

Abstract
Stable RNA, mainly comprised of rRNA and tRNA, accounts for the majority of cellular RNA. Although normally stable under favorable growth conditions in the laboratory, these RNA species undergo extensive degradation responding to many environmental changes and stress conditions. Multiple ribonucleases and other enzymes may be involved in the decay of stable RNA. The onset and rate of degradation are probably determined by the status of the RNA as well as the availability of the degrading activities. The elucidation of pathways for stable RNA decay has been benefited by many biochemical and genetic approaches. These include purification of the enzymes and characterization of their substrate specificity in vitro, and studies of stable RNA decay by inactivating and over-expressing the degradation activities in vivo. Furthermore, RNA degradation intermediates have been characterized in detail, such as determining the sizes, the sequences, the 5′ and 3′ termini, etc. In this work, we describe the methods that are most commonly used in the study of the degradation and processing of stable RNA in E. coli. Most of them should be also useful in studies of other RNA species or RNA from other organisms.
1. Introduction
Stable RNAs constitute >95% of total RNA in an exponentially growing E. coli cell, of which approximately 80% are rRNAs and approximately 15% are tRNAs. In contrast to unstable mRNAs, stable RNAs are resistant to the cellular activities that degrade RNA under normal growth conditions. Their half-lives are usually longer than the doubling time of cells in culture, allowing their function to be maintained. Stable RNAs are protected from degradation through formation of highly stable secondary and tertiary structures, by complexing with proteins such as in the ribosome, and by amino acylation in the case of tRNA.
Under certain circumstances, stable RNAs may also undergo efficient decay (Deutscher, 2003). For example, ribosomes are extensively degraded during starvation, amounting to as much as 95% in some cases. This massive degradation seems to involve primarily ribosomal RNAs, not ribosomal proteins. Ribosome degradation begins with the conversion of polysomes to monosomes and then to ribosome subunits. Interestingly, once ribosome breakdown starts, degradation is quickly completed. The remaining ribosomes are intact and presumably functional. Similar ribosome decay may happen under stationary phase and slow growth conditions. This process seems to play a major role in bacterial adaptation to changing environments under natural conditions. Bacterial cells are able to survive under nutrient-limiting conditions solely from nutrients provided by the degradation of ribosomes and rRNAs. The residual ribosomes may keep cells viable and allow them to recover from unfavorable growth conditions. Despite its importance, our understanding of the activities and regulation of rRNA degradation during starvation, stationary phase, and slow growth conditions is still in its infancy.
When E. coli cultures are treated with certain chemical agents or antibiotics, cellular RNAs also undergo extensive breakdown (Deutscher, 2003). Many of these agents alter permeability of the cell membrane, resulting in loss of ions (including Mg++) from the cytoplasm. The reduction in [Mg++] may affect ribosome structure and render the rRNAs more accessible to RNases. RNase I, a nonspecific endoribonuclease present primarily in the periplasmic space, is largely responsible for RNA degradation in these cases, resulting in formation of 3′-mononucleotides. Presumably, membrane damage allows RNase I to enter the cytoplasm and destroy RNA. Loss of Mg++, an inhibitor of RNase I, and increased accessibility of rRNAs all contribute to the extensive RNA decay. It should be noted that certain toxin-antitoxin pairs, such as the hok/sok system encoded by R1 plasmids, work by a similar mechanism (Gerdes and Wagner, 2007). The activated toxin causes depolarization of the cellular membrane, resulting in RNase I influx and subsequent massive RNA degradation and cell death (Nielsen et al., 1991).
Another important aspect of stable RNA degradation was revealed by observations that stable RNA species with abnormal sequence or structure may be degraded. Such quality control of the RNA pool has recently attracted considerable attention. In one example, it was shown that a mutant tRNATrp is unstable and degraded at the precursor level (Li et al., 2002). Degradation involves the exoribonucleases, polynucleotide phosphorylase (PNPase), and RNase R and is promoted by polyadenylation by poly(A) polymerase (Deutscher, 2006; Li et al., 2002). In the absence of these enzymes, a precursor to the mutant tRNATrp accumulates to high levels. In many respects, degradation of the mutant tRNATrp is very similar to the pathway for mRNA decay (Deutscher, 2006). An additional example of stable RNA degradation occurs during misassembly of ribosomes. rRNA fragments generated in this process are normally degraded by PNPase and RNase R. When both enzymes are inactive, the RNA fragments accumulate to high levels, and the cells lose viability (Cheng and Deutscher, 2003). Apparently, the fragments need to be removed rapidly, because their presence interferes with ribosome assembly and leads to even more rRNA fragmentation. In contrast to the aforementioned RNA breakdown catalyzed by RNase I, this degradation is confined to misassembled ribosomes and is carried out by RNase activities that produce 5′-mononucleotides.
