe, to measure the contribution of the primary probe to the background. The slopes of the lines give the cycling cleavage rate of the secondary reaction, α2, and the intercepts of the y axis indicate the contribution of background, kb, to the total rate. Error bars indicate the standard deviations obtained from the quadruplicate measurements with each sample.
The experimental data in Fig. 2 show that in the absence and presence, respectively, of the primary probe, the α2 values are 32 and 30 min−1, and kb values are 0.013 and 0.28 nM⋅min−1. The slight decrease in α2 in the presence of the primary probe is apparently because of competition between the cleaved 5′ arm and the primary probe for the same binding region in the secondary probe. The more than 20-fold increase in the background constant kb in the presence of the primary probe is in agreement with the suggested critical role of the X-structure in background generation during SISAR.
Kinetics of the Serial Invasive Signal Amplification Reaction.
The kinetics of signal accumulation in SISAR, with both primary and secondary reactions running simultaneously, can be described by integrating Eqs. 1 and 3:
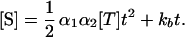 |
4 |
Eq. 4 predicts that the signal of SISAR, measured as the concentration of the cleaved secondary probe, is a quadratic function of time, t, and a linear function of the target concentration, [T]. However, the background level, defined as signal in the absence of the target ([T] = 0), should be a linear function of time. According to Eqs. 1 and 4, the coupling of the primary and secondary reactions into SISAR increases the signal amplification by a factor of ½α2t as compared with the primary reaction alone.
The SISAR kinetics measured at different concentrations of the target, [T], are shown in Fig. 3A. A subset of the data, corresponding to the initial 10% of the cleavage (Fig. 3B), was used to determine the kinetic parameters of SISAR. At zero target concentration, as predicted by Eq. 4, the kinetics fit a linear function whose slope gives a kb value of 0.29 nM⋅min−1. The independently determined kb value of the secondary reaction, 0.28 nM⋅min−1 (Fig. 2), is in excellent agreement with this number, confirming that the X-structure is the major source of SISAR background.
Figure 3.

Kinetics of the secondary probe cleavage in the serial invasive reaction. (A) Dependence of cleaved secondary probe concentration, [S], on time, t, of the reaction with (a) 0, (b) 0.01, (c) 0.03, (d) 0.1, (e) 0.3, (f) 1, and (g) 3 pM target strand. The dotted rectangle shows the data corresponding to the initial 10% of the cleavage as shown in B. (B) Initial kinetics of the secondary cleavage reaction (from A). Rate curve (a) was fit with a linear function, and rates for curves b–g were approximated by quadratic functions with the same linear term determined from (a); these data are summarized in Table 1.
In the presence of the target, the kinetics perfectly fit to the quadratic functions with the fixed linear term kb obtained from the no-target control experiment. The quadratic terms of the kinetic functions obtained for different target concentrations are listed in Table 1. According to Eq. 4, the quadratic term normalized for the target concentration is 1/2α1α2, which has an average value of 290 ± 17 min−2 determined from the data shown in Table 1. This value is in good agreement with individually determined values of 15 and 30 min−1 for α1 and α2, respectively.
Table 1.
Kinetic parameters of SISAR
| [T], pM | ½ α 1α2 [T], pM min−2 | α1 α2, min−2 |
|---|---|---|
| 0.01 | 3.20 × 10−03 | 640 |
| 0.03 | 8.38 × 10−03 | 558 |
| 0.1 | 3.03 × 10−02 | 606 |
| 0.3 | 8.50 × 10−02 | 566 |
| 1 | 2.77 × 10−01 | 554 |
| 3 | 8.49 × 10−01 | 566 |
Eq. 4 and knowledge of α1α2 allowed us to calculate the expected signal amplification, 1/2α1α2t2, of SISAR after incubation for a fixed time, t. For example, in a 200-min reaction by using an average α1α2 value of 580 min−2, the signal should be amplified by a factor of 1/2 × 580 × 2002 = 1.2 × 107. Thus, a reaction containing 100 target molecules would generate >109 free Fl molecules.
Application of the Serial Invasive Signal Amplification Reaction for DNA Detection and Polymorphism Identification.
