

Abstract
Mutation of the Rb tumor suppressor gene is strongly linked to osteosarcoma formation. This observation, and the documented interaction between the retinoblastoma protein (pRb) and Runx2, suggests that pRb is important in bone development. To assess this hypothesis, we used a conditional knockout strategy to generate pRb-deficient embryos that survive to birth. Analysis of these embryos shows that Rb-inactivation causes the abnormal development and impaired ossification of several bones, correlating with an impairment in osteoblast differentiation. We further show that Rb-inactivation acts to promote osteoblast differentiation in vitro and, through conditional analysis, establish that this occurs in a cell intrinsic manner. Although these in vivo and in vitro differentiation phenotypes appear paradoxical, we find that Rb-deficient osteoblasts have an impaired ability to exit the cell cycle both in vivo and in vitro that can explain the observed differentiation defects. Consistent with this observation, we show that the cell cycle and the bone defects in Rb-deficient embryos can be suppressed by deletion of E2f1, a known proliferation inducer that acts downstream of Rb. Thus, we conclude that pRb plays a key role in regulating osteoblast differentiation by mediating inhibition of E2F and consequently promoting cell cycle exit.
Keywords: Retinoblastoma, Bone, Osteoblast, Differentiation, Development
INTRODUCTION
The first tumor suppressor to be cloned was the retinoblastoma gene, Rb. Rb is mutated in approximately one-third of all sporadic human tumors, but there is strong correlation with certain tumor types. Specifically, Rb mutations are observed in almost all retinoblastomas (1) and also in a large percentage of osteosarcomas and small cell lung carcinomas. For patients who carry germline Rb mutations, osteosarcoma is the second most common tumor type after retinoblastoma (2). Overall, greater than 70% of osteosarcomas demonstrate a molecular change or mutation at the Rb locus (3,4).
The gene product, pRb, belongs to a family of proteins, including p107 and p130, termed the pocket proteins, although only pRb has been demonstrated to possess significant tumor suppressive properties (5). The best characterized role of pRb is its regulation of cell cycle progression. Overexpression of pRb causes G1 cell cycle arrest (6), while acute ablation of pRb induces cell cycle re-entry in quiescent cells (7). To execute its cell cycle inhibitory function, hypophosphorylated pRb binds to and inhibits the E2F family of transcription factors (8). During G1, pRb becomes hyperphosphorylated by the cyclin D - cdk4/6 complex and subsequently by cyclin E - cdk2. This phosphorylation releases the E2Fs from pRb to induce the transcription of cellular genes essential for S phase entry and cell division.
The analyses of in vivo mouse models and in vitro experiments demonstrate that pRb is required for the differentiation of specific tissues. In erythropoiesis the loss of Rb results in inefficient enucleation and incomplete terminal differentiation of erythroid cells (9,10). In skeletal muscle pRb is required for proper cell cycle exit and differentiation (11). Conditional deletion of Rb in the intestine causes increased proliferation and abnormal expression of differentiation markers (12,13). The loss of pRb affects the normal expression of differentiation genes, such as β- and γ-crystallins, in the lens (14). These deficiencies in differentiation appear to be due, at least partially, to a defect in cell cycle exit, a step believed to be required in most differentiation pathways. However, this does not rule out the possibility that pRb contributes to differentiation in a more distinct and specific manner. Notably, pRb binds to NRP/B, a protein upregulated during neuronal differentiation and involved in neuronal process formation (15). Relevant to this, other markers of neuronal differentiation are decreased in the Rb-deficient embryo (16). With respect to fat cells, pRb physically interacts with C/EBPβ, and the loss of this interaction inhibits adipocyte differentiation (17).
Several studies implicate a role for pRb in osteoblast differentiation. Simian virus 40-derived large T-antigen, which targets the pocket proteins, prevents the differentiation of stromal cell lines into osteoblasts (18). The adenoviral E1A 12S protein also represses osteoblast differentiation, and this is dependent on a functional E1A pocket protein binding domain (19). Most striking is the finding that in immortalized cell lines pRb physically interacts with Runx2/CBFA1, one of the transcription factors essential for osteoblast differentiation (20,21). This latter observation suggests that pRb may play a role in osteoblast differentiation that is independent of cell cycle regulation.
Determining the role of pRb in osteoblast differentiation in vivo may ultimately provide some important insights concerning the high prevalence of Rb mutations in osteosarcoma. However, murine embryos deficient for pRb die between embryonic day (e) 13.5 and 15.5 (22–24). This early lethality has thus far precluded the study of pRb in bone development, which primarily does not occur until e15.5 in mice. To circumvent this problem, we generated a conditional Rb mouse strain that allows pRb-deficient embryos to survive until birth. This mouse model has enabled us to perform in vitro and in vivo studies to determine the effects of pRb loss in osteoblast differentiation and bone development.
RESULTS
pRb-deficient embryos exhibit bone defects during development
The retinoblastoma gene, Rb, is mutated in a large proportion of osteosarcomas. In vitro studies suggest that Rb may play a direct role in bone development (18–20) but this has not been examined in vivo. The germline Rb−/− mice die in mid-gestation (between e13.5 and e15.5), prior to the formation of most bones. However, recent studies show that this mid-gestational lethality results from a placental defect (25,26). Thus, we generated a conditional mouse strain that allows Rb mutant embryos to develop in the presence of a wild-type placenta. Specifically, we crossed an Rb mutant mouse line with loxP sites flanking the third exon of Rb (Rbc/c; 7) with a Mox2-Cre transgenic line (Mox2+/Cre) that expresses the Cre recombinase in the embryo proper, but not in the placenta, beginning at approximately e6.5 (27). The resulting Mox2+/Cre conditionally null Rb embryos (Rbc−/c−) survive until birth, allowing us to assess pRb’s role in bone development. Importantly, we observed no difference between wild-type embryos or cells (Rb+/c;Mox2+/+) and heterozygous animals or cells (Rb+/c−;Mox2+/Cre) in any of our in vivo or in vitro experiments, and therefore have used wild-type animals as controls in our study.
Initially, we examined skeletons of wild-type and Rbc−/c− embryos at e17.5 by alizarin red staining of bone and alcian blue staining of cartilage. Compared to wild-type littermate controls, the e17.5 Rbc−/c− embryos displayed less ossification in a variety of bones (Figure 1). These include the frontal and parietal calvarial bones of the skull (Figure 1A) that arise through intramembraneous ossification and the hyoid bone (Figure 1B) that develops by endochondral ossification. These defects were partially penetrant, as 9 of 13 Rbc−/c− embryos exhibited the decreased ossification. Moreover, other bones in the Rbc−/c− embryos, including the pterygoid bone and palatine process in the head and the xiphoid process of the sternum, were appropriately ossified but demonstrated an abnormal structure (Figure 1C,D). These abnormal structures were observed in all 13 Rbc−/c− embryos examined. Finally, several other bones such as the long bones of the forelimbs and hindlimbs did not exhibit any differing phenotypes between the Rbc−/c− and wild-type embryos. It is possible that certain embryonic bones, such as the limbs, are less susceptible to the effects of Rb loss than others, perhaps due to the compensation effects of p107 and p130. Alternatively, the Mox2-Cre transgene may be less efficient in some settings.
Figure 1. Deletion of Rb causes defects in embryonic bone development.

