

Abstract
Although the dominant approach to drug development is the design of compounds selective for a given target, compounds targeting more than one biological process may have superior efficacy, or alternatively a better safety profile than standard selective compounds. Here, this possibility has been explored with respect to the endocannabinoid system and pain. Compounds inhibiting the enzyme fatty acid amide hydrolase (FAAH), by increasing local endocannabinoid tone, produce potentially useful effects in models of inflammatory and possibly neuropathic pain. Local increases in levels of the endocannabinoid anandamide potentiate the actions of cyclooxygenase inhibitors, raising the possibility that compounds inhibiting both FAAH and cyclooxygenase can be as effective as non-steroidal anti-inflammatory drugs but with a reduced cyclooxygenase inhibitory ‘load’. An ibuprofen analogue active in models of visceral pain and with FAAH and cyclooxygenase inhibitory properties has been identified. Another approach, built in to the experimental analgesic compound N-arachidonoylserotonin, is the combination of FAAH inhibitory and transient receptor potential vanilloid type 1 antagonist properties. Although finding the right balance of actions upon the two targets is a key to success, it is hoped that dual-action compounds of the types illustrated in this review will prove to be useful analgesic drugs.
Keywords: endocannabinoid, anandamide, fatty acid amide hydrolase, cyclooxygenase, non-steroidal anti-inflammatory drugs, transient receptor potential vanilloid type 1, inflammatory pain
Introduction
A standard approach to drug development has been the design of compounds that selectively affect a given target, with the aim of achieving a therapeutic effect with an acceptable safety profile. However, this approach has not always been successful, and indeed the efficacy of selective compounds may in some cases be less than suggested on the basis of published clinical trials (Turner et al., 2008). One way of both increasing efficacy and improving safety, long used in cancer treatment regimes, is the use of more than one drug. The drugs can be given either separately or in single tablets containing more than one active ingredient, such as combinations of angiotensin converting inhibitors and diuretics for the treatment of hypertension in patients where monotherapy is insufficient. While the former approach is highly flexible in terms of dosing, the drawback of both of these strategies is the potential for a large pharmacokinetic variability that is associated with the concomitant use of separate drugs.
An alternative that avoids this difficulty is the development of drugs that target more than one molecular mechanism (for a review of ‘designed multiple ligands’ from a pharmaceutical industrial standpoint, see Morphy and Rankovic, 2005). A recent example of a designed multiple ligand is the compound tapentadol [(-)-(1R,2R)-3-(3-dimethylamino-1-ethyl-2-methyl-propyl)-phenol hydrochloride], which both activates µ-opioid receptors and inhibits the reuptake of noradrenaline (Tzschentke et al., 2007). The rationale for the compound, that the two pharmacodynamic effects will improve efficacy (or the range of pain states that are amenable to treatment) and/or reduce the µ-opioid ‘load’ for a given degree of analgesia, was based upon the properties of tramodol, and tapentadol was found to be active in models of both inflammatory and neuropathic pain (Tzschentke et al., 2007). Most importantly, the development of tolerance to the analgesic effect of tapentadol in the chronic constriction injury model of neuropathic pain was slower than seen with an equi-analgesic dose of morphine (Tzschentke et al., 2007).
In the present review, the case is presented for the development of dual-action analgesic compounds, where one of the targets is the endocannabinoid metabolizing enzyme fatty acid amide hydrolase (FAAH).
Fatty acid amide hydrolase as the endocannabinoid target
It has been known since ancient times that extracts of cannabis have antinociceptive properties (see Reynolds 1890; Zias et al., 1993), and the buccal extract Sativex™ is now used in Canada for the treatment of pain in patients with multiple sclerosis. The main drawback of this approach is that the psychotropic actions of cannabis, which are mediated via activation of cannabinoid (CB)1 receptors, together with concerns about long-term effects of cannabis treatment, place a limitation upon the dose that can be given, and hence the pain relief obtained (review, see McCarberg and Barkin, 2007). CB1 receptors are part of the endocannabinoid system, which is defined here as comprising the endogenous CB ligands arachidonoylethanolamide (anandamide, AEA) and 2-arachidonoylglycerol (2-AG), their target receptors (CB1 and CB2 receptors) and their synthetic and degradative enzymes (review, see Pacher et al., 2006). This definition is restrictive, because it does not include candidate endocannabinoid ligands such as noladin ether and docosatetraenoylethanolamine or putative CB receptors such as GPR55 and the endothelial non-CB1 non-CB2 receptor, but it is sufficient for this review. Potential targets for the treatment of pain, where beneficial effects can be separated from the psychotropic effects resulting from central CB1 receptor activation include peripherally located CB1 receptors (see Agarwal et al., 2007), CB2 receptors (review see Guindon and Hohmann, 2008) and FAAH (see below).
