

Abstract
Simian Virus 40 (SV40) and Mouse Polyoma Virus (PY) are small DNA tumor viruses that have been used extensively to study cellular transformation. The SV40 early region encodes three tumor antigens, Large T (LT), small T (ST) and 17KT that contribute to cellular transformation. While PY also encodes LT and ST, the unique Middle T (MT) generates most of the transforming activity. SV40 LT mediated transformation requires binding to the tumor suppressor proteins Rb and p53 in the nucleus and ST binding to the protein phosphatase PP2A in the cytoplasm. SV40 LT also binds to several additional cellular proteins including p300, CBP, Cul7, IRS1, Bub1, Nbs1 and Fbw7 that contribute to viral transformation. PY MT transformation is dependent binding to PP2A and the Src family protein tyrosine kinases (PTK) and assembly of a signaling complex on cell membranes that leads to transformation in a manner similar to Her2/neu. Phosphorylation of MT tyrosine residues activates key signaling molecules including Shc/Grb2, PI3K and PLCγ1. The unique contributions of SV40 LT and ST and PY MT to cellular transformation have provided significant insights into our understanding of tumor suppressors, oncogenes and the process of oncogenesis.
Keywords: SV40, polyoma, T antigen, p53, retinoblastoma, Src, PI3 kinase, Shc
Introduction
Murine polyomavirus (PY), the founding member of the family Polyomaviridae was identified in the 1950s. As the name implies, PY causes a wide variety of tumors. SV40 was discovered in 1960 as a vacuolating virus contaminating rhesus monkey kidney cell cultures used to produce the poliovirus vaccine [1]. This discovery was quickly followed by reports that SV40 could form tumors in newborn rodents and transform primary human cells. These initial reports led to nearly 50 years of intense investigation into the biology and oncogenic potential of these viruses.
These viruses have been important models for study of DNA and RNA metabolism. Our appreciation of the compact and efficient SV40 promoter and origin of replication as well as mRNA splicing and polyadenylation signals led to their widespread use in many mammalian expression vectors. However, the greatest gains have come from studies on T antigen-mediated neoplastic transformation and tumorigenesis that apparently reflect side effects of viral signaling needed for infection and replication. Study of SV40 led to discovery of the tumor suppressor p53 and insight into the structure and function of the retinoblastoma tumor suppressor Rb. Similarly, the study of PY led to discovery of phosphotyrosine kinases (PTK) and phosphoinositide 3-kinase (PI3K) signaling.
Proteins encoded by the early region of the two viruses are responsible for transformation. Splicing of primary early transcripts takes place in both viruses, but in ways that generates different kinds of gene products. Importantly, each of the T antigens functions in different cellular compartments reflecting their unique functions to the virus life cycle and to cellular transformation.
The SV40 viral early region encodes three proteins LT, ST and 17KT expressed from the early promoter after differential splicing (Figure 1). The three T antigens share the N-terminal 82-residues that contains a DnaJ or J domain (Figure 1). ST continues in the same open reading frame to encode a 174-residue protein. LT and 17KT use the same intron that skips the ST unique region and share residues 1 to 131. LT continues to residue 708. 17KT has a second intron followed by a third exon that encodes just 4 additional residues to encode a 135-residue protein.
Figure 1. SV40 and Mouse Polyoma T antigen.

Large (LT), 17KT and Small (ST) of SV40 and PY as well as Middle (MT) T antigens from PY share coding regions. The N terminal 82 (SV40) and 79 (PY) residues for all T antigens are identical (magenta). SV40 LT and 17KT share residues 83 to 131 (blue). The last 4 residues of SV4017KT are unique (red). PY MT and ST share residues 80–191 (yellow). PY MT has a unique domain residue 192 to 421 (green).
The primary transcripts of the PY early region are also differentially spliced to produce LT, MT and ST with a common N-terminal, 79-residue, J domain (Fig. 1). ST shares an additional 112-residues with MT. The C-terminal region of each protein is unique and determines their subcellular location. Similar to SV40, PY LT is nuclear and ST is predominantly cytoplasmic. PY MT is anchored to membranes.
LTs from both viruses have essential roles in viral DNA replication. SV40 LT also provides most of the transforming activity. For example, while both SV40 and PY LT have a helicase domain, this SV40 LT domain also serves to bind to p53. PY MT provides nearly all of the transforming functions for PY and also plays a role in viral replication. PY MT activates PTK signaling in a manner similar to that induced by the cellular receptor tyrosine kinase Her2/neu. ST serves similar but not identical roles in these two viruses and cooperates with the other T antigens to transform cells.
It is not clear why SV40 and PY have such distinct transforming mechanisms. A similar species-specific dichotomy is seen in papillomavirus, where the bovine BPV E5 activates tyrosine kinase signaling and human HPV E6 inactivates p53 and not conversely. A reasonable hypothesis is that the viruses take advantage of unique species or tissues differences, such as transcription factors, for viral growth.
Dissociation of SV40 LT replication from transformation functions
Since SV40 LT provides essential functions for viral DNA replication that can ultimately cause lysis of the infected cell, these replication functions must be disabled for LT to promote long-term cellular transformation and tumorigenesis. Infection of permissive monkey cells typically results in rapid viral replication followed by lysis or vacuolation of the host cell and subsequent release of progeny. SV40 can infect non-permissive rodent cells and semi-permissive human cells, but the transformed state lasts for a few days before reverting to a normal phenotype in a process known as abortive transformation. Notably, primary human mesothelial cells that can support low levels of SV40 replication with persistence of episomal SV40 DNA and a transformed phenotype [2]. SV40 transformation can be readily studied in rodent and human cells by transfection of viral DNA or with retroviral vectors encoding LT and ST [3].
The ability of SV40 to transform cells reflects a summary of several distinct contributions. LT can immortalize primary cells, reduce serum requirements for growth, enable cells to overgrow a monolayer of cells and form foci, support anchorage independent and induce tumor formation when cells are implanted as xenografts in nude mice. There is a hierarchy of these transformed features with immortalization and growth in reduced serum as the least stringent and anchorage independent growth and tumor formation as more stringent. In general, immortality or unlimited growth in vitro is a prerequisite for malignant transformation or unlimited in vivo growth. There is often a high degree of correlation between anchorage independent growth and growth as xenografts [3].
The temperature sensitive SV40 LT mutant tsA58, containing the point substitution A438V, has been useful for defining the contribution of LT to transformation and immortalization. Stable expression of tsA58 LT at the permissive temperature (32–33.5°C) is necessary for continued proliferation of rodent and human fibroblasts [4]. Shifting cells to the restrictive temperature (> 39°C) reduces the stability of the tsA58 LT and eliminates its ability to bind to p53 resulting in growth arrest, senescence and crisis [5]. The tsA58 LT has also been used to generate several different transgenic mouse and rat strains enabling normal development at the restrictive temperature in vivo while permitting immortalization of various tissues when cultured at the permissive temperature in vitro [6].
Domains of SV40 LT
LT forms a hexameric structure with the oligomerization domain (residues 251–627) containing ATPase and helicase activities that participate in viral DNA replication [7]. The outside surface of each subunit of the LT hexamer serve to bind to p53 that in turn recruits p300 and CBP [8]. LT interaction with host replication proteins including DNA polymerase α and replication protein A is required for LT-mediated viral replication but not for LT-mediated transformation.
J domain
The N-terminal domain of SV40 LT and ST forms a J domain, a highly conserved structural domain present in the DnaJ/Hsp40 family of molecular chaperones. The J domain of SV40 LT binds specifically to Hsc70 and activates its ATPase activity (Fig. 2) [9, 10]. The HPDK residues in the J domain of LT are required for this activity. Conversely, a variety of LT mutants can bind to Hsc70 without activating the ATPase activity and may reflect their unfolded nature [11].
Figure 2. SV40 LT binding proteins.

SV40 LT binding proteins and their approximate binding regions in LT. Hsc70 binds specifically to the N-terminal DNA J domain. The Cullin ring ligase complex containing Cul7, Fbxw8 as well as Rbx1 and Skp1 (not shown) bind to residues between 69–102. Bub1 requires residues 89–98 and IRS1 binds to an N-terminal domain. The Rb-related proteins including p107 and p130 bind to the LXCXE motif encoded by residues 103–107. The E2F proteins and DP1 heterodimeric partner bind to LT by the Rb proteins. The MRN complex containing Mre11, Rad50 and Nbs1 binds to the DNA binding domain (DBD). The p53 tumor suppressor binds to the Helicase domain and requires residue 350 through 627. The F-box containing protein Fbw7 binds to the phosphorylated residue T701.
The J domain of LT contributes to two distinct functions of LT. On one hand, it facilitates efficient viral DNA replication in a manner not completely understood [11]. On the other hand, the J domain contributes to LT-mediated transformation by inactivation of p130 and p107 by promoting the release of the E2F4 and DP1 heterodimeric transcription factor and dissociation from E2F-dependent promoters [10, 12, 13]. Expression of the 17KT can restore the transformational potential of J domain mutant LT antigen in human fibroblast cells supporting the idea the 17KT has a functional J domain [14].
The SV40 LT and ST J domain contains 3 α-helices with the highly conserved HPDK residues present in a loop between the second and third α-helix. The structure of LT bound to Rb revealed a fourth α-helix encoded by LT residues 89 to 100 that makes contacts with the J domain as well as to Rb [15, 16]. Residues in the fourthα-helix are also necessary for LT binding to Cul7 and Bub1 [17–19]. It is not known how Cul7 and Bub1 bind to LT and if the binding is direct or through an intermediary.
Crystal structure analysis revealed that the ST J domain makes direct contacts with the ST unique domain that serve to stabilize the overall globular structure of ST bound to PP2A [20, 21]. The ST J domain may also contribute to directly inhibit the catalytic activity of the PP2A C subunit.
LXCXE motif
The LT LXCXE motif is conserved in polyoma viruses from several species including SV40 and PY and serves to bind to the Rb family (Fig. 2). The LXCXE motif was first recognized as part of the highly conserved region 2 (CR2) in adenovirus E1A 12S and 13S products required for cellular transformation as well as induction of cellular DNA synthesis. Similar to E1A, the LXCXE motif in SV40 LT is required for cellular transformation [22]. Substitution for the CR2 of E1A with the LXCXE motif from SV40 LT restored the E1A transforming activities [23]. The LXCXE is also present in many serotypes of the high-risk HPV E7 proteins.