(A–D) Alizarin red (bone) and alcian blue (cartilage) staining of embryos. e17.5 Rbc−/c− mice exhibit less ossification in the cranium (A) and hyoid bone (B). The difference in hyoid bone ossification at e17.5 is demonstrated by the bar in (B). Rbc−/c− embryos at e17.5 and e18.5 display aberrant formation of bones in the head (ventral view of head in C) and sternum (D). The aberrantly shaped or missing palatine process in the Rbc−/c− embryos is circled in (C). (E) Pregnant mothers were injected at e18 with calcein for 12 hours. Coronal sections of the frontal bone of e18.5 mice were analyzed for calcein incorporation. Rbc−/c− embryos incorporate less calcein than their wild-type littermates. 2X magnification shown. The distance from the front of calcein incorporation (arrow) to the midline of the suture was measured in 9 Rbc−/c− and 9 wild-type embryo sections. These data are represented in the histogram. Error bars signify one standard deviation. * denotes a statistically significant difference, p< 0.001. Abbreviations: fr, frontal bone; pa, parietal bone; pp, palatine process; pt, pterygoid bone; xp, xiphoid process. WT = Rb+/c;Mox2+/+, Rb = Rbc−/c−;Mox2+/Cre.
To further explore the defects that were observed in the Rbc−/c− embryos, we examined the skeletons of mutant embryos at other developmental stages. At earlier time points, e15.5 and e16.5, the Rbc−/c− embryos displayed all of the bone defects described above (data not shown). At the later timepoints, e18.5 and e19.5/birth, the phenotype was altered somewhat: we still observed aberrantly developed bones, such as the pterygoid, palatine process, and xiphoid process (Figure 1C,D; data not shown) with nearly complete penetrance (7 of 8 e18.5 Rbc−/c− embryos). However, we observed a similar alizarin red staining in the calvaria and hyoid bone of Rbc−/c− embryos versus wild-type littermate controls (Figure 1A,B; data not shown). We considered two explanations for this latter observation. The first possibility was that pRb-loss initially impaired or delayed bone differentiation, but this defect was then corrected by acceleration in the rate of bone deposition after e17.5. The second possibility was that pRb-loss impaired bone differentiation at all developmental stages, but this impairment was not apparent at later time points because the alizarin red detection method is more qualitative than quantitative. In other words, by e18.5 there was some ossification in the appropriate regions of the Rbc−/c− calvaria and hyoid bone but the level of deposited bone was still lower than in the wild-type controls. To distinguish between these two possibilities, we directly assessed the rate of new bone formation after e18.5 using calcein incorporation. Calcein is a fluorescent compound that can be injected into an animal and is then incorporated into newly forming bones. We analyzed the amount of calcein incorporation into the frontal bone of e18.5 embryos 12 hours after the calcein injection of pregnant females. Notably, the Rbc−/c− frontal bones incorporated significantly less calcein compared to wild-type littermates (Figure 1E). Similar results were obtained when calcein was injected approximately 12 hours prior to birth (data not shown). These data indicate that pRb loss does not cause an acceleration in frontal bone formation in the late stages of gestation. Instead, the rate of ossification remains considerably lower than that observed in wild-type embryos. Taken together, our data indicate that the loss of pRb causes a defect in the rate of ossification and/or proper formation of several bones throughout embryonic skeletal development.
The loss of pRb affects an early step in the differentiation of osteoblasts in vivo
Notably, pRb-loss impairs the development of bones that arise through two distinct mechanisms, termed endrochondral (e.g. the hyoid) and intramembraneous (e.g. the calvaria) ossification. The former is influenced by three cell types: chondrocytes, which form an essential cartilage template, osteoblasts, which differentiate to secrete the bone matrix, and osteoclasts, which oppose bone formation by degrading and reabsorbing bone. In contrast, intramembranous ossification is influenced by osteoblasts and osteoclasts but occurs in a cartilage-independent manner. This fact, along with the apparently normal development of the cartilage skeleton within Rbc−/c− embryos (Figure 1B–D; data not shown), suggests that a chondrocyte defect cannot fully account for the defective bone development. Therefore, we examined both osteoblast and osteoclast function. To assess osteoclast levels, we screened the frontal bones of e17.5 embryos for the presence of tartrate-resistant acid phosphatase (TRAP) activity, an osteoclast-specific marker. There were no active osteoclasts present in either the wild-type or the Rbc−/c− frontal bones (Supplementary Figure 1). Thus, the decreased ossification in Rbc−/c− embryos is likely not due to either cartilage defects or increased osteoclast activity.
Given these findings, we next screened e17.5 frontal bones for the presence of osteoblast-specific markers. Two early markers of differentiating osteoblasts are alkaline phosphatase (ALP) activity and Collagen1a1 (Col1) mRNA expression. The activity and expression, respectively, of these two markers were significantly decreased in the Rbc−/c− frontal bone compared to those in wild-type sections (Figure 2). Moreover, the expression levels of Osteopontin (OPN), an early to mid-differentiation marker, were also typically downregulated in the Rbc−/c− embryos relative to wild-type controls (data not shown). These data indicate that osteoblast differentiation is perturbed in Rbc−/c− embryos at the earliest stages of the pathway.
Figure 2. pRb-deficient frontal bones display decreased levels of osteoblast markers.
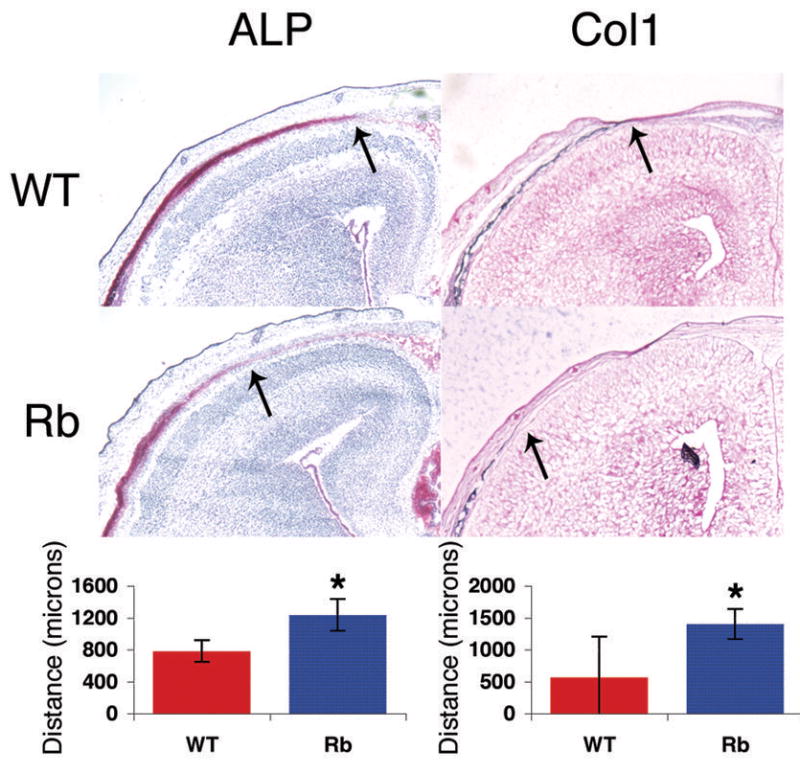
Coronal sections of frontal bones from e17.5 embryos were assessed by histochemical analysis of alkaline phosphatase activity (left column) and in situ analysis of Collagen1a1 mRNA (right column). Rbc−/c− frontal bone sections (bottom row) exhibit decreased levels of both markers compared to wild-type (top row). 2X magnification shown. The distance from the front of activity or expression (arrows) to the midline of the suture was measured in at least 8 embryo pairs for Col1 and 12 for ALP. These data are represented in the histogram. Error bars signify one standard deviation. * denotes a statistically significant difference, p< 0.01. WT = Rb+/c; Mox2+/+, Rb = Rbc−/c−;Mox2+/Cre.
