

Abstract
The capsid domain of the human immunodeficiency virus type 1 (HIV-1) Gag polyprotein is a critical determinant of virus assembly, and is therefore a potential target for developing drugs for AIDS therapy. Recently, a 12-mer α-helical peptide (CAI) was reported to disrupt immature- and mature-like capsid particle assembly in vitro; however, it failed to inhibit HIV-1 in cell culture due to its inability to penetrate cells. The same group reported the X-ray crystal structure of CAI in complex with the C-terminal domain of capsid (C-CA) at a resolution of 1.7 Å. Using this structural information, we have utilized a structure-based rational design approach to stabilize the α-helical structure of CAI and convert it to a cell-penetrating peptide (CPP). The modified peptide (NYAD-1) showed enhanced α-helicity. Experiments with laser scanning confocal microscopy indicated that NYAD-1 penetrated cells and colocalized with the Gag polyprotein during its trafficking to the plasma membrane where virus assembly takes place. NYAD-1 disrupted the assembly of both immature- and mature-like virus particles in cell-free and cell-based in vitro systems. NMR chemical shift perturbation analysis mapped the binding site of NYAD-1 to residues 169-191 of the C-terminal domain of HIV-1 capsid encompassing the hydrophobic cavity and the critical dimerization domain with an improved binding affinity over CAI. Furthermore, experimental data indicate that NYAD-1 most likely targets capsid at a post-entry stage. Most significantly, NYAD-1 inhibited a large panel of HIV-1 isolates in cell culture at low micromolar potency. Our study demonstrates how a structure-based rational design strategy can be used to convert a cell-impermeable peptide to a cell-permeable peptide that displays activity in cell-based assays without compromising its mechanism of action. This proof-of-concept cell-penetrating peptide may aid validation of capsid as an anti-HIV-1 drug target and may help in designing peptidomimetics and small molecule drugs targeted to this protein.
Keywords: cell-penetrating peptide, HIV-1 capsid, viral assembly, electron microscopy, nuclear magnetic resonance
Introduction
Assembly, a critical step in the human immunodeficiency virus type 1 (HIV-1) life-cycle,1-4 is generally thought to occur through the controlled polymerization of the Gag polyprotein, which is transported to the plasma membrane, where assembly takes place.5,6 Virus particles are then formed and bud out as spherical, immature non-infectious particles. Immediately after budding, the particles undergo a process known as maturation. During this step, the Gag polyprotein precursor is cleaved sequentially by the viral protease to matrix (MA), capsid (CA), nucleocapsid (NC), and p6 domains, as well as two spacer proteins, SP1 and SP2. This process triggers a dramatic change in the morphology of the particles, and an electron-dense core is formed. Formation of the mature core has a critical role in viral infectivity. Mutations in CA have been shown to lead to defects in viral assembly and core condensation,7-11 and CA has been proposed to serve as a determinant of lentivirus infectivity in non-dividing cells.12 It is evident that CA has an important role in HIV-1 assembly and virion maturation, and is therefore a potential target for developing a new generation of drugs for AIDS therapy. Identification of peptides and small molecular compounds that disrupt HIV-1 assembly has been reported.13-18 However, the mechanism of inhibition, especially for peptide inhibitors, has not been clearly demonstrated. The first breakthrough in identifying small-molecule inhibitors (CAP-1 and CAP-2) of CA was reported by Summers's group.16 Only CAP-1 showed dose-dependent inhibition of HIV-1 in viral infectivity assays, while CAP-2 was cytotoxic. Although the affinity of CAP-1 for the N-terminal domain of CA (N-CA) was low (Kd ~800 μM), it serves as a lead compound for more potent inhibitors against this target. The small-molecule inhibitor PA-457 that targets Gag processing, has been reported recently.18,19 Both these inhibitors interfere only with the maturation step of HIV-1. Recently, the 12-mer peptide CAI (Fig. 1a) was identified by phage-display techniques and shown to inhibit HIV-1 assembly in vitro by targeting the C-terminal domain of capsid, C-CA.20 CAI was the first peptide reported to disrupt the assembly of both immature-and mature-like particles in vitro. However, CAI could not inhibit HIV-1 in cell culture due to its lack of cell permeability and hence is not suitable as an antiviral drug.
Fig. 1.

Sequences, synthesis and primary and tertiary structures of NYAD-1. (a) The sequence of CAI; (b) synthesis steps for “hydrocarbon stapling” of NYAD-1; (c) CD spectra of CAI and NYAD-1 to measure secondary structures. CD spectra were measured at 20 °C in Tris-HCl buffer (20 mM Tris, pH 8.0) in the presence of 1%-15% (v/v) acetonitrile at a final concentration of peptide of 125-500 μM. NYAD-1 showed typical wavelength minima at 208 nm and 222 nm, whereas CAI showed a minimum at 205 nm; and (d) a three-dimensional model of NYAD-1 bound to C-CA was built manually on the basis of the CAI X-ray crystal structure bound to C-CA. The tertiary structure shows that the hydrocarbon-stapled area in NYAD-1 (purple) is located in a non-interfering site distant from the hydrophobic pocket. The binding site residues of NYAD-1 are indicated in yellow.
A high-resolution X-ray structure revealed details of the CAI structure in complex with C-CA.21 The peptide adopts an α-helical conformation and binds to a hydrophobic pocket formed by helices 1, 2 and 4 of C-CA. We utilized a structure-based rational design approach to convert CAI to a helically stable, cell-penetrating peptide (CPP). We reasoned that if we could modify the linear CAI peptide to a cell-penetrating peptide while preserving the critical residues of CAI that bind to the residues in the hydrophobic pocket of C-CA, then the antiviral potency of the modified CAI peptide in cell culture could be improved.
Major efforts have been underway for many years to overcome the problem of cell permeability and enhance the stability of peptide/protein-based drugs. Many techniques have been reported that enhance helix formation and metabolic stability of peptides.22-25 In most cases, the modifications improved the binding affinity in vitro. However, inhibitory potency in vivo or in cell-based assays is seldom reported, indicating that these modifications may not render the peptides permeable to cells. Recently, Schaffmeister et al.26 reported a method to stabilize the helical structures of peptides. This technique, termed hydrocarbon stapling, has been applied successfully by Walensky et al. to design stabilized helical peptides of BCL-2 (SAHBs) that mimic the BH3 domain in triggering apoptosis in tumor cells in vivo.27
We report here the successful application of hydrocarbon stapling in rationally designing a cell-penetrating peptide NYAD-1 (Fig. 1b) that binds to the critical dimerization domain of C-CA. NYAD-1 was also shown to colocalize with Gag during Gag trafficking to the plasma membrane, and to disrupt the formation of both immature- and mature-like virus particles in cell-free and cell-based in vitro assembly systems. In addition, NYAD-1 showed potent anti-HIV-1 activity in cell culture against a large panel of laboratory-adapted and primary HIV-1 isolates. NMR-based chemical shift perturbation assays mapped the binding site of NYAD-1 to a hydrophobic binding pocket identified previously in X-ray studies of C-CA complexed with CAI.21 NYAD-1 holds promise as a lead compound in the development of CA-targeted anti-HIV-1 drugs.
Results
Hydrocarbon stapling enhanced α-helicity of NYAD-1
We used circular dichroism (CD) to characterize the secondary structure of NYAD-1 and CAI in the uncomplexed state in solution. The CD spectrum of CAI did not show typical helix minima at 222 nm and 208 nm; instead, a strong minimum at 205 nm was observed, indicative of random-coil structure in solution. This supports a binding-induced conformational change of the CAI peptide in complex with C-CA. In contrast, the CD spectrum of NYAD-1 showed distinct minima at both 222 nm and 208 nm. The α-helicity of NYAD-1, calculated from the molar elipticity value at 222 nm, is ~80% (Fig. 1c). The results confirm our hypothesis that hydrocarbon stapling enhances the α-helicity of CAI.
NMR mapping of the binding site of NYAD-1
Chemical shift difference mapping was used to characterize the binding site for NYAD-1 on C-CA (W184A/M185A). The measurement of chemical shifts during the titration of NYAD-1 with C-CA revealed large changes in the amide hydrogen and nitrogen chemical shifts that have been mapped onto the structure of C-CA (Fig. 2a and c). Assignments in free protein and complexes were obtained as described in Materials and Methods. The most significant changes map to residues 169-190, which include helix-1 (161-174) and helix-2 (180-192) (Fig. 2b and c). These results are in complete agreement with the X-ray structure of CAI bound to wild-type protein and the NMR mapping studies of CAI bound to C-CA (W184A/M185A).20 The strong similarities in the chemical shift difference profiles of NYAD-1 and CAI bound to C-CA argue in favor of very similar binding modes.
Fig. 2.