The specificity of the 5′ nucleases is defined by the structure of the overlapping substrate rather than by its sequence. Thus, the assay can be applied to the analysis of targets with practically any sequence. Fig. 4A shows the SISAR kinetics by using a primary probe designed to recognize nucleotide C667 in the human MTHFR gene. Average net fluorescence signal (determined as the difference between the signals obtained in the presence and in the absence of the target DNA) was measured as a function of time with the kinetics of signal accumulation being approximated by a quadratic equation for each target level. The sensitivity of the method is sufficient to detect as few as 600 molecules (sample b) present in 2 ng of human genomic DNA homozygous for C667 in the MTHFR gene. Fig. 4B shows that both the net signal accumulated after 4 h and the quadratic term at 2 h of the reaction are linear functions with respect to the amount of target DNA, which is in agreement with Eq. 4.
Figure 4.

Quantitative analysis of human genomic DNA. (A) Kinetics of the average net relative fluorescence signal (rfu) accumulated in the serial invasive reaction with the primary probe specific for the C at position 667 of the human MTHFR gene with (a) 0, (b) 2, (c) 5, (d) 10, (e) 15, (f) 20, and (g) 25 ng of homozygous C667 human genomic DNA. The net signal was determined as the difference between the signals obtained in the presence and in the absence of the target DNA. (B) Linearity of the net signal (●) and the quadratic kinetic term (○) with respect to the amount of human genomic DNA used in the 20-μl reaction after 4 and 2 h incubations, respectively. Error bars indicate the standard deviations obtained from the quadruplicate measurements with each sample.
Fig. 5 shows the utility of SISAR for detection of a C/T polymorphism at position 667 in the MTHFR gene present in 20 ng of human genomic DNA. By using primary probes specific for either C667 or T667, all three possible genotypes, homozygous C667, homozygous T667, and heterozygous, were detected and differentiated, as shown in Fig. 5 A–C, respectively. The net signals generated by each probe with the heterozygous DNA is approximately 2-fold lower than the signal produced with the homozygous DNA, reflecting the reduced amount of each target. Also, the absolute levels of signal differ for C667 and T667, demonstrating that sequence can affect the efficiency of individual serial invasive reactions. Nevertheless, correct polymorphism identifications can be made, because both probes generated a significant amount of net signal even after a 90-min reaction, and both exhibit kinetics that have a quadratic component with respect to time.
Figure 5.

Identification of single-nucleotide polymorphisms in nonamplified human genomic DNA. Time courses of the net relative fluorescence signal (rfu) generated by SISAR with the primary probes specific for either the T677 (●) or C677 (○) polymorphism of the human MTHFR gene. The amount of human genomic DNA used in each reaction was 20 ng, equivalent to ≈6,000 copies of the gene. (A) homozygous C677, (B) homozygous T677, and (C) heterozygous alleles.
Discussion
Any new method of nucleic acids detection is inevitably compared with PCR technology (10), a current gold standard in genetic analysis and quantitation. The ability to amplify target sequences more then 12 orders of magnitude in just a few hours makes the PCR assay an indispensable tool in molecular biology and nucleic acids diagnostics. However, the use of exponential target amplification creates the possibility of amplicon crosscontamination, makes quantitative analysis complicated, and reduces the specificity with which small genetic variations can be detected. Although these shortcomings can be overcome, the solutions usually increase the complexity and cost of the assay, which make it less attractive for high throughput analysis.
The problems associated with PCR are not an issue for signal amplification methods that work by producing reporter molecules in response to the presence of a target, rather than by amplifying the target (1, 11–14). The amounts of reporter molecules generated by signal amplification methods are proportional to the amounts of target, leading to simple quantitative analysis. However, the signal generated by such methods are not sufficient for direct sample analysis, and thus, must be serially coupled to a secondary amplification step to obtain the sensitivity of PCR-based methods.
The invasive signal amplification assay described previously (1) is quantitative, simple, and very specific for target sequences. That results from the requirement for two, rather than one, analyte-specific oligonucleotides (the upstream oligonucleotide and the probe) and the high specificity of the 5′ nuclease, which discriminates by a factor of at least 100–1,000 between substrates that differ by one nucleotide (3, 4, 7). In contrast, mutation detection based solely on mismatch formation uses just a single analyte-specific probe and is limited by thermodynamic considerations to a discrimination factor of only about 10. The disadvantage of the single invasive signal detection method (1) was the low level of signal amplification, 3,000- to 10,000-fold, which required an additional step of chemiluminescence signal amplification. The serial coupling of two invasive reactions described here eliminates the need for an additional amplification step. Indeed, the ability of SISAR to amplify the signal more than seven orders of magnitude results in generation of more than 109 reporter molecules (Fl) in response to 100 target molecules, which is within the range of detection for conventional fluorescence plate readers.
