

Abstract
Signal transducing adaptor molecules (STAMs) are involved in growth factor and cytokine signaling as well as receptor degradation, and they form complexes with a number of endocytic proteins, including Hrs and Eps15. Here we demonstrate that STAM proteins also localize prominently to early exocytic compartments and profoundly regulate Golgi morphology. Upon STAM overexpression in cells the Golgi apparatus becomes extensively fragmented and dispersed, but when STAMs are depleted the Golgi becomes highly condensed. Under both scenarios, vesicular stomatitis virus G protein (VSVG)-GFP trafficking to the plasma membrane is markedly inhibited, and recovery of Golgi morphology after brefeldin A treatment is substantially impaired in STAM-depleted cells. Furthermore, STAM proteins interact with COPII proteins, probably at endoplasmic reticulum (ER) exit sites, and Sar1 activity is required to maintain the localization of STAMs at discrete sites. Thus, in addition to their roles in signaling and endocytosis, STAMs function prominently in ER-to-Golgi trafficking, most likely through direct interactions with the COPII complex.
Keywords: endocytosis, VHS domain, Golgi fragmentation, brefeldin A, VSV-G, Sec31
Introduction
Signal transducing adaptor molecules (STAMs) were initially identified as proteins highly phosphorylated on tyrosine residues in response to cytokine and growth factor stimulation, and they have been implicated in cytokine signaling (1–4) as well as surface receptor degradation (5–9). STAMs are evolutionarily conserved, with orthologs in S. cerevisiae, C. elegans, and D. melanogaster, and they are present within the cytoplasm as well as at early endosomes in heteromeric complexes with Hrs and Eps15 (8,10,11). STAM adaptor proteins have a characteristic domain organization comprising VHS (Vps27, Hrs, STAM homology), ubiquitin-interacting (UIM), SH3, and immunoreceptor-based tyrosine activation (ITAM) motifs. The ITAM domain partially overlaps with a coiled-coil region subsequently identified as a portion of a GAT domain (12).
The STAM family comprises STAM1 and STAM2 in mammals, which have overall amino acid identity of ~51%, but up to 89% identity within the functional domains (1), and studies evaluating knock-out mice have indicated that they are functionally redundant (13,14). Indeed, both STAM1 and STAM2 knock out mice appear morphologically normal at birth, but double mutations of STAM1 and STAM2 are embryonically lethal (13,14). Interestingly, despite the importance of the ITAM domain in signaling, tyrosine phosphorylation, and protein interactions, this domain is apparently absent in STAM proteins of invertebrate species such as S. cerevisiae, C. elegans, and D. melanogaster (5). Furthermore, thymocytes from T-cell specific knock-out mice lacking both STAM1 and STAM2 exhibit normal cytokine signaling but decreased viability, suggesting that STAMs have important cellular functions independent of their roles in signal transduction (13).
A key functional motif present at the N-terminus of STAMs is the ~140-residue VHS domain. While the function of this domain remains unclear, its presence in multiple proteins involved in endocytosis prefigures a role in endosomal trafficking. Furthermore, since several proteins with VHS domains localize to the Golgi apparatus, this domain may function in trafficking of proteins from the Golgi apparatus to other cellular compartments (15). In fact, some VHS-containing proteins have dual roles. For instance, GGA3 is a member of a family of monomeric clathrin adaptors, all of which harbor a VHS domain, that are involved in protein sorting at the trans-Golgi network (16–18). RNA interference studies have shown that loss of GGA3 results in endosomal enlargement and accumulation of EGF within these enlarged endosomes, suggesting that GGA3 functions at endosomes as well as at the trans-Golgi network (19).
Here we demonstrate that STAM adaptor proteins also have dual functions at different membrane compartments within the cell. In addition to partial co-localization with endosomal markers, we found co-localization of STAM proteins with markers for endoplasmic reticulum (ER) exit sites (ERES). Moreover, STAMs interact with COPII proteins and regulate Golgi morphology, recovery of Golgi structure after brefeldin A (BFA) treatment, and trafficking of vesicular stomatitis virus G protein (VSVG)-GFP to the plasma membrane.
Results
STAMs localize to the early exocytic pathway
We examined subcellular distributions of STAM proteins by confocal immunofluorescence microscopy. Previous reports had localized overexpressed STAM1 and STAM2 to endosomes and diffusely within the cytoplasm, while studies of endogenous STAMs identified a perinuclear area of enrichment that did not co-stain with antibodies against early endosomal antigen 1 (EEA1) (10). To characterize this area further, we immunostained HeLa cells using an anti-peptide antibody targeted against STAM1 as well as an anti-peptide antibody specific for STAM2. Both STAM antibodies partially co-localized with EEA1 (Figure 1A and not shown), and the STAM2 staining pattern revealed a more prominent perinuclear enrichment. The STAM2 labeling was abolished in cells deplete dof STAM2 using small interfering RNA (siRNA; Figure 5B), confirming the specificity of the immunostaining. We examined markers for several subcellular organelles, and the STAM2 perinuclear labeling co-localized to a moderate degree with several early exocytic markers, including the cis-Golgi marker GM130 and the vesicular tubular cluster (VTC) marker ERGIC-53 (Figure 1A and S1A). We did not observe co-localization with markers for other organelles such as lysosomes (LAMP1; Figure S1A). This STAM2 distribution was also very similar to the GM130 staining pattern in other human cell lines investigated (Figure S1B).
Figure 1. STAMs interact with proteins of the early secretory pathway.

A) HeLa cells were co-immunostained for endogenous STAM2 (green) and either EEA1 or GM130 (red), then visualized using confocal microscopy. Arrows in the upper panels identify an area of colocalization, and an arrowhead in a lower panel identifies an area enlarged in the insets. Bar, 10 μm. B) HeLa cell lysates were immunoprecipitated (IP) with anti-STAM1 antibodies or control IgG and immunoblotted with antibodies against endogenous STAM1, Sec31A, GGA3, GM130, AP1, or βCOP. Inputs represent 5% of the starting material. C) Lysates from cells expressing the indicated constructs were incubated with either GST or GST-STAM1 immobilized on glutathione-Sepharose beads and immunoblotted with anti-GFP antibodies. Inputs represent 5% of the starting material. D and E) HeLa cells were transfected with CFP-Sec31A (D) or Sec13-YFP (E) or else co-transfected with either construct and Myc-STAM2. Lysates were immunoprecipitated with anti-GFP antibodies and immunoblotted with antibodies against GFP or Myc-epitope. Both Sec31 and Sec13 co-precipitate with STAM2. Actin was probed to monitor specificity. An asterisk (*) in (D) denotes the IgG heavy chain. F) HeLa cells were transfected with Myc-STAM2 or else co-transfected with Myc-STAM2 and εCOP-YFP, then immunoprecipitated with anti-GFP antibodies. G) HeLa cells were singly transfected with CFP-Sec31A or co-transfected with CFP-Sec31A and HA-tagged STAM2 domains (schematic diagram on right). Cell lysates were immunoprecipitated with anti-GFP antibodies and immunoblotted with anti-GFP or anti-HA antibodies (left panels). Only the C-terminal region of STAM2 co-precipitates with Sec31 (arrowhead); asterisks (*) denote IgG heavy and light chains. HeLa cells were also singly transfected with Sec13-YFP or co-transfected with Sec13-YFP and HA-tagged STAM2 domain constructs, then immunoprecipitated with anti-GFP antibodies (middle panels). Both VHS-SH3 and C-terminal segments of STAM2 co-precipitate with Sec13 (arrowheads); asterisks (*) identify IgG heavy and light chains.
Figure 5. STAM2 depletion results in a highly condensed Golgi apparatus.

A) Lysates from HeLa cells transfected with either of two different siRNAs specific for STAM2 or else control siRNA were immunoblotted for STAM2 at 72 h post-transfection. Equal protein loading was monitored by immunoblotting for PLCγ. STAM1 levels are unchanged. B) Immunostaining of HeLa cells for STAM2 after transfection with control siRNA or STAM2 siRNA #1. Bar, 10 μm. C) HeLa cells transfected with control siRNA or STAM2 siRNA #1 were immunostained for GM130. STAM2 siRNA cells have a highly condensed Golgi apparatus and clustered ERES. Bar, 10 μm. D) Graphical representation of percentage of cells with Golgi condensation in STAM2 siRNA and control siRNA cells 72 h after transfection (n=3; 100 cells per condition; ±SD). *p <0.001. E) Graphical representation of the ratio of Golgi circumference to cell circumference in control versus STAM2 siRNA cells (n=3; 100 cells per condition). **p=0.02. F) Electron microscopy images of the Golgi complex in control cells (single asterisk; left panel) compared with that in STAM2 siRNA cells, where multiple Golgi cisternae are seen at the same magnification (asterisks; right panel). Bar, 500 nm.
STAMs interact with COPII and ERES proteins
Prompted by their co-localizations with early exocytic proteins, we assessed directly the interactions of STAMs with these proteins. Anti-STAM1 antibodies were more effective than anti-STAM2 antibodies in preliminary immunoprecipitation studies (not shown), and thus co-immunoprecipitations of endogenous HeLa cell proteins were performed using anti-STAM1 antibodies. As shown in Figure 1B, The COPII protein Sec31A clearly co-precipitated with anti-STAM1 antibodies while, in control experiments, non-immune immunoglobulin G (IgG) did not precipitate Sec31A. Additionally, the COPI protein βCOP and Golgi proteins including GM130, GGA3, and AP1 were not co-precipitated by the anti-STAM1 antibody (Figure 1B), indicating a selective interaction of STAMs with the COPII complex.