Binding-induced chemical shift changes in the 1H-15N HSQC spectra of mutant C-CA (W184A/M185A): (a) Selected region of the overlay of C-CA spectrum (blue) complexed with NYAD-1 at a ratio of 1:15 (red). The final concentration of protein was 75 μM. (b) Plot of the weighted chemical shift difference as a function of residue number. The difference was calculated using the relation .(c) Residues with ΔN 0.6 ppm (red) and 0.4 ppmb Δb 0.6 ppm (blue) are mapped on the X-ray structure (PDB code 1A8O).
CAI had been shown to form an amphipathic helix that makes important hydrophobic (helix 1) and N-terminal capping (helix 2) interactions within the binding pocket of C-CA.21 The C-terminal end of the CAI peptide is completely exposed to the solvent. Presumably, NYAD-1 binds in a similar fashion, since our design strategy did not alter the residues of CAI that are crucial for binding C-CA. The role of the bulky olefinic link was of some concern but appeared not to perturb the interactions at the binding site. As was the goal of the original design, the linker is on the solvent-exposed surface of the bound peptide (Fig. 1d).
The low solubility of NYAD-1 interfered with a reliable estimate of Kd by NMR. However, the slow exchange kinetics of binding monitored by NMR supports an upper bound on the Kd of ~10 μM. The binding of a highly soluble NYAD-1 analog, NYAD-13, is identical to that of NYAD-1 in all respects (Supplementary Data Fig. S1), and the binding of NYAD-13 to C-CA is slow on the NMR timescale (Fig. 3). The calculation from NMR data yields Kd ~1 μM. On the basis of the NMR chemical shift mapping studies, we conclude that hydrocarbon stapling of CAI does not alter the principal interactions in the binding site of C-CA, and the affinity is in the low micromolar range.
Fig. 3.
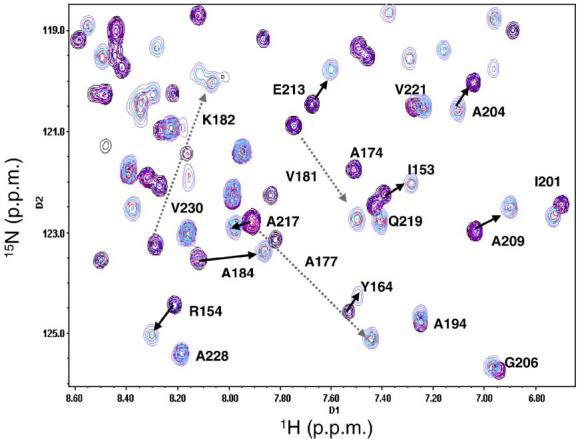
Overlay of 15N-HSQC spectra acquired at different ratios (0.19, 0.21, 0.75, 1.69, and 2.93) of NYAD-13 and C-CA during the NMR titration. The colors of the contour lines range from magenta for the free protein to blue for the peptide-bound fraction of the protein. The slow exchange results in the observation of both peaks at each point of the titration accompanied by a change in the relative intensities.
NYAD-1 penetrates cells efficiently
Although cell-penetrating peptides mostly contain positively charged residues, cells are generally less permeable to peptides containing charged residues. Increasing lipophilicity may enhance cell penetration. On the basis of this rationale and the reported improved cell penetration by stapled peptides,27 we modified CAI to NYAD-1. To show that NYAD-1 penetrates cells, we utilized a fluorescence-activated cell sorter (FACS) analysis using two different cell types, 293T and MT-2 cells. The data indicate that about 40% and 96% of 293T cells were stained positive for FITC-conjugated CAI and for FITC-conjugated NYAD-1, respectively. In contrast, none of the MT-2 cells was stained positive for FITC-conjugated CAI, whereas about 92% of MT-2 cells were stained positive for FITC-conjugated NYAD-1 (Fig. S2). It has been reported that FACS analysis may be unreliable in demonstrating the cellular uptake of peptides, since they bind to the cell membrane with fairly high affinity and cannot be dissociated with repeated washes.28 Therefore, we used confocal microscopy to demonstrate that NYAD-1 and NYAD-13 but not CAI, penetrate cells (Fig. 4).
Fig. 4.
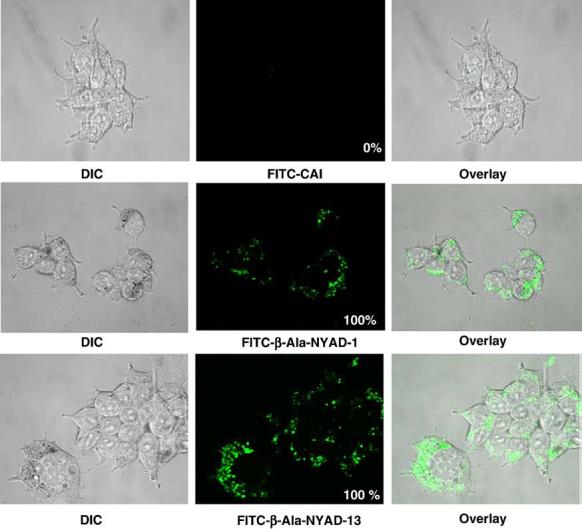
Cell penetration of NYAD-1 and NYAD-13 in 293T cells. Representative confocal microscopy images of 293T cells incubated for 20 h at 37 °C with FITC-conjugated peptides. Upper panel: Left, differential interference contrast (DIC) image of cells with FITC-CAI; center, FITC fluorescent image of the same cells with FITC-CAI; and right, overlay of DIC and FITC fluorescent images. Middle panel: Left, DIC image of cells with FITC-β-Ala-NYAD-1; center, FITC fluorescent image of the same cells with FITC-β-Ala-NYAD-1; and right, overlay of DIC and FITC fluorescent images. Lower panel: Left, DIC image of cells with FITC-β-Ala-NYAD-13; center, FITC fluorescent image of the same cells with FITC-β-Ala-NYAD-13; and right, overlay of DIC and FITC fluorescent images. A total of 200 cells were scored in each treatment with FITC-CAI, FITC-β-Ala-NYAD-1 or FITC-β-Ala-NYAD-13. The percentage of cells in the population that exhibited the internal staining is shown at the bottom right of each panel (P < 0.001 for FITC-CAI versus FITC-β-Ala-NYAD-1 or FITC-β-Ala-NYAD-13).
NYAD-1 colocalizes with HIV-1 Gag
The fact that NYAD-1 penetrates cells does not guarantee that it will colocalize and interact with the Gag polyproteins to inhibit viral assembly. To address this question, we performed a direct colocalization experiment using an HIV-1 Gag-mStrawberry fusion protein and FITC-conjugated NYAD-1. When Gag-mStrawberry-expressing cells were exposed to FITC-conjugated NYAD-1, a significant fraction colocalized inside cells (Fig. 5, data shown at two different angles). The colocalization data firmly establish the cell permeability of NYAD-1 and suggest interactions with the Gag polyproteins before they are transported to the plasma membrane.
Fig. 5.

Direct colocalization study of NYAD-1 and Gag by confocal microscopy. Representative images at two different angles are shown. (a and d) FITC-conjugated NYAD-1 (green). (b and e) Gag-mStrawberry (red). (c and f) Merged views demonstrate colocalization of FITC-NYAD-1 with Gag-mStrawberry. All images were obtained from living cells 24 h post transfection. Regions of colocalization are highlighted. A DIC image is shown in the upper left corner. Data were collected from 30 cells and colocalization was assessed utilizing Zeiss imaging software (weighted colocalization coefficient was 0.402 ± 0.051).
NYAD-1 disrupts the formation of both immature- and mature-like particles
Cell-free system
In order to verify whether NYAD-1 retains the ability to inhibit both immature and mature virus assembly we set up two in vitro assembly systems. We used full-length Gag proteins to form spherical immature-like particles (Fig. 6a(a)). The effect of inhibitors on immature particle assembly was studied by performing assembly reactions in the presence of varied doses of NYAD-1. A clear dose-dependent effect was observed (Fig. 6b). After incubation with 0.25-fold and 0.5-fold molar equivalent of NYAD-1, particle formation was reduced substantially (Fig. 6a (b) and (c)), whereas with a molar equivalent of NYAD-1, the particle assembly was disrupted almost completely (Fig. 6a(d)). We observed complete disruption with fourfold (data not shown) and fivefold molar equivalents of NYAD-1 (Fig. 6a(e)). For the mature-like particles, we expressed and purified CA protein and obtained tube-shaped particles (Fig. 6a (g)) similar to those reported in the literature.29,30 A similar dose-response effect was also observed (Fig. 6h-k). After incubation with a molar equivalent and fivefold molar equivalents of NYAD-1, the assembly of tube-shaped particles was blocked completely (Fig. 6a(j) and (k)). A control hydrocarbon-stapled peptide, NYAD-17, with the same amino acid sequence of NYAD-1 but scrambled (Supplementary Data, sequence in Table S1), showed no effect on the formation of virus-like particles (data not shown). A fivefold molar equivalent of CAI was used as positive control (Fig. 6a(f) and (l)). The rationale for using CA instead of CANC to form the mature-like particles was to confirm that NYAD-1 targets CA only.