The design of SISAR is relatively straightforward because the primary and secondary reactions use the same principles of invasive cleavage. However, to run SISAR isothermally, the optimal temperatures of each reaction should be close. This is easily accomplished because the sequences of the 5′ arm and the analyte-specific region of the primary probe are independent of each other. Thus, their melting temperatures can be adjusted to a standard reaction temperature (63°C in this work) by altering the length of the analyte-specific region and both the length and sequence of the 5′ arm. For optimal cleavage, the upstream oligonucleotide should remain bound to the target during the primary reaction, so it is designed to have a melting temperature 10–15°C higher than that of the primary probe's analyte-specific region. The sequence design of the upstream oligonucleotide and the probe can be done with a computer program by using nearest-neighbor thermodynamic parameters (15) (data not shown). The secondary reaction requires only one secondary probe to detect the 5′ arm that is released in the primary reaction. With FRET detection, the secondary probe is labeled with two dyes, so its synthesis and purification could be costly. However, the secondary probe is not analyte specific, so the same secondary probe can be used with practically any primary reaction; a scale-up in quantity can significantly reduce the cost of the assay.
The detection limit of any method is defined by its signal-to-noise ratio, and therefore analysis of the background variation or noise origin is important. The experiments described here indicate that the majority of background generated during the reaction can be attributed to target-independent cleavage of the X-structure formed by annealing of the uncut primary probe to the secondary probe (Fig. 1C). This result is consistent with our previous analysis of the cleavage rates of different nonspecific substrates that are likely to form during SISAR (7).
An important characteristic of the SISAR background is that it increases linearly with time. In contrast, signal generation follows quadratic kinetics (Fig. 3). Thus, real-time detection of SISAR provides the attractive capability of discriminating between the signal and background solely on the basis of quadratic vs. linear increases in signal over time. We find, however, that for most applications of single-nucleotide polymorphism analysis, a single time point measurement (e.g., 4 h) suffices and can be conveniently presented as the ratio of net signals generated by two signal probes in a simple bar graph.
A potentially troublesome source of background, which would increase with the square of the incubation time, is avoided by another specific attribute of the enzyme, its sensitivity to a 3′-PO4 in the upstream oligonucleotide (3). A 20-μl reaction used for polymorphism analysis of the MTHFR gene in this work contains approximately 5 × 1012 primary probes, of which only 2 × 106 molecules, or less then one millionth, are cleaved in a 4-h reaction with 600 target molecules (when α1 = 15 min−1). If under these conditions the rates of formation of abasic sites and the subsequent strand breakage at these abasic sites in a DNA molecule are ≈10−9 sec−1 (16, 17) and ≈5 × 10−5 sec−1 (18), respectively, thermal instability would generate ≈2 × 107 5′ arm molecules (19, 20), which is an order of magnitude higher than the number of specifically cleaved primary probes. If amplified in the secondary reaction, these breakdown products would make target detection at the level of 600 molecules almost impossible. Fortunately, thermal degradation predominately makes products with 3′-PO4 ends (ref. 21 and data not shown), whereas the 5′ nuclease generates only products with 3′-OH termini (9). Because the 3′-PO4 group greatly inhibits enzymatic activity (3), these thermal degradation products are not efficiently amplified in the secondary reaction.
In conclusion, the serial invasive signal amplification reaction described here conforms well to a simple kinetic analysis of two coupled cleavage reactions by using the product of one reaction to drive the second one. Compared with the previously described invasive signal amplification assay (1), SISAR yields more than three orders of magnitude higher signal amplification, has an almost 100-fold better limit of detection, and uses a simpler homogeneous FRET readout. These improvements make this sensitive homogeneous and isothermal assay suitable for automated high throughput analysis of single-nucleotide polymorphisms and mutation detection.
Acknowledgments
This work was supported by Cooperative Agreements 70NANB5H1030 and 70NANB7H3015 from the Department of Commerce Advanced Technology Program to Lance Fors.