To investigate further the STAM1-COPII interactions, we performed a series of GST-STAM1 pull-downs. GST-STAM1 and GST fusion proteins immobilized on glutathione-Sepharose were incubated with lysates of HeLa cells overexpressing CFP-Sec31A, Sec13-YFP, GFP-LAMP1, or εCOP-YFP. As a positive control, there was very robust pull down of the GFP-Hrs protein (Figure 1C). Of the other proteins examined, only Sec31A was specifically retained on immobilized GST-STAM1. At longer exposure times, a faint interaction was also identified with Sec13, which may be indirect. The negative controls εCOP and LAMP1 exhibited no interactions with GST-STAM1 even at prolonged exposure times (Figure 1C and not shown).
We next examined interactions of the STAM1 paralog STAM2 with CFP-Sec31A and Sec13-YFP overexpressed in HeLa cells. Lysates from HeLa cells transfected with CFP-Sec31A alone or else co-transfected with Myc-STAM2 were immunoprecipitated with anti-GFP antibodies and then immunoblotted with anti-Myc antibodies, revealing that STAM2 co-precipitates with Sec31A (Figure 1D). Similar experiments were carried out using Sec13-YFP, which also co-immunoprecipitated with Myc-STAM2 (Figure 1E), while there was no interaction between Myc-STAM2 and εCOP-YFP (Figure 1F). Thus, both STAM1 and STAM2 interact with the Sec13/Sec31A COPII cage.
To identify which domains of STAM2 are required for interaction with the COPII cage proteins Sec13 and Sec31, co-transfections as described above were carried out using various STAM2 domain-specific deletion constructs. Of all the domains co-expressed with Sec31A, only the C-terminal region of STAM2 co-precipitated with Sec31A (Figure 1G). We were unable to narrow down further the interaction domain within the C-terminal region of STAM2, as smaller STAM2 C-terminal fragments were unstable (not shown). Similar experiments were conducted to determine regions of STAM2 that interact with Sec13. In addition to an interaction with the STAM2 C-terminal region, as seen with Sec31A, Sec13 also interacted with the VHS-SH3 segment of STAM2. Interestingly, the VHS domain alone was not sufficient to co-precipitate either Sec13 or Sec31A (Figure 1G). Non-interacting portions of Myc-STAM2 also serve as additional negative controls for the interactions identified between STAM2 and both Sec13 and Sec31A.
STAM overexpression results in Golgi fragmentation
To establish the functional role of STAMs in the ER-to-Golgi pathway, we overexpressed either STAM1 or STAM2 and assessed the Golgi apparatus for any morphological changes. In many HeLa cells overexpressing STAM1 or STAM2, the Golgi apparatus as identified with GM130 and giantin staining was highly fragmented and dispersed throughout the cell, though this phenotype was most prominent in STAM2-expressing cells (Figure 2A, B). In fact, expression of a control vector resulted in fragmentation in only 7% of cells, possibly attributable to normal disruption of Golgi structure that occurs during mitosis, whereas overexpression of Myc-STAM1 caused the Golgi apparatus to fragment in 33% of cells (p=0.0012); a stronger phenotype was seen upon overexpression of Myc-STAM2 (74% of cells with fragmentation; p<0.001). These studies were repeated using markers for several other well-characterized Golgi-associated proteins (AP1, CIMPR and GGA3), with similar results (not shown). Interestingly, the VTC marker ERGIC-53 was redistributed to the ER in STAM2-overexpressing cells (Figure 2A). There was, however, no significant difference in LAMP1 distribution upon STAM2 overexpression (Figure 2A).
Figure 2. Overexpression of STAMs causes dispersion of the Golgi complex.

A) Overexpression of Myc-STAM2 (left panels) in HeLa cells disperses the Golgi markers GM130 and giantin, and causes ERGIC-53 to be retained in the ER, but does not affect LAMP1 distribution (right panels). Golgi apparatus and VTCs exhibit normal, perinuclear localization patterns in untransfected cells. Arrows identify transfected cells. Bar, 10 μm. B) Quantification of Golgi fragmentation in cells overexpressing Myc-STAM1, Myc-STAM2, or empty vector (Control) (n=3; 100 cells per experiment; ±SD). *p=0.0012; **p<0.001. C) Electron microscopy images showing typical Golgi morphology in control cells (asterisk; top panel), but much smaller stacked Golgi complexes in STAM2-overexpressing cells (asterisk; bottom panel). Images are at the same magnification. Bar, 500 nm. D) Immunogold localization of GM130 in control cells (1a), with higher magnification of the boxed area in (1b). Immunogold localization of GM130 in STAM2-overexpressing cells (2a), with the boxed region enlarged in (2b). Bar, 500 nm.
We examined the fragmented Golgi apparatus in STAM2-overexpressing cells using electron microscopy. Most cells overexpressing STAM2 had no detectable Golgi stacks, in marked contrast to control cells. However, in the few STAM2-overexpressing cells with an identifiable Golgi complex, the Golgi comprised “mini stacks,” where the length of the complex was significantly shortened (Figure 2C). The distribution of GM130 was assessed using immunogold electron microscopy. In control cells, GM130 localized to the cis-Golgi (Figure 2D1). However, in cells overexpressing STAM2, gold particles mostly decorated membranes that likely represent tubules of smooth ER, since they are more elongated than mini-stacks and lack the characteristic stacked cisternae of the Golgi apparatus. Even so, we cannot completely exclude the possibility that they represent isolated cisternae of an atypical Golgi mini stack (Figure 2D2).
STAM2 domains responsible for targeting and Golgi fragmentation
To identify domains of STAM2 that are involved in its localization to early exocytic membranes and maintenance of Golgi morphology, we expressed deletion constructs comprising various STAM2 domains in HeLa cells, then depleted cytosolic proteins with saponin prior to fixation. The full-length STAM2 expression pattern resembled an ER-like lattice intertwined with fragmented Golgi; this was most evident in regions where the Golgi appeared less fragmented, as assessed using GM130 (Figure 3A).
Figure 3. Effects of STAM2 deletions on subcellular distribution and Golgi fragmentation.

A) HeLa cells transfected with full-length Myc-STAM2, HA-STAM2VHS, or HA-STAM2C Term were treated with saponin to deplete cytosolic proteins and then co-stained for Myc- or HA-epitope and GM130 or, Sec61α. HeLa cells overexpressing full-length STAM2 or the C-terminal fragment display the lattice-like staining adjacent to GM130 puncta, with areas of juxtaposed staining indicated with arrows (upper panels). Colocalization of thhe Myc-STAM2 lattice-like staining with the ER protein Sec61α is shown in the middle panels. In cells expressing HA-tagged STAM2VHS, the juxtapositioned pattern with GM130 is more evident. Boxed areas are enlarged in the insets. B) Schematic diagram of STAM2 deletion constructs. Amino acid numbers are indicated. C) HeLa cells were transfected with control vector, HA-STAM2VHS, HA-STAM2VHS-SH3, HA-STAM2C Term, or full-length Myc-STAM2 (FL), co-stained with GM130 and either Myc- or HA-epitope antibodies, then assessed for Golgi fragmentation (100 cells per condition; n=3; ±SD). *p<0.01; **p<0.005; ***p<0.001.
To establish whether overexpressed STAM2 localized to ER, we co-stained for the endogenous ER protein Sec61α, which exhibited significant co-localization (Figure 3A). STAM2VHS-SH3 and STAM2C Term fragments had expression patterns similar to full-length STAM2 (Figure 4A, B, and not shown). However, when STAM2VHS was overexpressed, punctate STAM2VHS labeling was prominently juxtaposed to fragmented Golgi, consistent with co-localization of ERES and Golgi proteins (20). This co-distribution was most evident in cells with less dispersion of Golgi fragments (Figure 3A). Taken together, these results indicate that the VHS domain is sufficient to target STAM2 to a location, potentially representing ERES, adjacent to the Golgi remnants, but not for stabilization of the interaction (as shown by the co-immunoprecipitation results in Figure 1G). A likely explanation for this apparent discrepancy is that the VHS-SH3, C Terminal, and full-length constructs of STAM2 are able to interact with COPII proteins, thus acting as dominant-negatives and displacing COPII proteins from ERES. This would lead to decreased co-localization by immunofluorescence. Since the VHS domain does not act as a dominant-negative, its co-localization with COPII proteins is potentially much greater. In other words, STAMs may be recruited to ERES before Sec31A and Sec13, and while STAM2VHS may interact with a protein already at ERES, the other STAM2 domains could be important for stabilizing interactions with Sec13/Sec31A.
Figure 4. STAM proteins colocalize with and redistribute COPII proteins.

A) Full-length Myc-STAM2 and Sec13-YFP expressed in HeLa cells colocalize at multiple discrete puncta (arrows; upper panels). Sec13-YFP and HA-STAM2VHS puncta also co-localize (arrows, middle panels), sometimes juxtaposed to Golgi fragments as revealed by GM130 staining (arrowheads). The boxed area is enlarged in the lower panels. B) Overexpression of Myc-STAM2 as revealed by staining for Myc-epitope in HeLa cells results in decreased intensity of punctate staining for the COPII proteins Sec31A and Sec24C, but overexpression of STAM2 only slightly alters the Sec16L staining pattern. C) Overexpression of Myc-STAM2 has no effect on mitochondrial morphology or apoptosis as assessed with cytochrome c (Cyto c) staining (arrow indicates transfected cell). Bars, 10 μm.
To identify effects of these isolated domains on Golgi fragmentation, they were overexpressed individually, and cells were co-stained with GM130. While STAM2VHS overexpression caused considerable Golgi fragmentation in comparison to control cells (p=0.0024), the effect was not as large as that seen with full-length STAM2 (Figure 3C), STAMVHS-SH3, or STAM2C Term, indicating that other domains are functionally important. Thus, overexpression of the STAM2 deletion constructs that interact with COPII (Figure 1G) are able to function in a dominant-negative manner and disrupt the Golgi complex, but to a lesser degree.