Fig. 6.

Inhibition of in vitro assembly by NYAD-1. (a) Negatively stained EM images of immature- and mature-like particles resulting from in vitro assembly of Gag (25 μM) and CA proteins (50 μM), respectively, in the presence of no peptide (a and g, control), 0.25-fold (b and h), 0.5-fold (c and i), a molar equivalent (d and j), and fivefold molar equivalent of NYAD-1 (e and k) and CAI (f and l). A dose-response effect was observed with NYAD-1. The integrity of the mature-like particles is shown in the insets. Gag in vitro assembly was conducted by dialyzing against 50 mM Na2HPO4, pH 8.0 containing 0.1 M NaCl in the presence of 5% total E. coli RNA (RNA/protein = 1:20, w/w). The CA assembly reaction was initiated at a final concentration of 1.2 M NaCl. (b) Dosage-dependent inhibition of Gag assembly; 20 fields under EM were screened and the number of VLPs was plotted against the ratio of concentration of NYAD-1 to Gag proteins in the assembly reaction.
Cell-based system
To determine whether NYAD-1 disrupts virus assembly in cell culture, we transfected 293T cells with the full-length HIV-1 molecular clone pNL4-3 and treated transfected cells with NYAD-1 for 16-20 h. Cells were then metabolically labeled with [35S]Met/Cys; cell- and virus-associated proteins were immunoprecipitated with HIV-Ig. The results indicated that HIV-1 release efficiency was reduced in a concentration-dependent manner (Fig. 7a and b). Interestingly, we also observed a dose-dependent accumulation of Pr55Gag in cells treated with NYAD-1 (Fig. 7a and c). These results demonstrate that NYAD-1 binding to CA inhibited both the assembly and processing of HIV-1 Gag. To examine whether the inhibition of virus release mediated by NYAD-1 is specific to HIV-1, we analyzed the release of another lentivirus, equine infectious anemia virus (EIAV) in 293T cells. The release of EIAV particles was not inhibited by NYAD-1 (Fig. 7d and e), indicating that the effect of NYAD-1 on HIV-1 particle production was not the result of non-specific effects such as cytotoxicity.
Fig. 7.

NYAD-1 inhibits HIV-1 but not EIAV particle production. 293T cells transfected with either pNL4-3 (a-c) or pPRE-Gag (d and e) were treated with the indicated concentration of NYAD-1. Peptide-treated cells were metabolically labeled with [35S]Met/Cys for 2 h (HIV-1) or 7-8 h (EIAV). Cells were lysed and virions were pelleted by ultracentrifugation. Cell and virus lysates were immunoprecipitated with HIV-Ig or anti-EIAV antiserum and subjected to SDS-PAGE (a and d); data were quantified by phosphorimager analysis. HIV-1 release efficiency was calculated as the amount of virion-associated p24 relative to total (cell + virion) Gag (b). Accumulation of HIV-1 Pr55Gag in cells was measured by calculating the ratio of Pr55Gag to total Gag in cells (Pr55Gag + p24) (c). EIAV release efficiency was calculated as the amount of virion-associated Pr55Gag to total Pr55Gag in cells and virions (e). For HIV-1 and EIAVassays, n = 4 and n = 3, respectively, with means + SD. The percentages were normalized to untreated levels.
Electron microscopic (EM) analysis of the untreated 293T cells transfected with a Gag expression vector showed distinct immature-like particles (Fig. 8a(a)). However, when the cells were treated with 6.25 μM or 50 μM NYAD-1, a majority of the particles displayed an aberrant shape (Fig. 8a(b) and (c)) similar to that reported by Mariani et al. upon expression of HIV-1 Gag in murine cells.31 A large number of mature particles containing electron-dense core structures were found in untreated cells transfected with a Gag-Pol expression vector (Fig. 8a(d)). When cells expressing Gag-Pol were treated with 6.25 μM or 50 μM NYAD-1, formation of electron-dense core structures was markedly inhibited (Fig. 8a(e) and (f), and b). Taken together, these data confirm that NYAD-1 targets Gag and impairs proper particle assembly and maturation in Gag-expressing cells.
Fig. 8.
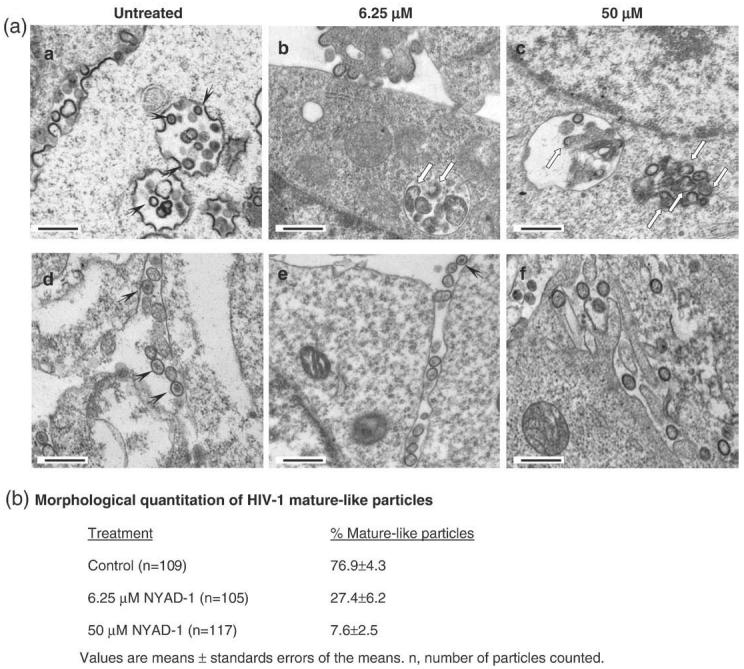
Electron microscopic analysis of HIV-1 virus-like particles produced in the presence of 6.25 μM and 50 μM NYAD-1. (a) 293T cells expressing Gag (upper panel) or Gag-Pol(lower panel) were incubated with 2 ml of culture medium containing no peptide or 6.25 μMor50 μM NYAD-1 4 h post transfection with vectors encoding Gag or Gag-Pol. At 24 h post transfection, cells were processed and examined under the electron microscope. The scale bar represents 500 nm; (b) the morphological quantification of mature-like particles.
NYAD-1 inhibits single-cycle HIV-1 infection post entry
We wanted to determine whether NYAD-1 could inhibit single-cycle HIV-1 infectivity. AZT was used as a positive control in the infectivity assays. We used an envelope-defective luciferase reporter virus complemented with an X4 envelope. We observed that NYAD-1 inhibited infectivity when virus was incubated with peptides for 30 min or 4 h (Fig. 9a and b). Since the data presented above demonstrate that NYAD-1 is a cell-penetrating peptide and it targets CA in both cell-free and cell-based assays, we hypothesize that it binds to CA at the pre-entry and/or post entry stage. In order to determine further whether NYAD-1 inhibits HIV-1 by disrupting entry or post-entry events, we performed time-of-addition experiments in which compounds were added at various lengths of time after infection. NYAD-1 and AZT both showed inhibition when added after 8 h, but activity dropped when added after 16 h, whereas the entry inhibitor T-20 lost most of its activity when added after 4 h (Fig. 9c).
Fig. 9.
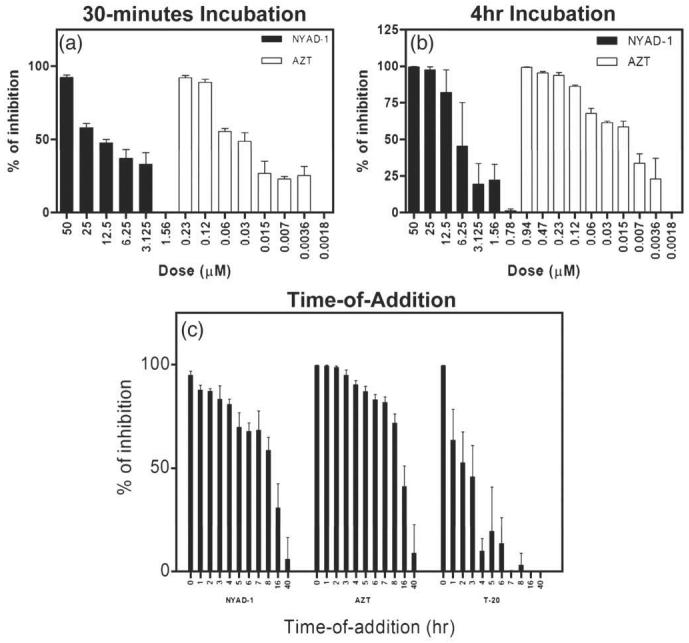
Inhibitory effects of NYAD-1 in a single-cycle viral infection assay. (a and b) Dose-dependent HIV-1 inhibitory effects of NYAD-1 and reference compound, AZT. Env-pseudotyped virus particles were incubated in the presence of serially diluted NYAD-1 or AZT for 30 min or 4 h and then U-87-T4-CXCR4 cells were infected with the treated virus. Virus infectivity was determined by luciferase assay three days post-infection. (c) Time-of-addition experiments using the above-described single-cycle viral infection assay. U87-T4-CXCR4 cells were infected in the presence of 50 μM NYAD-1, 3.75 μM AZT or 222 nM T-20. The compounds were applied at different times after infection. The concentrations of compound correspond to the final concentrations of compounds when the cells were exposed to pseudoviruses.