The LXCXE motif consists of alternating conserved and non-conserved residues. The X-ray crystal structure of Rb with the LXCXE motif from HPV E7, E1A and LT each demonstrated that the Leucine, Cysteine and Glutamate side-chain residues make direct contact with a highly conserved ridge in the B domain of Rb while the non-conserved X side-chains are directed away from the binding surface [15, 24]. This ridge is conserved in p107 and p130 as well as in the Rb homologs from other species. Despite the high degree of conservation in the Rb binding cleft and the numerous reports of cellular proteins with LXCXE motifs, it is not certain what, if any, cellular proteins bind specifically to the LXCXE binding cleft of Rb. Alternatively, it has been proposed that the C-terminus of Rb makes an intra-molecular bond to the LXCXE binding region.
LT DNA binding domain
Although the ability of LT DNA binding domain (DBD) to bind to the origin of replication is not required for transformation, the DBD can bind to the Mre11, Rad50 and Nbs1 (MRN) complex (Fig. 2) [25, 26]. MRN binds to double stranded DNA breaks and recruits additional factors for repair by homologous recombination. LT binding to the MRN complex bypasses DNA damage signaling and this may contribute at least indirectly to transformation.
LT C-terminus
The SV40 LT C-terminal residues 627–708 contain a host range domain that is conserved with JCV LT and BKV LT but not PY LT. In addition, SV40 LT is specifically phosphorylated on T701 and binds to Fbw7, a substrate specificity factor for the E3 ubiquitin Ligase Cul1 (Fig. 2). The LT C-terminus is also acetylated on K697 that is dependent on CBP/p300 [27]. While deletion of the C-terminus does not diminish the transforming activity of LT, this does not preclude a contributing role of phosphorylation of T701, acetylation of K697 and the Host range/Adeno helper functions.
SV40 LT binding proteins
SV40 LT-mediated transformation of primary human and rodent cells is dependent on binding to the Rb and p53 tumor suppressor. Mutations that render LT unable to bind to Rb or p53 disable its ability to transform primary rodent and human cells. In addition to Rb and p53, LT binds to several additional cellular proteins that contribute to its transforming potential.
Rb, p107, and p130
The product of the retinoblastoma susceptibility gene Rb1 is a negative regulator of cell proliferation. Rb and the two Rb related proteins, p130 and p107, repress the E2F transcription factors that, in turn, regulate the expression of genes required for entry into and progression through the cell cycle. Rb binds specifically to the activating E2F1, E2F2 and E2F3a while p130 and p107 bind to the repressing E2F4 and E2F5 (Fig. 2). Phosphorylation of the Rb proteins by Cyclin dependent kinases (Cdk) serves to release the Rb proteins from E2Fs and thereby permit expression of E2F-regulated genes.
The LXCXE motif of LT binds directly to Rb, p107 and p130 and is required to inactivate the function of all three proteins. For example, the LT LXCXE motif is required for transformation of Rb1-null MEFs as well as p107/p130 double knockout MEFs [13, 28, 29]. LT binds preferentially to the hypo- or under-phosphorylated form of Rb thereby perturbing its function during the G0/G1 phase of the cell cycle. Rb function is disrupted in nearly every human cancer by mutation of the Rb1 gene itself or by functional inactivation by phosphorylation by Cyclin D and Cdk4/6. Although, mutations of p107 and p130 have been rarely found in human cancers, overexpression of Cyclin D, loss of p16Ink4a, or expression of LT serves to inactivate all three Rb family members.
The LT J domain cooperates with Hsc70 to specifically dissociate p107 and p130 from E2F4 [13]. In addition, the LT J domain cooperates with the LXCXE motif to promote the dephosphorylation of p130 and p107 that may, in turn, contribute to inactivation of their function [13]. In a transgenic model where LT is expressed in the intestinal epithelium, the J domain was required for LT induced enterocyte proliferation and intestinal hyperplasia. This result suggests that p130 and p107 contribute specifically to enterocyte growth regulation and that LT specifically targets p130 and p107 in this cell type [30].
p53
The p53 tumor suppressor was initially identified as a cellular protein in association with SV40 LT [31, 32]. In response to a variety of cellular stresses, p53 serves as a sequence specific transcription factor that promotes expression of many genes that induce cell cycle arrest or apoptosis. The p53 DNA binding domain resides between residues 102 and 292 and oncogenic mutations typically disrupt its ability to bind to DNA. The levels of p53 are regulated by a negative feedback loop with Mdm2. Activation of p53 results in binding to the Mdm2 promoter and activating its expression. Mdm2 is an ubiquitin ligase that specifically binds to p53 and promotes its ubiquitination and degradation by the proteosome. The transcriptional activity of p53 is dependent on its interaction with the CBP and p300 co-activators [33]. The activity of p53 is highly regulated by post-translational modifications including phosphorylation and acetylation.
LT binds to the DBD of p53 and directly interferes with its ability to act as a DNA sequence specific transcription factor. LT binding to p53 results in decreased Mdm2 expression and reduced ubiquitination of p53 resulting in its stabilization. p53 bound to LT is very stable with a half-life of at least 24 hours and comparable to LT. Notably, many oncogenic mutations that inactivate p53 function disrupt p53’s ability to bind to LT.
In primary MEFs, LT binding to p53 is required for immortalization [34]. This requirement for LT binding to p53 for immortalization is tightly linked to its transforming potential and is only relieved in cells that have inactivated by p53 by mutation or functionally, for example, as in p19 Arf-null MEFs [35]. Notably in MEFs with a triple knockout loss of Rb1, Rbl1 and Rbl2 are also spontaneously immortal apparently independent of the wild type p53 status [36]. Immortalization of primary human cells has been linked to LT binding to both Rb and p53. LT can cooperate with stable expression of the telomerase catalytic subunit hTERT to bypass senescence and avoid crisis. Continuous expression of LT is required for proliferation of primary human fibroblasts and endothelial cells immortalized by telomerase.
p300 and CBP
While LT binding inactivates p53’s ability to bind specifically to DNA, it does not interfere with p53’s ability to bind to CBP and p300. Indeed, LT binds to p300 and CBP in a p53-dependent manner (Fig. 2) [37]. The interaction of CBP and p53 with LT results in specific acetylation of LT on residue K697 [27]. The LT-p53 interaction may serve as a gain-of-function by binding to p300 and CBP that, in turn, contributes to cellular transformation. Given that LT binds to both Rb and p53, LT could recruit the transcriptional co-activating activity of p300/CBP to Rb dependent promoters and activate gene expression. Conversely, LT could recruit Rb and associated repression activity to promoters activated by p300/CBP and repress gene expression. While there is only limited data to support this model, it remains an intriguing possibility with more work needed to describe the specific interactions and functional consequences [38, 39].
Cul7
Cul7 is an E3 ubiquitin ligase complex suspected to be involved in protein degradation. Cul7 forms a cullin RING ligase (CRL) complex with Fbxw8 (Fbx29, Fbw6), Rbx1 and Skp1 [40, 41]. Cul7 also binds to Parc, p53 and Glmn (Glomulin, FAP68) [42]. The 3-M short stature syndrome results from homozygous inactivating mutations in Cul7 [43]. Notably mice with homozygous mutations in Cul7 or Fbxw8 have features similar to the 3-M syndrome with severe pre-natal and post-natal retardation [40, 41]. MEFS with homozygote loss of Cul7 or Fbxw8 have significant growth retardation that is dependent on p53 [40, 41, 44]. The Cul7-Fbxw8 CRL has been reported to target Cyclin D1 and the Insulin receptor substrate-1 (IRS1) for ubiquitination and degradation [44, 45]. Inactivation of Cul7/Fbxw8 may result in high levels of IRS1 that serve to inhibit growth in a negative feedback loop.
Cul7 was originally identified as a 185 kDa LT associated protein [46]. LT binds specifically to the Cul7 complex containing Fbxw8, Skp1 and Rbx1 [17, 18]. Residues 69 to 81 and 98–99 in LT are required for binding to Cul7 and Fbxw8 (Fig. 2). LT mutants that are unable to bind to Cul7/Fbxw8 are transformation defective [17, 18]. Conversely, certain LT mutants defective for Cul7 binding are capable of transforming Cul7-null MEFs suggesting that LT serves to inactivate at least one function of Cul7. Although this result appears to be inconsistent with the requirement for Cul7 in normal growth of mice and humans, it is possible that LT interferes with a Cul7-mediated negative feedback loop. Alternatively, LT binding to Cul7 may have a gain-of-function by recruitment of the ubiquitin ligase to promote the degradation of the MRN complex [26].
IRS1
SV40 LT has been reported to bind to IRS1 (Fig. 2) [47]. IRS1 is a cytoplasmic protein that transduces signaling from the Insulin like growth factor receptor 1 (IGFR1). In addition, IRS1 provides negative feedback to IGFR1 and can promote resistance to insulin and IGF1 signaling. Significantly, LT is unable to transform MEFs null for IGFR1 implying a role for IGF1 signaling in LT transformation [48].
IRS1 binding to LT may serve to activate AKT. Notably, the K1 (E107K) mutation in the LXCXE motif that disrupts binding to Rb reduces LT binding to IRS1 and reduces AKT phosphorylation relative to wild type LT [49]. LT binding to Cul7 may also impact on IRS1 signaling given the possibility that Cul7 can degrade IRS1 [44]. LT could serve to interfere with Cul7-mediated degradation of IRS1 to promote increased IRS1-PI3K-AKT signaling.
Fbxw7
Fbxw7 (Fbw7, Cdc4) serves as a substrate recognition factor for Cul1 to form a cullin RING ligase. Fbxw7 binds to Cyclin E to promote its ubiquitination and subsequent degradation by the proteosome. Fbxw7 also targets Myc, Notch and Jun for ubiquitination. Fbxw7 is mutated in a variety of human cancers resulting in elevated expression of Cyclin E and chromosomal instability.