STAM2 impacts COPII protein recruitment to ERES
Golgi architecture is highly influenced by the organization and activity of the ERES. To establish whether the fragmented Golgi resulting from overexpression of STAMs represents Golgi remnants retained at ERES, we investigated the co-localization of STAMs with COPII proteins and their relationship to GM130 puncta. HeLa cells were co-transfected with Myc-STAM2 and Sec13-YFP, one of the first proteins to appear at the site of the nascent Golgi to mediate vesicle budding and also a marker for ER exit sites (21). Unfortunately, a majority of co-transfected cells had cytoplasmic displacement of Sec13, with subsequent loss of protein following the saponin treatment used to diminish the intense cytosolic expression of STAMs (not shown). Co-expressing cells with lower STAM2 levels treated with saponin exhibited less protein displacement, and multiple STAM2-positive puncta co-localized with Sec13-YFP puncta (Figure 4A). However, this paradigm may have limited co-localization of STAM2 with some ERES. When STAM2VHS was co-expressed with Sec13-YFP, co-localization was more apparent, most likely due to decreased dominant-negative effects of this STAM2 deletion construct. Additionally, some puncta were adjacent to Golgi fragments, as shown by GM130 co-staining (Figure 4A), consistent with localization of STAM2 to ERES.
We sought to determine the effects of STAMs on distributions of COPII and ERES proteins. Cells were transfected with full-length STAM2 and immunostained for endogenous Sec31A, Sec24, and Sec16L (Sec16 comprises both Sec16L and Sec16S isoforms). Cells over-expressing STAM2 exhibited decreased intensity of Sec31A puncta as well as cytoplasmic redistribution of Sec24, when compared to untransfected cells (Figure 4B). On the other hand, no significant differences were apparent in the Sec16L staining pattern (Figure 4B). Given these effects of STAM2 overexpression, we investigated whether STAM2 had effects on other cellular organelles to ensure that the Golgi fragmentation phenotype in STAM-overexpressing cells was not secondary to apoptosis induction. We assessed mitochondrial structure and initiation of the apoptotic pathway by staining for cytochrome c. All cells overexpressing STAM2 had a normal mitochondrial reticulum, without fragmentation or cytochrome c release (Figure 4C).
Depletion of STAMs causes Golgi condensation
To assess further the role of STAMs on Golgi morphology, we determined the effects of STAM depletion on the Golgi apparatus using siRNA oligonucleotides. While both STAM2-specific oligonucleotides tested significantly knocked down STAM2 expression, siRNA oligonucleotide #1 depleted protein levels the most at 72 h (Figure 5A, B) and thus was used for subsequent studies.
Golgi morphology was examined using GM130 immunostaining at 48 h (not shown) and 72 h after transfection with STAM2 siRNA (Figure 5C). In a majority of siRNA-transfected cells, the normal ribbon-like Golgi structure had collapsed into a highly condensed, ball-like structure. A similar effect was seen when examining the Golgi markers GGA3 and CIMPR, but there was no apparent change in cellular distributions of tubulin or actin (Figure S2). Our criteria for assessing Golgi condensation were strictly set as a diameter ≤5 μm in all three dimensions, and there was a substantial difference when STAM2 siRNA cells were compared to control cells (p=0.0084; Figure 5D). Similarly, prominent Golgi condensation was observed upon specific knockdown of STAM1 (Figure 6A–C). We attempted a double knock down (DKD) of both STAM1 and STAM2 using siRNA. Though we were able to achieve effective depletion of STAM2, only about 50% of STAM1 was depleted (Figure 6A). Other conditions that increased depletion of STAM1 concominantly increased cell death (not shown). Under STAM DKD conditions, an even greater degree of Golgi condensation was observed (Figure 6B, C).
Figure 6. Knock down and overexpression of STAM1 confirms some functional redundancy with STAM2.

A) Left panel, Extracts from HeLa cells transfected with any of three siRNA oligonucleotides specific for STAM1 or else with control siRNA were immunoblotted at 72 h post-transfection. Levels of STAM2 were unaltered. Right panel, Extracts from HeLa cells transfected with control siRNA or cotransfected with siRNAs for both STAM1 and STAM2 (STAM DKD) were immunoblotted with the indicated antibodies. Equal protein loading was monitored using PLCγ. B) HeLa cells were transfected with STAM1 siRNA or co-transfected with siRNAs for STAM1 and STAM2 (STAM double knock down [DKD]) or else with control siRNA. Percentages of cells with Golgi condensation (≤5 μm diameter in all dimensions) in STAM1 siRNA, STAM DKD, and control siRNA groups (n=3; 100 cells per experiment) are presented graphically (±SD). *p=0.0042; **p<0.0001. C) HeLa cells transfected with control, STAM1, or STAM DKD siRNAs were immunostained with anti-GM130 antibodies. As observed for STAM2 siRNA knock down cells, STAM1 siRNA cells also have a highly condensed Golgi apparatus. Bar, 10 μm. D) Re-transfection of HeLa cells with Myc-STAM1 after transfection with STAM2 siRNA suppressed the condensed Golgi phenotype (arrow). Bar, 10 μm.
Importantly, upon depletion of STAM2 we also noted an overall decrease in cell size. To determine whether the Golgi appeared condensed simply as a result of decreased cell size, we compared the ratio of Golgi size to cell size between the control and STAM2 siRNA groups. A significant difference was noted between the two groups, indicating that the Golgi size had decreased to a greater extent than cell size (p= 0.02; Figure 5E). Morphological changes in the Golgi complex were further assessed ultrastructurally using electron microscopy (Figure 5F). When comparing control and STAM2 siRNA cells at the same magnification in equivalent fields, we found that under control conditions an average of 1.8 Golgi stacks were clearly visible as compared to 5.9 stacks present in STAM2 siRNA cells (Figure 5F; n=25 cells per condition), demonstrating that the Golgi complex is condensed into a smaller area with retention of its cisternal organization.
STAM2 siRNA phenotype is rescued by STAM overexpression
Given the marked morphological changes in the Golgi apparatus upon STAM siRNA treatment, we sought to confirm that these effects could be reversed. We created a Myc-tagged form of STAM2 (STAM2′) with two silent point mutations in the region of STAM2 siRNA #1, rendering it siRNA-resistant without altering the primary amino acid sequence. We compared STAM2 levels in control siRNA cells, STAM2 siRNA cells, and STAM2 siRNA cells overexpressing Myc-STAM2′. There was a marked reduction of endogenous STAM2 protein in the STAM2 siRNA cells as compared to controls, and a significant increase in STAM2 levels in the STAM2 siRNA plus Myc-STAM2′ cells. This observed increase was due to production of the Myc-STAM2′ protein, as confirmed by immunoblotting with anti-Myc antibodies (Figure S3A).
Subsequently, we performed immunocytochemical analysis to detect any suppression of the loss-of-function, condensed Golgi phenotype seen in STAM2 siRNA cells. Cells overexpressing STAM2′ had an extensively fragmented Golgi apparatus, as expected (Figure S3B, C). In addition, the highly-condensed Golgi phenotype in STAM2 siRNA cells was suppressed by STAM2′ overexpression. In STAM2 siRNA-transfected cells with a relatively low expression of STAM2′, the Golgi was normal in appearance, while higher STAM2′ levels resulted in reversal of the condensed Golgi phenotype to one of fragmentation, consistent with a dose-dependent effect (Figure S3B). Lastly, STAM1 overexpression in STAM2 siRNA cells also suppressed the condensed Golgi morphology phenotype (Figure 6D), consistent with some functional redundancy between the STAMs.
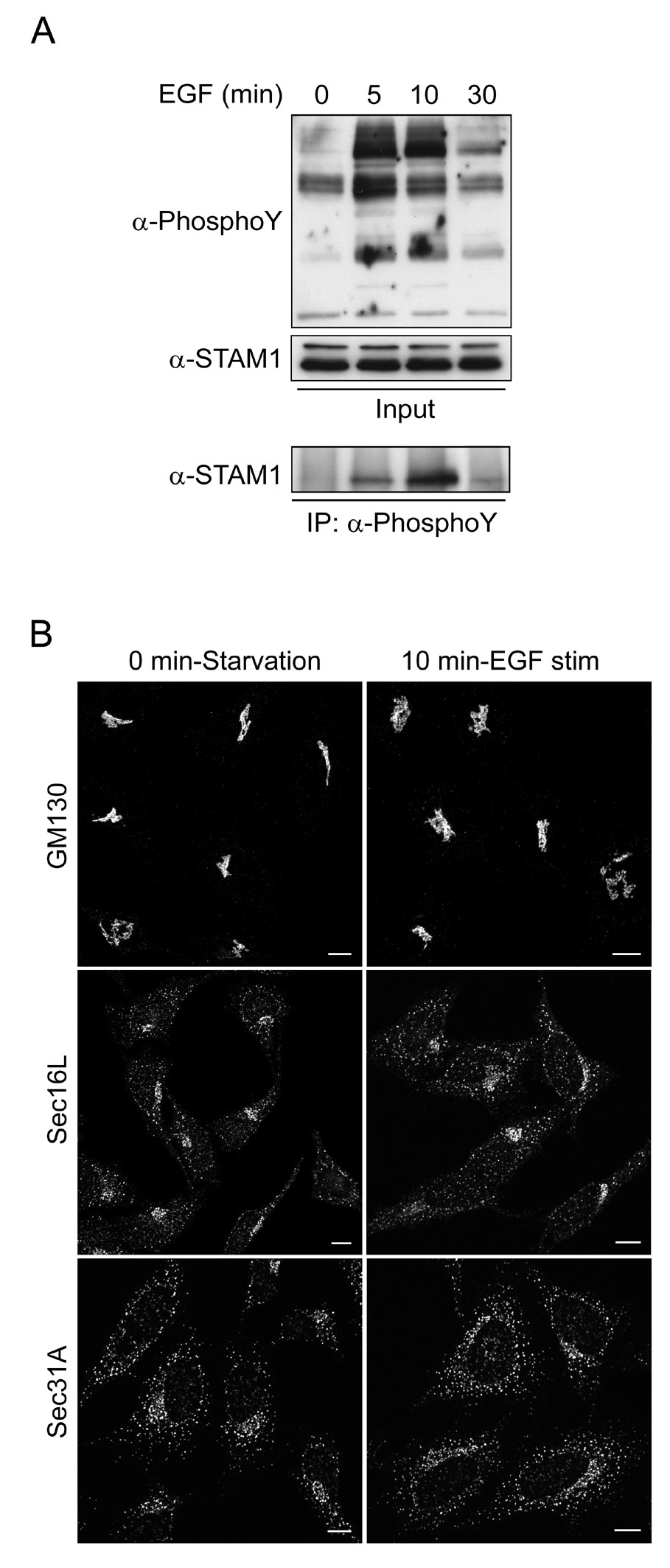
Given the known role of STAMs in cytokine and growth factor signaling, we asked whether changes in Golgi and ERES distribution correlated with the tyrosine phosphorylation state of STAMs. We stimulated serum-starved HeLa cells with 100 ng EGF, and fixed them at 0–30 min [(22) and Figure S4]. Since peak tyrosine phosphorylation occurred at ~10 min (Figure S4A), consistent with previous reports (3), we immunostained cells just before, and 10 min after, EGF stimulation for GM130, Sec16L, or Sec31A (Figure S4B). There were no differences in GM130, Sec16L, or Sec31A staining patterns upon EGF stimulation, suggesting that the effects of STAMs on ERES are independent of their tyrosine phosphorylation state.