pRb-deficient osteoblasts differentiate to a greater extent than wild-type cells in vitro
Our in vivo data demonstrate that an early step in osteoblast differentiation is affected. One possibility is that pRb regulates osteoblast differentiation directly. For example, it has been reported previously that pRb can interact with and coactivate Runx2/CBFA1, one of the transcription factors essential for osteoblast differentiation (20,21). To further dissect the role of pRb in osteoblast differentiation, we utilized a well-defined and often used in vitro osteoblast differentiation system. Specifically, primary cells were isolated from the calvaria of wild-type and Rbc−/c− embryos and expanded. 250,000 cells were plated to three-cm tissue culture dishes and then induced to differentiate upon confluency. In this system, bone-like calcium deposits are secreted by fully differentiated osteoblasts and can be analyzed by alizarin red staining. Based on our in vivo data and previous in vitro differentiation studies with fibroblasts (20), we anticipated that Rbc−/c− osteoblasts would differentiate to a lesser extent than wild-type cells. Contrary to this hypothesis, however, the Rbc−/c− osteoblasts secreted a greater number of calcium deposits than wild-type osteoblasts based on the alizarin red staining (Figure 3A).
Figure 3. Rbc−/c− primary osteoblasts differentiate to a greater extent than wild-type.

(A) Terminal differentiation of primary calvarial osteoblasts was determined by alizarin red staining of secreted calcium deposits from 0 to 35 days. Rbc−/c− osteoblasts (bottom row) secrete a greater number of calcium deposits than wild-type osteoblasts (top row). (B) Quantitative RT-PCR results of bone marker expression levels from wild-type (red bars) and Rbc−/c− (blue bars) osteoblasts during differentiation. Rbc−/c− osteoblasts express greater mRNA levels of Runx2, Osterix, Osteopontin, and Osteocalcin but not Alkaline Phosphatase or Collagen1a1 compared to wild-type osteoblasts. Ubiquitin was used as an internal control to normalize for RNA levels within the samples. Each time point is an average of four reactions. Error bars signify one standard deviation. Graphs shown depict results from a representative littermate pair. WT = Rb+/c;Mox2+/+, Rb = Rbc−/c−;Mox2+/Cre. (C) Rbc/c primary calvarial osteoblasts were infected with adenovirus expressing either the Cre recombinase enzyme or GFP two days prior to differentiation. Terminal differentiation was assessed by alizarin red staining. Rbc/c osteoblasts acutely ablated for pRb (bottom row) secrete a greater number of calcium deposits than control infected osteoblasts (top row). (D) Quantitative RT-PCR analysis, performed as described in (B). Osteoblasts acutely ablated for pRb (blue bars) express greater mRNA levels of Runx2, Osterix, Osteopontin, and Osteocalcin but not Alkaline Phosphatase or Collagen1a1 compared to control infected osteoblasts (red bars).
We then used quantitative RT-PCR to analyze the mRNA levels of several osteoblast markers during the differentiation of these cells. Although the transcriptional levels of Alp and Col1 were unchanged, the Rbc−/c− osteoblasts exhibited significantly greater levels of expression for several other osteoblast genes compared to the wild-type cells (Figure 3B). Notably, Runx2 and Osterix (OSX), two transcription factors that are necessary to induce osteoblast differentiation (28–30), were upregulated in the Rbc−/c− cells from the earliest stages of the differentiation process (Figure 3B). Runx2 and OSX have been shown to induce the transcription of downstream osteoblast differentiation genes (28,30,31). In accordance with these findings, we observed the increased expression of the early/mid- and late-differentiation markers, Osteopontin (OPN) and Osteocalcin (OC) respectively, in the Rbc−/c− osteoblasts. Together, these data suggest that osteoblasts deficient for pRb differentiate to a greater extent than wild-type cells in vitro, and this correlates with the increased transcriptional levels of Runx2, OSX, and their downstream targets.
Acute ablation of pRb promotes the differentiation of osteoblasts in vitro
The wild-type and Rbc−/c− osteoblasts were prepared at e17.5, when there was a significant difference in the degree of calvarial differentiation (Figure 1A). This raised the possibility that the increased in vitro differentiation of the Rbc−/c− versus wild-type cells simply reflected the presence of a larger pool of progenitor osteoblasts in the Rbc−/c− versus wild-type calvaria. To address this hypothesis, we isolated conditional Rbc/c osteoblasts. These cells were brought to confluence and then infected with either a control adenovirus containing GFP (Adeno-GFP) or one expressing the Cre recombinase gene (Adeno-Cre). This strategy yielded parallel populations of control and Rbc−/c− osteoblasts that had identical starting numbers of progenitors. Consistent with previous studies (7), we found that the Adeno-Cre was sufficient to acutely ablate pRb within two days of infection (data not shown). Therefore, two days post-infection (denoted Day 0 in Figures 3C,D and 4D–F) we placed the confluent wild-type and Rbc−/c− cells in differentiation media. The acutely ablated Rbc−/c− osteoblasts differentiated to a greater extent than the control infected Rbc/c cells, just as we had observed with the germline Rbc−/c− osteoblasts (compare Figures 3C and 3A). Moreover, the acutely ablated Rbc−/c− cells expressed increased levels of Runx2, OSX, OPN, and OC relative to the Adeno-GFP infected cells in a comparable manner to that observed in the germline Rbc−/c− osteoblasts (compare Figures 3D and 3B). These data show that loss of pRb acts in an intrinsic manner to increase the differentiation of primary osteoblast cultures in vitro.
Figure 4. Confluent osteoblasts in vitro exhibit excess proliferation upon loss of pRb.

(A) Immunofluorescence analysis of BrdU incorporation in differentiating osteoblasts. Wild-type (top two rows) and Rbc−/c− (bottom two rows) osteoblasts were treated with BrdU (green) for 24 hours at the indicated time points during differentiation in vitro. Nuclei are stained with DAPI (blue). 20X magnification shown. (B) Quantitation of the immunofluorescence analysis in (A). A minimum of 250 cells was counted from each of three or more separate images for each sample. A greater percentage of Rbc−/c− osteoblasts incorporate BrdU compared to wild-type cells at all time points. (C) Quantitative RT-PCR analysis was performed as described in Figure 3. Rbc−/c− osteoblasts (blue bars) express greater mRNA levels of cyclin E and cyclin A relative to wild-type osteoblasts (red bars) during in vitro differentiation. (D) Nuclei of mock infected (top two rows) and acutely ablated (bottom two rows) Rbc/c osteoblasts were stained for BrdU (green) and DAPI (blue). 20X magnification shown. (E) Quantitation of the immunofluorescence analysis in (D). A greater percentage of Rbc/c osteoblast nuclei acutely ablated for pRb (blue bars) stain positively for BrdU incorporation than control nuclei (red bars). (F) Quantitative RT-PCR demonstrates that acutely ablated Rbc/c osteoblasts (blue bars) express greater mRNA levels of cyclin E and cyclin A compared to mock infected osteoblasts (red bars). All error bars signify one standard deviation. * denotes a statistically significant difference, p<0.05. WT = Rb+/c;Mox2+/+, Rb = Rbc−/c−;Mox2+/Cre.