LT binds specifically to Fbxw7 and interferes with its ability to target Cyclin E [50]. Phosphorylation of LT residue T701 is required for Fbxw7 binding (Fig. 2). Notably, expression of ST can activate Cyclin E/Cdk2 complexes and could conceivably cooperate with the LT binding to Fbxw7 to increase the amount and activity of Cyclin E/Cdk2 [51].
Bub1
Bub1 is a kinase functioning in the mitotic spindle checkpoint. Mutation of Bub1 leads to aneuploidy and human cancer. Bub1 was identified as a LT interacting protein in a yeast 2-hybrid screen and subsequently demonstrated to co-precipitate (Fig. 2) [19]. LT may interfere with Bub1’s role in the spindle checkpoint leading to cellular aneuploidy [52]. LT binding to Bub1 promotes phosphorylation of p53 on Ser15 [53].
SV40 ST
SV40 ST alone cannot transform cells but can cooperate with LT to fully transform cells especially when LT expression is limiting. ST has positive effects on transcription and can transactivate the Cyclin A and D1 promoters [54, 55]. ST can also cooperate with overexpression of Cyclin E to induce cell cycle entry, proliferation and foci formation in otherwise normal human fibroblasts by promoting the phosphorylation of Cdk2 on activating residue T160 [51].
One contribution of SV40 ST to transformation could be its ability to alter cellular morphology and perturb the actin cytoskeleton [56]. Co-expression of ST with LT can alter the spectrum of tumors in animals when virus is injected into animals or in transgenic mouse models [57]. Expression of ST alone in an MMTV transgenic mouse model blocked breast development, led to apoptosis of breast epithelial during lactation, and induced a low frequency of tumors [58].
The major target protein of SV40 ST is serine-threonine protein phosphatase A (PP2A) [59]. PP2A is perhaps the most important cellular serine/threonine protein phosphatase. PP2A is a heterotrimeric protein made from 2 structural A subunits, 2 catalytic C subunits and 17 regulatory B subunits. PP2A is ordinarily found as a trimeric ABC complex. The A subunit is a scaffold that binds the regulatory B and catalytic C subunit. SV40 ST as well as PY ST and MT binding to PP2A occur in a manner that either displaces B subunits or prevents them from binding (Fig. 3). PY MT binds both isoforms Aα and Aβ of the scaffolding A subunit, while SV40 ST only binds Aα [60]. Recent structural analysis showed that SV40 ST binds the Aα subunit in an area involved in B binding [20, 21]. PP2A functions as a tumor suppressor. Mutations in Aβ are associated with lung and colon cancer. Cancer-associated Aβ mutants fail to regulate the RalA GTPase [61].
Figure 3. MT Replaces B Subunits in PP2A Complexes.
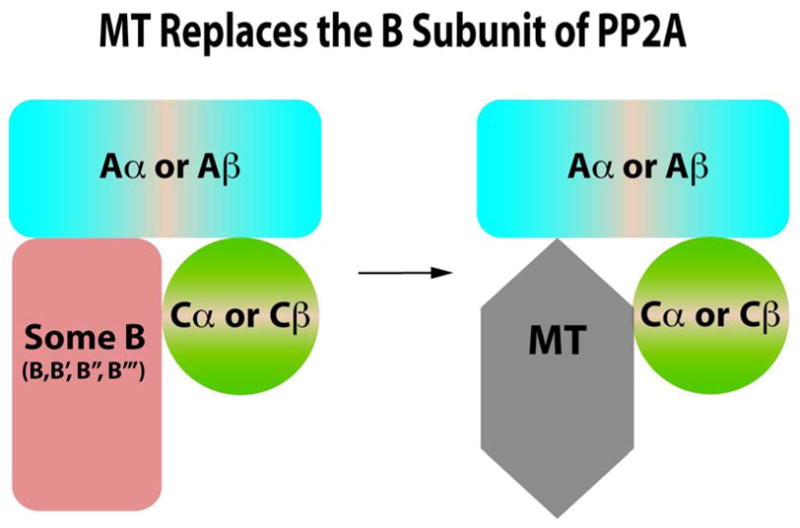
PP2A exists as heterotrimeric ABC complexes. There are two different A isoforms that act as scaffolds. There are also two different catalytic C isoforms. There are many different B family regulatory subunits. MT complexes with A and C in place of B subunits.
SV40 ST interaction with PP2A leads to activation of a number of signaling pathways that may be different than those activated by PY ST and MT [62]. SV40 ST can activate AKT signaling through activation of PI3K [63, 64]. SV40 ST contributes to stabilization of Myc by inhibiting PP2A-dependent dephosphorylation of Serine residue 62 thereby blocking Myc degradation by Fbxw7 [65]. ST also interferes with dephosphorylation of 4E-BP1 in a PP2A-dependent manner resulting in inhibition of cap-dependent translation [66].
PY MT Functions in Both Transformation and Productive Infection
Here we will discuss the role of MT in both PY infection and in tumorigenesis. For more detailed views see [67] or Fluck and Schaffhausen (in prep.).
MT is key to PY transformation and tumorigenesis, since it is necessary and frequently sufficient for transformation in tissue culture [68, 69]. When MT is not sufficient, complementation can be provided by other polyoma or non-polyoma oncogenes. For example, in REF52 cells, complementation by ST is observed using a mechanism that controls p53 [70].
Three different kinds of experiments illuminate the role of MT in tumor induction. Historically, PY infection of newborn mice has been used. To cite one example [71], infection of 259 mice with PTA4 virus led to 967 tumors at a variety of epithelial and mesenchymal sites. Studies of PY mutants show the importance of MT for induction of tumors by PY [71–75]. MT can be expressed from cDNA or using a retrovirus. Such experiments showed that MT alone induces tumors with some exceptions. In a recent elegant approach ALSV retrovirus was employed to target MT expression to tissues engineered to express the retroviral receptor. Mammary tumors [76] as well as pancreatic and liver induced by MT have been examined in this way. In a third approach, MT as a transgene causes tumors in a range of tissues. The MMTV MT transgenic model efficiently induces metastatic mammary carcinomas [77] and has been used to study a wide range of issue in mammary cancer.
Polyoma MT [75] operates at several levels in viral infection. Viruses that lack MT are defective in viral assembly in some cell types, because the pattern of the VP1 capsid protein modification is altered. MT also participates in regulating both viral DNA replication and RNA transcription [78, 79]. The polyoma enhancer, which contains PEA3 (ETS family) and AP1 binding sites, controls both viral gene expression and DNA replication. MT leads to the activation of both AP1 and PEA3 [80]. The result is a feedback loop (MT → AP-1+PEA3 → MT) that governs PY transcription and DNA replication.
While important for viral replication, this cycle of transcriptional activation has important consequences for transformation. Many genes associated with tumorigenesis use the same transcription factors. This is perhaps not surprising since ETS/PEA3 and AP-1 are linked to oncogenesis by their discovery as retroviral oncogenes. In addition, PY provides an important clue. Having AP1 and ETS bindings juxtaposed together is much better than just these sites in isolation. Since many tumor-associated genes show juxtaposed ETS/AP1 sites, synergy is probably occurring at those promoters as well. How would this occur? Most ETS family members bind DNA with difficulty at single sites [81]. AP1 facilitates binding of ETS proteins and activity. There is an additional issue. When MAP kinase signaling is strong, Fra-1 replaces Fos. However, Fra1 lacks the strongest fos-family activation domain. This is an apparent conundrum since these promoters are highly active under these conditions. Interactions between Fra/Jun and ETS family members could synergize and provide the transactivation. One could hypothesize that Fra1/Jun is especially suited to recruit ETS proteins, since tethering them to the Jun DNA binding domain overcomes the DNA binding inhibition of most ETS proteins seen in the absence of palindromic ETS binding sites.
MT – The Protein
Simply viewed, MT is a catalytically inactive 421-residue protein that establishes signaling complexes on membranes (Figure 4). Membrane association is required for transformation and tumorigenesis. Sequence comparisons between hamster and PY MTs suggest that MT has two parts. Sequences through residue 190 are substantially conserved, whereas more C-terminal sequences are quite different, although some elements such as the C-terminal membrane anchor can be discerned.
Figure 4. MT: Its Interactions and Signaling.

MT, its principal phosphorylation sites, and its interaction with cellular proteins are shown. Each signal transducer shown below MT is known to be important for transformation. PP2A = protein phosphatase 2A, PTK = src family tyrosine kinase (src, yes, fyn), PI3K = phosphoinositide 3-kinase, PLCg = phospholipase Cg1. PPP represents the proline rich/E349K sequence important for transformation. Generic versions of signaling pathways downstream of Y250, Y315 and Y322 are illustrated.
MT and PP2A
MT binding to PP2A occurs in a manner that either displaces B subunits or prevents them from binding [59]. Residues from almost the N-terminus through residues near 190 are important for the PP2A binding.
PP2A is required for association of MT with PTKs [82, 83]. Since phosphatase activity is not required [83], PP2A is likely to serve a structural role. One can imagine that MT could reduce cellular PP2A activity or alter the patterns of B subunit containing complexes like SV40 ST. The effect of SV40 ST in transformation can be mimicked by knockdown of specific B subunits, while over-expression of B subunits reverses ST action some of the effect of SV40ST. Another possibility is that MT targets PP2A to particular substrates. SV40 ST “activates” PP2A towards some substrates. Recent work shows that Lipin, a target of insulin signaling, is dephosphorylated because of its interaction with MT (B. Schaffhausen et al., unpublished).
The MT J domain
The N-terminus sequence of MT is a J domain that binds hsc70 [82]. The role of hsc70 is uncertain since only when MT is expressed at high levels, or is unable to bind PP2A is the association with hsc70 evident. Moreover, the HPDK loop sequence required for hsc70 binding can be deleted with no effect of MT’s transforming ability [82].
The MT J domain sequences do function in binding other proteins including PP2A and PTK. In addition, MT amino acid residues 2–4 bind the WW domain of TAZ [84]. TAZ regulates transcription and protein degradation through a SCFβ-Trcp E3 ligase complex. TAZ contributes to mesenchymal stem cell differentiation, cell proliferation as well as tumorigenesis of breast cancer cells suggesting that its role for MT requires careful examination. Unpublished data indicate a TAZ-binding defective mutant virus is unable to transform or induce tumors. Whether this mutation affects MT, ST or LT function needs to be determined.