Loss of STAM2 inhibits Golgi recovery after brefeldin A treatment
To dissect the role of STAMs in regulating Golgi morphology, we used BFA, a drug commonly used to block transport of secretory cargo from the ER to Golgi by inhibiting the Arf1 GTPase. In BFA-treated HeLa cells, endogenous STAM2 remained adjacent to the Golgi marker GM130 even after Golgi membrane dispersal, and STAM2 puncta co-localized with the COPII protein Sec31A (Figure 7A). Thus, this co-localization is independent of Arf1 activity and, consequently, recruitment of STAM2 to this perinuclear region occurs upstream of Arf1 activity, suggesting that STAMs are targeted to ERES.
Figure 7. Localization of STAM2 to ERES in BFA-treated cells and delayed recovery of Golgi morphology in STAM2 siRNA cells following BFA.

A) HeLa cells were treated with BFA and co-stained for STAM2 (green) and GM130 or Sec31A (red). STAM2 puncta are adjacent to GM130 puncta, and STAM2 puncta colocalize with Sec31A puncta. B) HeLa cells transfected with control or STAM2 siRNAs were treated with BFA and assessed after wash-out. Most control cells (upper panels) display normal Golgi morphology as assessed by GM130 staining after 1 h of recovery, whereas most STAM2 siRNA-treated cells (lower panels) have not recovered even after 3 h. Bar, 10 μm. C) Graphical presentation of the percentage of cells with Golgi reconstitution (lacking tubular Golgi) after transfection with STAM2 siRNA and control siRNA (n=3; 100 cells per experiment; ±SD).
We investigated whether loss of STAM2 altered the cellular response to BFA and whether STAM2 was necessary for reconstitution of the Golgi, as assessed by GM130 staining, after BFA wash out. Upon BFA treatment of control and STAM2 siRNA cells, no significant differences in disruption of the Golgi could be detected (not shown). Unexpectedly, once cells were returned to growth media after BFA treatment, STAM2-depleted cells exhibited a substantial impairment in Golgi reconstitution as compared to control cells. While almost all control siRNA cells showed full recovery of normal Golgi morphology 1 h after wash out, a majority of STAM2 siRNA cells still had severely disrupted Golgi membranes even after 3 h (Figure 7B, C), with many comprising punctate structures with tubular networks en route to the microtubule-organizing center (MTOC). Taken together, these results suggest that STAM2 is involved in reassembly of the Golgi apparatus after disruption with BFA and is important in efficient trafficking of Golgi proteins from the ER to the Golgi complex.
VSVG trafficking is impaired in STAM2-depleted cells
To assess whether STAM2 knock down impairs trafficking through the Golgi apparatus, we used VSVG ts045 as a reporter. Two days after transfection of HeLa cells with either control or STAM2 siRNA, both cell groups were re-transfected with VSVG-GFP and placed overnight at a temperature (40 °C) non-permissive for VSVG exiting the ER. Cells were then moved to a permissive temperature (32 °C) and examined at several time points. STAM2 siRNA-treated cells showed a substantial impairment in the ability of VSVG to traffic to the plasma membrane (Figure 8A). In control cells, VSVG was observed at the plasma membrane as early as 60 min after temperature change. However, at as late as 180 min the majority of the VSVG was present at the Golgi in the STAM2 siRNA cells, with VSVG barely detectable at the plasma membrane (Figure 8A). These results suggest that the presence of STAM2 is critical for efficient trafficking in the VSVG secretory pathway.
Figure 8. VSVG trafficking is impaired in cells lacking or overexpressing STAM2.

A) HeLa cells were transfected with control siRNA (top panels) or STAM2 siRNA (bottom panels), and then re-transfected with ts045 VSVG-GFP. VSVG trafficked to the plasma membrane as early as 60 min after moving to a permissive temperature in control cells, while trafficking to plasma membrane was substantially reduced in the STAM2 siRNA cells. B) HeLa cells were singly transfected with VSVG-GFP (top panels) or else co-transfected with Myc-STAM2 (bottom panels; Myc staining not shown), then assessed at different time points after moving to a temperature permissive for VSVG trafficking. VSVG is present at the plasma membrane in control cells as soon as 90 min after the temperature change, whereas it is retained within the fragmented Golgi of STAM2-overexpressing cells even at 180 min. C) VSVG and GM130 are present in the same fragmented Golgi compartments in HeLa cells overexpressing STAM2 180 min after temperature change. The enlarged image in the inset shows VSVG staining (green) adjacent to GM130 puncta (red). Bar, 10 μm.
Overexpression of STAM2 inhibits VSVG trafficking
We investigated whether VSVG trafficking was affected by the Golgi dispersal resulting from overexpression of STAMs. Cells were transfected with VSVG-GFP or else co-transfected with Myc-STAM2 and VSVG-GFP, and both groups were placed overnight at 40 °C, as described above, and then assessed at different time points after moving to a permissive temperature (Figure 8B). In control cells, VSVG could be seen at the plasma membrane as soon as 60 min after the temperature change, and in all cells at 180 min, consistent with previous reports (23). In HeLa cells overexpressing STAM2, however, VSVG was visualized within fragmented structures dispersed throughout the cell. Furthermore, VSVG appeared retained within these punctate structures even at 180 min, with little detectable localization at the plasma membrane.
To ensure that these punctate VSVG structures coincided with the dispersed Golgi fragments reported earlier, cells were co-stained with GM130 (Figure 8C). We observed that the fragmented Golgi structures seen upon overexpression of STAM2 were adjacent to VSVG, consistent with the proteins being retained at ERES. These findings suggest that not only does STAM2 overexpression disrupt Golgi morphology, but also that it markedly impairs trafficking from ER to Golgi.
Distribution of STAM depends on Sar1, but not on Sec31A
We asked whether the localization of STAMs to ERES was dependent on the presence of Sec31A, Sec16, or else Sar1 activity. Effects of depletion of Sec31A on STAM2 recruitment to ERES were examined using siRNA. All three Sec31A-specific siRNAs tested significantly knocked down Sec31A expression (Figure 9A), and STAM2 and Sec31A staining patterns were then examined (Figure 9B). There was no change in STAM2 distribution, indicating that Sec31A is not required for the STAM2 perinuclear localization, presumably at ERES. By contrast, Sec16L depletion using any of three different siRNAs clearly caused a shift in STAM2 staining to a more punctate, dispersed pattern that does not co-localize with the punctate GM130 structures normally present in this Sec16 depletion paradigm (Figure S5). However, since direct interaction of STAM2 with Sec16L has not been established, mechanisms underlying this change remain unclear, and thus represent a potentially important area for future study.
Figure 9. STAM2 recruitment to the early secretory pathway is dependent on Sar1 activity, but not on Sec31.

A) Lysates from HeLa cells transfected with any of three different Sec31A siRNAs or else control siRNA were immunoblotted for Sec31A 72 h post-transfection. Equal protein loading was monitored using PLCγ. B) HeLa cells were co-stained with antibodies against STAM2 and Sec31A after transfection with control or Sec31A siRNAs; there is no change in STAM2 distribution upon Sec31A depletion. C) Control HeLa cells were stained for STAM2 or Sec31A (left panels). Cells transfected with HA-Sar1 [H79G] (HA-epitope staining in insets) show changes in distribution patterns for both proteins, with STAM2 localizing to large punctate structures and Sec31A labeling becoming more condensed (middle panels). Cells expressing HA-Sar1 [T39N] (HA-epitope staining in insets) show decreased staining intensity of puncta and dispersal of immunoreactivity for both STAM2 and Sec31A (right panels).