Study of stable RNA decay has benefited tremendously from mutants that display altered degradation. These include mutants that lead to increased membrane permeability, altered ribosome assembly, misfolded RNA structure, and inactive RNases. At least 17 ribonucleases have been identified in E. coli. Most of them have been extensively studied for their role in RNA degradation with genetic and biochemical approaches (Li and Deutscher, 2004). Although the function of many RNases in mRNA decay is well understood (Kushner, 2002), relatively little is known about how these enzymes might degrade stable RNAs (Deutscher, 2003). Early work suggested that stable RNA degradation, similar to mRNA decay, is initiated by endonucleolytic cleavages followed by 3′ → 5′ exonucleolytic action on the resulting RNA fragments (Kaplan and Apirion, 1975). As discussed previously, RNase I also participates in nonspecific RNA decay under certain unusual conditions, resulting in extensive loss of cellular RNA and possibly cell death. However, it is unlikely that RNase I plays any role in stable RNA decay under physiologic conditions of starvation, stationary phase, and slow growth, or in the surveillance of defective RNA. In fact, breakdown of stable RNAs under these conditions seems to be more controlled by use of many of the same enzymes that are involved in mRNA turnover (Deutscher, 2006). It is likely that poly(A) tails are added to the 3′-ends of both mRNAs and stable RNAs, or their fragments, to facilitate binding of the processive exoribonucleases that are needed for degradation of highly structured RNAs (Cohen, 1995; Hajnsdorf et al., 1995; Li et al., 2002; Vincent and Deutscher, 2006).
Many ribonucleases participate in stable RNA degradation. RNase E is an essential endoribonuclease involved in the processing of rRNA and tRNA and degradation of mRNA. Numerous studies have shown that RNase E cleavages are responsible for initiating degradation of many mRNAs. RNase E is also the central component of the degradosome, a multienzyme complex also containing PNPase, RNA helicase B, and enolase. It is believed that such organization ensures that mRNA degradation will proceed to completion efficiently. However, it remains to be determined whether RNase E and/or the degradosome is also responsible for breakdown of stable RNAs. Other endoribonucleases, such as RNase III, RNase P, and the RNase E homolog, RNase G, are also likely candidates for this process. Some protein toxins, such as MazF and RelE, have been shown to possess endoribonuclease activities and to cleave and inactivate mRNA in response to stress conditions (Gerdes et al., 2005). Whether any of the RNase toxins are involved in breakdown of stable RNA is not known. The exoribonucleases PNPase, RNase II, and RNase R progressively degrade RNA in the 3′ to 5′ direction. Two of them, PNPase and RNase R, have been shown to participate in degradation of rRNA fragments and unstable tRNA precursors, RNAs that contain a large amount of structured regions. Polyadenylation and RNA helicase activities also are probably required for efficient digestion (Cheng and Deutscher, 2005; Deutscher, 2006; Khemici and Carpousis, 2004).
Study of stable RNA decay requires quantification and characterization of the stable RNA species, their degradation intermediates, and their end products. The amount of intact RNAs and of their decay products provides a measure of the extent of degradation, whereas the decay intermediates provide important information on how degradation proceeds. Depending on the RNase(s) responsible for degradation, RNA products may contain a 3′-hydroxyl or a 3′-phosphate, whereas the released nucleotides may be 3′ or 5′-monophosphates or 5′-diphosphates. The length of RNA decay intermediates is determined by the positions of the endonucleolytic cleavages and by stop sites for exonucleolytic trimming. Modified nucleotides are abundant in rRNAs and tRNAs and may play a role in the degradation of these RNAs (see later).
Many of the methods used to determine the level, half-life, and end products during mRNA decay are also applicable to the study of stable RNA degradation. Methods specifically useful for detection and characterization of stable RNAs and their degradation intermediates are discussed in the following.
2. Preparation of Stable RNA Substrates for In Vitro Degradation Assays
Various stable RNA substrates have been used extensively to identify and characterize degradation activities of purified RNases or in cell extracts. Sometimes, extracts from cells lacking specific enzymes (Li and Deutscher, 1994) or overexpressing certain activities (Li et al., 1999) are used to study the function of relevant activities. Because of the high abundance of rRNAs and tRNAs in preparations of total RNA, it is relatively easy to isolate these RNA species to sufficient purity for in vitro assays. Frequently, the RNA is labeled by radioisotopes to simplify detection of products. Cellular RNA can be labeled with 32P by the addition of carrier-free 32P orthophosphate to a culture growing in low-phosphate medium. Low-phosphate TB medium (Gegenheimer et al., 1977) is easy to prepare and provides good labeling of RNA. Typically, incubation of an exponentially growing culture for 1 h in the presence of 20 μCi/ml 32Pi results in a specific radioactivity of ~50,000 cpm per μg RNA. Alternatively, RNA can be labeled in vivo with 3H-uridine by including the radioactive nucleoside in a rich medium (Li et al., 1999). Finally, purified RNA can also be labeled in vitro (see the following).
rRNAs are usually prepared from isolated ribosomes. For this purpose, cell extracts should be made under conditions that preserve ribosome structure such as by grinding or by French press treatment. After removing cell debris by centrifugation at 30,000g, ribosomes in the supernatant fraction (S30) are pelleted at 100,000g. Ribosomes in the pellet P100 are often pure enough for most purposes and sometimes are used directly as substrates in RNA processing or degradation reactions. In these cases, ribosomes may be washed with high concentrations of KCl or NH4Cl (0.5 or 1 M) and repelleted.