MT is associated with membranes
MT is tightly bound to membranes [85–87] through a hydrophobic stretch in the C-terminus. Truncations in the hydrophobic sequence render MT non-transforming [68]. The hydrophobic sequence is not uniquely required for MT transformation, since its replacement by the C-terminal lipid modification motif from H-Ras restores transforming activity [88]. Nonetheless, there is some specificity since mutations within the hydrophobic domain or substitution with some other membrane localization sequences yielded MTs that failed to transform.
MT binds both plasma and intracellular membranes. The amount of MT reported for each location has varied, with biochemical approaches emphasizing the plasma membrane and immunolocalization favoring intracellular membranes. Within the membrane, MT associates with membrane skeleton and cytoskeleton elements. Since MT/PTK complexes are enriched in these skeletal fractions [86], they are likely to be functionally important.
MT and the Src Family of Tyrosine Kinases
Signaling via activation of Src-family PTKs is critical to MT functions. Protein tyrosine kinase activity is associated with wild type, but not transformation defective MT, [89]. The demonstration of tyrosine phosphorylation led to a whole new research area. Tyrosine kinase activity came from Src associated with MT [90]. Some other Src family members, such as Yes and Fyn, but not Lck or Hck can bind MT. Tyrosine phosphorylation is essential for MT transformation. MT mutants have consistently revealed a strong correlation between tyrosine phosphorylation and transformation [68, 89, 91]. Moreover, the level of MT-PTK activity correlated to the strength of the transformed phenotype [92]. Transformation could be reduced using antisense to Src. Crossing MMTV-MT mice with Src knockout mice showed that Src is specifically important for MT-induced mammary carcinomas [93].
The association of MT with PTKs has been explored in some detail. No mutants exist that clearly distinguish different Src family members. MT must be membrane-associated for the association to occur [68]. The primary binding site for Src on MT lies from residues 185–210 [94]. This is may be why hamster MT, which has a difference sequence there, prefers to bind Fyn. Other sequences are also needed for formation of the MT/PTK complex. Most importantly, MT must recruit protein phosphatase 2A (PP2A) in order to bind PTKs [82, 83]. Curiously, PP2A enzymatic activity is not required for MT/Src interaction [83].
Src sequences at the C-terminal tail and the catalytic domain N-terminal to it bind MT. Mutations in the C-terminal region, such as Src truncated at 516 or v-Src with its truncated C-terminus, abolish MT binding. The Src family is regulated by C-terminal region phosphorylation, Y527 for Src, which results in an interaction with the upstream SH2 domain to give a closed configuration with restricted activity. MT seems to target an open configuration. For example, Hck did not associate with MT in a closed configuration, but could bind in an open one. Similarly, Src with an SH2 domain deletion is unable to assume a closed configuration and has enhanced MT binding.
Src bound to MT is activated [95]. The Src in the MT complex is not phosphorylated at Y527 and therefore in the active configuration [96]. A second, activating, Src phosphorylation at Y416 is found in MT complexes. What are the cellular substrates for MT/Src tyrosine phosphorylation? Unlike v-Src transformants, MT transformants do not show a broad increase in cellular phosphotyrosine. Very few proteins (e.g. Stat3) become tyrosine phosphorylated unless, like PI3K, they bind MT.
One obvious substrate is MT itself. MT is phosphorylated on Y250, Y315 and Y322. Additional phosphorylation is seen on Y297 and possibly Y258 or Y288 but its role remains to be uncovered. The effects of mutating single tyrosine phosphorylation sites are much less dramatic for lytic infection than for transformation. Mutations of single tyrosine have showed little effect on gene expression and genome replication in tissue culture or in mice. In the absence of ST, even triply mutated Y250/315/322F MT retained substantial activity in lytic function [78]. However, function was lost when three additional tyrosines (Y258, Y288 and Y297) were mutated as well. In the most stringent test - persistence at one month post neonatal infection- persistence of the sextuple mutant was 100 fold less than wild type, while the triple mutant Y250/315/322F was down only eight fold (Wang, X and Fluck, M., unpublished).
There remain puzzling aspects to the MT/PTK association. There must be some unappreciated regulatory aspect to the association since the interaction is limited to small fraction (~10%) of each protein even when either partner is over-expressed. Furthermore, binding of Src to MT takes several hours [97].
MT Y250 and Adaptors
Y250 is phosphorylated in the MT/Src complexes. Its importance to transformation depends on the assay system. Mutation of Y250 can have a dire effect on transforming ability [98, 99]. The Y250 mutant virus give a different tumor profile with, for example, a dramatic decrease in kidney tumors [73] Expressed as a transgene, Y250F is much less, if at all, tumorigenic in the mammary gland than wild type [100]. In contrast, Y250F has no effect on transformation of human mammary epithelial cells and fibroblasts in vitro [99]. Also unlike the transgene result, Y250S virus infection showed little difference from wild type in mammary tumorigenesis [73]. Sometimes loss of 250 signaling even seemed to enhance tumorigenesis. Y250S virus caused larger hair follicle tumors and more frequent penile papillomas than wild type. The striking difference between transgenic and virus approaches in the mammary gland may reflect viral expression of LT and ST and/or a difference in cell type giving rise to the tumors.
Adaptors of the ShcA family bind MT phosphorylated on Y250 [101, 102]. Three overlapping ShcA isoforms, p46, p52 and p66 have N-terminal PTB and C-terminal SH2 domains that can bind phosphotyrosine sequences and a central proline-rich CH1 region. The ShcA PTB recognition motif is ΨXNPXpY. This N-terminal specificity differentiates it from SH2 domains using residues C-terminal to the phosphotyrosine. Genetic evidence shows that MT uses the PTB domain, since mutation of residues N-terminal to Y250 abrogate Shc binding and transformation. Ectopic expression of the Shc PTB reduces MT transformation of NIH3T3 cells. Some contribution from the SH2 cannot be excluded. MT binds the Shc SH2 in vitro [102] and P248H, which should not interact with the PTB, induces some Shc phosphorylation not seen in Y250F [101].
When MT is expressed, Shc is tyrosine-phosphorylated [101, 102]. Since much more p52 ShcA is phosphorylated than is bound to MT [101], there is presumably turnover of ShcA in MT complexes. Blotting with site-specific phospho-Shc antibodies (Jiang, Zhu & Schaffhausen, unpublished observations) shows that both Y317 (Y313 in mouse) and the double phosphorylation site at Y239/are targeted. Expression of Y239F/Y240F Shc, but not Y317F, changes the phenotype of MT transformed cells.
Association of Shc and MT drives Shc association with Grb2 [101–103]. Grb2 is an adaptor that allows recruitment of additional proteins such as the exchange factor SOS that activates Ras [103]. This is consistent with the earlier observation that MT activates Ras. Grb2 contributes to MT tumorigenesis. Grb2 knockout reduced MT mammary carcinogenesis. Conversely, overexpression of Grb2 (or Shc) reduced tumor latency of Y250F expressing mice. Experiments have been performed to determine whether pY250 is merely recruiting Shc so that Grb2 can be recruited. Substitution of the 250 sequence in MT with the ShcA sequences that bind Grb2 (Y317/Y313 and Y239/Y240) yields MT that can transform Rat-2 cells to some extent [103]. Since the substituted MTs bound Grb2 like wild type, the decreased transforming activity suggests that Shc binding to MT does something in addition to simply recruiting Grb2. However, when strong Grb2 SH2 binding sequences (YVNQ/YYND) were introduced [98], substituted MT produced hemangiomas like wild type.
Grb2 mediates recruitment of another adaptor, Gab1 [98, 103]. Gab1 is involved in signaling for growth factor and cytokine receptors. Gab1 provides another way to activate PI3K. Gab1 should bind SHP-2 tyrosine phosphatase and the adaptor Crkl, although these have not been confirmed in the MT complex.
MT Y315 and PI3K
Y315 is a major MT phosphorylation site. Similar to Y250, mutagenesis of Y315 resulted in a range of effects in vitro [91] and confirmed its importance in vivo [71]. Y315F was much less tumorigenic at sites such as kidney, salivary, mammary or adrenal glands. However, tumors at some sites, including thymus, hair follicle and subcutaneous connective tissue, did not show differences compared to wild type. Conversely, Y315F caused more bone or lung tumors than wild type. Comparison of Y315/Y322F to wild MMTV-MT transgenic mice confirmed the importance of Y315 in mammary carcinogenesis [100].
The primary role for pY315 is the recruitment of PI3K. Phosphatidylinositol kinase activity in MT immunoprecipitates was tightly correlated with MT transforming ability [104]. Later work showed this enzymatic activity phosphorylated the inositol ring on the 3 position (hence PI3K). PI3K activity tracked with an 85-kDa polypeptide with MT and activated receptor tyrosine kinases (RTKs).
The PI3Ks associated with MT are the Class 1A enzymes, which include three catalytic subunits (α, βand δ). Knockout of p110α blocks MT transformation in MEFs [99]. This is consistent with observations showing that activating p110α mutations are found in cancers, such as colon, breast, prostate, liver, and brain and that p110α is critical for RTK signaling.
The purified PI3Ks associated with MT and RTKs also have a p85 adaptor subunit. p85 regulatory subunits have multiple functions. They contain SH2 domains that couple PI3K to RTKs or MT through specific motifs such as the MT pY315MPM. This allows MT to recruit PI3K to membranes and stimulate PI3K activity. P85α and p85βare similar isoforms that have different roles in development. Although MT binds both forms [97], isoform-specific functions are not known.
Class IA PI3Ks produce PIP3 that recruits proteins to membranes by direct interaction with specific pleckstrin homology (PH) domains. Critical targets include the kinase PDK1 and Serine/Threonine kinase PKB/Akt that is activated by PDK1 phosphorylation and by binding PIP3. MT activates Akt ([105]. This allows MT to block apoptosis and prevent cell cycle withdrawal. In the MMTV MT transgenic mice, apoptosis observed in Y315/322F mutant mice [100] was reversed by co-expression of activated Akt. There are three Akt isoforms. Knockout of Akt1 dramatically inhibited induction of mammary carcinomas, while Akt2 knockout enhanced tumorigenesis [106]. Akt phosphorylates many substrates, such as GSK3, TSC1/2, MDM2 and IKK that participate in many processes. How such phosphorylations contribute to MT function has not yet been explored.