FAAH hydrolyses AEA and related N-acylethanolamines to their corresponding fatty acids and at least in some tissues, such as the rat paw and periaqueductal grey, contributes significantly to the hydrolysis of 2-AG (Schmid et al., 1985; Deutsch and Chin, 1993; Cravatt et al., 1996; Jhaveri et al., 2006; Maione et al., 2006). In 1999, Walker et al. reported that inflammatory pain produced by formalin injection into the paw produced a release of AEA in the periaqueductal grey. The authors concluded that the ‘release of anandamide in a pain suppression circuit suggests that drugs that inhibit the reuptake of anandamide or block its degradation may form the basis of a modern pharmacotherapy for pain’ (Walker et al. 1999). Consistent with this conclusion, mice lacking FAAH show increased brain and spinal cord AEA levels, reduced pain-related behaviour in response to intraplantar administration of either formalin or carrageenan, a reduced sensitivity to thermal pain and a hypoalgesic profile in a model of visceral pain, but do not show a hypoalgesic response in the chronic constriction injury model of neuropathic pain (Cravatt et al., 2001; 2004; Lichtman et al., 2004a; Naidu and Lichtman, 2007). Further studies using mice lacking peripheral FAAH alone indicated that the reduced sensitivity to thermal pain was mediated centrally (Cravatt et al., 2004). In contrast, the reduced oedema response to carrageenan seen in the FAAH−/− mice (Lichtman et al., 2004a) was a peripherally mediated effect (Cravatt et al., 2004).
A variety of selective inhibitors of FAAH have been described in the literature, and, judging from the patent literature and recent publications of high-throughput screening strategies for the enzyme (see e.g. Kage et al., 2007 for a method with a capacity >100 000 compounds per day), more compounds are on the way (for a recent review see Di Marzo, 2008). Selective FAAH inhibitors also produce increased levels of brain and spinal cord AEA (see e.g. Kathuria et al., 2003; Lichtman et al., 2004b) and show useful analgesic properties. With respect to inflammatory pain, Jayamanne et al. (2006) reported that the potent selective FAAH inhibitor URB597 (cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester) reduced mechanical allodynia and thermal hyperalgesia produced by the injection 24 h previously of complete Freund's adjuvant. Motor performance on the rotorod test was not affected (Jayamanne et al., 2006), nor other markers of a general central CB1 receptor activation (Kathuria et al., 2003). This effect of URB597 in the models of inflammatory pain was blocked completely by a combination of the CB1 receptor antagonist AM251 [N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide] and SR144528 (N-[(1S)-endo-1,3,3-trimethyl bicyclo [2.2.1] heptan-2-yl]-5-(4-chloro-3-methylphenyl)-1-(4-methylben-zyl)-pyrazole-3-carboxamide). The latter compound was used ostensibly to block CB2 receptors, although it also has an off-target action upon peroxisome proliferator-activated receptor α (PPARα) (LoVerme et al., 2006). Given that palmitoylethanolamide, which has analgesic actions, is also a substrate for FAAH (Schmid et al., 1985) and can activate PPARα (LoVerme et al., 2006), antagonist effects of SR144528 cannot be ascribed with certainty to actions upon CB2 receptors. URB597 has also been found to potentiate non-opioid stress-induced analgesia by a mechanism involving CB1 receptors (Hohmann et al., 2005), and to show antinociceptive effects in models of visceral pain (Naidu and Lichtman, 2007; see also Haller et al., 2006). Finally, URB597 has been shown to increase the levels of 2-AG in the paws of sham-operated rats (Jhaveri et al., 2006). 2-AG is antinociceptive when given intraplantarly (Guindon et al., 2007). Interestingly, prostaglandin E2 1-glyceryl ester [the cyclooxygenase (COX)-2-catalysed product of 1-AG] produces mechanical allodynia and thermal hyperalgesia when given by this route (Hu et al., 2008). Increased 2-AG levels have also been seen following administration of URB597 into the periaqueductal grey, an administration that produces both analgesia and hyperalgesia, dependent upon the dose used (Maione et al., 2006).