Sar1 is a GTPase whose activity is critical for recruitment of COPII proteins to ERES, and it is required to maintain the discrete localization of Sec16 to ER membranes (24). We investigated whether STAM recruitment was dependent on Sar1 activity using two mutant forms: HA-Sar1 [H79G] is constitutively active, and HA-Sar1 [T39N] is inactive. In cells overexpressing Sar1 [H79G], STAM2 was dispersed throughout the cell and in large punctate structures; however, Sec31 was condensed within the perinuclear region, reminiscent of its redistribution upon STAM depletion. When Sar1 [T39N] was overexpressed, both STAM2 and Sec31A were dispersed and exhibited decreased staining intensity, with fewer and smaller puncta (Figure 9C). Our earlier results had suggested that localization of STAMs to ERES likely occurs upstream of Arf1 activity and recruitment of Sec31A. However, since STAM distributions are clearly influenced by Sar1 expression in a manner distinct from that of Sec31A, STAMs likely act at the level of Sar1, perhaps through interactions with these proteins that do not depend on COPII proteins such as Sec31A.
Discussion
In this study, we have identified a critical function of STAM adaptor proteins in ER-to-Golgi trafficking and Golgi morphogenesis. This finding was unexpected, as STAMs have been widely studied for their effects on cytokine and growth factor signaling and receptor degradation (1,3,4,7,10). On the other hand, there have been several reports identifying dual localizations and functions for other endocytic proteins such as GGA3 (19), Eps15 (25), and mammalian Bet3p (mBet3p; (26–28)). In fact, mBet3p is present both on VTCs as well as early endosomes (26–28), reminiscent of the two distinct STAM localizations that we identify here. The Rab5 GTPase also has a role in ER morphogenesis independent of its functions during endocytosis (29), consistent with the notion that the effects of STAM proteins on the exocytic pathway are likely not secondary to endocytic defects.
Our data demonstrate that STAMs are present at early exocytic compartments, likely at ERES, and they co-localize and interact with the COPII complex. Although STAM2 appears to exhibit partial co-localization with markers for VTCs and the cis-Golgi, this could be due to the close proximity of ERES to these structures (20). Even so, we cannot rule out the presence of STAMs at these membranes as well. Additionally, because endogenous STAMs co-immunoprecipitate with COPII proteins, but not with COPI or Golgi proteins, we focused on its potential localization at ERES. To address differences in staining patterns between endogenous and overexpressed STAM2, we would emphasize that one consequence of STAM overexpression is disruption of VTCs and Golgi membranes, which can have effects on the distribution pattern of central ERES.
Here we provide evidence supportive of a direct effect of STAM proteins on ER-to-Golgi transport and Golgi morphogenesis. Overexpression of STAMs results in dispersed GM130 puncta rather than the usual ribbon-like distribution pattern, and these apparent Golgi remnants likely represent GM130 retained at ER tubules, probably at ERES (Figure 2D). Furthermore, we observe a clear redistribution of ERGIC-53 to the ER upon STAM overexpression. These effects parallel findings reported for cells overexpressing Sec16 (24,30–32). In addition, the impairment in ER-to-Golgi transport associated with the presence of Golgi remnants that we observe upon STAM overexpression is similar to that seen with BFA treatment or expression of dominant-negative Arf1T31N, indicating that STAMs may act upstream or at the same time as Arf1, but most likely downstream of Sar1 activation based on the ability of GM130 to be recruited to ERES in STAM-overexpressing cells (20). In fact, our VSVG studies indicate that protein trafficking is halted at ERES of STAM-overexpressing cells, with protein retained in the ER. While it remains possible that VSVG is retained in VTCs, this is not likely since we clearly see ERGIC-53 redistribute to ER membranes upon overexpression of STAMs as well as GM130 gold particles retained in presumptive ER tubules. Upon overexpression of STAMs, there is minimal redistribution of Sec16L. However, endogenous Sec31A recruitment to exit sites, and to a lesser degree Sec24, is substantially decreased, indicating that recruitment of these COPII proteins to ERES is blocked, likely by interactions of STAMs with these proteins outside of the proper cellular context. Taken together, these findings suggest that STAMs act at the ER as part of a protein scaffold to recruit COPII proteins.
Despite considerable evidence supporting a direct interaction of STAMs with COPII proteins, we also considered the possibility that since STAMs are involved in signal transduction, overexpression may result in initiation of mitosis, which also results in Golgi fragmentation (33). Importantly, this is very likely not the cause of the fragmentation that we observe here, since the region responsible for signal transduction in STAMs, the ITAM domain, is present in the C-terminal region, and we have shown that overexpression of the N-terminal fragment STAMVHS-SH3 that does not harbor this domain caused Golgi fragmentation at levels similar to full-length STAM2. Additionally, STAM overexpression and depletion phenotypes are not dependent on the phosphorylation state of STAM. Thus, functions of STAMs in ER-to-Golgi trafficking are very likely independent of their role in cellular signaling.
SiRNA depletion studies of both STAM1 and STAM2 support of our contention that STAMs function prominently in Golgi morphogenesis. The condensed Golgi phenotype resulting from loss of STAM proteins has previously been identified by other investigators to correlate with a given protein’s importance in Golgi function as well as its cellular localization (34,35). Although the Golgi condensation we observed could conceivably be secondary to actin depolymerization, we saw no significant difference in actin staining patterns between control and STAM siRNA conditions. Ultrastructural changes in Golgi morphology upon STAM depletion do not represent loss of total membrane or collapse of the cisternae, but rather condensation of the normally extended Golgi structure into a more compact area within the cell. Moreover, STAM-depleted cells exhibit impaired recovery of Golgi morphology after washout of BFA, similar to effects reported upon loss of Sec16 or Sec12, suggesting that STAMs play a role in protein trafficking out of ERES (31). Thus, this impairment in Golgi reconstitution most likely results from delayed assembly of the COPII complex or else export of proteins from the ER. Indeed, VSVG trafficking in STAM siRNA cells is markedly impaired, as also reported for Sec16-depleted cells (24,31,32).
Although we found it curious that the loss of one STAM would have such a profound effect while the other isoform is still present, similar effects have also been reported upon knock down of either Sec16 isoform (31). In addition, we observed suppression of the STAM2 siRNA phenotype upon STAM1 overexpression, consistent with at least partial functional redundancy. Thus, our STAM knock down and overexpression results likely reflect the sensitivity of cells to even moderate changes in overall STAM protein levels. This notion could also explain the lack of significant phenotype of either STAM1 or STAM2 knock-out mice at birth (13,14). In these animals, levels of the other STAM isoform may be up-regulated, minimizing effects of the loss of the other isoform, while our acute knock down paradigm may permit us to see more prominent effects. Also consistent is the fact that loss of both STAM proteins is embryonically lethal in mice (13), and we also found an increase in cell death during our attempts to deplete both STAMs. Still, the STAMs may have slight differences in functions or interactions, though none been reported to date even for their well-studied roles in cytokine and growth factor signaling and receptor degradation.
We demonstrate a clear association of STAMs with ERES by co-localization as well as co-immunoprecipitation and GST pull down studies with several ERES proteins. Importantly, we show that anti-STAM antibodies co-precipitate the COPII component Sec31A, indicating that they exist together in a protein complex endogenously. Importantly, in assessing STAM interactions we did not detect any association with Golgi proteins involved in the lysosomal degradation pathway, consistent with previous results (36). As described previously, the COPII complexes that co-stained for STAMs had a slightly offset “co-distribution” pattern with GM130 at these sites, and this pattern is also seen in cells immunostained after BFA treatment (20). Although we can not rule out the possibility that STAM-COPII interactions occur in the cytoplasm, the interaction most likely occurs at ERES since STAM and COPII puncta co-localize adjacent to Golgi remnants (Figure 4).
While STAM recruitment does not rely on Sec31A within the cell, it is highly regulated by Sar1 activity (24). In cells overexpressing a constitutively-active, GTP-bound form of Sar1, STAMs form large punctate structures, and when cells express the inactive GDP-bound form of Sar1, STAMs show a nearly complete loss of punctate staining. The paradoxical effects of constitutively-active Sar1 on STAMs, in contrast to other COPII proteins, may reflect the fact that overactive Sar1 may not require STAMs to stabilize COPII components, and it may even act as a dominant-negative for STAMs at ERES.
In future studies, it will be important to determine whether other known STAM-interacting proteins such as Hrs and Eps15 function in ER-to-Golgi trafficking as well. Indeed, Audhya et al (29) have suggested that effects of the endocytic protein Rab5 on ER morphogenesis might derive from Rab5 on endosomes interacting in trans with effectors at the ER. Similar mechanisms could conceivably be involved in STAM interactions in the early secretory pathway as well. There are also interesting parallels between the COPII and clathrin cages. Both clathrin and Sec13/Sec31 COPII cage proteins are able to self-assemble into a molecular scaffold to generate a coat polymer, and Sec31 of the COPII cage, much like clathrin heavy chains, harbors both N-terminal WD40 domains and C-terminal α-solenoid domain motifs, and structurally these proteins have similar scaffold components. In fact, it has been suggested that the general contacts that mediate the assembly of clathrin cages are similar to those for COPII cages (37). Thus, since STAMs are also associated with clathrin-coated pits (10), STAMs may interact with structurally similar components of the COPII cage.
Materials and Methods
DNA Constructs
The STAM2 (GenBank accession number NM_005843) coding sequence was cloned into the XmaI site of the mammalian expression vector pGW1-Myc, which contains an N-terminal Myc-epitope tag (38). The full STAM1 coding sequence (GenBank accession number NM_003473) was cloned into the EcoRI site of the bacterial fusion protein vector pGEX4T-1 (GE Healthcare). A siRNA-resistant Myc-STAM2′ was produced by creating two silent point mutations, A1335G and A1341T (nucleotide numbering from start of coding sequence) using QuikChange (Stratagene). STAM2 cDNA fragments were PCR amplified using PfuTurbo (Stratagene) and cloned into the XmaI site of pGW1-HA, which contains an N-terminal HA tag; HA-STAM2VHS comprises residues 1-151, HA-STAM2VHS-SH3 residues 1–258, and HA-STAM2 C Term residues 257–525. The Myc-STAM1 construct was described previously (38). Sec13-YFP, εCOP-YFP, ts045 VSVG-GFP, HA-Sar1 [T39N], and HA-Sar1 [H79G] constructs were provided by Dr. J. Lippincott-Schwartz (39, 40). The CFP-Sec31A construct was from Dr. R. Pepperkok (41). The GFP-LAMP1 construct was from Dr. E. Dell’Angelica (42). The GFP-Hrs construct (43) was provided by Dr. Sylvie Urbé.