The small GTPase Rac is activated by PIP3 activation of PH domain-containing nucleotide exchange factors. MT uses PI3K signaling to increase the fraction of Rac in the active GTP-bound state (Denis and Schaffhausen, unpublished observations). Dominant-negative Rac reduces MT transformation [107]. S6 kinase and, interestingly, PI3K are Rac effectors in endothelial cells. MT also uses Rac activation to activate c-fos.
MT Y322 and PLCγ1
Phosphorylation of Y322 mediates MT binding and phosphorylation of PLCγ1 [108]. Consistent with PLCγ1 activation, MT increased the amount of inositol trisphosphate (IP3), an effector of intracellular calcium. Except at low serum, mutation of Y322 has modest effects on transformation. Y322 signaling could be important for aspects of tumorigenesis not tested in culture. Expression of dominant-negative PLCγ1 markedly decreased lung metastases from MT mammary carcinomas [109].
MT S257 and 14-3-3 proteins
Serine 257 phosphorylation allows MT to bind 14-3-3 proteins [72]. While binding of 14-3-3 seems unimportant for transformation in vitro or for tumor induction at many sites, virus bearing a mutation that prevents 14-3-3 binding induces no salivary gland tumors and causes few fibrosarcomas.
The MT Proline-Rich Region: A Genetic Puzzle
The transformation-defective MT mutant dl1015 lacks a proline-rich region (residues 339–347). Deletion of prolines 336–338 causes a striking defect in kidney, salivary gland, or thymic tumors [74]. The basis for this defect in transformation is unknown. Dl1015 has normal tyrosine kinase activity associated with it. A point mutant E349K that mimics the defect suggests that PI3K is blocked from signaling to downstream targets such as Akt, while still a recruited to membranes, allowing recruitment of the enzyme to membranes (Schaffhausen & Denis, unpublished).
The MMTV MT transgenic model has been used extensively to examine many breast cancer issues
When expressed in mammary glands using the MMTV promoter, MT induces metastatic carcinomas with high frequency and short latency [77]. Investigators concerned with virtually every aspect of breast cancer have intensively used the MMTV-MT system. MMTV-MT presents a relatively good model for the human breast cancer [110, 111] although there are some differences. For example, MMTV-MT tumors metastasize to the lung, not more broadly as human tumors do.
The MMTV-MT transgenic line FVB/N Tg(MMTV-PyMT634Mul) (#634) [77] has been used in most experiments. Tumors appear in a quasi-synchronous way with a latency of 34 days in females. Early multifocal lesions involve prepubertal, nipple proximal buds at the ends of growing ducts (end buds/TDLU) surrounding the main collecting duct. Early preneoplastic lesions are characterized as MIN: mammary intraepithelial neoplasia, proliferating lumina with cytological atypia (nuclear morphological abnormalities). The nipple proximal MIN generates the main tumor that becomes invasive [77, 110]. 94% of females have metastatic lung disease by 3 months of age. Src activation [93] and the MT Y315-PI3K and Y250-Shc/Grb2 pathways [100] are very important in this system.
An issue is whether MT expression is sufficient to generate metastatic carcinomas or whether other genetic changes are also required. Although high frequency, short latency and the homogeneous pattern of altered gene expression in independently derived tumors [112] argue for sufficiency, MINs with varied properties have been isolated [111]. For example, their transplantation outgrowths are marked by different gene expression patterns, different genetic aberrations and different progression characteristics. These neoplasias eventually progress in transplantation tests. The variability is difficult to interpret since the amount of MT/PTK activity is uncertain. Another question is whether the same progenitor cells are being compared. There is a gradient of focal atypical lesions along the duct. Distal foci that arise at a stage of estrogen-dependent growth might have different properties.
Metastasis in the MMTV-MT Model
A traditional view of metastasis suggests a series of interrelated steps including invasion, entering blood vessels (intravasation), dissemination and escape from blood vessels (extravasation), leading to colony formation at the metastatic site. MMTV-MT-634 is contributing to an apparent paradigm shift in our view of metastasis. There is now evidence that MT-mammary cells can already be disseminated in very early (ADH or DCIS) stage tumors [113]. Even more strikingly “normal” mammary cells carrying an inducible MT to produce tumors could colonize lungs when introduced into the blood stream prior to MT expression [114].
MT induces factors required for cell migration and invasion. Osteopontin (OPN) is one example. Comparison of a low metastasis mutant MT cell line to a highly metastatic one (Met) showed the Met line produced OPN and suppression of OPN blocked its metastasis [115]. Expression of OPN does not affect growth, but contributes to migration [115, 116]. MT regulates the OPN transcription through PEA3 sites [116]. Thrombospondin-1 is a matricellular protein, which like OPN, is controlled by MT and appears to promote migration and metastasis.
MT mammary tumors are enriched in proteases such as the matrix metalloproteinases or uPA that are important for tissue remodelling [117]. Proteases are clearly important to the metastatic phenotype since the broad spectrum metalloproteinase inhibitor or specific knockout of urokinase reduced metastasis. However, protease overexpression seems to be more a story of stromal cells than tumor cells.
Consistent with the role of AP1/ETS transcription factors mentioned earlier, many Ets genes are upregulated in MMTV-MT634 tumors [118] and disruption of coactivators such as AIB1 or Src1 that are connected to ETS family members including PEA3 reduces MT metastases.
Stromal-epithelial cell interactions play a key role in tumor progression
The importance of interactions between tumor cells and stroma is well known. Transplantation experiments make this obvious. For example, transplant of MMTV-MT #634 derived tumor cells into mice with altered ETS2 expression strongly affects tumor development [119]. The MMTV-MT-634 model has been used to examine stromal-carcinoma interactions, especially with respect to macrophages. Many aspects of the mutual epithelial-macrophage interactions have been confirmed or established in this model system.
Macrophages are found at the surface and close to perivascular regions of MMTV-MT#634 tumors. CSF-1 produced by epithelial cells is a chemoattractant and growth factor for macrophages. CSF-1, overexpressed in many breast tumors, is considered a marker for poor prognosis. In CSF-null mice, #634 tumors showed a relative absence of macrophages. Further although early tumor development is normal, invasion and metastases are both compromised [120]. In the reverse experiment, overexpression of CSF-1 speeds progression to invasive carcinoma. What do the macrophages do? The interplay between tumor-generated CSF-1 and macrophage-produced EGF is important for migration. Synergistic co-migration of macrophages and tumor cells towards either chemoattractant was observed; inhibition of either CSF or EGF signals decreased migration of both cell types [121]. Chemo-attracted tumor cells were more invasive, showed less apoptosis, but a reduced proliferative index. Macrophages also accelerate the intravasation of tumor cells. Furthermore, the load of tumor cells in the bloodstream depended on CSF-1.
Mouse Background and MT Tumorigenesis
There is considerable interest in individualized therapy using pharmacogenomic approaches for cancer. Studies of MT demonstrate the complexity of these issues. From analysis of the MT MMTV model, it is evident that the genetic background of the mouse affects production of breast tumors and their ability to metastasize (see [122]). The original MT-MMTV transgenics produced in FVB/N animals [77] were bred to females of different inbred mouse strains [123]. Some such as ST/J showed significantly increased latency (20 days slower), while other crosses such as I/LnJ yielded tumors 21 days faster than FVB/N. Although the lung remained the metastatic destination, there was a wide range in metastatic index for different strains.
Conclusion
The small DNA tumor viruses have provided many insights into the signaling of cell transformation and tumorigenesis. Not only have they generated new leads, but they have also enabled our understanding of many of the molecular mechanisms involved in the induction of tumors. SV40 LT has identified several important tumor suppressors and confirmed the relevance of their role in suppressing growth and transformation. PY MT has provided insights and discovery into PTK and PI3K signaling. SV40 and PY ST have revealed specific roles for PP2A signaling in oncogenesis. The genetics of these T antigens show clearly that new discoveries await further study. The MT E349K mutant, for example, indicates there are crucial aspects of PI3K signaling that are not yet understood. The ability of these T antigens to induce tumors in many tissues and the increasing power of mouse genetics should combine to illuminate the roles of both the signaling pathways themselves and the genetic background in which they operate.
Acknowledgments
Supported in parts by Public Health Service NIH grants PO1-CA50661 (JAD and BSS), RO1-CA34722 (BSS), RO1-CA63113 (JAD), R01-CA93804 (JAD) and R01-CA29270 (MF).