OL135 (1-oxo-1[5-(2-pyridyl)-2-yl]-7-phenylheptane), is a potent FAAH inhibitor reported to show less off-target actions than URB597, and with a reversible mode of action in contrast to the irreversible action of the latter (Lichtman et al., 2004b). OL135 was efficacious in both phases of the formalin test in mice in a manner blocked by the CB1 receptor antagonist rimonabant, but not by SR144528 (Lichtman et al., 2004b). Other selective FAAH inhibitors have also been reported to be active in the formalin test in a manner blocked by rimonabant (e.g. Sit et al., 2007), and so it can be considered as a class effect. In a rat model of peripheral tissue damage (mild thermal injury), OL135 reduced the tactile allodynia, but this was blocked by the opioid antagonist naloxone rather than by rimonabant and SR144528 (Chang et al., 2006). Because no effect of OL135 upon the allodynia was seen in FAAH−/− mice (in contrast to wild-type mice), the simplest explanation for these results is that the compound produces a mobilization of endogenous opioids secondary to inhibition of FAAH (Chang et al., 2006).
Effects of FAAH inhibitors in models of neuropathic pain are less clear-cut. URB597 was reported initially not to reduce allodynia in rats with partial sciatic nerve ligations, in contrast to the marked effects of the CB1/CB2 receptor agonist HU210 [(6aR)-trans-3-(1,1-dimethylheptyl)-6a,7,10,10a-tetrahyd ro-1-hydroxy-6,6-dimethyl-6H-dibenzo[b,d]pyran-9-methanol] (Jayamanne et al., 2006). A subsequent paper, however, reported that the repeated oral administration of URB597 to mice following chronic constriction injury reduced allodynia in a manner blocked by rimonabant, and to a similar extent as seen with gabapentin (Russo et al., 2007). Local administration of URB597 into the paw of rats with allodynia subsequent to spinal nerve ligations was without effect upon the response of wide-dynamic range spinal neurons to mechanical stimulation, whereas the compound was efficacious when given spinally, in a manner reduced by spinally pre-administered AM251 (Jhaveri et al., 2006). OL135 given i.p. (intraperitoneally) showed efficacy, in a manner blocked by SR144538 or by naloxone, but not by rimonabant, in the spinal nerve ligation model of neuropathic pain (Chang et al., 2006) and repeated treatment with the compound also reduced mechanical allodynia and thermal hyperalgesia in rats with chronic constriction injury, whereas the effects of URB597 were restricted to thermal analgesia alone at the dose used (Maione et al., 2007). Other novel compounds (bisarylimidazole and benzyl piperidine derivatives) with nmol·L−1 potency towards FAAH have also been reported to be efficacious in reducing tactile allodynia in spinally ligated rats (Sit et al., 2007; Timmons et al., 2008). Thus, the efficacies of FAAH inhibitors per se in neuropathic pain are dependent upon the compound, mode of administration and/or the animal model used.
Choice of the second target
From the above discussion, FAAH is a good target for the development of novel drugs aimed at the treatment of inflammatory pain, whereas the potential of FAAH inhibitors in the treatment of neuropathic pain is less clear. The choice of the second target for a dual-action drug can be based upon a number of considerations, such as the potential to provide synergistic or additional effects, a profile of unwanted actions (for targets with drugs already on the market) that can be reduced rather than potentiated by the FAAH inhibitory component of the drug, and last but not least the availability of lead compounds with which to develop drugs with the desired profiles. The ability of AEA to interact both with FAAH and with the pain-related targets COX-2 and transient receptor potential vanilloid type 1 (TRPV1) (see Figure 1 for a schematic), would suggest that these targets are reasonable ones to consider as examples of potential second targets.
Figure 1.