Abbreviations
- FRET
fluorescence resonance energy transfer
- MTHFR
methylene tetrahydrofolate reductase
- SISAR
serial invasive signal amplification reaction: Fl, fluorescein
- HBV
hepatitis B virus
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.140225597.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.140225597
References
- 1.Lyamichev V, Mast A L, Hall J G, Prudent J R, Kaiser M W, Takova T, Kwiatkowski R W, Sander T J, de Arruda M, Arco D A, et al. Nat Biotechnol. 1999;17:292–296. doi: 10.1038/7044. [DOI] [PubMed] [Google Scholar]
- 2.Lyamichev V, Brow M A, Varvel V E, Dahlberg J E. Proc Natl Acad Sci USA. 1999;96:6143–6148. doi: 10.1073/pnas.96.11.6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaiser M W, Lyamicheva N, Ma W, Miller C, Neri B, Fors L, Lyamichev V I. J Biol Chem. 1999;274:21387–21394. doi: 10.1074/jbc.274.30.21387. [DOI] [PubMed] [Google Scholar]
- 4.Kwiatkowski R W, Lyamichev V, de Arruda M, Neri B P. Mol Diagn. 1999;4:353–364. doi: 10.1016/s1084-8592(99)80012-5. [DOI] [PubMed] [Google Scholar]
- 5.Richards E G. In: Handbook of Biochemistry and Molecular Biology. Fasman G D, editor. Vol. 1. Cleveland, OH: CRC; 1975. p. 597. [Google Scholar]
- 6.Okamoto H, Tsuda F, Sakugawa H, Sastrosoewignjo R I, Imai M, Miyakawa Y, Mayumi M. J Gen Virol. 1988;69:2575–2583. doi: 10.1099/0022-1317-69-10-2575. [DOI] [PubMed] [Google Scholar]
- 7.Lyamichev, V. I., Kaiser, M. W., Lyamicheva, N. E., Vologodskii, A. V., Hall, J. G., Ma, W.-P., Allawi, H. T. & Neri, B. (2000) Biochemistry, in press. [DOI] [PubMed]
- 8.Frosst P, Blom H J, Milos R, Goyette P, Sheppard C A, Matthews R G, Boers G J, den Heijer M, Kluijtmans L A, van den Heuvel L P, et al. Nat Genet. 1995;10:111–113. doi: 10.1038/ng0595-111. [DOI] [PubMed] [Google Scholar]
- 9.Lyamichev V, Brow M A, Dahlberg J E. Science. 1993;260:778–783. doi: 10.1126/science.7683443. [DOI] [PubMed] [Google Scholar]
- 10.Mullis K B, Faloona F A. Methods Enzymol. 1987;155:335–350. doi: 10.1016/0076-6879(87)55023-6. [DOI] [PubMed] [Google Scholar]
- 11.Urdea M S, Running J A, Horn T, Clyne J, Ku L L, Warner B D. Gene. 1987;61:253–264. doi: 10.1016/0378-1119(87)90189-2. [DOI] [PubMed] [Google Scholar]
- 12.Stollar B D, Rashtchian A. Anal Biochem. 1987;161:387–394. doi: 10.1016/0003-2697(87)90467-2. [DOI] [PubMed] [Google Scholar]
- 13.Duck P, Alvarado-Urbina G, Burdick B, Collier B. BioTechniques. 1990;9:142–148. [PubMed] [Google Scholar]
- 14.Copley C G, Boot C. BioTechniques. 1992;13:888–892. [PubMed] [Google Scholar]
- 15.Allawi H T, SantaLucia J., Jr Biochemistry. 1997;36:10581–94. doi: 10.1021/bi962590c. [DOI] [PubMed] [Google Scholar]
- 16.Lindahl T, Nyberg B. Biochemistry. 1972;11:3610–3618. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- 17.Lindahl T, Karlstrom O. Biochemistry. 1973;12:5151–5154. doi: 10.1021/bi00749a020. [DOI] [PubMed] [Google Scholar]
- 18.Lindahl T, Andersson A. Biochemistry. 1972;11:3618–3623. doi: 10.1021/bi00769a019. [DOI] [PubMed] [Google Scholar]
- 19.Mendes P. Comput Appl Biosci. 1993;9:563–571. doi: 10.1093/bioinformatics/9.5.563. [DOI] [PubMed] [Google Scholar]
- 20.Mendes P. Trends Biochem Sci. 1997;22:361–363. doi: 10.1016/s0968-0004(97)01103-1. [DOI] [PubMed] [Google Scholar]
- 21.Doetsch P W, Cunningham R P. Mutat Res. 1990;236:173–201. doi: 10.1016/0921-8777(90)90004-o. [DOI] [PubMed] [Google Scholar]