Constraints of space permit discussion of only some issues and citation of just a small fraction of the relevant literature.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sweet BH, Hilleman MR. The vacuolating virus, S.V. 40. Proc Soc Exp Biol Med. 1960;105:420–7. doi: 10.3181/00379727-105-26128. [DOI] [PubMed] [Google Scholar]
- 2.Bocchetta M, Di Resta I, Powers A, Fresco R, Tosolini A, Testa JR, et al. Human mesothelial cells are unusually susceptible to simian virus 40-mediated transformation and asbestos cocarcinogenicity. Proc Natl Acad Sci U S A. 2000;97:10214–9. doi: 10.1073/pnas.170207097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hahn WC, Dessain SK, Brooks MW, King JE, Elenbaas B, Sabatini DM, et al. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol. 2002;22:2111–23. doi: 10.1128/MCB.22.7.2111-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jat PS, Sharp PA. Cell lines established by a temperature-sensitive simian virus 40 large-T-antigen gene are growth restricted at the nonpermissive temperature. Mol Cell Biol. 1989;9:1672–81. doi: 10.1128/mcb.9.4.1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ray S, Anderson ME, Tegtmeyer P. Differential interaction of temperature-sensitive simian virus 40 T antigens with tumor suppressors pRb and p53. J Virol. 1996;70:7224–7. doi: 10.1128/jvi.70.10.7224-7227.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jat PS, Noble MD, Ataliotis P, Tanaka Y, Yannoutsos N, Larsen L, et al. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci U S A. 1991;88:5096–100. doi: 10.1073/pnas.88.12.5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li D, Zhao R, Lilyestrom W, Gai D, Zhang R, DeCaprio JA, et al. Structure of the replicative helicase of the oncoprotein SV40 large tumour antigen. Nature. 2003;423:512–8. doi: 10.1038/nature01691. [DOI] [PubMed] [Google Scholar]
- 8.Lilyestrom W, Klein MG, Zhang R, Joachimiak A, Chen XS. Crystal structure of SV40 large T-antigen bound to p53: interplay between a viral oncoprotein and a cellular tumor suppressor. Genes Dev. 2006;20:2373–82. doi: 10.1101/gad.1456306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sawai ET, Butel JS. Association of a cellular heat shock protein with simian virus 40 large T antigen in transformed cells. J Virol. 1989;63:3961–73. doi: 10.1128/jvi.63.9.3961-3973.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sullivan CS, Gilbert SP, Pipas JM. ATP-dependent simian virus 40 T-antigen-Hsc70 complex formation. J Virol. 2001;75:1601–10. doi: 10.1128/JVI.75.4.1601-1610.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campbell KS, Mullane KP, Aksoy IA, Stubdal H, Zalvide J, Pipas JM, et al. DnaJ/hsp40 chaperone domain of SV40 large T antigen promotes efficient viral DNA replication. Genes Dev. 1997;11:1098–110. doi: 10.1101/gad.11.9.1098. [DOI] [PubMed] [Google Scholar]
- 12.Zalvide J, Stubdal H, DeCaprio JA. The J domain of simian virus 40 large T antigen is required to functionally inactivate RB family proteins. Mol Cell Biol. 1998;18:1408–15. doi: 10.1128/mcb.18.3.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stubdal H, Zalvide J, Campbell KS, Schweitzer C, Roberts TM, DeCaprio JA. Inactivation of pRB-related proteins p130 and p107 mediated by the J domain of simian virus 40 large T antigen. Mol Cell Biol. 1997;17:4979–90. doi: 10.1128/mcb.17.9.4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boyapati A, Wilson M, Yu J, Rundell K. SV40 17KT antigen complements dnaj mutations in large T antigen to restore transformation of primary human fibroblasts. Virology. 2003;315:148–58. doi: 10.1016/s0042-6822(03)00524-5. [DOI] [PubMed] [Google Scholar]
- 15.Kim HY, Ahn BY, Cho Y. Structural basis for the inactivation of retinoblastoma tumor suppressor by SV40 large T antigen. Embo J. 2001;20:295–304. doi: 10.1093/emboj/20.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee C, Chang JH, Lee HS, Cho Y. Structural basis for the recognition of the E2F transactivation domain by the retinoblastoma tumor suppressor. Genes Dev. 2002;16:3199–212. doi: 10.1101/gad.1046102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ali SH, Kasper JS, Arai T, DeCaprio JA. Cul7/p185/p193 binding to simian virus 40 large T antigen has a role in cellular transformation. J Virol. 2004;78:2749–57. doi: 10.1128/JVI.78.6.2749-2757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kasper JS, Kuwabara H, Arai T, Ali SH, DeCaprio JA. Simian virus 40 large T antigen’s association with the CUL7 SCF complex contributes to cellular transformation. J Virol. 2005;79:11685–92. doi: 10.1128/JVI.79.18.11685-11692.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cotsiki M, Lock RL, Cheng Y, Williams GL, Zhao J, Perera D, et al. Simian virus 40 large T antigen targets the spindle assembly checkpoint protein Bub1. Proc Natl Acad Sci U S A. 2004;101:947–52. doi: 10.1073/pnas.0308006100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y, Xu Y, Bao Q, Xing Y, Li Z, Lin Z, et al. Structural and biochemical insights into the regulation of protein phosphatase 2A by small t antigen of SV40. Nat Struct Mol Biol. 2007;14:527–34. doi: 10.1038/nsmb1254. [DOI] [PubMed] [Google Scholar]
- 21.Cho US, Morrone S, Sablina AA, Arroyo JD, Hahn WC, Xu W. Structural basis of PP2A inhibition by small t antigen. PLoS Biol. 2007;5:e202. doi: 10.1371/journal.pbio.0050202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen S, Paucha E. Identification of a region of simian virus 40 large T antigen required for cell transformation. J Virol. 1990;64:3350–7. doi: 10.1128/jvi.64.7.3350-3357.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moran E. A region of SV40 large T antigen can substitute for a transforming domain of the adenovirus E1A products. Nature. 1988;334:168–70. doi: 10.1038/334168a0. [DOI] [PubMed] [Google Scholar]
- 24.Lee JO, Russo AA, Pavletich NP. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature. 1998;391:859–65. doi: 10.1038/36038. [DOI] [PubMed] [Google Scholar]
- 25.Wu X, Avni D, Chiba T, Yan F, Zhao Q, Lin Y, et al. SV40 T antigen interacts with Nbs1 to disrupt DNA replication control. Genes Dev. 2004;18:1305–16. doi: 10.1101/gad.1182804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao X, Madden-Fuentes RJ, Lou BX, Pipas JM, Gerhardt J, Rigell CJ, et al. Ataxia telangiectasia-mutated damage-signaling kinase- and proteasome-dependent destruction of Mre11-Rad50-Nbs1 subunits in Simian virus 40-infected primate cells. J Virol. 2008;82:5316–28. doi: 10.1128/JVI.02677-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poulin DL, Kung AL, DeCaprio JA. p53 targets simian virus 40 large T antigen for acetylation by CBP. J Virol. 2004;78:8245–53. doi: 10.1128/JVI.78.15.8245-8253.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zalvide J, DeCaprio JA. Role of pRb-related proteins in simian virus 40 large-T-antigen-mediated transformation. Mol Cell Biol. 1995;15:5800–10. doi: 10.1128/mcb.15.10.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Christensen JB, Imperiale MJ. Inactivation of the retinoblastoma susceptibility protein is not sufficient for the transforming function of the conserved region 2-like domain of simian virus 40 large T antigen. J Virol. 1995;69:3945–8. doi: 10.1128/jvi.69.6.3945-3948.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rathi AV, Saenz Robles MT, Pipas JM. Enterocyte proliferation and intestinal hyperplasia induced by simian virus 40 T antigen require a functional J domain. J Virol. 2007;81:9481–9. doi: 10.1128/JVI.00922-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278:261–3. doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- 32.Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17:43–52. doi: 10.1016/0092-8674(79)90293-9. [DOI] [PubMed] [Google Scholar]
- 33.Lill NL, Grossman SR, Ginsberg D, DeCaprio J, Livingston DM. Binding and modulation of p53 by p300/CBP coactivators. Nature. 1997;387:823–7. doi: 10.1038/42981. [DOI] [PubMed] [Google Scholar]
- 34.Tevethia MJ, Pipas JM, Kierstead T, Cole C. Requirements for immortalization of primary mouse embryo fibroblasts probed with mutants bearing deletions in the 3′ end of SV40 gene A. Virology. 1988;162:76–89. doi: 10.1016/0042-6822(88)90396-0. [DOI] [PubMed] [Google Scholar]
- 35.Chao HH, Buchmann AM, DeCaprio JA. Loss of p19(ARF) eliminates the requirement for the pRB-binding motif in simian virus 40 large T antigen-mediated transformation. Mol Cell Biol. 2000;20:7624–33. doi: 10.1128/mcb.20.20.7624-7633.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, et al. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 2000;14:3037–50. doi: 10.1101/gad.843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eckner R, Ludlow JW, Lill NL, Oldread E, Arany Z, Modjtahedi N, et al. Association of p300 and CBP with simian virus 40 large T antigen. Mol Cell Biol. 1996;16:3454–64. doi: 10.1128/mcb.16.7.3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singhal G, Kadeppagari RK, Sankar N, Thimmapaya B. Simian virus 40 large T overcomes p300 repression of c-Myc. Virology. 2008;377:227–32. doi: 10.1016/j.virol.2008.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bocchetta M, Eliasz S, De Marco MA, Rudzinski J, Zhang L, Carbone M. The SV40 large T antigen-p53 complexes bind and activate the insulin-like growth factor-I promoter stimulating cell growth. Cancer Res. 2008;68:1022–9. doi: 10.1158/0008-5472.CAN-07-5203. [DOI] [PubMed] [Google Scholar]
- 40.Arai T, Kasper JS, Skaar JR, Ali SH, Takahashi C, DeCaprio JA. Targeted disruption of p185/Cul7 gene results in abnormal vascular morphogenesis. Proc Natl Acad Sci U S A. 2003;100:9855–60. doi: 10.1073/pnas.1733908100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsutsumi T, Kuwabara H, Arai T, Xiao Y, Decaprio JA. Disruption of the Fbxw8 gene results in pre- and postnatal growth retardation in mice. Mol Cell Biol. 2008;28:743–51. doi: 10.1128/MCB.01665-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Skaar JR, Florens L, Tsutsumi T, Arai T, Tron A, Swanson SK, et al. PARC and CUL7 form atypical cullin RING ligase complexes. Cancer Res. 2007;67:2006–14. doi: 10.1158/0008-5472.CAN-06-3241. [DOI] [PubMed] [Google Scholar]