Depletion of pRb in progenitor osteoblasts causes cell cycle exit defects in vitro
We aimed to understand the molecular changes that accompanied this increased differentiation. One possibility is that pRb possesses a cell cycle-independent repressive function in osteoblast differentiation. In this manner, loss of pRb would allow for the deregulated increase in osteoblast genes such as Runx2 and OSX. We have attempted several experiments to test the potential contribution of this interaction, including conducting chromatin immunoprecipitations (ChIP) of Runx2 at osteoblast-specific promoters in wild-type, Rbc−/c−, and Rbc−/c−;E2f1−/− calvarial preparations (data not shown). These studies did not yield any evidence that Rb-loss altered Runx2 promoter-binding activity. Moreover, we did not detect any pRb binding to the Runx2 and OSX promoters. This latter, negative ChIP result is not particularly informative, since pRb ChIP works poorly in murine cells. However, the Runx2 and OSX promoter both lack conventional E2F binding sites. Thus, while these observations do not rule out a direct, repressive role for pRb in osteoblast differentiation in vitro, we have no data to support this model.
A second potential cause of the observed increase in osteoblast differentiation in vitro upon pRb loss may be related to cell cycle defects. Notably, the increased density of osteoblast cultures is known to enhance their differentiation (32,33). We hypothesized that loss of pRb may affect the normal confluence arrest of the calvarial cells, leading to an increase in proliferation and consequently, an increase in cell density. Thus, we compared the proliferation of wild-type versus germline Rbc−/c− cells throughout the differentiation process. At all time points, we found that a higher proportion of the Rbc−/c− osteoblast nuclei incorporated BrdU than the wild-type controls (Figure 4A,B). In agreement with these findings, the Rbc−/c− osteoblasts showed elevated levels of cyclin A and cyclin E mRNAs (Figure 4C). Finally, total cell counts during the initiation of differentiation demonstrated an increase in the total number of cells present in Rbc−/c− confluent cultures compared to wild-types (Table 2). Similar results in all of these assays were observed in the analyses of osteoblasts acutely ablated for pRb (Figure 4D–F; Table 2). Thus, we conclude that pRb-loss increases the proliferation and, consequently, the density of confluent osteoblast cultures, thereby leading to an increase in primary calvarial osteoblast differentiation in vitro. Notably, the increased proliferation in Rbc−/c− cultures is not perpetual, as the percent of proliferating cells does decrease to almost zero by day 35 (Figure 4, data not shown). This suggests that compensatory mechanisms, perhaps through the pocket proteins p107 and p130, exist to eventually enable cell cycle exit in the osteoblasts.
Table 2.
Cell numbers at Day 0 of differentiation*
| Germline | Conditional | |||
|---|---|---|---|---|
| Genotype | Wild-type | Rbc−/c− | Adeno-GFP | Adeno-Cre |
| Cell Count (X1000) | 481 ±16.5 | 656 ± 14.1 | 483 ± 24.5 | 579 ± 17.6 |
250,000 cells were plated onto a three-cm tissue culture dish and allowed to reach confluency (typically four days later). For “Germline” cells, this confluency arrest constituted Day 0 of differentiation, and the number of cells was ascertained. For “Conditional” cells (Rbc/c) at confluence, adenovirus containing either GFP or Cre recombinase was added to the media. Two days after adenovirus addition (designated as Day 0 of differentiation) cells were counted. Average cell counts from at least 3 separate experiments ± standard deviation are shown.
The loss of Rb prevents osteoblasts from properly exiting the cell cycle in vivo
Having established a likely basis for the increased differentiation of pRb-deficient osteoblasts in vitro, we wished to determine whether a similar mechanism could explain the impaired bone development in vivo. Specifically, since appropriate cell cycle exit is important for the early stages of osteoblast differentiation in vivo, we hypothesized that pRb loss might impair cell cycle exit in vivo and cause a negative impact on bone formation. Thus, to assess cell cycle progression in vivo, we analyzed coronal sections of e17.5 frontal bones for 5-Bromo-2-deoxyuridine (BrdU), which incorporates into newly synthesized DNA during S phase. Embryos deficient for pRb exhibited a significantly greater percentage of osteoblast nuclei that incorporated BrdU compared to the wild-type embryos (Figure 5A). We also tested frontal bone sections for protein expression of PCNA, a known proliferation marker. Consistent with our BrdU data, we observed a greater number of Rbc−/c− osteoblast nuclei that stained positively for PCNA compared to wild-type nuclei (Figure 5B). Interestingly, at the apex of the frontal bone (the midline of the skill) where most of the osteoprogenitors were still proliferating, we did not observe a difference in BrdU or PCNA staining between the wild-type and Rbc−/c− embryos. (data not shown). This would indicate that the loss of pRb does not affect the proliferation rate of osteoprogenitors but does affect their ability to properly exit the cell cycle and remain exited from the cell cycle. We did not observe any proliferative differences between Rbc−/c− and wild-type forelimbs (data not shown), corresponding with our finding that there was no difference in the forelimbs based on alizarin red staining.
Figure 5. pRb-deficient osteoblasts do not properly exit the cell cycle in vivo.
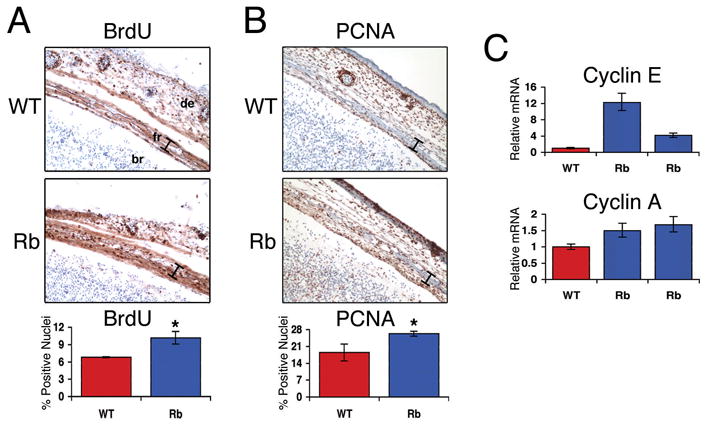
(A,B) Immunohistochemical analysis of BrdU incorporation (A) or PCNA protein expression (B) in coronal sections of frontal bones from e17.5 embryos. Pregnant females were injected with BrdU for two hours. Rbc−/c− frontal bones (bottom) exhibit a greater number of nuclei positively staining for BrdU or PCNA than wild-type littermates (top). 20X magnification shown. Frontal bones are marked with the bar. Quantified results from 4 pairs of Rbc−/c− and wild-type frontal bone sections are represented in the histograms. Error bars signify one standard deviation. * denotes a statistically significant difference, p<0.05. (C) Quantitative RT-PCR analysis of cyclin E (top) and cyclin A (bottom) mRNA levels from Rbc−/c− and control littermates. mRNA was isolated from the calvaria of e16.5 embryos. Analysis performed as described in Figure 3. Rbc−/c− calvaria (blue bars) express increased levels of cyclin A and cyclin E relative to wild-type littermates (red bars). Error bars signify one standard deviation. Abbreviations: br, brain; de, dermis; fr, frontal bone. WT = Rb+/c;Mox2+/+, Rb = Rbc−/c−;Mox2+/Cre.
We also extracted RNA from the calvaria of Rbc−/c− and wild-type embryos to examine the transcript levels of cyclin A and cyclin E. Like PCNA, these transcripts are specifically induced in proliferating cells. Rbc−/c− calvaria typically expressed greater mRNA levels of cyclin A and cyclin E than wild-type skulls (Figure 5C). Importantly, the unrestricted cell cycle progression in Rbc−/c− frontal bones was not associated with an apoptotic response, as determined by TUNEL staining (data not shown). These data suggest that pRb-deficiency impairs osteoblasts from exiting the cell cycle in vivo at the appropriate developmental stage.