Antibodies
Rabbit polyclonal antibodies were used against the following proteins: STAM1 and STAM2 (Calbiochem), Sec16L (Bethyl Laboratories), Sec24C (from Dr. D. Stephens), HA-epitope (Abcam), giantin (Abcam), and GFP (Invitrogen). Mouse monoclonal antibodies were used against the following proteins: GM130 (IgG1, clone 35; BD Transduction Laboratories), Myc-epitope (IgG1, clone 9E10; Santa Cruz Biotechnology), mannose-6-phosphate receptor (IgG2a, clone 2G11; Abcam), γ-adaptin (AP-1; IgG2b, clone 100/3; Sigma-Aldrich), GGA3 (IgG2a, clone 40; BD Transduction Laboratories), tubulin (IgG1, clone DM1A; Sigma-Aldrich), ERGIC-53 (IgG1, clone G1/93; Axxora), PLCγ (Upstate Biotechnology), EEA1 (IgG1, clone 14; BD Transduction Laboratories), Sec31A (IgG1, clone 32; BD Transduction Laboratories), LAMP1 (IgG1, clone H4A3; Southern Biotechnology Associates), cytochrome c (IgG1, clone 6H2.B4; BD Biosciences Pharmingen), and phosphotyrosine (IgG2b, clone 4G10; Upstate Biotechnology). Actin distribution was assessed using rhodamine phalloidin (Invitrogen). Goat polyclonal anti-Myc-epitope antibodies were from Bethyl Laboratories. The goat polyclonal anti-Sec61α antibody (G-20) was from Santa Cruz Biotechnology. Alexa Fluor secondary antibodies were from Invitrogen.
Cell culture and transfection
HeLa cells were maintained in Dulbecco’s MEM/10% fetal bovine serum, plated on coverslips, and transfected with 1 μg of plasmid DNA using Lipofectamine (Invitrogen). Twenty-four hours later, cells were washed in phosphate-buffered saline (PBS; pH 7.4) and fixed using 4% formaldehyde or methanol. Cells used for immunoblot analysis were washed with PBS and collected in 0.1% Triton X-100 in PBS. For saponin treatment, HeLa cells were treated the day following transfection with 0.05% saponin in 80 mM PIPES (pH 6.8)/5 mM EGTA/1 mM MgCl2 for 4 min, washed in PBS, fixed in 4% formaldehyde, and then processed for immunocytochemical analysis.
For siRNA transfections, HeLa cells were plated at 50% confluency and transfected the next day with 100 nM siRNA oligonucleotides for 4 h using Oligofectamine (Invitrogen). Cells were scraped for immunoblot analysis or fixed for immunocytochemical analysis 48 or 72 h after transfection. STAM2-specific siRNA oligonucleotides tagged with Alexa 647 fluorophore (Qiagen) were targeted against the following cDNA sequences: #1, CTGCTCAAACTTCATATTTAA; and #2, ATGGTACTTGATCTACATTTA. The control siRNA was from Ambion. STAM1 siRNAs (Invitrogen) were as follows: #1, UAACUUGGUAUAUAAGGAAAGGGCC; #2, AUACAUGGAAUACAUCGGAUCUUCG; #3, UUAAUACGUUGCUUACUUCACUAGC. Sec31A siRNAs (Invitrogen) were as follows: #1, CCAUAGCAGGUGGACAAGAACUCUU; #2, GCUCAGCAAUUGGAUGCAACAUUUA; #3, CCAGGCCAAUAAGCUGGGUGUCUAA. Sec16L siRNAs (Invitrogen) were as follows: #1, GGGCGCAAAGUGAGCUGCCAGAUUU; #2, CCGUCCCAUUCUGACAGCCUCGCUU; #3, CCCUGCCUAGUUUCCAGGUGUUUAA.
Immunoprecipitation and immunoblotting
HeLa cells were scraped with 0.5% NP-40 in PBS, centrifuged (16,200 × g, 15 min) and incubated with anti-STAM1 antibodies or else control IgG (5 μg) for 2 h at 4 °C for endogenous immunoprecipitation studies. For co-precipitation of overexpressed proteins, anti-GFP antibodies (5 μg) were used. Protein A-Sepharose beads were then added for 1 h. Beads were washed four times with decreasing concentrations of NP-40 in PBS, with the final wash in PBS alone. Samples were subjected to SDS-PAGE, transferred to PVDF membrane (Millipore), and immunoblotted.
GST fusion protein pull-downs
GST and GST-STAM1 fusion proteins were prepared and purified using glutathione-Sepharose beads as described previously (38). HeLa cells were singly transfected as described previously, and cell lysates were incubated with either GST beads or with GST-STAM1 beads for 90 min at 4°C. Beads were again washed vigorously with binding buffer three times and finally once with PBS, then immunoblotted.
Confocal microscopy
Cells were imaged using a Zeiss LSM510 confocal microscope with a 63× 1.4 NA Plan-APOCHROMAT lens. Acquisition was performed using LSM 510 version 3.2 SP2 software (Carl Zeiss Microimaging). Data were processed using Adobe Photoshop 7.0 and Adobe Illustrator CS2 software. For quantification studies, at least 100 cells were counted per experimental group, and experiments were conducted at least three times unless otherwise indicated.
To determine the ratio of Golgi size to cell size, projected images of cells from either control or STAM2 siRNA groups were analyzed using MetaMorph software (Molecular Devices). Cells were analyzed individually to obtain the ratio of the Golgi circumference (as assessed by GM130 staining) to cell size (determined using differential interference contrast microscopy). One hundred cells were assessed from each group, with n=3 for a total of 300 cells per group.
Electron microscopy
For structural assessment of Golgi, HeLa cells were fixed for 1 h in 4% glutaraldehyde in 0.1M sodium cacodylate buffer. For GM130 immunogold staining, cells were fixed in 2% paraformaldehyde and 0.1% glutaraldehyde in PBS for 1 h. Cells were blocked and permeabilized in 5% goat serum and 0.1% saponin in PBS for 1 h then placed in blocking buffer with the primary antibody for 1 h. Subsequent preparation of cells for electron microscopy has been described previously (44).
BFA treatment and VSVG-GFP trafficking
HeLa cells plated on coverslips in 6-well plates were placed in growth media containing 5 μg/ml BFA (Epicentre) for 1 h, then washed 3 times with PBS and placed in growth media for recovery of Golgi structure, then fixed at various times points.
For VSVG-GFP trafficking studies, HeLa cells were transfected with the VSVG-GFP construct and placed at 40 °C. Sixteen hours after transfection, HeLa cells were moved to 32°C to permit VSVG trafficking from the ER, and then fixed and processed for immunocytochemical analysis 0–180 min after the temperature change.
Statistical analysis
Statistical significance was assessed using two-tailed, unpaired Student’s t-tests, assuming unequal variance.
Supplementary Material
Figure S1: Localization of endogenous STAM proteins. A) HeLa cells were co-stained for endogenous STAM2 (green; left panels) and either the VTC marker ERGIC-53 (red; top) or the lysosomal marker LAMP1 (red; bottom). STAM shows a distribution pattern similar to that of ERGIC-53, but not LAMP1. An arrowhead identifies the region enlarged in the inset. B) Human embryonic kidney 293 cells (upper panels) and human astrocytes (bottom panels) were co-stained for endogenous STAM2 (green) and GM130 (red). The enlarged images (insets) reveal some co-localization, but staining patterns are not identical and appear juxtaposed. Bar, 10 μm.
{kind=link}
Figure S2: Depletion of STAMs alters distribution patterns of Golgi markers. HeLa cells were transfected with control (upper panels) or STAM2 (lower panels) siRNA and co-stained with Golgi markers CIMPR and GGA3 or else the cytoskeletal markers actin (as assessed using phalloidin) or tubulin. As for GM130 (Figure 5C), the distribution pattern of each of these markers is much more condensed in STAM2 siRNA-transfected cells than in control cells. Actin and tubulin localizations are unchanged in STAM2 siRNA cells as compared to control siRNA cells. Bar, 10 μm.
{kind=link}
Figure S3: Suppression of STAM2 siRNA-induced, condensed Golgi phenotype by STAM2 overexpression. A) HeLa cells were transfected with control or STAM2-specific siRNAs, or else transfected with STAM2 siRNA and then re-transfected with a modified, “knock down resistant” form of Myc-STAM2 (STAM2′), then immunoblotted for STAM2 and Myc-epitope. Equal protein loading was monitored by immunoblotting for PLCγ. B) HeLa cells transfected with either control or STAM2 siRNA were then transfected with Myc-STAM2′. As observed for Myc-STAM2, overexpression of Myc-STAM2′ causes fragmentation of the Golgi apparatus in control cells (left panels). Lower levels of STAM2′ expression suppress the STAM2 siRNA-induced condensed Golgi phenotype in cells (low; middle panels); higher expression levels not only suppress the condensed Golgi phenotype, but also cause Golgi fragmentation (high; right panels). C) The percentage of cells with condensed Golgi was quantified in control siRNA, STAM2 siRNA, and STAM2 siRNA plus Myc-STAM2′ transfected cells; the proportion of cells with condensed Golgi in the siRNA recovery group (siRNA + STAM2′) was similar to that observed in control siRNA cells (n=3; 100 cells per experiment; ±SD).