- 43.Huber C, Dias-Santagata D, Glaser A, O’Sullivan J, Brauner R, Wu K, et al. Identification of mutations in CUL7 in 3-M syndrome. Nat Genet. 2005;37:1119–24. doi: 10.1038/ng1628. [DOI] [PubMed] [Google Scholar]
- 44.Xu X, Sarikas A, Dias-Santagata DC, Dolios G, Lafontant PJ, Tsai SC, et al. The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1 for ubiquitin-dependent degradation. Mol Cell. 2008;30:403–14. doi: 10.1016/j.molcel.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Okabe H, Lee SH, Phuchareon J, Albertson DG, McCormick F, Tetsu O. A critical role for FBXW8 and MAPK in cyclin D1 degradation and cancer cell proliferation. PLoS ONE. 2006;1:e128. doi: 10.1371/journal.pone.0000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kohrman DC, Imperiale MJ. Simian virus 40 large T antigen stably complexes with a 185-kilodalton host protein. J Virol. 1992;66:1752–60. doi: 10.1128/jvi.66.3.1752-1760.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fei ZL, D’Ambrosio C, Li S, Surmacz E, Baserga R. Association of insulin receptor substrate 1 with simian virus 40 large T antigen. Mol Cell Biol. 1995;15:4232–39. doi: 10.1128/mcb.15.8.4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sell C, Rubini M, Rubin R, Liu JP, Efstratiadis A, Baserga R. Simian virus 40 large tumor antigen is unable to transform mouse embryonic fibroblasts lacking type 1 insulin-like growth factor receptor. Proc Natl Acad Sci U S A. 1993;90:11217–21. doi: 10.1073/pnas.90.23.11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu Y, Alwine JC. Interaction between simian virus 40 large T antigen and insulin receptor substrate 1 is disrupted by the K1 mutation, resulting in the loss of large T antigen-mediated phosphorylation of Akt. J Virol. 2008;82:4521–6. doi: 10.1128/JVI.02365-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Welcker M, Clurman BE. The SV40 large T antigen contains a decoy phosphodegron that mediates its interactions with Fbw7/hCdc4. J Biol Chem. 2005;280:7654–8. doi: 10.1074/jbc.M413377200. [DOI] [PubMed] [Google Scholar]
- 51.Sotillo E, Garriga J, Kurimchak A, Grana X. Cyclin E and SV40 small T antigen cooperate to bypass quiescence and contribute to transformation by activating CDK2 in human fibroblasts. J Biol Chem. 2008;283:11280–92. doi: 10.1074/jbc.M709055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gjoerup OV, Wu J, Chandler-Militello D, Williams GL, Zhao J, Schaffhausen B, et al. Surveillance mechanism linking Bub1 loss to the p53 pathway. Proc Natl Acad Sci U S A. 2007;104:8334–9. doi: 10.1073/pnas.0703164104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hein J, Boichuk S, Wu J, Cheng Y, Freire R, Jat PS, et al. Simian Virus 40 Large T Antigen Disrupts Genome Integrity and Activates a DNA Damage Response via Bub1 Binding. J Virol. 2009;83:117–27. doi: 10.1128/JVI.01515-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Porras A, Bennett J, Howe A, Tokos K, Bouck N, Henglein B, et al. A novel simian virus 40 early-region domain mediates transactivation of the cyclin A promoter by small-t antigen and is required for transformation in small-t antigen-dependent assays. J Virol. 1996;70:6902–8. doi: 10.1128/jvi.70.10.6902-6908.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Watanabe G, Howe A, Lee RJ, Albanese C, Shu IW, Karnezis AN, et al. Induction of cyclin D1 by simian virus 40 small tumor antigen. Proc Natl Acad Sci U S A. 1996;93:12861–6. doi: 10.1073/pnas.93.23.12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nunbhakdi-Craig V, Craig L, Machleidt T, Sontag E. Simian virus 40 small tumor antigen induces deregulation of the actin cytoskeleton and tight junctions in kidney epithelial cells. J Virol. 2003;77:2807–18. doi: 10.1128/JVI.77.5.2807-2818.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Matthews BJ, Levine AS, Dixon K. Deletion mutations in the small t antigen gene alter the tissue specificity of tumors induced by simian virus 40. J Virol. 1987;61:1282–5. doi: 10.1128/jvi.61.4.1282-1285.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goetz F, Tzeng YJ, Guhl E, Merker J, Graessmann M, Graessmann A. The SV40 small t-antigen prevents mammary gland differentiation and induces breast cancer formation in transgenic mice; truncated large T-antigen molecules harboring the intact p53 and pRb binding region do not have this effect. Oncogene. 2001;20:2325–32. doi: 10.1038/sj.onc.1204355. [DOI] [PubMed] [Google Scholar]
- 59.Pallas DC, Shahrik LK, Martin BL, Jaspers S, Miller TB, Brautigan DL, et al. Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell. 1990;60:167–76. doi: 10.1016/0092-8674(90)90726-u. [DOI] [PubMed] [Google Scholar]
- 60.Zhou J, Pham HT, Ruediger R, Walter G. Characterization of the Aalpha and Abeta subunit isoforms of protein phosphatase 2A: differences in expression, subunit interaction, and evolution. Biochem J. 2003;369:387–98. doi: 10.1042/BJ20021244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sablina AA, Chen W, Arroyo JD, Corral L, Hector M, Bulmer SE, et al. The tumor suppressor PP2A Abeta regulates the RalA GTPase. Cell. 2007;129:969–82. doi: 10.1016/j.cell.2007.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rodriguez-Viciana P, Collins C, Fried M. Polyoma and SV40 proteins differentially regulate PP2A to activate distinct cellular signaling pathways involved in growth control. Proc Natl Acad Sci U S A. 2006;103:19290–5. doi: 10.1073/pnas.0609343103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao JJ, Gjoerup OV, Subramanian RR, Cheng Y, Chen W, Roberts TM, et al. Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell. 2003;3:483–95. doi: 10.1016/s1535-6108(03)00088-6. [DOI] [PubMed] [Google Scholar]
- 64.Boehm JS, Hession MT, Bulmer SE, Hahn WC. Transformation of human and murine fibroblasts without viral oncoproteins. Mol Cell Biol. 2005;25:6464–74. doi: 10.1128/MCB.25.15.6464-6474.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004;6:308–18. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- 66.Yu Y, Kudchodkar SB, Alwine JC. Effects of simian virus 40 large and small tumor antigens on mammalian target of rapamycin signaling: small tumor antigen mediates hypophosphorylation of eIF4E-binding protein 1 late in infection. J Virol. 2005;79:6882–9. doi: 10.1128/JVI.79.11.6882-6889.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schaffhausen BS, Roberts TM. Lessons from polyoma middle T antigen on signaling and transformation: A DNA tumor virus contribution to the war on cancer. Virology. 2008 doi: 10.1016/j.virol.2008.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carmichael G, Schaffhausen B, Dorsky D, Oliver D, Benjamin T. The role of the carboxy terminus of middle T antigen of polyoma virus. Proc Natl Acad Sci USA. 1982;79:3579–83. doi: 10.1073/pnas.79.11.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Treisman R, Novak U, Favaloro J, Kamen R. Transformation of rat cells by an altered polyoma virus genome expressing only the middle T protein. Nature. 1981;292:595–600. doi: 10.1038/292595a0. [DOI] [PubMed] [Google Scholar]
- 70.O’Shea CC, Fried M. Modulation of the ARF-p53 pathway by the small DNA tumor viruses. Cell Cycle. 2005;4:449–52. doi: 10.4161/cc.4.3.1555. [DOI] [PubMed] [Google Scholar]
- 71.Freund R, Dawe CJ, Carroll JP, Benjamin TL. Changes in frequency, morphology, and behavior of tumors induced in mice by a polyoma virus mutant with a specifically altered oncogene. Am J Pathol. 1992;141:1409–25. [PMC free article] [PubMed] [Google Scholar]
- 72.Cullere X, Rose P, Thathamangalam U, Chatterjee A, Mullane KP, Pallas DC, et al. Serine 257 phosphorylation regulates association of polyomavirus middle T antigen with 14-3-3 proteins. J Virol. 1998;72:558–63. doi: 10.1128/jvi.72.1.558-563.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bronson R, Dawe C, Carroll J, Benjamin T. Tumor induction by a transformation-defective polyoma virus mutant blocked in signaling through shc. Proc Nat Acad Sci USA. 1997;94:7954–8. doi: 10.1073/pnas.94.15.7954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yi X, Freund R. Deletion of proline-rich domain in polyomavirus T antigens results in virus partially defective in transformation and tumorigenesis. Virology. 1998;248:420–31. doi: 10.1006/viro.1998.9246. [DOI] [PubMed] [Google Scholar]
- 75.Freund R, Sotnikov A, Bronson RT, Benjamin TL. Polyoma virus middle T is essential for virus replication and persistence as well as for tumor induction in mice. Virology. 1992;191:716–23. doi: 10.1016/0042-6822(92)90247-m. [DOI] [PubMed] [Google Scholar]
- 76.Du Z, Podsypanina K, Huang S, McGrath A, Toneff MJ, Bogoslovskaia E, et al. Introduction of oncogenes into mammary glands in vivo with an avian retroviral vector initiates and promotes carcinogenesis in mouse models. Proc Natl Acad Sci U S A. 2006;103:17396–401. doi: 10.1073/pnas.0608607103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–61. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen L, Wang X, Fluck MM. Independent contributions of polyomavirus middle T and small T to the regulation of early and late gene expression and DNA replication. J Virol. 2006;80:7295–307. doi: 10.1128/JVI.00679-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen L, Fluck MM. Role of middle T-small T in the lytic cycle of polyomavirus: control of the early-to-late transcriptional switch and viral DNA replication. J Virol. 2001;75:8380–9. doi: 10.1128/JVI.75.18.8380-8389.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wasylyk C, Flores P, Gutman A, Wasylyk B. PEA3 is a nuclear target for transcription activation by non-nuclear oncogenes. EMBO J. 1989;8:3371–8. doi: 10.1002/j.1460-2075.1989.tb08500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sharrocks AD. The ETS-domain transcription factor family. Nat Rev Mol Cell Biol. 2001;2:827–37. doi: 10.1038/35099076. [DOI] [PubMed] [Google Scholar]