To isolate rRNAs from ribosome preparations, ribosomal proteins are denatured by the addition of sodium dodecyl sulfate (SDS, 0.5%) followed by extraction with phenol and chloroform. Protein-free rRNAs recovered in the aqueous phase are precipitated by ethanol. The resulting mixture of rRNAs can be used directly. Alternatively, the three rRNA components (23S, 16S, 5S) can be separated in agarose gels and purified by extraction (Cheng and Deutscher, 2002; Li et al., 1999). Up to 10 mg of rRNA can be obtained from 1 g of exponentially growing cells.
A rapid procedure may be used to obtain small amounts of rRNA directly from cell lysates (Li et al., 1999). This can be important when large-scale isolation is not feasible or necessary. Cells are pelleted from several milliliters of culture and resuspended in a lysis buffer (10 mM Tris-Cl, pH 7.4, 10 mM Na2EDTA, pH 7.4, 1% SDS, 40% glycerol, 0.1% DEPC, and 0.1% bromphenol blue, as described in Gegenheimer et al., [1977]). After boiling to release RNA, the lysate is directly applied to an agarose gel for electrophoretic separation. The 23S and 16S rRNAs are then individually extracted from the gel (Li et al., 1999b). A few micrograms of 23S and 16S rRNAs can be obtained from 1 ml of an exponential phase culture. This method is not suitable for isolation of 5S rRNA, because it is not separated well from tRNAs on an agarose gel.
tRNA can be prepared in large amounts by direct phenol extraction of cells followed by isopropanol fractionation (Deutscher and Hilderman, 1974). Cells are first suspended in a small volume of saline solution and are extracted with an equal volume of 88% phenol. Because the extraction procedure does not result in complete disruption of cells, large RNAs are only partially released. The RNA in the aqueous phase is then recovered by ethanol precipitation. RNAs are further purified by isopropanol fractionation, which separates on the basis of their sizes. RNA is dissolved in 0.3 M sodium acetate, pH 7.0, at room temperature. Larger RNAs are precipitated with 0.54 volumes of isopropanol and small RNAs with 0.98 volumes. The tRNA preparations are normally devoid of any high-molecular-weight material but can be contaminated with a small amount of 5S RNA (Deutscher and Hilderman, 1974). Two to three milligrams of tRNA can be isolated from 1 g of log-phase cells (Deutscher and Hilderman, 1974).
As noted previously, rRNA and tRNA preparations also can be labeled after they are isolated. Labeling of the 5′ end of a dephosphorylated RNA can be accomplished with polynucleotide kinase and γ-32P-ATP. The 3′-end can be labeled by addition of [32P]pCp with RNA ligase. In the case of tRNA, the 3′-terminal A residue can be removed by periodate treatment and β-elimination. This process causes oxidation of the terminal nucleoside which contains a vicinal 2′,3′-hydroxyl, and in the presence of an amine, eliminates the oxidized residue from the phosphate. The resulting phosphate at the 3′-end of the tRNA is subsequently removed with alkaline phosphatase. This process can be repeated to remove the C residues at the second and third positions from the 3′-end. The resulting truncated tRNAs are treated with tRNA nucleotidyltransferase (the CCA enzyme) and CTP or ATP to add back radioactive C or A residues (Deutscher and Ghosh, 1978). Such substrates are useful in tRNA degradation experiments, especially for study of degradation of the 3′-termini.
Specific rRNA and tRNA species also can be easily prepared by in vitro transcription and have been widely used for RNA processing and degradation assays (e.g., Li and Deutscher [1994] and Tuohy et al. [1994]). RNA transcripts can be uniformly labeled by including one or more radioactive ribonucleoside triphosphates in the reaction, or they can be end labeled as described previously. If a radioactive nucleotide is used in the in vitro transcription reaction, the same nonradioactive nucleotide sometimes is added at a low concentration to reach a sufficient concentration for efficient labeling. Approximately 1 μg of RNA transcript can be produced in a 25-μl reaction mixture containing 60 μM of the radioactive ribonucleoside triphosphate. If each ribonucleoside triphosphate is provided at 0.5 mM in a 25-μl reaction mixture, approximately 5 μg of RNA is normally produced. The RNA is often gel-purified before enzyme treatment (Li and Deutscher, 1999). It should be noted that tRNA or rRNA prepared by in vitro transcription lacks the modifications present in the corresponding cellular RNAs.