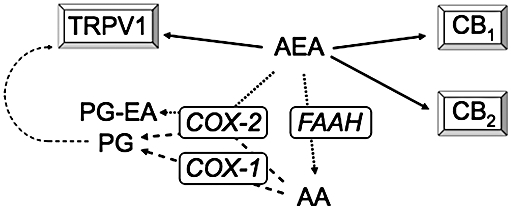
Interaction of AEA with CB and TRPV1 receptors and its metabolism by FAAH and COX-2. Normally, AEA is an order of magnitude more potent as an agonist at CB receptors than at TRPV1 receptors. However inflammatory conditions sensitize TRPV1 receptors to AEA (Singh Tahim et al., 2005 and references therein). AEA is primarily metabolized by FAAH to produce AA, but can also act as a substrate for COX-2 to produce PG-EAs. It is not known whether these metabolites, which are biologically active, are involved in inflammatory pain, but the COX-2 pathway represents an alternative metabolic route for AEA when FAAH is inhibited (review, see Fowler, 2007). Other metabolic pathways, such as lipoxygenase and P450 oxidative metabolism, are not shown in the figure for reasons of simplicity. AA, arachidonic acid; AEA, anandamide; CB, cannabinoid; COX, cyclooxygenase; FAAH, fatty acid amide hydrolase; PG, prostaglandin; PG-EA, prostaglandin ethanolamide; TRPV1, transient receptor potential vanilloid 1.
Combined FAAH/COX inhibitors
Non-steroidal anti-inflammatory drugs (NSAIDs, comprising both non-selective and COX-2-selective inhibitors) are a standard and effective treatment for inflammatory pain. The long-term use of non-selective NSAIDs is associated with an unacceptably high incidence of gastrointestinal complications, although this has been reduced by concomitant medication with proton pump inhibitors (Ray et al., 2007). A second issue, noted originally for the COX-2 selective compound rofecoxib, is an increased risk for cardiovascular events due at least in part to the pressor effects of NSAIDs (White, 2007). These unwanted actions remain an obstacle to long-term NSAID use, and novel compounds are sorely needed (for a review on possible approaches targeting components of the prostaglandin system other than COX, see Zeilhofer and Brune, 2006). There is evidence to support the contention that dual-action FAAH/COX inhibitors may be useful in this respect. Guindon et al. (2006a) reported that the intraplantar administration of AEA and the NSAID ibuprofen reduced the inflammatory pain response in the formalin model in an additive manner, and that the effects of the combined administration of the two were blocked by AM251. A subsequent study demonstrated that the COX-2 inhibitor rofecoxib also acted additively with AEA in this model, and that the combination of AEA with either ibuprofen or rofecoxib produced increases in the tissue levels of AEA and the related N-acylethanolamines oleoylethanolamide and palmitoylethanolamide that were greater than seen after administration of either AEA or NSAID alone (Guindon et al., 2006b). The ability of ibuprofen to inhibit FAAH (Fowler et al., 1997) may contribute to this effect. More recently, synergistic effects were reported between systemically administered URB597 and the NSAID diclofenac in a model of visceral pain in the mouse. The ED50 values for the two compounds given together in a 1:1 ratio were approximately ninefold lower than their corresponding ED50 values when given separately (Naidu and Lichtman, 2007).
On the basis of the discussion above, a case can be made that compounds with inhibitory actions towards both FAAH and COX can provide the same degree of efficacy in inflammatory pain as NSAIDs while reducing the COX-inhibitory ‘load’ and hence incidence of gastrointestinal disturbances and potential cardiovascular complications. Cannabinoids have long been known to have actions on the cardiovascular system (Ashton and Smith, 2007). With respect to FAAH, old (28–31 months) FAAH−/− mice show better haemodynamics (such as a higher stroke work and cardiac output) than old FAAH+/+ mice, while no differences were seen for 2–3 month-old animals (Bátkai et al., 2007). Young FAAH−/− mice are more sensitive to the hypotensive actions of AEA than FAAH+/+ mice (Pacher et al., 2005). URB597 reduces the mean arterial pressure of spontaneously and angiotensin II-induced hypertensive rats without affecting the blood pressure in normotensive animals (Bátkai et al., 2004). In vitro, relaxation of mesenteric arteries by AEA is potentiated both by URB597 and by COX-2 inhibitors (Ho and Randall, 2007). AEA can also act as a pulmonary vasoconstrictor as a result of its metabolism by FAAH and then COX-2 (Wahn et al., 2005). Thus, FAAH inhibitors are unlikely to compound, and may even negate, the pressor effects of COX inhibitors, although this suggestion requires further investigation in vivo.