NYAD-1 does not prevent entry of HIV-1 into cells
We have used GFP-Vpr-labeled HIV-1 virions to assess the ability of NYAD-1 to prevent entry of the virus into human Sup-T1 cells using various concentrations. The results indicate (Fig. 10) that, even at a concentration of 100 μM, NYAD-1 failed to prevent the entry of GFP-Vpr-labeled virus into cells, whereas a 222 nM T-20 prevented the entry of the virus almost completely.
Fig. 10.

Flow cytometric analysis of human SupT1 cells incubated with GFP-Vpr-labeded HIV-1 virions in the presence of reference compound, T-20 or NYAD-1. SupT1 cells were incubated with GFP-Vpr-labeded HIV-1 virions (300 ng of p24 CA) together with various concentrations of T-20 or NYAD-1 at 37 °C for 3 h. The cells were then washed once with PBS, fixed with 1% (w/v) paraformaldehyde, and washed again before FACS analysis.
NYAD-1 shows broad-spectrum anti-HIV-1 activity
NYAD-1 showed inhibition of both immature- and mature-like particle assemblies in cell-free as well as cell-based assembly systems. We next wished to measure its anti-HIV-1 activity in a cell-based assay using several laboratory-adapted and primary isolates in MT-2 cells and peripheral blood mononuclear cells (PBMC), respectively. The inhibition of p24 production in MT-2 cells by NYAD-1 was measured over a range of concentrations and the concentration required to inhibit 50% (IC50) and 90% (IC90) of the p24 production was calculated. The results given in Table 1 indicate that NYAD-1 inhibited a broad range of HIV-1 strains, representing different subtypes, which use R5, X4 or R5X4 coreceptors. NYAD-1 inhibited the laboratory strains with low micromolar potency (IC50 ~4-15 μM), and both R5- and X4-tropic viruses were inhibited with similar potency. We also tested one X4-tropic RT-resistant (AZT-R) strain in MT-2 and one dual-tropic (R5X4) RT-resistant (AR17) strain in PBMC, and NYAD-1 inhibited the dual tropic-resistant virus with slightly higher potency. We have also tested the linear peptide and a control hydrocarbon-stapled peptide, NYAD-17. None of these peptides showed any antiviral activity when tested up to 200 dose. In addition, we have tested several modified analogs of NYAD-1 against the laboratory strain HIV-1 IIIB (Supplementary Data, Table S1). The soluble analog, NYAD-13, showed antiviral activity similar to NYAD-1 against HIV-1 IIIB; however, it showed pronounced cytotoxicity. When we selected different i and i+4 residues as targets for possible hydrocarbon stapling, the resultant peptides NYAD-14 and NYAD-16 did not show any antiviral activity. When we mutated several residues of NYAD-1 and generated NYAD-15, it showed improved antiviral activity; however, it was more cytotoxic than the parent NYAD-1. Nevertheless, this structure-activity study indicates that NYAD-1 can be optimized to inhibitors that are more efficacious.
Table 1.
Antiviral activity of the constrained peptide, NYAD-1, in laboratory-adapted and primary HIV-1 isolates
| HIV-1 Virus | Subtype | Cell Type | Coreceptor use | IC50 (μM)±SD* | IC90 (μM)±SD* |
|---|---|---|---|---|---|
| Laboratory-Adapted | |||||
| IIIB | B | MT-2 | X4 | 6.22±0.75 | 23.83±1.74 |
| MN | B | MT-2 | X4 | 6.79±0.65 | 13.12±2.46 |
| RF | B | MT-2 | X4 | 4.29±0.42 | 9.86±2.08 |
| V32 | B | MT-2 | X4 | 7.91±0.70 | 23.12±1.94 |
| BaL | B | PBMC | R5 | 6.47±0.85 | 15.46±3.71 |
| SF162 | B | PBMC | R5 | 15.44±3.23 | 60.64±8.13 |
| RT-Resistant (AZT) | |||||
| AZT-R | B | MT-2 | X4 | 16.28±2.79 | 61.50±2.12 |
| A17 | B | PBMC | R5X4 | 10.55±1.56 | 38.46±0.62 |
| Primary Isolates | |||||
| 92RW008 | A | PBMC | R5 | 12.12±1.64 | 43.90±5.53 |
| 92UG029 | A | PBMC | X4 | 13.85±1.34 | 36.51±19.39 |
| 92US657 | B | PBMC | R5 | 10.54±2.78 | 64.77±6.31 |
| 93IN101 | C | PBMC | R5 | 16.48±0.47 | 63.30±6.17 |
| 93MW959 | C | PBMC | R5 | 16.49±2.83 | 54.23±4.41 |
| 92UG001 | D | PBMC | R5X4 | 9.14±0.27 | 37.30±3.67 |
| CMU02 | EA | PBMC | X4 | 10.03±0.81 | 14.92±1.34 |
| 93TH051 | E | PBMC | R5X4 | 20.50±1.90 | 40.88±13.20 |
| 93BR020 | F | PBMC | R5X4 | 6.60±1.60 | 17.27±2.72 |
| RU570 | G | PBMC | R5 | 9.79±2.49 | 39.97±9.65 |
| BCF02 | (Group O)** | PBMC | R5 | 21.60±3.04 | 59.14±11.58 |
The linear peptide CAI and a scrambled peptide NYAD-17 did not show any activity up to 200 μM dose level. The CC50 value in MT-2 cells was > 135 μM; and in PBMC cells were > 300 μM. SD, standard deviation.
All other HIV-1 strains belong to Group M.
We also tested the inhibition of NYAD-1 against a panel of primary HIV-1 isolates in PBMC representing mostly group M (subtypes from A to G) with diverse coreceptor usage. NYAD-1 showed inhibition against all primary isolates tested including one from group O (Table 1). However, the IC50 values against this virus (BCF02) as well as one from clade E (93TH051) were slightly higher. The inhibitory activities against this diverse range of primary isolates were similar, indicating its effectiveness against a wide range of HIV-1 isolates.
The cytotoxicity of the peptides was assessed by the XTT method in either MT-2 cells or in both MT-2 cells and PBMC. Cytotoxicity assays were performed in parallel with the HIV-1 inhibition assays. The concentrations of inhibitor required to produce 50% cytotoxicity (CC50) of NYAD-1 in MT-2 cells and PBMC were >135 μM and >300 μM, respectively.
Discussion
In this study, we have demonstrated the application of structure-based design in converting a cell-impermeable peptide, CAI, to a cell-permeable peptide, NYAD-1, by hydrocarbon stapling. The biophysical data confirm the α-helical structure of NYAD-1 in solution, whereas CAI exists as a random coil, typical of small linear peptides <20 amino acid residues in length. The X-ray structure of CAI-C-CA indicates that the α-helical structure of CAI is induced upon binding to C-CA, and this structure is essential for its binding to residues in the hydrophobic pocket created by helices 1, 2 and 4 of C-CA (PDB code 2Buo). The enhanced, stable α-helical structure of NYAD peptides in solution may have contributed to the marked improvement of the dissociation constant Kd of the NYAD class (<1 μM) over CAI (~15 μM). A similar improvement in cell penetration has been reported for a BID BH3 peptide, an antagonist of BCL-2 activity. Before stapling, the measured α-helicity was low (16%) and Kd was 269 nM. Hydrocarbon stapling induced structure in the BID BH3 peptide (87% α-helical) and enhanced the affinity to 38.8 nM.27 The possibility that the olefinic linker could alter the binding mode of the NYAD-1 peptide compared to that of CAI complexed with C-CA was addressed by the NMR mapping studies. The chemical shift perturbation studies localized the putative binding site of NYAD-1 to residues 169-191 of C-CA, which superimposes with the site identified in the CAI complex structure.21 A detailed structural characterization is in progress and is expected to shed further light on the nature of the modified peptide-protein interactions. The binding site of NYAD-1 encompasses a part of the dimerization interface of CA spanning residues 169-189.32,33 Mutation studies have demonstrated that these sites are critical for HIV-1 assembly and infectivity. For example, the Y169C mutation drastically reduced viral infectivity but did not produce grossly defective viruses.34 Similarly, mutations introduced near the dimer interface also showed deleterious effects on virus assembly. For example, mutation of W184, M185, L189 and L190 induced defects in virus assembly and release.10,11,32 A recent study showed that a mutant created by deleting residues 178-191, which spans almost the entire α-helix 2 at the dimer interface,32 had detrimental effects on both virus assembly and Gag-Pol incorporation.8 We demonstrated in this study that NYAD-1 inhibited Gag-mediated viral assembly efficiently in both cell-free and cell-based assembly systems. The specificity of NYAD-1 for HIV-1 was demonstrated by the lack of an assembly/release defect for the equine lentivirus EIAV. EM results indicate that even at a low concentration (6.25 μM) NYAD-1 affected the formation of spherical and cone-shaped particles in Gag- and GagPol-expressing 293T cells, respectively. Similar EM results were reported with the L189A mutant.11 NYAD-1 also showed detrimental effects on the morphology of mature-like particles obtained from HIV-1 capsid when these particles were incubated overnight at 4 °C (data not shown), indicating possible post-entry effect by binding to capsid. Furthermore, NYAD-1 did not show any activity in cell fusion assays,35 suggesting that this modified peptide does not inhibit entry of the virus into cells (data not shown). Taken together, our NMR and experimental data indicate that NYAD-1 most likely does not block entry; however, it impairs viral assembly by binding directly to critical residues of CA and/or indirectly affecting the conformation of the dimer interface. As a result, it affects the formation of immature- and mature-like particles, and reduces viral infectivity. In addition to these effects on particle assembly and maturation, NYAD-1 may affect the uncoating process by binding to capsid, as indicated by the single-cycle inhibition and time-of-addition assays.