3. Detection of Degradation Products In Vitro
All eight exoribonucleases identified in E. coli degrade RNA in the 3′ to 5′ direction, generating 5′-mononucleotides. The endoribonuclease RNase I cleaves RNA nonspecifically, producing short RNAs and 3′-mononucleotides. These products are soluble in cold trichloroacetate (TCA), whereas longer RNAs are precipitated. Quantification of acid-soluble products from radioactively labeled RNA substrates has been widely used to study the activity of these RNases in vitro. To study the rate of stable RNA decay, radioactively labeled stable RNAs are treated with RNase preparations, followed by addition of carrier yeast RNA to 0.5% and cold TCA to 10% final concentration (Cheng and Deutscher, 2002). Long RNAs precipitate quickly in 10 min on ice and are present in the pellet after centrifugation. The supernatant fractions are counted in a scintillation counter to quantify the release of acid-soluble material. By controlling the amount of RNA substrate, the amount of enzyme preparation, and the length of incubation, conversion to acid-soluble products can be kept low (<20%), ensuring a nearly linear rate of enzyme action.
Degradation intermediates of stable RNA frequently accumulate because of their resistance to RNases. This resistance may be conferred by highly stable structures, nucleotide modifications, and, in many cases, association with RNA-binding proteins. These intermediates are readily detectable by gel electrophoresis. Depending on the size of the RNAs to be detected, one may use an agarose gel (for RNAs from hundreds to thousands of nucleotides) (Lalonde et al., 2007) (Fig. 2.1) or a denaturing polyacrylamide gel (for RNAs of 1 to hundreds of nucleotides) (Li and Deutscher, 1994, 1995, 1996; Li et al., 1998a, 1998b). A polyacrylamide gel containing 0.2% SDS and 3% glycerol is also useful in separating large rRNAs (Gegenheimer et al., 1977; Li et al., 1999). RNA labeled with 32P is detected by autoradiography. Nonlabeled RNAs can be stained by fluorescent dyes. The dye SYBR Gold (Invitrogen, Carlsbad, CA) stains single-stranded nucleic acids with at least 10 times the sensitivity of the double-strand specific dye, ethidium bromide.
Figure 2.1.
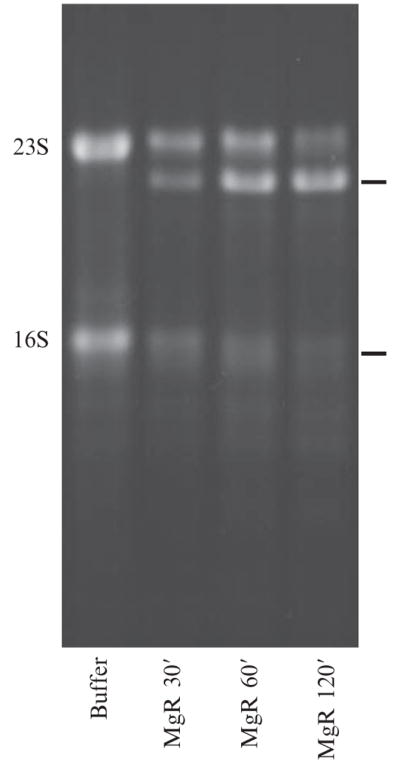
Degradation of E. coli 23S and16S rRNAsby RNase R from Mycoplasma genitalium (MgR). rRNAs isolated from ribosomes (see text) were incubated with buffer alone for 120 min, or with 0.5 μg of MgR in a 10-μl reaction for 30, 60, or 120 min under conditions described previously (Lalonde et al., 2007). The resulting products were separated in a 1.5% agarose gel, stained with SYBR Gold, and visualized under UV light. The starting 23S and 16S RNAs are labeled on the left. Prominent degradation intermediates are labeled with bars on the right.
When nonlabeled RNA is used as substrate, Northern blotting is useful for detection of degradation intermediates derived from specific RNAs. Because of the high abundance of stable RNAs and their decay intermediates, a very sensitive probe is usually not necessary. For instance, oligonucleotide DNA probes labeled at their 5′ end with 32P are usually sufficient to detect the degradation products of tRNA (Li et al., 2002) or rRNA (Cheng and Deutscher, 2003). A carefully performed Northern blot can achieve single-nucleotide resolution for products of stable RNAs in the range from tens to more than 300 nucleotides in length (Li and Deutscher, 1995, 1996; Li et al., 1998a, 1998b).