Deletion of E2f1 suppresses the cell cycle and ossification defects in Rbc−/c− embryos
The cell cycle regulatory activity of pRb is known to be at least partially dependent on its ability to suppress the E2F transcription factors and prevent the activation of genes such as PCNA, cyclin A and cyclin E that control cell cycle progression. E2F1 is an archetypal member of the E2F family. It is bound to and inhibited by pRb in arrested cells, and it contributes to the activation of target genes once pRb is inactivated by either mitogenic signaling in wild-type cells or genetic lesions in tumor cells. Previous work has demonstrated that the loss of E2F1 can suppress the ectopic cell cycles arising from the loss of Rb in other tissues (34). We found that Rb and E2f1 are both expressed in the calvaria (Supplemental Figure 2). Thus, we crossed a mouse possessing a deletion of E2f1 into our conditional Rb model, and we then examined the compound mutant embryos to determine if E2F activity contributes to the excess proliferation and ossification defects arising in the Rbc−/c− embryos.
First, we assessed the level of cellular proliferation in the embryonic osteoblasts through analysis of both BrdU incorporation and PCNA expression in frontal bone sections from e17.5 embryos (Figure 6A). These two assay methods yielded highly concordant results. First, there was no significant difference in the levels of either BrdU- or PCNA-positive nuclei in the wild-type versus the E2f1−/− osteoblasts. Thus, loss of E2f1 alone appears insufficient to perturb osteoblast proliferation. Second, consistent with our prior analysis, proliferating osteoblasts were present at significantly higher levels in the Rbc−/c− frontal bone compared to the wild-type and E2f1−/− controls. Finally, the deletion of E2f1 was sufficient to almost fully suppress the excess proliferation arising in the Rbc−/c− embryos. The loss of E2f1 on its own or in the Rbc−/c− background did not affect proliferation at the apex of the frontal bone (data not shown), suggesting that the rate of progenitor proliferation remained unaffected. Therefore, we conclude that inappropriate activation of E2F1 contributes to the inability of pRb-deficient osteoprogenitors to properly exit the cell cycle in vivo.
Figure 6. Deletion of E2f1 suppresses the bone defects due to the loss of pRb.
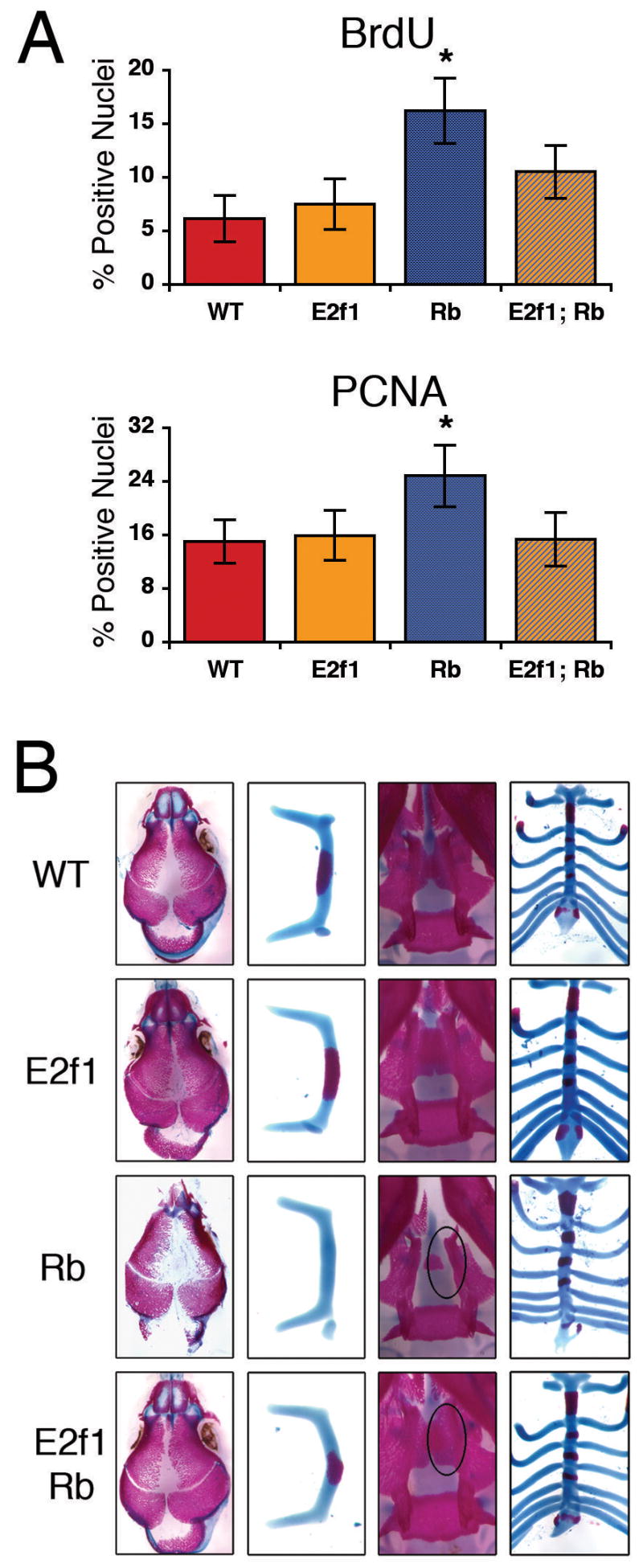
(A) Immunohistochemical analysis of BrdU incorporation (top) or PCNA protein expression (bottom) in coronal sections of frontal bones from e17.5 embryos, performed with 4 to 6 samples of each genotype. Deletion of E2f1 suppresses the increased BrdU incorporation and PCNA expression observed in Rbc−/c− frontal bone osteoblasts. (B) Skeletal staining of e17.5 embryos as described in Figure 1. Deletion of E2f1 suppresses the decreased ossification found in the Rbc−/c− calvaria (first column) and hyoid bone (second column). Deletion of E2f1 also suppresses the aberrant formation of the palatine process and pterygoid bone (third column) and xiphoid process (fourth column) observed in Rbc−/c− skeletons. An aberrant palatine process in the Rbc−/c− and a suppressed palatine process in the double mutant are circled in the respective third column figures. Error bars signify one standard deviation. * denotes a statistically significant difference between Rbc−/c− and wild-type, E2f1−/−, or Rbc−/c−;E2f1−/−, p < 0.05. WT = Rb+/c;Mox2+/+;E2f1+/+, E2f1 = Rb+/c;Mox2+/+;E2f1−/−, Rb = Rbc−/c−;Mox2+/Cre;E2f1+/+, RbE2f1 = Rbc−/c−;Mox2+/Cre;E2f1−/−.
We then assessed whether E2f1-inactivation modulated the Rbc−/c− embryonic skeletal defects observed at e17.5 (Fig 7B). Consistent with the absence of any proliferation defects, the deletion of E2f1 alone did not cause any detectable defects in skeletal development. As observed previously, Rb deficiency caused decreased ossification in the skull and hyoid and aberrant formation of the xiphoid process, palatine process, and pterygoid bone. Notably, in almost all E2f1−/−;Rbc−/c− double mutant embryos (12 of 13), the reduced ossification was partially or completely ameliorated (Figure 6B, first two columns). Moreover, approximately 40% (5 of 13) of the double mutants exhibited normal formation of the palatine process, pterygoid bone, and the xiphoid process was completely normal (Figure 6B, latter two columns). Taken together, these data demonstrate that deletion of Rb causes defects in embryonic skeletal development that are due, at least in part, to the inappropriate release of E2F1.