We also demonstrated the antiviral efficacy of NYAD-1 in cell culture in two different cell lines using six laboratory-adapted and 11 clinical HIV-1 isolates. Furthermore, NYAD-1 was tested against two RT-resistant HIV-1 isolates. To rule out the possibility of an inhibitory effect of NYAD-1 originating from hydrocarbon stapling, we have tested a hydrocarbon-stapled peptide, NYAD-17, having the scrambled sequence of NYAD-1. This peptide did not show any antiviral activity up to a concentration of 200 μM. In addition, our initial structure-activity analysis with several NYAD-1 analogs (Supplementary Data, Table S1) indicates that the selection of amino acid positions for hydrocarbon stapling in the linear peptides is critical as we hypothesized that the fourth (i)and eighth (i+4) positions are best suited for the stapling. Selecting other positions may interfere with the required hydrophobic and other critical interactions for binding of these peptides to C-CA and eliciting antiviral activity.
The data indicated that NYAD-1 has broad-spectrum activity against HIV-1, irrespective of the subtype, coreceptor use and drug-resistance status. These data were not surprising to us, since our independent analysis and data reported by others indicate that the binding site residues at the hydrophobic pocket are highly conserved among HIV-1, HIV-2 and SIV.20,21 For example, we have tested NYAD-1 against six laboratory strains and ten primary isolates all belonging to group M. The Gag sequences of four of these laboratory strains and six of the primary isolates are available. Alignment of these sequences revealed that the binding site of CAI and NYAD-1 is highly conserved (70% identity and 93% similarity).
This study provides evidence that NYAD-1 penetrates cells without the help of a carrier protein, disrupts formation of both immature- and mature-like HIV-1 particles and effectively inhibits HIV-1 infection in cell cultures. NYAD-1 should be considered as a proof-of-concept peptide-based cell-penetrating HIV-1 inhibitor, which binds to the C-terminal domain of capsid with relatively high affinity, and shows potential to be optimized as a new class of drugs for the treatment of AIDS. Our preliminary data on the NMR structure of NYAD-1 complexed with the C-CA (unpublished results) indicate that there are opportunities to optimize the fit of hydrophobic side-chains from NYAD-1 within the binding pocket of C-CA. Attempts will also be made to impart additional hydrogen bonding interactions to generate new peptides with improved affinity and antiviral activity. Furthermore, unraveling the mechanism of anti-HIV-1 activity may aid in designing new class of peptidomimetics and small-molecule drugs for AIDS therapy. The cell permeability of this short peptide also provides hope that it may cross the blood-brain barrier, which is a critical obstacle in treating virus infection in the central nervous system, especially in the brain. This possibility may have broader consequences in anti-HIV-1 drug design and development for AIDS therapy, including neuro-AIDS.
Materials and Methods
Reagents
AZT (Roche cat no. 3485), T-20, fusion inhibitor (Roche cat no. 9845), MT-2 cells (Dr D. Richman), Sup-T1 cells (Dr James Hoxie), laboratory-adapted and primary HIV-1 strains, U87-T4-CXCR4 cells were obtained through the NIH AIDS Research and Reference Reagent Program. The 293T cells were obtained from the American Type Culture Collection. PBMC were processed from blood obtained from the New York Blood Center.
Plasmids
pET14b or pET28a plasmids encode Gag-derived proteins from the HIV-1NL4-3 strain. The full-length Gag gene was obtained from the NIH AIDS Research and Reference Reagent Program. The CA coding region was obtained by PCR and inserted into the pET28a vector. The C-CA DNA fragment was provided by Drs Ming Luo and Peter E Prevelige Jr. The C-CA DNA fragment was subcloned into the pET14b vector. The C-CA mutant (W184A/M185A) was generated from pET14b-C-CA by overlapping PCR. pRSET-BmStrawberry was provided by Dr Roger Tsien. Codon-optimized Gag or Gag-Pol gene was synthesized by DNA 2.0 Inc. based on pNL4-3. Plasmids expressing HIV-1 Gag, Gag-Pol and Gag-mStrawberry proteins, namely pEF6A-Gag, pEF6A-Gag-pol and pEF6A-Gag-mStrawberry, respectively, were used to express virus-like particles or to track the Gag proteins in human cell lines by confocal microscopy. DNA pNL4-3.luc.R-E- (Dr Nathaniel Landau) and pHXB2 env were obtained through the NIH AIDS Research and Reference Reagent Program. The full-length, infectious HIV-1 molecular clone pNL4-3,36 and the EIAV Gag expression plasmid pPRE-Gag have been described.37,38
Synthesis of stapled peptides
Asymmetric synthesis of (S)-Fmoc-2-(2′-pentenyl) alanine was conducted with the Ala-Ni(II)-BPB-complex method.39 NYAD-1 was generated by replacing two natural amino acids at the 4 (i)and 8(i+4) positions of the CAI sequence by a non-natural amino acid (S)-Fmoc-2-(2′-pentenyl) alanine and was synthesized by the method described below. The rationale for selecting the fourth and eighth residues of CAI was based on the X-ray crystal structure of CAI complexed with the C-CA of capsid. The structure revealed that residues 4 (E) and 8 (D) are located on the opposite side of the binding pocket of CAI. We hypothesized that stapling residues in these positions would not affect the binding of the modified peptide to the hydrophobic pocket; instead, it might enhance the α-helicity and cell permeability of the peptide. To validate our hypothesis, we synthesized NYAD-14 and NYAD-16 by stapling at eight (i) and 12 (i+4), and at three (i) and seven (i+4) positions, respectively (Supplementary Data Table S1). Stapling at positions 1 and 5 were not tried because residue 1 of CAI was deeply buried in the hydrophobic pocket in the crystal structure. In addition, we synthesized NYAD-15, an analog of NYAD-1, by substituting several non-critical amino acids to initiate a preliminary structure-activity study. We have designed and synthesized NYAD-13, a soluble analog of NYAD-1, for biophysical studies including NMR. These peptides were (NYAD-1 was shown as an example) synthesized (Fig. 1b) manually by Fmoc solid-phase synthesis using Rink amide MBHA resin (0.33 mmol/g). For the normal amino acids, the couplings were performed with a fourfold excess of amino acids. Fmoc-amino acids were activated using a ratio of 1:1:1:2 for Fmoc-amino acid:HBTU:HOBt:DIEA. For (S)-Fmoc-2-(2′-pentenyl) alanine, the coupling was performed with a two-fold excess of amino acids, which was activated with DIC: HOAt (1:1). For peptide olefin metathesis, peptide resin with the N terminus protected by an Fmoc group was treated with degassed 1,2-dichloroethane containing the Grubbs catalyst bis(tricyclohexylphosphine)benzylidineruthenium(IV) dichloride (10 mM) at room temperature for 2 h, and the reaction was repeated once for completion. After de-Fmoc, the resin-bound peptide was cleaved using standard protocols (95% (v/v) TFA, 2.5% (v/v) water, 2.5% (v/v) triisopropylsilane). The cleaved peptide was purified by RP-HPLC using 0.1% (v/v) TFA/water and 0.1% (v/v) TFA/acetonitrile, and its purity and mass were confirmed by mass spectroscopy.
For the fluorescently labeled peptides FITC-β-Ala-NYAD-1 and FITC-β-Ala-NYAD-13, the N-terminal residues of NYAD-1 and NYAD-13, respectively, were further derivatized with β-Ala and FITC (DMF/DIEA) on the resin before cleavage. The other cleavage, purification, and confirmation steps were performed as described above.