4. Examination of Stable RNA Decay In Vivo
As discussed previously, stable RNA molecules are broken down by complicated processes involving both endoribonucleases and exoribonucleases, and possibly regulatory proteins as well (Deutscher, 2003, 2006). Mutants affecting various RNases have proven extremely useful for determining pathways of stable RNA decay in vivo. Such mutants may harbor interrupted and/or deleted genes or may contain temperature-sensitive alleles. For studies of essential enzymes, temperature-sensitive mutants are most useful. In addition, introduction of cloned genes that overexpress a particular activity or that rescue a missing activity in a mutant strain provide additional important information regarding the function of the genes and their products.
In the absence of a particular RNase, specific degradation intermediates may accumulate to high levels, which would suggest a key role of that RNase in the removal of the intermediate. Roles for other proteins such as poly(A) polymerase and RNA helicases may be assessed in a similar manner (Deutscher, 2006). Certain steps of stable RNA degradation are often carried out by multiple activities with overlapping functions. In these situations, degradation intermediates may only accumulate when most or all of the overlapping activities are inactivated. For instance, mutant E. coli cells devoid of RNase R or PNPase show little defect in rRNA degradation or in cell growth. In contrast, mutant cells lacking both activities accumulate degradation intermediates of 23S and 16S rRNAs to high levels and lose viability (Cheng and Deutscher, 2003), and these could be identified by Northern blotting.
Mutant forms of stable RNA species have been proven equally important in studying stable RNA decay. A mutant tRNATrp containing a single-nucleotide change in the acceptor stem renders this tRNA temperature-sensitive and exceedingly sensitive to degradation such that it is present at only ~15% the level of wild-type tRNATrp (Li et al., 2002). Degradation of this mutant tRNA is impeded in cells lacking PNPase and/or poly(A) polymerase. In the absence of both enzymes, the amount of mutant tRNATrp increases to 60% of the wild-type level, and much of it is present as the tRNA precursor (Li et al., 2002). These data suggest that both enzymes and RNase R as well (Deutscher, 2006) are responsible for the degradation of this mutant tRNA. Moreover, mutations affecting the stability of an RNA greatly facilitate elucidation of RNA stability determinants. For example, it was noted that most stable RNA species in E. coli undergo exonucleolytic trimming to form their mature 3′-ends (Li and Deutscher, 2004; Li et al., 1998a). Remarkably, a base-paired structure, formed between the 3′ and 5′-terminal sequences, is present in each of these stable RNAs. Disruption of this structure may result in a marked decrease in the RNA’s stability as shown in the case of the tRNATrp.
5. Determination of the 3′ and 5′ Termini of Intermediates during the Processing and Degradation of Stable RNAs
The processing of stable RNA precursors and the degradation of stable RNAs produce intermediates and breakdown products that often contain defined 5′ and 3′ termini. Because a 5′ to 3′-exonucleolytic activity has not yet been identified in E. coli, the 5′ ends of these intermediates are most likely generated by endonucleolytic cleavages (Li and Deutscher, 2004). The 3′-end, however, can be formed by the action of either an endoribo-nuclease or exoribonuclease. Knowledge of the 5′ and 3′ termini of intermediates often helps to explain RNA sequence and structural features that govern endonucleolytic cleavage sites or that impede exonucleolytic digestion. The methods discussed in the following generally apply to determination of the termini of a specific stable RNA present in a total RNA preparation. When other RNA species might interfere with detection of specific RNA products, isolation of the specific RNA under study may be required before the analysis (Li et al., 1999).
Primer extension has been widely used to determine the 5′ termini of stable RNA products (Li and Deutscher, 1995, 1996; Li et al., 1998a). A cDNA is synthesized from a labeled oligonucleotide primer that is annealed to a site on the target RNA. From the length of the cDNA product and the site of the primer, it is possible to determine exactly the 5′-end of the RNA. Mapping with S1 nuclease is often used to determine 3′-ends of RNAs. S1 nuclease digests single-stranded nucleic acids much more efficiently than double-stranded molecules. A single-stranded DNA probe hybridized to the RNA of interest is protected from S1 digestion in the region in which it is complementary to the stable RNA, whereas the rest is digested. This results in a shortened, labeled probe whose size indicates the 3′-terminus of the stable RNA. Although used routinely, S1 nuclease also often cleaves at weak double-stranded regions of the hybrid. Moreover, the “breathing” at the end of the double-stranded region leads S1 to generate staggered ends. As a consequence, S1 mapping may not be an ideal method for precise determination of 3′-termini and of their relative abundance.