{kind=link}
Figure S4: EGF stimulation has no effect on Golgi morphology or ERES distribution in HeLa cells. A) HeLa cells were serum starved for 16 h, then stimulated with 100 ng/ml EGF and harvested at the indicated time points. Cell extracts were immunoprecipitated with anti-phosphotyrosine antibodies, then immunoblotted with either anti-phosphotyrosine or anti-STAM1 antibodies. Peak STAM1 tyrosine phosphorylation occurred at about 10 min. B) HeLa cells were serum starved for 16 h and then stimulated with 100 ng/ml EGF for 0 or 10 min prior to fixation. Cells were immunostained for endogenous GM130, Sec16L, and Sec31A. There were no significant differences in cellular distributions of these proteins.
{kind=link}
Figure S5: Effects of Sec16L depletion on STAM2 distribution. A) Lysates from HeLa cells transfected for 72 h with any of three different Sec16L siRNAs or else control siRNA were immunoblotted for Sec16L. Equal protein loading was monitored by immunoblotting for PLCγ. B and C) HeLa cells were stained with antibodies against Sec16L (B) or else STAM2 (C) and GM130 (red; bottom panel) 72 h after transfection with control or Sec16L-specific siRNAs. The STAM2 distribution pattern in Sec16L-depleted cells is more punctate, and these puncta do not colocalize with the GM130 puncta (inset).
{kind=link}
Acknowledgments
We thank James Nagle and Deborah Kauffman (NINDS DNA Sequencing Facility) for DNA sequencing, Carolyn Smith for assistance with confocal microscopy, Jung-Hwa Tao-Cheng for assistance with electron microscopy, and Chuang-Rung Chang for helpful discussions. This work was supported by the Intramural Research Programs of the NINDS and NHLBI, National Institutes of Health.
Abbreviations list
- BFA
brefeldin A
- DKD
double knock down
- EEA1
early endosomal antigen 1
- ER
endoplasmic reticulum
- ERES
ER exit sites
- IgG
immunoglobulin G
- MTOC
microtubule-organizing center
- PBS
phosphate-buffered saline
- siRNA
small interfering RNA
- STAM
signal transducing adaptor molecule
- VSVG
vesicular stomatitis virus G protein
- VTC
vesicular tubular cluster
References
- 1.Endo K, Takeshita T, Kasai H, Sasaki Y, Tanaka N, Asao H, Kikuchi K, Yamada M, Chenb M, O’Shea JJ, Sugamura K. STAM2, a new member of the STAM family, binding to the Janus kinases. FEBS Lett. 2000;477:55–61. doi: 10.1016/s0014-5793(00)01760-9. [DOI] [PubMed] [Google Scholar]
- 2.Pandey A, Fernandez MM, Steen H, Blagoev B, Nielsen MM, Roche S, Mann M, Lodish HF. Identification of a novel immunoreceptor tyrosine-based activation motif-containing molecule, STAM2, by mass spectrometry and its involvement in growth factor and cytokine receptor signaling pathways. J Biol Chem. 2000;275:38633–38639. doi: 10.1074/jbc.M007849200. [DOI] [PubMed] [Google Scholar]
- 3.Takeshita T, Arita T, Asao H, Tanaka N, Higuchi M, Kuroda H, Kaneko K, Munakata H, Endo Y, Fujita T, Sugamura K. Cloning of a novel signal-transducing adaptor molecule containing an SH3 domain and ITAM. Biochem Biophys Res Commun. 1996;225:1035–1039. doi: 10.1006/bbrc.1996.1290. [DOI] [PubMed] [Google Scholar]
- 4.Takeshita T, Arita T, Higuchi M, Asao H, Endo K, Kuroda H, Tanaka N, Murata K, Ishii N, Sugamura K. STAM, signal transducing adaptor molecule, is associated with Janus kinases and involved in signaling for cell growth and c-myc induction. Immunity. 1997;6:449–457. doi: 10.1016/s1074-7613(00)80288-5. [DOI] [PubMed] [Google Scholar]
- 5.Lohi O, Lehto V-P. STAM/EAST/Hbp adapter proteins--integrators of signalling pathways. FEBS Lett. 2001;508:287–290. doi: 10.1016/s0014-5793(01)03079-4. [DOI] [PubMed] [Google Scholar]
- 6.Bilodeau PS, Urbanowski JL, Winistorfer SC, Piper RC. The Vps27p Hse1p complex binds ubiquitin and mediates endosomal protein sorting. Nat Cell Biol. 2002;4:534–539. doi: 10.1038/ncb815. [DOI] [PubMed] [Google Scholar]
- 7.Kanazawa C, Morita E, Yamada M, Ishii N, Miura S, Asao H, Yoshimori T, Sugamura K. Effects of deficiencies of STAMs and Hrs, mammalian class E Vps proteins, on receptor downregulation. Biochem Biophys Res Commun. 2003;309:848–856. doi: 10.1016/j.bbrc.2003.08.078. [DOI] [PubMed] [Google Scholar]
- 8.Mizuno E, Kawahata K, Kato M, Kitamura N, Komada M. STAM proteins bind ubiquitinated proteins on the early endosome via the VHS domain and ubiquitin-interacting motif. Mol Biol Cell. 2003;14:3675–3689. doi: 10.1091/mbc.E02-12-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu J, Wittekind SG, Barr MM. STAM and Hrs down-regulate ciliary TRP receptors. Mol Biol Cell. 2007;18:3277–3289. doi: 10.1091/mbc.E07-03-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bache KG, Raiborg C, Mehlum A, Stenmark H. STAM and Hrs are subunits of a multivalent ubiquitin-binding complex on early endosomes. J Biol Chem. 2003;278:12513–12521. doi: 10.1074/jbc.M210843200. [DOI] [PubMed] [Google Scholar]
- 11.Mizuno E, Kawahata K, Okamoto A, Kitamura N, Komada M. Association with Hrs is required for the early endosomal localization, stability, and function of STAM. J Biochem (Tokyo) 2004;135:385–396. doi: 10.1093/jb/mvh046. [DOI] [PubMed] [Google Scholar]
- 12.Prag G, Watson H, Kim YC, Beach BM, Ghirlando R, Hummer G, Bonifacino JS, Hurley JH. The Vps27/Hse1 complex is a GAT domain-based scaffold for ubiquitin-dependent sorting. Dev Cell. 2007;12:973–986. doi: 10.1016/j.devcel.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamada M, Ishii N, Asao H, Murata K, Kanazawa C, Sasaki H, Sugamura K. Signal-transducing adaptor molecules STAM1 and STAM2 are required for T-cell development and survival. Mol Cell Biol. 2002;22:8648–8658. doi: 10.1128/MCB.22.24.8648-8658.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamada M, Takeshita T, Miura S, Murata K, Kimura Y, Ishii N, Nose M, Sakagami H, Kondo H, Tashiro F, Miyazaki JI, Sasaki H, Sugamura K. Loss of hippocampal CA3 pyramidal neurons in mice lacking STAM1. Mol Cell Biol. 2001;21:3807–3819. doi: 10.1128/MCB.21.11.3807-3819.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lohi O, Poussu A, Mao Y, Quiocho F, Lehto V-P. VHS domain -- a longshoreman of vesicle lines. FEBS Lett. 2002;513:19–23. doi: 10.1016/s0014-5793(01)03287-2. [DOI] [PubMed] [Google Scholar]
- 16.Dell’Angelica EC, Puertollano R, Mullins C, Aguilar RC, Vargas JD, Hartnell LM, Bonifacino JS. GGAs: a family of ADP ribosylation factor-binding proteins related to adaptors and associated with the Golgi complex. J Cell Biol. 2000;149:81–94. doi: 10.1083/jcb.149.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirst J, Lui WW, Bright NA, Totty N, Seaman MN, Robinson MS. A family of proteins with gamma-adaptin and VHS domains that facilitate trafficking between the trans-Golgi network and the vacuole/lysosome. J Cell Biol. 2000;149:67–80. doi: 10.1083/jcb.149.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takatsu H, Yoshino K, Nakayama K. Adaptor γ ear homology domain conserved in γ-adaptin and GGA proteins that interact with γ-synergin. Biochem Biophys Res Commun. 2000;271:719–725. doi: 10.1006/bbrc.2000.2700. [DOI] [PubMed] [Google Scholar]
- 19.Puertollano R, Bonifacino JS. Interactions of GGA3 with the ubiquitin sorting machinery. Nat Cell Biol. 2004;6:244–251. doi: 10.1038/ncb1106. [DOI] [PubMed] [Google Scholar]
- 20.Ward TH, Polishchuk RS, Caplan S, Hirschberg K, Lippincott-Schwartz J. Maintenance of Golgi structure and function depends on the integrity of ER export. J Cell Biol. 2001;155:557–570. doi: 10.1083/jcb.200107045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ho HH, He CY, de Graffenried CL, Murrells LJ, Warren G. Ordered assembly of the duplicating Golgi in Trypanosoma brucei. Proc Natl Acad Sci U S A. 2006;103:7676–7681. doi: 10.1073/pnas.0602595103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Row PE, Clague MJ, Urbé S. Growth factors induce differential phosphorylation profiles of the Hrs-STAM complex: a common node in signalling networks with signal-specific properties. Biochem J. 2005;389:629–636. doi: 10.1042/BJ20050067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirschberg K, Miller CM, Ellenberg J, Presley JF, Siggia ED, Phair RD, Lippincott-Schwartz J. Kinetic analysis of secretory protein traffic and characterization of golgi to plasma membrane transport intermediates in living cells. J Cell Biol. 1998;143:1485–1503. doi: 10.1083/jcb.143.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watson P, Townley AK, Koka P, Palmer KJ, Stephens DJ. Sec16 defines endoplasmic reticulum exit sites and is required for secretory cargo export in mammalian cells. Traffic. 2006;7:1678–1687. doi: 10.1111/j.1600-0854.2006.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kent HM, McMahon HT, Evans PR, Benmerah A, Owen DJ. γ-adaptin appendage domain: structure and binding site for Eps15 and γ-synergin. Structure. 2002;10:1139–1148. doi: 10.1016/s0969-2126(02)00801-8. [DOI] [PubMed] [Google Scholar]