The solubility of NYAD-1 and NYAD-13 in DMSO was ~100 mM. NYAD-1 was practically insoluble in water, whereas NYAD-13 was soluble (~10 mM).
CD spectroscopy
CD spectra were obtained on a Jasco J-715 spectropola-rimeter (Jasco Inc, Japan) at 20 °C using the standard measurement parameters in Tris-HCl buffer (20 mM Tris, pH8.0) in the presence of 1-15% (v/v) acetonitrile at a final concentration of 125-500 μM. In all samples, the final concentrations of peptides and salt were always the same, and the spectra were corrected by subtracting the CD spectrum of the appropriate reference solvent. The percentage α-helix was calculated from the molar ellipticity [Θ] value at 222 nm.40
NMR samples
Uniformly 15N-enriched and 15N/13C-enriched protein samples of mutated C-CA (W184A/M185A) were produced by expressing the pET14b plasmid encoding the mutant C-CA gene in Escherichia coli BL21 (DE3) cells cultured in M9 minimal medium containing 15NH4Cl (Cambridge Isotope Laboratories) and [13C6]glucose as sole nitrogen and carbon source, respectively. Recombinant proteins were isolated from bacteria as described,1 and the integrity of the samples was confirmed by mass spectrometry. The NMR samples were prepared in a buffer containing 100 mM ammonium acetate (pH 7.0), 95% H2O/5% 2H2O and 2-10 mM DTT. The protein concentrations were determined from measurement of the UV absorbance at 280 nm using an extinction coefficient of 2980 M-1cm-1.
NMR resonance assignments
Backbone assignments of C-CA in the absence of peptide were made with a 380 μM U-15N,13C-labeled sample. Triple resonance experiments HNCA, HN(CO)CA, HNCACB, CBCA(CO)NH, HNCO and HN(CA)CO41 were acquired at 25 °C on a Bruker AVANCE 700 MHz spectrometer equipped with a Z-axis gradient TXI CryoProbe.
Owing to the poor solubility of NYAD-1, triple resonance experiments of the complex with C-CA were not feasible. However, the 1H-15N-heteronuclear single quantum coherence (HSQC) spectra of C-CA bound to NYAD-1 and NYAD-13 are nearly identical (Supplementary Data Fig. S3) and we decided to pursue the assignments of the latter bound to C-CA and transfer the assignments to the complex with NYAD-1. A sample of [U-15N,13C]C-CA in the presence of unlabeled NYAD-13 peptide was prepared at a molar ratio of 1:1 under identical buffer conditions. The final concentration of protein in this sample was ~1.9 mM. The backbone experiments used for assignment were acquired at 25 °C on a Bruker AVANCE 500 MHz spectrometer equipped with a Z-axis gradient TXI CryoProbe.
All data were processed in Topspin 1.3 and analyzed using CARA1.5.42 The backbone chemical shifts of 79 out of 84 (five Pro) residues were assigned in the free and NYAD-13 complexed states of C-CA.
NMR perturbation assay with NYAD-13
The NMR-based titrations were carried out at 25 °C on a Bruker AVANCE 900 MHz spectrometer equipped with a Z-axis gradient TCI CryoProbe. Aliquots of 5-10 μl of 3.77 mM unlabeled NYAD-13 in identical buffer conditions were added to 500 μl of 256 μM [U-15N]C-CA sample in NH4Ac buffer (pH 7.0), 5% 2H2O, and 10 mM DTT. The final peptide/protein ratio was 1:1.8. A 2D 1H15N-HSQC spectrum was acquired after each addition. The data were processed in Topspin 1.3 and analyzed in NMRViewJ v6.12.43
A second perturbation assay was carried out by adding 6 μl of 100 mM NYAD-1 in 100% DMSO to 500 μl of 76 μM [U-15N]C-CA sample in ammonium acetate buffer (pH 7.0), 95% H2O/5% 2H2O, 10 mM DTT.
Calculation of NYAD-13 binding constant from NMR data
Owing to the poor solubility of NYAD-1, the binding constant could be calculated only for NYAD-13 from the NMR titration data. The binding of the peptide to C-CA was slow on the NMR timescale, and at each peptide to protein ratio two sets of peaks were observed for the bound and free protein, respectively. The fraction of bound protein was calculated from the change in relative intensities of the two peaks through the titration and fitted to a standard equation to yield a Kd value of 1.2 ± 0.6 μM.
A second method used the linewidth at half height of the bound protein to calculate the koff rate.44 A Kd value of 0.2 ± 0.1 μM was calculated from the ratio of the experimentally determined koff and a diffusion limited kon of ~108 M-1 s-1. (Details of these methods are given as Supplementary Data).
FACS analysis
293T and MT-2 cells were maintained in RPMI 1640 (Invitrogen), 10% (v/v) fetal bovine serum, 100 U/ml of penicillin, 100 μg/ml of streptomycin, 2 mM glutamine, 50 mM Hepes (pH 7), 50 mM β-mercaptoethanol. Cells (2 × 104/well) were seeded into a 24-well plate on the day before treatment with FITC-conjugated peptides. After two washes with PBS, cells were incubated with 5 μM FITC-conjugated peptide in serum-free medium for 4 h at 37 °C, and then washed three times with PBS and digested with 0.25% (w/v) trypsin for 30 min at 37 °C. After one more wash with PBS, resuspended cells were subjected to FACS analysis (Becton Dickinson). The GFP-Vpr-labeled HIV-1 entry assay was as described.45
Confocal microscopy
For the cell penetration study, 293T and MT-2 cells were seeded in four-well chamber plates and incubated at 37 °C with FITC-conjugated peptides in serum-free medium for 4 h and/or an additional 16 h in the medium containing serum. After three washes with PBS, live cells were examined and imaged under a Zeiss LSM510 laser scanning confocal microscope (Zeiss).
A direct colocalization study was performed by transfecting 293T cells with pEF6A-Gag-mStrawberry for 4 h and then washing cells once with PBS. A serum-free or serum-containing medium containing FITC-conjugated peptide was added for another 20 h of culture. After three washes, the cells were examined and imaged as described above.
Electron microscopy to study inhibition of in vitro assembly by peptides
Cell-free system
In vitro assembly systems were set up essentially as described,1,10,46,47 but with minor modification. We used 50 mM Na2HPO4, pH 8.0 as the dialysis buffer. The buffer used for assembly studies also contained 0.1~2 M NaCl, and 500 Da cutoff dialysis tubes (Spectra/Por) were used for the dialysis of peptides. Briefly, stock proteins were adjusted to the appropriate concentration (25 μÌ Gag proteins and 50 μÌ CA proteins) with the Na2HPO4 buffer at pH 8.0. After addition of 5% total E. coli RNA (RNA/protein 1:20, w/w), incubation with or without fivefold equivalent of CAI or various doses of NYAD-1 for 30 min at 4 °C, the samples were dialyzed overnight in Na2HPO4 buffer at pH 8.0 containing 100 mM NaCl at 4 °C. For assembly of CA mature-like particles, the addition of 5% total E. coli RNA was omitted. Negative staining was used to check the assembly. Carbon-coated copper grids (200 pore size; EM Sciences) were treated with 20 μl of poly-L-lysine (1 mg/ml; Sigma) for 2 min. Then 20 μl of reaction solution was placed onto the grid and left for 2 min, then stained with 30 μl of 2% (w/v) uranyl acetate solution for 2 min. Excess stain was removed, and grids were air-dried. Specimens were examined with a Philips EM410 electron microscope.
Cell-based system
To analyze the impact of NYAD-1 on VLP release and morphology, electron microscopy was conducted one day post transfection with plasmids encoding Gag or Gag-Pol. 4 × 105 293 T cells were seeded per well in a six-well plate on the day before transfection. Cells were washed twice 4 h post transfection and incubated with complete culture medium in the presence or in the absence of NYAD-1 at different concentrations for another 20 h. Cells were fixed in 3% (v/v) glutaraldehyde in 100 mM sodium cacodylate for 1 h and post-fixed in 1% (w/v) OsO4 in 100 mM sodium cacodylate for another 1 h. Specimens were then dehydrated through a graded series of ethanol solutions and embedded in EPON. After staining with 4% (w/v) uranyl acetate and Reynold's lead citrate, ultra-thin sections were examined under a Philips EM410 electron microscope at 80 Kv.