Site-directed RNase H cleavage is another method that has proven useful for determining both the 5′ and 3′-ends of stable RNA products (Li et al., 1999a,b). The endoribonuclease RNase H nonspecifically cleaves the RNA in an RNA/DNA duplex. For this method, a chimeric complementary oligonucleotide containing a short stretch of DNA and flanking 2-O-methyl RNA regions is used to force RNase H cleavage to occur at only a single position on the RNA (Fig. 2.2A). The number, position(s), and efficiency of RNase H cleavages depend on the length and sequence of the DNA in the chimera and on the source of the RNase H (Lapham et al., 1997). A DNA stretch of 3 to 4 nt usually results in a single cleavage. Sometimes different RNase H enzymes need to be tested with a particular chimeric oligonucleotide to find the best match that cleaves efficiently at a single position on target RNA (Zhongwei Li, Shilpa Pandit, and Murray P. Deutscher, unpublished observation). The complementary chimera is usually designed to anneal to the target RNA at a position close to its 5′ or 3′-end. After RNase H treatment, the resulting shortened RNA fragments are separated to single-nucleotide resolution in a denaturing polyacrylamide gel and are detected either by autoradiography for 32P-labeled RNAs (Li et al., 1999) or by Northern blotting for nonlabeled RNA (Li et al., 1999). On the basis of the sizes of the RNA fragments, the position of the 5′ or 3′-terminus can be determined. Site-directed RNase H cleavage works better than primer extension or S1 mapping to eliminate nonspecific products and to determine the relative abundance of different products. Figure 2.2B shows the heterogeneous 3′-termini of 23S rRNA revealed by a site-directed RNase H cleavage experiments. These 3′-ends were generated by polyadenylation in a mutant strain deficient in multiple exoribonucleases (Li et al., 1999).
Figure 2.2.

Analysis of RNA termini by site-directed RNase H cleavage. (A) Illustration of the method. A chimeric oligonucleotide containing a stretch of DNA (underlined sequence) is used to direct RNase H cleavage at a position close to the 3′ end of 23S rRNA. (B) The 3′ ends of precursor to 23S RNA revealed by site-directed RNase H cleavage (adapted from Li et al., [1999] with permission). The products were separated in a denaturing polyacrylamide gel and were detected by Northern blotting with a probe specific for the 3′ terminal sequence of 23S RNA. A small amount of precursor with eight extra nucleotides at the 3′ end is present in 23S RNA from wild-type cells. In a mutant strain deficient in the exoribonucleases, RNase T, PH, D, and BN (T−PH−D−BN−), a precursor containing 9 or more 3′ extra residues was found. These longer products are formed by polyadenylation. Removal of poly(A) polymerase from the multi-RNase–deficient strain (T−PH−D−BN−PAP−) restored the precursor to +8nt size. Introduction of the plasmid pJL89 harboring the poly(A) polymerase gene into the PAP− background (T−PH−D−BN−PAP−/pJL89) resulted in formation of the polyadenylated species.
The 3′ end of a stable RNA often is sufficiently homogeneous that its identity can be determined by a modification of rapid amplification of cDNA 3′ ends (3′ RACE). This method requires ligation of a short oligo-nucleotide linker to the 3′ end of the RNA, followed by reverse transcription from an oligonucleotide primer complementary to the linker. The cDNA for the RNA of interest is then specifically amplified by polymerase chain reactions (PCR) with the same primer originally used for cDNA synthesis and a second primer specific for the RNA of interest. The PCR product can then be directly sequenced with the RNA-specific primer (Fig. 2.3) and the 3′ end of the RNA determined.
Figure 2.3.

Explanation of RNA 3′ terminal sequences by 3′ RACE and cloning. The sequence of RNA is converted to that of cDNA by ligation of a linker to the 3′ end of the RNA and reverse transcription. Specific RNA sequences are amplified by PCR. The resulting PCR product can be directly sequenced (option 1) if a homogeneous 3′ end is expected (Lalonde et al., 2007). Alternately, multiple individual clones can be constructed and sequenced (option 2) if heterogeneous 3′ ends are analyzed (Li et al.,1998b).
By use of this method, degradation of E. coli 23S rRNA by RNase R from Mycoplasma genitalium was shown to stop 1-nt downstream of two close by ribose-methylation sites (shorter 23S products in Fig. 2.1), demonstrating the sensitivity of this particular RNase R to such RNA modifications (Lalonde et al., 2007). Although RNA linkers are often used (Li et al., 1998b, 2002), DNA linkers work equally well (Lalonde et al., 2007). Linkers should be phosphorylated at their 5′ ends, and to prevent self-ligation, linkers should be blocked at their 3′ end by phosphorylation or other modifications. The sequence of the linker may include useful restriction sites, enabling subsequent digestion of the PCR products to remove the linkers, to eliminate unwanted cDNA products, or to generate cloning sites (see following). In addition to overcome occasional difficulties of the site-directed RNase H cleavage method, 3′ RACE can provide sequence information that may reveal posttranscriptional changes in the RNA sequence (see following).