- 82.Campbell KS, Auger KR, Hemmings BA, Roberts TM, Pallas DC. Identification of regions in polyomavirus middle T and small t antigens important for association with protein phosphatase 2A. Journal of Virology. 1995;69:3721–8. doi: 10.1128/jvi.69.6.3721-3728.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ogris E, Mudrak I, Mak E, Gibson D, Pallas DC. Catalytically inactive protein phosphatase 2A can bind to polyomavirus middle tumor antigen and support complex formation with pp60(c-src) J Virol. 1999;73:7390–8. doi: 10.1128/jvi.73.9.7390-7398.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tian Y, Li D, Dahl J, You J, Benjamin T. Identification of TAZ as a binding partner of the polyomavirus T antigens. J Virol. 2004;78:12657–64. doi: 10.1128/JVI.78.22.12657-12664.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ito Y. Polyoma virus-specific 55K protein isolated from plasma membrane of productively infected cells is virus-coded and important cell transformation. Virology. 1979;98:261–6. doi: 10.1016/0042-6822(79)90545-2. [DOI] [PubMed] [Google Scholar]
- 86.Schaffhausen BS, Dorai H, Arakere G, Benjamin TL. Polyoma virus middle T antigen: relationship to cell membranes and apparent lack of ATP-binding activity. Mol Cell Biol. 1982;2:1187–98. doi: 10.1128/mcb.2.10.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dilworth SM, Hansson HA, Darnfors C, Bjursell G, Streuli CH, Griffin BE. Subcellular localisation of the middle and large T-antigens of polyoma. EMBO J. 1986;5:491–9. doi: 10.1002/j.1460-2075.1986.tb04238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Elliott J, Jones MD, Griffin BE, Krauzewicz N. Regulation of cytoskeletal association by a basic amino acid motif in polyoma virus middle T antigen. Oncogene. 1998;17:1797–806. doi: 10.1038/sj.onc.1202083. [DOI] [PubMed] [Google Scholar]
- 89.Eckhart W, Hutchinson MA, Hunter T. An activity phosphorylating tyrosine in polyoma T antigen immunoprecipitates. Cell. 1979;18:925. doi: 10.1016/0092-8674(79)90205-8. [DOI] [PubMed] [Google Scholar]
- 90.Courtneidge S, Smith AE. Polyoma virus transforming protein associates with the product of the c-src cellular gene. Nature. 1983;303:435–9. doi: 10.1038/303435a0. [DOI] [PubMed] [Google Scholar]
- 91.Carmichael G, Schaffhausen BS, Mandel G, Liang TJ, Benjamin TL. Transformation by polyoma virus is drastically reduced by substitution of phenylalanine for tyrosine at residue 315 of middle-sized tumor antigen. Proc Natl Acad Sci USA. 1984;81:679–83. doi: 10.1073/pnas.81.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Raptis L, Lamfrom H, Benjamin TL. Regulation of cellular phenotype and expression of polyoma virus middle T antigen in rat fibroblasts. Mol Cell Biol. 1985;5:2476–85. doi: 10.1128/mcb.5.9.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Guy CT, Muthuswamy SK, Cardiff RD, Soriano P, Muller WJ. Activation of the c-Src tyrosine kinase is required for the induction of mammary tumors in transgenic mice. Genes & Development. 1994;8:23–32. doi: 10.1101/gad.8.1.23. [DOI] [PubMed] [Google Scholar]
- 94.Glover HR, Brewster CE, Dilworth SM. Association between src-kinases and the polyoma virus oncogene middle T-antigen requires PP2A and a specific sequence motif. Oncogene. 1999;18:4364–70. doi: 10.1038/sj.onc.1202816. [DOI] [PubMed] [Google Scholar]
- 95.Bolen JB, Thiele CJ, Israel MA, Yonemoto W, Lipsich LA, Brugge JS. Enhancement of cellular src gene product associated tyrosyl-kinase activity following polyoma virus infection and transformation. Cell. 1984;38:767–77. doi: 10.1016/0092-8674(84)90272-1. [DOI] [PubMed] [Google Scholar]
- 96.Courtneidge SA. Activation of the pp60c-src kinase by middle T antigen binding or by dephosphorylation. EMBO J. 1985;4:1471–7. doi: 10.1002/j.1460-2075.1985.tb03805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cohen B, Liu YX, Druker B, Roberts TM, Schaffhausen BS. Characterization of pp85, a target of oncogenes and growth factor receptors. Mol Cell Biol. 1990;10:2909–15. doi: 10.1128/mcb.10.6.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ong SH, Dilworth S, Hauck-Schmalenberger I, Pawson T, Kiefer F. ShcA and Grb2 mediate polyoma middle T antigen-induced endothelial transformation and Gab1 tyrosine phosphorylation. Embo J. 2001;20:6327–36. doi: 10.1093/emboj/20.22.6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Utermark T, Schaffhausen BS, Roberts TM, Zhao JJ. The p110alpha isoform of phosphatidylinositol 3-kinase is essential for polyomavirus middle T antigen-mediated transformation. J Virol. 2007;81:7069–76. doi: 10.1128/JVI.00115-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Webster M, Hutchinson J, Rauh M, Muthuswamy S, Anton M, Tortorice C, et al. Requirement for both Shc and phosphatidylinositol 3′ kinase signaling pathways in polyomavirus middle T-mediated mammary tumorigenesis. Mol Cell Biol. 1998;18:2344–59. doi: 10.1128/mcb.18.4.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Campbell KS, Ogris E, Burke B, Su W, Auger KR, Druker BJ, et al. Polyoma middle T antigen interacts with SHC via the NPTY motif in middle T. Proc Natl Acad Sci USA. 1994;91:6344–8. doi: 10.1073/pnas.91.14.6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dilworth SM, Brewster CE, Jones MD, Lanfrancone L, Pelicci G, Pelicci PG. Transformation by polyoma virus middle T-antigen involves the binding and tyrosine phosphorylation of Shc. Nature. 1994;367:87–90. doi: 10.1038/367087a0. [DOI] [PubMed] [Google Scholar]
- 103.Nicholson PR, Empereur S, Glover HR, Dilworth SM. ShcA tyrosine phosphorylation sites can replace ShcA binding in signalling by middle T-antigen. Embo J. 2001;20:6337–46. doi: 10.1093/emboj/20.22.6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kaplan DR, Whitman M, Schaffhausen B, Raptis L, Garcea RL, Pallas D, et al. Phosphatidylinositol metabolism and polyoma-mediated transformation. Proc Natl Acad Sci USA. 1986;83:3624–8. doi: 10.1073/pnas.83.11.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dahl J, Jurczak A, Cheng L, Baker D, Benjamin T. Evidence of a role for phosphatidylinositol 3-kinase activation in the blocking of apoptosis by polyomavirus middle T antigen. J Virol. 1998;72:3221–6. doi: 10.1128/jvi.72.4.3221-3226.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Maroulakou IG, Oemler W, Naber SP, Tsichlis PN. Akt1 ablation inhibits, whereas Akt2 ablation accelerates, the development of mammary adenocarcinomas in mouse mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer Res. 2007;67:167–77. doi: 10.1158/0008-5472.CAN-06-3782. [DOI] [PubMed] [Google Scholar]
- 107.Urich M, Senften M, Shaw PE, Ballmer-Hofer K. A role for the small GTPase Rac in polyomavirus middle-T antigen-mediated activation of the serum response element and in cell transformation. Oncogene. 1997;14:1235–41. doi: 10.1038/sj.onc.1200982. [DOI] [PubMed] [Google Scholar]
- 108.Su W, Liu W, Schaffhausen B, Roberts T. Association of polyomavirus middle tumor antigen with phospholipase C-gamma 1. J Biol Chem. 1995;270:12331–5. doi: 10.1074/jbc.270.21.12331. [DOI] [PubMed] [Google Scholar]
- 109.Shepard CR, Kassis J, Whaley DL, Kim HG, Wells A. PLC gamma contributes to metastasis of in situ-occurring mammary and prostate tumors. Oncogene. 2007;26:3020–6. doi: 10.1038/sj.onc.1210115. [DOI] [PubMed] [Google Scholar]
- 110.Lin EY, Jones JG, Li P, Zhu L, Whitney KD, Muller WJ, et al. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163:2113–26. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Maglione JE, Moghanaki D, Young LJ, Manner CK, Ellies LG, Joseph SO, et al. Transgenic Polyoma middle-T mice model premalignant mammary disease. Cancer Res. 2001;61:8298–305. [PubMed] [Google Scholar]
- 112.Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, et al. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13:58–68. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 114.Podsypanina K, Du YC, Jechlinger M, Beverly LJ, Hambardzumyan D, Varmus H. Seeding and propagation of untransformed mouse mammary cells in the lung. Science. 2008;321:1841–4. doi: 10.1126/science.1161621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jessen KA, Liu SY, Tepper CG, Karrim J, McGoldrick ET, Rosner A, et al. Molecular analysis of metastasis in a polyomavirus middle T mouse model: the role of osteopontin. Breast Cancer Res. 2004;6:R157–69. doi: 10.1186/bcr768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Whalen KA, Weber GF, Benjamin TL, Schaffhausen BS. Polyomavirus middle T antigen induces the transcription of osteopontin, a gene important for the migration of transformed cells. J Virol. 2008;82:4946–54. doi: 10.1128/JVI.02650-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pedersen TX, Pennington CJ, Almholt K, Christensen IJ, Nielsen BS, Edwards DR, et al. Extracellular protease mRNAs are predominantly expressed in the stromal areas of microdissected mouse breast carcinomas. Carcinogenesis. 2005;26:1233–40. doi: 10.1093/carcin/bgi065. [DOI] [PubMed] [Google Scholar]
- 118.Galang CK, Muller WJ, Foos G, Oshima RG, Hauser CA. Changes in the expression of many Ets family transcription factors and of potential target genes in normal mammary tissue and tumors. J Biol Chem. 2004;279:11281–92. doi: 10.1074/jbc.M311887200. [DOI] [PubMed] [Google Scholar]
- 119.Man AK, Young LJ, Tynan JA, Lesperance J, Egeblad M, Werb Z, et al. Ets2-dependent stromal regulation of mouse mammary tumors. Mol Cell Biol. 2003;23:8614–25. doi: 10.1128/MCB.23.23.8614-8625.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–40. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004;64:7022–9. doi: 10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- 122.Winter SF, Hunter KW. Mouse Modifier Genes in Mammary Tumorigenesis and Metastasis. J Mammary Gland Biol Neoplasia. 2008 doi: 10.1007/s10911-008-9089-1. [DOI] [PubMed] [Google Scholar]
- 123.Lifsted T, Le Voyer T, Williams M, Muller W, Klein-Szanto A, Buetow KH, et al. Identification of inbred mouse strains harboring genetic modifiers of mammary tumor age of onset and metastatic progression. Int J Cancer. 1998;77:640–4. doi: 10.1002/(sici)1097-0215(19980812)77:4<640::aid-ijc26>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]