Virus assembly and release assays
293T cells were transfected with the HIV-1 molecular clone pNL4-336 and 6 h post transfection were treated with the indicated concentrations of NYAD-1 for 16-20 h. Cells were metabolically labeled with [35S]Met/Cys, and cell and virus lysates were immunoprecipitated and subjected to SDS-PAGE. Protein band intensities were quantified by phosphorimager analysis. The virus release efficiency was calculated as the amount of virion-associated Gag as a fraction of total (cell-plus virion-associated) Gag. Analysis of EIAV particle production was performed as described above with the EIAV Gag expression construct pPRE-Gag.37,38
Env-pseudotyped virus infectivity and luciferase assay
Pseudotyped virus was prepared by minor modifications of the method reported.48 The virus-containing supernatants were harvested two days later. One day pre-infection, 100 μl of U87-T4-CXCR4 at 105 cells/ml was seeded in a 96-well tissue culture plate. After 30 min or 4 h incubation with 50 μl of serially diluted NYAD-1, AZT or complete culture medium with 50 μl virus stocks, the mixtures were added to the cells and co-cultured for another three days. After 72 h, cells were washed with PBS and luciferase lysis buffer (Promega), followed by one freeze-thaw cycle. The sample was assayed with the Promega Luciferase Assay System (Promega) and a multifunctional microplate reader (ULTRA, Tecan, Research Triangle Park, NC). NYAD-1 was dissolved in 1% (v/v) DMSO. However, 1% DMSO did not show any inhibition in viral infectivity assays.
Time-of-addition study
Experiments were done by using a single-cycle viral infectivity assay described above. U87-T4-CXCR4 cells were infected in the presence of 50 μM NYAD-1, 3.75 μM AZT or 222 nM T-20. The compounds were applied at different lengths of time after infection. The percentage of inhibition of virus by inhibitors was calculated by quantifying luciferase activity (Promega).
Measurement of HIV-1 infectivity
The inhibitory activity of NYAD-1 on infection by laboratory-adapted HIV-1 strains was determined essentially as described,49 but with minor modification. In brief, 104MT-2 cells were infected with HIV-1 at 100 TCID50 (50% tissue culture infective dose) (0.01 MOI) in 200 μl of RPMI 1640 medium containing 10% FBS in the presence or in the absence of peptides at graded concentrations overnight. The culture supernatants were then removed and fresh medium containing freshly prepared test peptide were added. On the fourth day post infection, 100 μl of culture supernatant was collected from each well, mixed with equal volume of 5% (v/v) Triton X-100 and tested for p24 antigen by ELISA.
The inhibitory activity of peptides on infection by primary HIV-1 isolates was determined as described.35 PBMCs were isolated from the blood of healthy donors at the New York Blood Center by standard density-gradient centrifugation using Histopaque-1077 (Sigma-Aldrich). The cells were cultured at 37 °C for 2 h. Non-adherent cells were collected and resuspended at 5 × 106 cells/ml in RPMI-1640 medium containing 10% (v/v) fetal bovine serum, 5 μg/ml of phytohemagglutinin, and 100 U/ml of IL-2 (Sigma-Aldrich), followed by incubation at 37 °C for three days. The phytohemagglutinin-stimulated cells (5 × 104 cells/ml) were infected with primary HIV-1 isolates at 500 TCID50 (0.01 MOI) in the absence or in the presence of peptide inhibitor at graded concentrations. Culture media were changed every three days and replaced with fresh medium containing freshly prepared inhibitor. The supernatants were collected seven days post-infection and tested for p24 antigen by ELISA. The percentage inhibition of p24 production, IC50 and IC90 values were calculated with GraphPad Prism software (GraphPad Software Inc.).
Cytotoxicity assay
Cytotoxicity of peptides in MT-2 cells and PBMC was measured by the XTT ((sodium 3′-(1-(phenylamino)-carbonyl)-3,4-tetrazolium-bis(4-methoxy-6-nitro) bezene-sulfonic acid hydrate)) method as previously.35 Briefly, for MT-2 cells, 100 μl of a peptide at graded concentrations was added to an equal volume of cells (105 cells/ml) in 96-well plates followed by incubation at 37 °C for four days, which ran parallel with the neutralization assay in MT-2 (except medium was added instead of virus). In the case of PBMC, 5 × 105 cells/ml were used and the cytotoxicity was measured after seven days. After addition of XTT (Poly-Sciences, Inc.), the soluble intracellular formazan was quantified colorimetrically at 450 nm 4 h later with a reference at 620 nm. The percentage cytotoxicity and the CC50 values were calculated as described above.
Acknowledgements
This work was supported by intramural funding from the New York Blood Center. We thank Lyudmil Angelov, Yelena Oksov and Drs James Farmar and Mousumi Ghosh for their technical help. NMR studies were supported by NIH GM-66354, the Keck Foundation, and the member institutions of NYSBC. This work was supported, in part, by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, NIH.
Abbreviations used
- HIV-1
human immunodeficiency virus type 1
- MA
matrix
- CA
capsid
- NC
nucleocapsid
- C-CA
C-terminal domain of capsid
- CPP
cell-penetrating peptide
- FACS
fluorescence-activated cell sorter
- EIAV
equine infectious anemia virus
- EM
electron microscope
- HSQC
heteronuclear single quantum coherence
- PBMC
peripheral blood mononuclear cells
Footnotes
Supplementary Data Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jmb.2008.02.066
References
- 1.Huseby D, Barklis RL, Alfadhli A, Barklis E. Assembly of human immunodeficiency virus precursor gag proteins. J. Biol. Chem. 2005;280:17664–17670. doi: 10.1074/jbc.M412325200. [DOI] [PubMed] [Google Scholar]
- 2.Gottlinger HG. The HIV-1 assembly machine. AIDS. 2001;5(Suppl):S13–S20. doi: 10.1097/00002030-200100005-00003. [DOI] [PubMed] [Google Scholar]
- 3.Freed EO. HIV-1 gag proteins: diverse functions in the virus life cycle. Virology. 1998;251:1–15. doi: 10.1006/viro.1998.9398. [DOI] [PubMed] [Google Scholar]
- 4.Lanman J, Sexton J, Sakalian M, Prevelige PE., Jr Kinetic analysis of the role of intersubunit interactions in human immunodeficiency virus type 1 capsid protein assembly in vitro. J. Virol. 2002;76:6900–6908. doi: 10.1128/JVI.76.14.6900-6908.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jouvenet N, Neil SJ, Bess C, Johnson MC, Virgen CA, Simon SM, Bieniasz PD. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 2006;4:e435. doi: 10.1371/journal.pbio.0040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finzi A, Orthwein A, Mercier J, Cohen EA. Productive human immunodeficiency virus type 1 assembly takes place at the plasma membrane. J. Virol. 2007;81:7476–7490. doi: 10.1128/JVI.00308-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abdurahman S, Hoglund S, Goobar-Larsson L, Vahlne A. Selected amino acid substitutions in the C-terminal region of human immunodeficiency virus type 1 capsid protein affect virus assembly and release. J. Gen. Virol. 2004;85:2903–2913. doi: 10.1099/vir.0.80137-0. [DOI] [PubMed] [Google Scholar]
- 8.Chien AI, Liao WH, Yang DM, Wang CT. A domain directly C-terminal to the major homology region of human immunodeficiency type 1 capsid protein plays a crucial role in directing both virus assembly and incorporation of Gag-Pol. Virology. 2006;348:84–95. doi: 10.1016/j.virol.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 9.Douglas CC, Thomas D, Lanman J, Prevelige PE., Jr Investigation of N-terminal domain charged residues on the assembly and stability of HIV-1 CA. Biochemistry. 2004;43:10435–10441. doi: 10.1021/bi049359g. [DOI] [PubMed] [Google Scholar]
- 10.Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI. Assembly properties of the human immunodeficiency virus type 1 CA protein. J. Virol. 2004;78:2545–2552. doi: 10.1128/JVI.78.5.2545-2552.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joshi A, Nagashima K, Freed EO. Mutation of dileucine-like motifs in the human immunodeficiency virus type 1 capsid disrupts virus assembly, gag-gag interactions, gag-membrane binding, and virion maturation. J. Virol. 2006;80:7939–7951. doi: 10.1128/JVI.00355-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamashita M, Emerman M. Capsid is a dominant determinant of retrovirus infectivity in non-dividing cells. J. Virol. 2004;78:5670–5678. doi: 10.1128/JVI.78.11.5670-5678.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niedrig M, Gelderblom HR, Pauli G, Marz J, Bickhard H, Wolf H, Modrow S. Inhibition of infectious human immunodeficiency virus type 1 particle formation by Gag protein-derived peptides. J. Gen. Virol. 1994;75:1469–1474. doi: 10.1099/0022-1317-75-6-1469. [DOI] [PubMed] [Google Scholar]