6. Detection of Poly(A) Tails on Products Generated by Processing or Degradation of Stable RNA
In a mutant strain lacking multiple exoribonucleases, unexpected elongated 3′ sequences were found on precursors of some RNAs. This observation led to the discovery of polyadenylation of these RNA species (Li et al., 1998b). Poly(A) tails were found on all stable RNA precursors examined in this mutant strain, and to a much less extent, also in wild-type cells. In the case of mutant tRNATrp, poly(A) tails were detected when PNPase was inactivated (Li et al., 2002). Polyadenylation seems to promote degradation of stable structures in RNA and participates in mRNA turnover and stable RNA quality control (Deutscher, 2006). The poly(A) tails on stable RNAs were identified by cloning the 3′ RACE products (see earlier) followed by DNA sequencing of individual clones (Fig. 2.3) (Li et al., 1998b, 2002). Because of the heterogeneous nature of the 3′ ends of these RNAs, multiple clones were sequenced for each stable RNA species. The results revealed RNA molecules containing all possible 3′ ends, including the normal mature end, encoded sequences shorter or longer than mature, and polyadenylated ends. For example, in a mutant lacking multiple RNases, approximately half of the precursors to 23S rRNA contain 1 to 5 adenylates at their 3′ ends (Fig. 2.2B) (Li et al., 1998b). Because of the laborious clone selection and sequencing, such an approach is only used when the population of termini needs to be clearly identified.
7. Concluding Remarks
Degradation of stable RNAs is of fundamental importance for bacteria to adapt to environmental changes and to maintain a functional RNA reservoir. Various biochemical and genetic approaches have been applied to study stable RNA decay in E. coli. The methods described here have been used to study processing and degradation of a variety of stable RNA species. Many of the methods can characterize RNA at single-nucleotide resolution or can provide well-defined sequence information. The use of mutants and of overexpressed and purified enzymes has enabled identification of activities responsible for specific steps of degradation pathways. These powerful approaches will eventually lead to the explanation of detailed molecular mechanisms of stable RNA decay. It should be noted that many features contribute to the normally high stability of the stable RNAs. Changes in these RNA features and alteration of the cellular environment may affect accessibility of degradation activities to RNA, triggering rapid breakdown of stable RNAs (Deutscher, 2003). Moreover, stable RNA decay may be regulated by cell metabolism and stress. Therefore, processes such as signal transduction and multiple molecular interactions may regulate stable RNA decay. If so, other methods that address these additional features may also be useful in future studies.
Acknowledgments
We thank Maureen S. Lalonde and Xin Gong for providing Fig. 2.1 for this work. This work was supported by NIH Grant GM16317 to M. P. D. and NIH grant S06 GM073621 and Florida Atlantic University Startup Fund to Z. L.
References
- Cheng ZF, Deutscher MP. Purification and characterization of the Escherichia coli exoribonuclease RNase R. Comparison with RNase II. J Biol Chem. 2002;277:21624–21629. doi: 10.1074/jbc.M202942200. [DOI] [PubMed] [Google Scholar]
- Cheng ZF, Deutscher MP. Quality control of ribosomal RNA mediated by polynucleotide phosphorylase and RNase R. Proc Natl Acad Sci USA. 2003;100:6388–6393. doi: 10.1073/pnas.1231041100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng ZF, Deutscher MP. An important role for RNase R in mRNA decay. Mol Cell. 2005;17:313–318. doi: 10.1016/j.molcel.2004.11.048. [DOI] [PubMed] [Google Scholar]
- Cohen SN. Surprises at the 3′ end of prokaryotic RNA. Cell. 1995;80:829–832. doi: 10.1016/0092-8674(95)90284-8. [DOI] [PubMed] [Google Scholar]
- Deutscher MP. Degradation of stable RNA in bacteria. J Biol Chem. 2003;278:45041–45044. doi: 10.1074/jbc.R300031200. [DOI] [PubMed] [Google Scholar]
- Deutscher MP. Degradation of RNA in bacteria: Comparison of mRNA and stable RNA. Nucleic Acids Res. 2006;34:659–666. doi: 10.1093/nar/gkj472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutscher MP, Ghosh RK. Preparation of synthetic tRNA precursors with tRNA nucleotidyltransferase. Nucleic Acids Res. 1978;5:3821–3829. doi: 10.1093/nar/5.10.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutscher MP, Hilderman RH. Isolation and partial characterization of Escherichia coli mutants with low levels of transfer ribonucleic acid nucleotidyltransferase. J Bacteriol. 1974;118:621–627. doi: 10.1128/jb.118.2.621-627.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegenheimer P, Watson N, Apirion D. Multiple pathways for primary processing of ribosomal RNA in Escherichia coli. J Biol Chem. 1977;252:3064–3073. [PubMed] [Google Scholar]