DISCUSSION
The Rb locus is mutated or altered in greater than 70% of all osteosarcomas (3,4). Moreover, several in vitro studies implicate pRb and the pocket proteins in osteoblast differentiation (18–21). Given these observations, we used the Mox2+/Cre transgene to conditionally inactivate Rb in the Rbc/c embryo proper, but not in the placenta, and thereby generate pRb-deficient embryos that survive until birth. This conditional strategy allows us to assess pRb’s role in bone development in vivo and primary osteoblast differentiation in vitro. Our analyses reveal a role for pRb in the promotion of osteogenesis via the regulation of proper cell cycle exit.
In the developing embryo, the loss of pRb impaired bone formation in a manner that caused two types of defects. Some bones, such as the pterygoid bone, palatine process and xiphoid process, developed abnormally and were misshapen, whereas the skull and hyoid bone exhibited decreased bone formation. The decreased ossification in the Rbc−/c− frontal bone was accompanied by reduced alkaline phosphatase activity and decreased levels of Col1 and OPN mRNA. Previous studies have shown that deletion of the pRb-related proteins, p107 and p130, or overexpression of E2F1 affect chondrocyte differentiation and development (35–37). Although our data do not rule out a role for pRb in cartilage development, they clearly show that pRb plays a role in bone development that is independent of chondrocytes. Specifically, Rbc−/c− skeletons did not show any apparent defects in cartilage formation, and several of the affected bones form via intramembranous ossification, a process that does not involve chondrocytes. Moreover, the bone defects in the Rbc−/c− frontal bone, and presumably in other affected bones, were not the result of increased osteoclast activity or apoptosis. Therefore, our data suggest that the loss of Rb impairs osteoblast differentiation in vivo at the earliest stages of the pathway.
One caveat of the in vivo studies is that they do not prove that pRb’s requirement for osteoblast differentiation is cell autonomous. To address this issue, we determined how the loss of pRb affects the differentiation of primary osteoblasts in vitro. Given our in vivo defects and the prior observation that pRb-deficient MEFs were impaired in their ability to undergo osteogenesis (20), we anticipated that primary osteoblasts isolated from Rbc−/c− embryos would display an impaired differentiation phenotype in vitro. However, the exact opposite was observed: the Rbc−/c− osteoblasts differentiated to a greater extent than the wild-type controls. Importantly, we found that the acute ablation of Rb in confluent osteoblasts was sufficient to trigger increased differentiation. These data show that loss of pRb acts in a cell autonomous manner to promote osteoblast differentiation in vitro.
Our study shows that two distinct molecular changes accompany the improved in vitro differentiation upon loss of pRb. First, we observe a dramatic upregulation of osteoblast genes, such as Runx2 and OSX, in differentiating pRb-deficient osteoblasts to levels that are sometimes not reached by wild-type cells. At this time, we do not know if the extreme upregulation in Rbc−/c− cultures is due to an increased ability of individual cells to induce osteoblast genes, an increased percentage of terminally differentiated cells in the culture, or both. Interestingly, in these in vitro assays, pRb loss clearly induces some (e.g Runx2 and OSX) but not all (ALP and Col1) osteoblast genes. The reason for this differential response is unclear. However, we note that, even prior to the induction of differentiation, the ALP and Col1 mRNAs are present at much higher levels in the cultured osteoblasts than in the endogenous calveria. This suggests that in vitro culture somehow induces ALP and Col1 expression or that it selects for a subpopulation of the calverial cells that are committed to the osteoblast lineage and therefore have high ALP and Col1 expression.
The second molecular change that accompanies the improved in vitro differentiation of pRb-deficient osteoblasts is an increase in the fraction of cells that are proliferating and the sustained presence of proliferating cells at later timepoints in the differentiation process. Since the density of osteoblasts has been reported to correlate positively with their ability to differentiate in vitro (32,33), we believe that the increased proliferation of the pRb-deficient osteoblasts contributes to their improved differentiation by increasing the density of the confluent cells. We tried two distinct approaches to directly test this model. First, we attempted to maintain the Rbc/c osteoblasts in the presence of anti-proliferative drugs prior to the ablation of pRb. However, the experiment requires several days of drug treatment to which the cells faired poorly. Second, since our in vivo data indicate that deletion of E2f1 suppresses excess proliferation due to the loss of Rb, we analyzed the consequence of E2f1 deficiency in acutely ablated and germline deleted Rbc−/c− osteoblasts. Unfortunately, the loss of E2f1 did not suppress the cell cycle defects of osteoblasts in this in vitro setting. Thus, we have been unable to prove that a cell cycle exit defect can account for the increased differentiation of Rb-depleted osteoblasts in vitro. Despite this limitation, our in vivo studies do provide strong support for this model. Specifically, we find that osteoblasts of the Rbc−/c− frontal bone fail to exit the cell cycle at the appropriate stage of development, and we can completely suppress both the proliferation defect and the decreased ossification of the skull and hyoid bones through inactivation of E2f1, a known pRb target and proliferation inducer.
If a cell cycle exit defect is the major underlying cause of both the in vitro and in vivo defects, how does this account for the apparently opposing effects on bone differentiation seen in the two settings? One possibility is that this is an aberrant consequence of the in vitro culture that somehow enables the Rb-deficient cells to overcome their differentiation defect. The alternative possibility, which we favor, is that pRB-loss affects cells at early and late stages of osteoblast differentiation in a differential manner, and the in vivo and the in vitro studies highlight the defects in these distinct populations. Specifically, we hypothesize that pRB loss leads to ectopic proliferation that prevents early progenitors from entering osteoblast differentiation but concomitantly enhances the differentiation of late stage osteoblasts. In this model, the in vitro cultures could favor analysis of the late stage osteoblasts, thereby showing that pRb loss promotes osteoblast differentiation. In contrast, the in vivo phenotype would be more complex. Specifically, our data clearly show ectopic proliferation of Rbc−/c− cells in the developing frontal bone, but we cannot know whether these represent uncommitted early progenitor cells or differentiating osteoblasts that are proliferating inappropriately. In fact, we believe that both populations co-exist. In this event, at early timepoints in the bone differentiation process, the shortage of committed osteoprogenitors would initially impair bone formation–exactly as we observe in the late stage embryos. However, as the committed osteoblasts accumulate, their increased proliferation would eventually allow, and perhaps ultimately enhance, bone differentiation as we observe in the in vitro assays. Unfortunately, because the Rbc−/c− animals die at birth, we cannot determine whether their osteoblast density and bone deposition ultimately exceeds that seen in wildtype animals.
There is considerable evidence to suggest that pRb plays a direct role in regulating the transcriptional programs that control osteoblast differentiation. Most compelling is the finding that pRb can positively regulate Runx2 in vitro (20,21). Our findings do not discount the possibility that pRb plays a direct role in bone differentiation through Runx2, or some other mechanism, or that this might contribute to the bone defects we observe in vivo. However, they argue that the primary role of pRb in bone differentiation is to inhibit E2F1 and thereby facilitate cell cycle exit. Given that Rb-inactivation is observed in a large proportion of osteosarcomas, it will be important to develop additional models that allow comparison of the mechanisms by which loss of Rb affects bone development versus osteosarcoma formation.
MATERIALS AND METHODS
Animal maintenance and histological preparations
The generation of Rbc/c and Mox2-Cre mice has been described previously (7,27). Rbc/c and E2f1−/− mice were provided by Tyler Jacks. Mox2-Cre mice were purchased from Jackson Laboratories. Gestation was dated by detection of a vaginal plug. Pregnant mice were injected with 10 μl/gm body weight of 5 mg/ml 5-Bromo-2′-deoxyuridine (BrdU) in phosphate buffered solution (PBS) two hours prior to tissue collection. For calcein incorporation, pregnant mice were injected with 10 μl/gm body weight of 2.5 mg/ml calcein 12 or 24 hours prior to tissue collection. Collected embryonic tissue was fixed in 4% paraformaldehyde (PFA) and embedded in OCT. Frozen sections were cut at 6–8 microns except those for in situ analysis, cut at 10–12 microns. The morphology of the brain and presphenoid bone were used to ensure that equivalent planes of the frontal bone were analyzed in all samples.