- 14.Hoglund S, Su J, Reneby SS, Vegvari A, Hjerten S, Sintorn IM, et al. Tripeptide interference with human immunodeficiency virus type 1 morphogenesis. Antimicrob. Agents Chemother. 2002;46:3597–3605. doi: 10.1128/AAC.46.11.3597-3605.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garzon MT, Lidon-Moya MC, Barrera FN, Prieto A, Gomez J, Mateu MG, Neira JL. The dimerization domain of the HIV-1 capsid protein binds a capsid protein-derived peptide: a biophysical characterization. Protein Sci. 2004;13:1512–1523. doi: 10.1110/ps.03555304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang C, Loeliger E, Kinde I, Kyere S, Mayo K, Barklis E, et al. Antiviral inhibition of the HIV-1 capsid protein. J. Mol. Biol. 2003;327:1013–1020. doi: 10.1016/s0022-2836(03)00289-4. [DOI] [PubMed] [Google Scholar]
- 17.Sakalian M, McMurtrey CP, Deeg FJ, Maloy CW, Li F, Wild CT, Salzwedel K. 3-O-(3′,3′-dimethysuccinyl) betulinic acid inhibits maturation of the human immunodeficiency virus type 1 Gag precursor assembled in vitro. J. Virol. 2006;80:5716–5722. doi: 10.1128/JVI.02743-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li F, Goila-Gaur R, Salzwedel K, Kilgore NR, Reddick M, Matallana C, et al. PA-457: a potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc. Natl Acad. Sci. USA. 2003;100:13555–13560. doi: 10.1073/pnas.2234683100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou J, Yuan X, Dismuke D, Forshey BM, Lundquist C, Lee KH, et al. Small-molecule inhibition of human immunodeficiency virus type 1 replication by specific targeting of the final step of virion maturation. J. Virol. 2004;78:922–929. doi: 10.1128/JVI.78.2.922-929.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sticht J, Humbert M, Findlow S, Bodem J, Muller B, Dietrich U, et al. A peptide inhibitor of HIV-1 assembly in vitro. Nature Struct. Mol. Biol. 2005;12:671–677. doi: 10.1038/nsmb964. [DOI] [PubMed] [Google Scholar]
- 21.Ternois F, Sticht J, Duquerroy S, Krausslich HG, Rey FA. The HIV-1 capsid protein C-terminal domain in complex with a virus assembly inhibitor. Nature Struct. Mol. Biol. 2005;12:678–682. doi: 10.1038/nsmb967. [DOI] [PubMed] [Google Scholar]
- 22.Phelan JC, Skelton NJ, Braisted AC, McDowell RS. A general method for constraining short peptides to an α-helical conformation. J. Am. Chem. Soc. 1997;119:455–460. [Google Scholar]
- 23.Leduc AM, Trent JO, Wittliff JL, Bramlett KS, Briggs SL, Chirgadze NY, et al. Helix-stabilized cyclic peptides as selective inhibitors of steroid receptor-coactivator interactions. Proc. Natl Acad. Sci. USA. 2003;100:11273–11278. doi: 10.1073/pnas.1934759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang B, Liu D, Huang Z. Synthesis and helical structure of lactam bridged BH3 peptides derived from pro-apoptotic Bcl-2 family proteins. Bioorg. Med. Chem. Lett. 2004;14:1403–1406. doi: 10.1016/j.bmcl.2003.09.101. [DOI] [PubMed] [Google Scholar]
- 25.Wang D, Liao W, Arora PS. Enhanced metabolic stability and protein-binding properties of artificial helices derived from a hydrogen-bond surrogate: application to Bcl-xL. Angew Chem. Int. Edit. 2005;44:6525–6529. doi: 10.1002/anie.200501603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schafmeister CE, Po J, Verdine GL. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J. Am. Chem. Soc. 2000;122:5891–5892. [Google Scholar]
- 27.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, et al. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003;278:585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 29.Li S, Hill CP, Sundquist WI, Finch JT. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature. 2000;407:409–413. doi: 10.1038/35030177. [DOI] [PubMed] [Google Scholar]
- 30.Gross I, Hohenberg H, Krausslich HG. In vitro assembly properties of purified bacterially expressed capsid proteins of human immunodeficiency virus. Eur. J. Biochem. 1997;249:592–600. doi: 10.1111/j.1432-1033.1997.t01-1-00592.x. [DOI] [PubMed] [Google Scholar]
- 31.Mariani R, Rutter G, Harris ME, Hope TJ, Krausslich HG, Landau NR. A block to human immunodeficiency virus type 1 assembly in murine cells. J. Virol. 2000;74:3859–3870. doi: 10.1128/jvi.74.8.3859-3870.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gamble TR, Yoo S, Vajdos FF, von Schwedler UK, Worthylake DK, Wang H, et al. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science. 1997;278:849–853. doi: 10.1126/science.278.5339.849. [DOI] [PubMed] [Google Scholar]
- 33.Lanman J, Lam TT, Barnes S, Sakalian M, Emmett MR, Marshall AG, Prevelige PE., Jr Identification of novel interactions in HIV-1 capsid protein assembly by high-resolution mass spectrometry. J. Mol. Biol. 2003;325:759–772. doi: 10.1016/s0022-2836(02)01245-7. [DOI] [PubMed] [Google Scholar]
- 34.McDermott J, Farrell L, Ross R, Barklis E. Structural analysis of human immunodeficiency virus type 1 Gag protein interactions, using cysteine-specific reagents. J. Virol. 1996;70:5106–5114. doi: 10.1128/jvi.70.8.5106-5114.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang S, Lu H, Liu S, Zhao Q, He Y, Debnath AK. N-substituted pyrrole derivatives as novel human immunodeficiency virus type 1 entry inhibitors that interfere with the gp41 six-helix bundle formation and block virus fusion. Antimicrob. Agents Chemother. 2004;48:4349–4359. doi: 10.1128/AAC.48.11.4349-4359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patnaik A, Chau V, Li F, Montelaro RC, Wills JW. Budding of equine infectious anemia virus is insensitive to proteasome inhibitors. J. Virol. 2002;76:2641–2647. doi: 10.1128/JVI.76.6.2641-2647.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shehu-Xhilaga M, Ablan S, Demirov DG, Chen C, Montelaro RC, Freed EO. Late domain-dependent inhibition of equine infectious anemia virus budding. J. Virol. 2004;78:724–732. doi: 10.1128/JVI.78.2.724-732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qiu W, Soloshonok VA, Cai C, Tang X, Hruby VJ. Convenient, large-scale assymetric synthesis of enantiometrically pure trans-cinnamylglycine and α-alanine. Tetrahedron. 2000;56:2577–2582. [Google Scholar]
- 40.Morrow JA, Segall ML, Lund-Katz S, Phillips MC, Knapp M, Rupp B, Weisgraber KH. Differences in stability among the human apolipoprotein E isoforms determined by the amino-terminal domain. Biochemistry. 2000;39:11657–11666. doi: 10.1021/bi000099m. [DOI] [PubMed] [Google Scholar]
- 41.Cavanagh J, Fairbrother WJ, Palmer AG, III, Skelton NJ. Protein NMR Spectroscopy: Principles and Practice. Academic Press; New York: 1996. [Google Scholar]
- 42.Keller R. The Computer Aided Resonance Assignment Tutorial. Cantina Verlag; Zurich, Switzerland: 2004. [Google Scholar]
- 43.Johnson BA, Blevins RA. NMRView: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 44.Sandstrom C, Berteau O, Gemma E, Oscarson S, Kenne L, Gronenborn AM. Atomic mapping of the interactions between the antiviral agent cyanovirin-N and oligomannosides by saturationtransfer difference NMR. Biochemistry. 2004;43:13926–13931. doi: 10.1021/bi048676k. [DOI] [PubMed] [Google Scholar]
- 45.Schaeffer E, Soros VB, Greene WC. Compensatory link between fusion and endocytosis of human immunodeficiency virus type 1 in human CD4 T lymphocytes. J. Virol. 2004;78:1375–1383. doi: 10.1128/JVI.78.3.1375-1383.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ehrlich LS, Liu T, Scarlata S, Chu B, Carter CA. HIV-1 capsid protein forms spherical (immature-like) and tubular (mature-like) particles in vitro: structure switching by pH-induced conformational changes. Biophys. J. 2001;81:586–594. doi: 10.1016/S0006-3495(01)75725-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gross I, Hohenberg H, Krausslich HG. In vitro assembly properties of purified bacterially expressed capsid proteins of human immunodeficiency virus. Eur. J. Biochem. 1997;249:592–600. doi: 10.1111/j.1432-1033.1997.t01-1-00592.x. [DOI] [PubMed] [Google Scholar]
- 48.Tomaras GD, Lacey SF, McDanal CB, Ferrari G, Weinhold KJ, Greenberg ML. CD8+ T cell-mediated suppressive activity inhibits HIV-1 after virus entry with kinetics indicating effects on virus gene expression. Proc. Natl Acad. Sci. USA. 2000;97:3503–3508. doi: 10.1073/pnas.070521097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang SB, Lin K, Neurath AR. Enhancement of human immunodeficiency virus type 1 infection by antisera to peptides from the envelope glycoproteins gp120/gp41. J. Expt. Med. 1991;174:1557–1563. doi: 10.1084/jem.174.6.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]