- Gerdes K, Christensen SK, Løbner-Olesen A. Prokaryotic toxin-antitoxin stress response loci. Nat Rev Microbiol. 2005;3:371–382. doi: 10.1038/nrmicro1147. [DOI] [PubMed] [Google Scholar]
- Gerdes K, Wagner EG. RNA antitoxins. Curr Opin Microbiol. 2007;10:117–124. doi: 10.1016/j.mib.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Hajnsdorf E, Braun F, Haugel-Nielsen J, Régnier P. Polyadenylylation destabilizes the rpsO mRNA of Escherichia coli. Proc Natl Acad Sci USA. 1995;92:3973–3977. doi: 10.1073/pnas.92.9.3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan R, Apirion D. Decay of ribosomal ribonucleic acid in Escherichia coli cells starved for various nutrients. J Biol Chem. 1975;250:3174–3178. [PubMed] [Google Scholar]
- Khemici V, Carpousis AJ. The RNA degradosome and poly(A) polymerase of Escherichia coli are required in vivo for the degradation of small mRNA decay intermediates containing REP-stabilizers. Mol Microbiol. 2004;51:777–790. doi: 10.1046/j.1365-2958.2003.03862.x. [DOI] [PubMed] [Google Scholar]
- Kushner SR. mRNA decay in Escherichia coli comes of age. J Bacteriol. 2002;184:4658–4665. doi: 10.1128/JB.184.17.4658-4665.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde MS, Zuo Y, Zhang J, Gong X, Wu S, Malhotra A, Li Z. Exoribonuclease R in Mycoplasma genitalium can carry out both RNA processing and degradative functions and is sensitive to RNA ribose methylation. RNA. 2007;13:1957–1968. doi: 10.1261/rna.706207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapham J, Yu YT, Shu MD, Steitz JA, Crothers DM. The position of site-directed cleavage of RNA using RNase H and 29-O-methyl oligonucleotides is dependent on the enzyme source. RNA. 1997;3:950–951. [PMC free article] [PubMed] [Google Scholar]
- Li Z, Deutscher MP. The role of individual exoribonucleases in processing at the 3′ end of Escherichia coli tRNA precursors. J Biol Chem. 1994;269:6064–6071. [PubMed] [Google Scholar]
- Li Z, Deutscher MP. The tRNA processing enzyme RNase T is essential for maturation of 5S RNA. Proc Natl Acad Sci USA. 1995;92:6883–6886. doi: 10.1073/pnas.92.15.6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Deutscher MP. Maturation pathways for E. coli tRNA precursors: A random multienzyme process in vivo. Cell. 1996;86:503–512. doi: 10.1016/s0092-8674(00)80123-3. [DOI] [PubMed] [Google Scholar]
- Li Z, Deutscher MP. Exoribonucleases and endoribonucleases. In: Curtiss R III, editor. EcoSal-Escherichia coli and Salmonella: Cellular and Molecular Biology. Chapter 4.6.3. ASM Press; Washington, DC: 2004. ([online] http://www.ecosal.org) [Google Scholar]
- Li Z, Pandit S, Deutscher MP. 3′ Exoribonucleolytic trimming is a common feature of the maturation of small, stable RNAs in Escherichia coli. Proc Natl Acad Sci USA. 1998a;95:2856–2861. doi: 10.1073/pnas.95.6.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Pandit S, Deutscher MP. Polyadenylation of stable RNA precursors in vivo. Proc Natl Acad Sci USA. 1998b;95:12158–12162. doi: 10.1073/pnas.95.21.12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Pandit S, Deutscher MP. Maturation of 23S ribosomal RNA requires the exoribonuclease RNase T. RNA. 1999a;5:139–146. doi: 10.1017/s1355838299981669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Pandit S, Deutscher MP. RNase G (CafA protein) and RNase E are both required for the 5′ maturation of 16S ribosomal RNA. EMBO J. 1999b;18:2878–2885. doi: 10.1093/emboj/18.10.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Reimers S, Pandit S, Deutscher MP. RNA quality control: Degradation of defective transfer RNA. EMBO J. 2002;21:1132–1138. doi: 10.1093/emboj/21.5.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen AK, Thorsted P, Thisted T, Wagner EG, Gerdes K. The rifampicin-inducible genes srnB from F and pnd from R483 are regulated by antisense RNAs and mediate plasmid maintenance by killing of plasmid-free segregants. Mol Microbiol. 1991;5:1961–1973. doi: 10.1111/j.1365-2958.1991.tb00818.x. [DOI] [PubMed] [Google Scholar]
- Tuohy TMF, Li Z, Atkins JF, Deutscher MP. A functional mutant of tRNAArg2 with 10 extra nucleotides in its TFC arm. J Mol Biol. 1994;235:1369–1376. doi: 10.1006/jmbi.1994.1093. [DOI] [PubMed] [Google Scholar]
- Vincent HA, Deutscher MP. Substrate recognition and catalysis by the exoribonuclease RNase R. J Biol Chem. 2006;281:29769–29775. doi: 10.1074/jbc.M606744200. [DOI] [PubMed] [Google Scholar]