- 26.Cai H, Zhang Y, Pypaert M, Walker L, Ferro-Novick S. Mutants in trs120 disrupt traffic from the early endosome to the late Golgi. J Cell Biol. 2005;171:823–833. doi: 10.1083/jcb.200505145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sacher M, Barrowman J, Wang W, Horecka J, Zhang Y, Pypaert M, Ferro-Novick S. TRAPP I implicated in the specificity of tethering in ER-to-Golgi transport. Mol Cell. 2001;7:433–442. doi: 10.1016/s1097-2765(01)00190-3. [DOI] [PubMed] [Google Scholar]
- 28.Yu S, Satoh A, Pypaert M, Mullen K, Hay JC, Ferro-Novick S. mBet3p is required for homotypic COPII vesicle tethering in mammalian cells. J Cell Biol. 2006;174:359–368. doi: 10.1083/jcb.200603044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Audhya A, Desai A, Oegema K. A role for Rab5 in structuring the endoplasmic reticulum. J Cell Biol. 2007;178:43–56. doi: 10.1083/jcb.200701139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Connerly PL, Esaki M, Montegna EA, Strongin DE, Levi S, Soderholm J, Glick BS. Sec16 is a determinant of transitional ER organization. Curr Biol. 2005;15:1439–1447. doi: 10.1016/j.cub.2005.06.065. [DOI] [PubMed] [Google Scholar]
- 31.Bhattacharyya D, Glick BS. Two mammalian Sec16 homologues have nonredundant functions in endoplasmic reticulum (ER) export and transitional ER organization. Mol Biol Cell. 2007;18:839–849. doi: 10.1091/mbc.E06-08-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iinuma T, Shiga A, Nakamoto K, O’Brien MB, Aridor M, Arimitsu N, Tagaya M, Tani K. Mammalian Sec16/p250 plays a role in membrane traffic from the endoplasmic reticulum. J Biol Chem. 2007;282:17632–17639. doi: 10.1074/jbc.M611237200. [DOI] [PubMed] [Google Scholar]
- 33.Acharya U, Mallabiabarrena A, Acharya JK, Malhotra V. Signaling via mitogen-activated protein kinase kinase (MEK1) is required for Golgi fragmentation during mitosis. Cell. 1998;92:183–192. doi: 10.1016/s0092-8674(00)80913-7. [DOI] [PubMed] [Google Scholar]
- 34.Gougeon P-Y, Prosser DC, Da-Silva LF, Ngsee JK. Disruption of Golgi morphology and trafficking in cells expressing mutant prenylated Rab acceptor-1. J Biol Chem. 2002;277:36408–36414. doi: 10.1074/jbc.M205026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gough LL, Fan J, Chu S, Winnick S, Beck KA. Golgi localization of Syne-1. Mol Biol Cell. 2003;14:2410–2424. doi: 10.1091/mbc.E02-07-0446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puertollano R, Aguilar RC, Gorshkova I, Crouch RJ, Bonifacino JS. Sorting of mannose 6-phosphate receptors mediated by the GGAs. Science. 2001;292:1712–1716. doi: 10.1126/science.1060750. [DOI] [PubMed] [Google Scholar]
- 37.Gurkan C, Stagg SM, Lapointe P, Balch WE. The COPII cage: unifying principles of vesicle coat assembly. Nat Rev Mol Cell Biol. 2006;7:727–738. doi: 10.1038/nrm2025. [DOI] [PubMed] [Google Scholar]
- 38.Blackstone C, Roberts RG, Seeburg DP, Sheng M. Interaction of the deafness-dystonia protein DDP/TIMM8a with the signal transduction adaptor molecule STAM1. Biochem Biophys Res Commun. 2003;305:345–352. doi: 10.1016/s0006-291x(03)00767-8. [DOI] [PubMed] [Google Scholar]
- 39.Hammond AT, Glick BS. Dynamics of transitional endoplasmic reticulum sites in vertebrate cells. Mol Biol Cell. 2000;11:3013–3030. doi: 10.1091/mbc.11.9.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- 41.Forster R, Weiss M, Zimmermann T, Reynaud EG, Verissimo F, Stephens DJ, Pepperkok R. Secretory cargo regulates the turnover of COPII subunits at single ER exit sites. Curr Biol. 2006;16:173–179. doi: 10.1016/j.cub.2005.11.076. [DOI] [PubMed] [Google Scholar]
- 42.Falcón-Peréz JM, Nazarian R, Sabatti C, Dell’Angelica EC. Distribution and dynamics of Lamp1-containing endocytic organelles in fibroblasts deficient in BLOC-3. J Cell Sci. 2005;118:5243–5255. doi: 10.1242/jcs.02633. [DOI] [PubMed] [Google Scholar]
- 43.Urbé S, Sachse M, Row PE, Preisinger C, Barr FA, Strous G, Klumperman J, Clague MJ. The UIM domain of Hrs couples receptor sorting to vesicle formation. J Cell Sci. 2003;116:4169–4179. doi: 10.1242/jcs.00723. [DOI] [PubMed] [Google Scholar]
- 44.Zhu P-P, Patterson A, Lavoie B, Stadler J, Shoeb M, Patel R, Blackstone C. Cellular localization, oligomerization, and membrane association of the hereditary spastic paraplegia 3A (SPG3A) protein atlastin. J Biol Chem. 2003;278:49063–49071. doi: 10.1074/jbc.M306702200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Localization of endogenous STAM proteins. A) HeLa cells were co-stained for endogenous STAM2 (green; left panels) and either the VTC marker ERGIC-53 (red; top) or the lysosomal marker LAMP1 (red; bottom). STAM shows a distribution pattern similar to that of ERGIC-53, but not LAMP1. An arrowhead identifies the region enlarged in the inset. B) Human embryonic kidney 293 cells (upper panels) and human astrocytes (bottom panels) were co-stained for endogenous STAM2 (green) and GM130 (red). The enlarged images (insets) reveal some co-localization, but staining patterns are not identical and appear juxtaposed. Bar, 10 μm.
Figure S2: Depletion of STAMs alters distribution patterns of Golgi markers. HeLa cells were transfected with control (upper panels) or STAM2 (lower panels) siRNA and co-stained with Golgi markers CIMPR and GGA3 or else the cytoskeletal markers actin (as assessed using phalloidin) or tubulin. As for GM130 (Figure 5C), the distribution pattern of each of these markers is much more condensed in STAM2 siRNA-transfected cells than in control cells. Actin and tubulin localizations are unchanged in STAM2 siRNA cells as compared to control siRNA cells. Bar, 10 μm.
Figure S3: Suppression of STAM2 siRNA-induced, condensed Golgi phenotype by STAM2 overexpression. A) HeLa cells were transfected with control or STAM2-specific siRNAs, or else transfected with STAM2 siRNA and then re-transfected with a modified, “knock down resistant” form of Myc-STAM2 (STAM2′), then immunoblotted for STAM2 and Myc-epitope. Equal protein loading was monitored by immunoblotting for PLCγ. B) HeLa cells transfected with either control or STAM2 siRNA were then transfected with Myc-STAM2′. As observed for Myc-STAM2, overexpression of Myc-STAM2′ causes fragmentation of the Golgi apparatus in control cells (left panels). Lower levels of STAM2′ expression suppress the STAM2 siRNA-induced condensed Golgi phenotype in cells (low; middle panels); higher expression levels not only suppress the condensed Golgi phenotype, but also cause Golgi fragmentation (high; right panels). C) The percentage of cells with condensed Golgi was quantified in control siRNA, STAM2 siRNA, and STAM2 siRNA plus Myc-STAM2′ transfected cells; the proportion of cells with condensed Golgi in the siRNA recovery group (siRNA + STAM2′) was similar to that observed in control siRNA cells (n=3; 100 cells per experiment; ±SD).
Figure S4: EGF stimulation has no effect on Golgi morphology or ERES distribution in HeLa cells. A) HeLa cells were serum starved for 16 h, then stimulated with 100 ng/ml EGF and harvested at the indicated time points. Cell extracts were immunoprecipitated with anti-phosphotyrosine antibodies, then immunoblotted with either anti-phosphotyrosine or anti-STAM1 antibodies. Peak STAM1 tyrosine phosphorylation occurred at about 10 min. B) HeLa cells were serum starved for 16 h and then stimulated with 100 ng/ml EGF for 0 or 10 min prior to fixation. Cells were immunostained for endogenous GM130, Sec16L, and Sec31A. There were no significant differences in cellular distributions of these proteins.
Figure S5: Effects of Sec16L depletion on STAM2 distribution. A) Lysates from HeLa cells transfected for 72 h with any of three different Sec16L siRNAs or else control siRNA were immunoblotted for Sec16L. Equal protein loading was monitored by immunoblotting for PLCγ. B and C) HeLa cells were stained with antibodies against Sec16L (B) or else STAM2 (C) and GM130 (red; bottom panel) 72 h after transfection with control or Sec16L-specific siRNAs. The STAM2 distribution pattern in Sec16L-depleted cells is more punctate, and these puncta do not colocalize with the GM130 puncta (inset).