Histological analyses
Enzymatic ALP assays were performed on unfixed frozen sections. Briefly, 0.06g sodium nitrite was dissolved into 1.5 ml of water and added to 600 μl of 50 mg/ml of new fuchsin (Sigma) in 2M HCl. This solution was added to 210 ml Tris buffer (pH 9.0). Finally, 1.8 ml of 83.3 mg/ml Naphthol AS-Bi-Phosphate (Sigma) in DMF (Sigma) was added. Sections were incubated with this overall solution for 15 minutes, washed in PBS and counterstained with hematoxylin. Immunohistochemical analyses were performed using antibodies against BrdU (1:50 347580, BD Biosciences) and PCNA (1:2000 sc56, Santa Cruz) as described (38). For collagen1a1 in situs, digoxigenin-11-UTP-labeled single-strand ribo-probe was prepared (probe was a gift from B. Olsen), and hybridization was carried out overnight in 50% formamide at 55°C. Washing, detection, staining and mounting of slides were carried out as described previously (39). Statistical significance was determined using the two sample Student’s T Testwith two-tailed distribution and unequal variance.
Skeletal staining
Embryos were sacrificed, skinned, and eviscerated. The remaining tissue was fixed in 95% ethanol for 4 days, transferred to acetone for 3 days, and subsequently transferred to staining solution (final volume of 0.015% alcian blue 8GX (Sigma), 0.005% alizarin red S (Sigma) and 5% glacial acetic acid in ethanol) at 37°C for two days and room temperature for a third day. Tissue was cleared in 1% potassium hydroxide for several days and ultimately stored in glycerol.
Calvarial preparations and culture
Calvaria from e17.5 embryos were removed, treated with several rounds of collagenase/trypsin digests at 37°C, and plated onto 6-well plates. Cells were grown and expanded in αMEM with 10% fetal bovine serum and Pen/Strep. For differentiation, 250,000 cells were plated onto 3cm tissue culture plates. Upon reaching confluence, calvarial osteoblasts were treated with media supplemented with 50 μg/ml of ascorbic acid and 10 mM β-glycerol-phosphate. Adenovirus (U. of Iowa Gene Transfer Vector Core) was added to the media at 100 plaque-forming units per cell and washed away 24 hours later. To assay for calcium deposits, plates were stained with 1% alizarin red S solution (pH 5.0).
Immunofluorescence
For BrdU incorporation, osteoblasts were plated onto coverslips prior to achieving confluence. BrdU was added to the media (final concentration of 10 μM) and incubated for 24 hours prior to 4% PFA fixation. Antigen was detected using antibody against BrdU (1:50 347580, BD Biosciences) with Texas Red-X goat anti-mouse secondary (1:1000, Invitrogen). Statistical significance was determined using the Student’s T Test.
Quantitative real-time PCR
RNA was isolated from differentiation plates using the Qiagen RNEasy kit. First-strand cDNA was transcribed from 1 μg of RNA using Superscript III Reverse Transcriptase (Invitrogen) following product instructions. Quantitative RT-PCR with 20–100 ng cDNA was performed using SYBR Green (Applied Biosystems). Reactions were run on the ABI Prism 7000 Sequence Detection System and analyzed using the 7000 SDS software. Primers are listed in Table 1.
Table 1.
Quantitative RT-PCR Primer Pairs
| Gene | Primer Sequence |
|---|---|
| Alkaline Phosphatase | For: TCT CCA GAC CCT GCA ACC TC |
| Rev: CAT CCT GAG CAG ACC TGG TC | |
| Collagen1a1 | For: CGA GTC ACA CCG GAA CTT GG |
| Rev: GCA GGC AGG GCC AAT GTC TA | |
| Cyclin A | For: AGT TTG ATA GAT GCT GAC CC |
| Rev: TAG GTC TGG TGA AGG TCC | |
| Cyclin E | For: TGT TTT TGC AAG ACC CAG ATG A |
| Rev: GGC TGA CTG CTA TCC TCG CT | |
| Osteocalcin | For: CTC TGT CTC TCT GAC CTC ACA G |
| Rev: CAG GTC CTA AAT AGT GAT ACC G | |
| Osteopontin | For: TGC TTT TGC CTG TTT GGC AT |
| Rev: TTC TGT GGC GCA AGG AGA TT | |
| Osterix | For: GCA AGG CTT CGC ATC TGA AA |
| Rev: AAC TTC TTC TCC CGG GTG TGA | |
| Runx2 | For: TGA GAT TTG TGG GCC GGA |
| Rev: TCT GTG CCT TCT TGG TTC CC | |
| Ubiquitin | For: TGG CTA TTA ATT ATT CGG TCT GCA T |
| Rev: GCA AGT GGC TAG AGT GCA GAG TAA |
Supplementary Material
Acknowledgments
We are grateful to the U. of Iowa Gene Transfer Vector Core (GTVC) for providing the adenovirus, Tyler Jacks for the Rbc/c and E2f1 mutant strains, Eric Olsen for in situ probes and also members of the Lees lab for helpful discussion throughout this work. We also thank Phil Iaquinta for assistance with ChIP and bioinformatics research. This project was supported by grants to J.A.L. (GM53204, CA121921) from the National Cancer Institute. S.B. is a David H. Koch Graduate Fellow and J.A.L. is a Ludwig Scholar at MIT.
References
- 1.Weinberg RA. The retinoblastoma gene and gene product. Cancer Surv. 1992;12:43–57. [PubMed] [Google Scholar]
- 2.Gurney JG, Severson RK, Davis S, Robison LL. Incidence of cancer in children in the United States. Sex-, race-, and 1-year age-specific rates by histologic type. Cancer. 1995 Apr 15;75(8):2186–95. doi: 10.1002/1097-0142(19950415)75:8<2186::aid-cncr2820750825>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 3.Belchis DA, Meece CA, Benko FA, Rogan PK, Williams RA, Gocke CD. Loss of heterozygosity and microsatellite instability at the retinoblastoma locus in osteosarcomas. Diagn Mol Pathol. 1996 Sep;5(3):214–9. doi: 10.1097/00019606-199609000-00011. [DOI] [PubMed] [Google Scholar]
- 4.Feugeas O, Guriec N, Babin-Boilletot A, et al. Loss of heterozygosity of the RB gene is a poor prognostic factor in patients with osteosarcoma. J Clin Oncol. 1996 Feb;14(2):467–72. doi: 10.1200/JCO.1996.14.2.467. Erratum in: J Clin Oncol 1996 Aug:14(8):2411. [DOI] [PubMed] [Google Scholar]
- 5.Lipinski MM, Jacks T. The retinoblastoma gene family in differentiation and development. Oncogene. 1999 Dec 20;18(55):7873–82. doi: 10.1038/sj.onc.1203244. [DOI] [PubMed] [Google Scholar]
- 6.Huang HJ, Yee JK, Shew JY, et al. Suppression of the neoplastic phenotype by replacement of the RB gene in human cancer cells. Science. 1988 Dec 16;242(4885):1563–6. doi: 10.1126/science.3201247. [DOI] [PubMed] [Google Scholar]
- 7.Sage J, Miller AL, Pérez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 2003 Jul 10;424(6945):223–8. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- 8.Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002 Jan;3(1):11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- 9.Clark AJ, Doyle KM, Humbert PO. Cell-intrinsic requirement for pRb in erythropoiesis. Blood. 2004 Sep 1;104(5):1324–6. doi: 10.1182/blood-2004-02-0618. [DOI] [PubMed] [Google Scholar]
- 10.Spike BT, Dirlam A, Dibling BC, et al. The Rb tumor suppressor is required for stress erythropoiesis. EMBO J. 2004 Oct 27;23(21):4319–29. doi: 10.1038/sj.emboj.7600432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huh MS, Parker MH, Scimè A, Parks R, Rudnicki MA. Rb is required for progression through myogenic differentiation but not maintenance of terminal differentiation. J Cell Biol. 2004 Sep 13;166(6):865–76. doi: 10.1083/jcb.200403004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haigis K, Sage J, Glickman J, Shafer S, Jacks T. The related retinoblastoma (pRb) and p130 proteins cooperate to regulate homeostasis in the intestinal epithelium. J Biol Chem. 2006 Jan 6;281(1):638–47. doi: 10.1074/jbc.M509053200. [DOI] [PubMed] [Google Scholar]
- 13.Yang HS, Hinds PW. pRb-mediated control of epithelial cell proliferation and Indian hedgehog expression in mouse intestinal development. BMC Dev Biol. 2007 Jan 26;7:6. doi: 10.1186/1471-213X-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morgenbesser SD, Williams BO, Jacks T, DePinho RA. p53-dependent apoptosis produced by Rb-deficiency in the developing mouse lens. Nature. 1994 Sep 1;371(6492):72–4. doi: 10.1038/371072a0. [DOI] [PubMed] [Google Scholar]
- 15.Kim TA, Lim J, Ota S, et al. NRP/B, a novel nuclear matrix protein, associates with p110(RB) and is involved in neuronal differentiation. J Cell Biol. 1998 May 4;141(3):553–66. doi: 10.1083/jcb.141.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee EY, Hu N, Yuan SS, et al. Dual roles of the retinoblastoma protein in cell cycle regulation and neuron differentiation. Genes Dev. 1994 Sep 1;8(17):2008–21. doi: 10.1101/gad.8.17.2008. [DOI] [PubMed] [Google Scholar]
- 17.Chen PL, Riley DJ, Chen Y, Lee WH. Retinoblastoma protein positively regulates terminal adipocyte differentiation through direct interaction with C/EBPs. Genes Dev. 1996 Nov 1;10(21):2794–804. doi: 10.1101/gad.10.21.2794. [DOI] [PubMed] [Google Scholar]
- 18.Feuerbach D, Loetscher E, Buerki K, Sampath TK, Feyen JH. Establishment and characterization of conditionally immortalized stromal cell lines from a temperature-sensitive TAg transgenic mouse. J Bone Miner Res. 1997 Feb;12(2):179–90. doi: 10.1359/jbmr.1997.12.2.179. [DOI] [PubMed] [Google Scholar]
- 19.Beck GR, Jr, Sullivan EC, Moran E, Zerler B. Relationship between alkaline phosphatase levels, osteopontin expression, and mineralization in differentiating MC3T3-E1 osteoblasts. J Cell Biochem. 1998 Feb 1;68(2):269–80. doi: 10.1002/(sici)1097-4644(19980201)68:2<269::aid-jcb13>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 20.Thomas DM, Carty SA, Piscopo DM, et al. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol Cell. 2001 Aug;8(2):303–16. doi: 10.1016/s1097-2765(01)00327-6. [DOI] [PubMed] [Google Scholar]
- 21.Luan Y, Yu XP, Xu K, et al. The retinoblastoma protein is an essential mediator of osteogenesis that links the p204 protein to the Cbfa1 transcription factor thereby increasing its activity. J Biol Chem. 2007 Jun 8;282(23):16860–70. doi: 10.1074/jbc.M610943200. [DOI] [PubMed] [Google Scholar]
- 22.Clarke AR, Maandag ER, van Roon M, et al. Requirement for a functional Rb-1 gene in murine development. Nature. 1992 Sep 24;359(6393):328–30. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- 23.Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992 Sep 24;359(6393):295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 24.Lee EY, Chang CY, Hu N, et al. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992 Sep 24;359(6393):288–94. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 25.Wenzel PL, Wu L, de Bruin A, et al. Rb is critical in a mammalian tissue stem cell population. Genes Dev. 2007 Jan 1;21(1):85–97. doi: 10.1101/gad.1485307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu L, de Bruin A, Saavedra HI, et al. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature. 2003 Feb 27;421(6926):942–7. doi: 10.1038/nature01417. [DOI] [PubMed] [Google Scholar]
- 27.Tallquist MD, Soriano P. Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis. 2000 Feb;26(2):113–5. doi: 10.1002/(sici)1526-968x(200002)26:2<113::aid-gene3>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 28.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997 May 30;89(5):747–54. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 29.Otto F, Thornell AP, Crompton T, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997 May 30;89(5):765–71. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 30.Nakashima K, Zhou X, Kunkel G, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002 Jan 11;108(1):17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 31.Wang X, Kua HY, Hu Y, et al. p53 functions as a negative regulator of osteoblastogenesis, osteoblast-dependent osteoclastogenesis, and bone remodeling. J Cell Biol. 2006 Jan 2;172(1):115–25. doi: 10.1083/jcb.200507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerber I, ap Gwynn I. Influence of cell isolation, cell culture density, and cell nutrition on differentiation of rat calvarial osteoblast-like cells in vitro. Eur Cell Mater. 2001 Jul 24;2:10–20. doi: 10.22203/ecm.v002a02. [DOI] [PubMed] [Google Scholar]
- 33.Purpura KA, Aubin JE, Zandstra PW. Sustained in vitro expansion of bone progenitors is cell density dependent. Stem Cells. 2004;22(1):39–50. doi: 10.1634/stemcells.22-1-39. [DOI] [PubMed] [Google Scholar]
- 34.Tsai KY, Hu Y, Macleod KF, Crowley D, Yamasaki L, Jacks T. Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol Cell. 1998 Sep;2(3):293–304. doi: 10.1016/s1097-2765(00)80274-9. [DOI] [PubMed] [Google Scholar]
- 35.Cobrinik D, Lee MH, Hannon G, et al. Shared role of the pRB-related p130 and p107 proteins in limb development. Genes Dev. 1996 Jul 1;10(13):1633–44. doi: 10.1101/gad.10.13.1633. [DOI] [PubMed] [Google Scholar]
- 36.Rossi F, MacLean HE, Yuan W, et al. p107 and p130 Coordinately regulate proliferation, Cbfa1 expression, and hypertrophic differentiation during endochondral bone development. Dev Biol. 2002 Jul 15;247(2):271–85. doi: 10.1006/dbio.2002.0691. [DOI] [PubMed] [Google Scholar]
- 37.Scheijen B, Bronk M, van der Meer T, Bernards R. Constitutive E2F1 overexpression delays endochondral bone formation by inhibiting chondrocyte differentiation. Mol Cell Biol. 2003 May;23(10):3656–68. doi: 10.1128/MCB.23.10.3656-3668.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Danielian PS, Bender Kim CF, Caron AM, Vasile E, Bronson RT, Lees JA. E2f4 is required for normal development of the airway epithelium. Dev Biol. 2007 May 15;305(2):564–76. doi: 10.1016/j.ydbio.2007.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Böhme K, Li Y, Oh PS, Olsen BR. Primary structure of the long and short splice variants of mouse collagen XII and their tissue-specific expression during embryonic development. Dev Dyn. 1995 Dec;204(4):432–45. doi: 10.1002/aja.1002040409. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.