One way of identifying a useful lead compound capable of inhibiting both FAAH and COX-2 is to start with a compound with these actions, but where one predominates, and thereafter optimize the compound with respect to the weaker action. AM404 [N-(4-hydroxyphenyl) arachidonylamide, a compound that inhibits the cellular accumulation of AEA] could be considered in this respect, because it affects both inflammatory and neuropathic pain (La Rana et al., 2006) and is both an FAAH inhibitor (by acting as a competing substrate) and a COX inhibitor (Lang et al., 1999; Högestätt et al., 2005). Intriguingly, AM404 is a metabolite of paracetamol (Högestätt et al., 2005). Whether or not this metabolic pathway is sufficient in terms of capacity to explain the CB1 receptor antagonist-sensitive actions of paracetamol in some pain models (Ottani et al., 2006; Dani et al., 2007) is as yet unclear. With respect to AM404 itself, the compound has off-target effects including agonism at TRPV1 receptors and cell-toxic actions over the same concentration range (Zygmunt et al., 2000; De Lago et al., 2006), which limits its usefulness. An alternative is to start with a COX inhibitor like ibuprofen or indomethacin, because both compounds inhibit FAAH, particularly at low pH, such as is found in inflamed tissue (Fowler et al., 1997; 2003a; see Figure 2). It is unlikely in our view that the FAAH-inhibitory components of these compounds are sufficient to contribute to the actions of these drugs when given orally to patients. However, FAAH inhibition may play a role when the drug is given locally. In this respect, Gühring et al. (2002) reported that the effects of indomethacin (9 µmol·L−1, given by spinal microdialysis) on the pain response to formalin was not reversed by prostaglandin E2, but was blocked by AM251. Furthermore, the effect of indomethacin was not seen in CB1−/− mice (Gühring et al., 2002).
Figure 2.
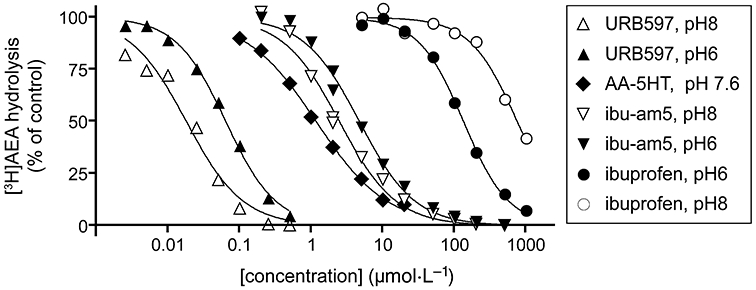
Inhibition of 2 µmol·L−1 AEA hydrolysis in rat brain (minus cerebellum) homogenates by URB597, AA-5-HT, ibu-am5 and ibuprofen. With the exception of AA-5-HT, the compounds were preincubated with the enzyme for 10 min prior to addition of substrate. The values (means), which were all obtained in the same laboratory to facilitate comparison, are redrawn from the raw data of Fowler et al. (2003b), Paylor et al. (2006) and Holt et al. (2007). The pH dependencies of URB597 and ibuprofen (and lack thereof for ibu-am5) are also seen in intact cells (Holt & Fowler, 2003; Paylor et al., 2006; Holt et al., 2007). AA-5-HT shows very little pH dependency for inhibition of AEA hydrolysis by rat brain homogenates (Holt et al., 2001). The inhibition of FAAH by URB597 is time-dependent, and the pH dependency shown in the figure is primarily due to differences in the rate constants for the covalent binding phase (Paylor et al., 2006). AA-5-HT also shows a time-dependent inhibition of FAAH (Bisogno et al., 1998) whereas ibuprofen and ibu-am5 do not (Fowler et al., 1997; Holt et al., 2007). The pH dependencies of URB597 and ibuprofen may have relevance in vivo given that the pH of inflamed tissue is lower than normal tissue (Häbler, 1929). AEA, anandamide; AA-5-HT, N-arachidonoylserotonin; ibu-am5, N-(3-methylpyridin-2-yl)-2-(4′-isobutylphenyl)propionamide; URB597, cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester.
In 2003, Cocco et al. reported the synthesis and actions of a series of heteroaromatic ibuprofen amides in a model of visceral pain in the rat. One compound, N-(3-methylpyridin-2-yl)-2-(4′-isobutylphenyl)propionamide (‘ibu-am5’) was significantly more efficacious than the same dose (20 mg·kg−1 i.p.) of ibuprofen. The efficacy of ibu-am5 is also seen in the mouse (Figure 3). The compound also showed a much lower ulcerogenic potency than ibuprofen (Cocco et al., 2003). Subsequently, it was found that ibu-am5 was approximately two orders of magnitude more potent as an FAAH inhibitor than ibuprofen, while the potencies of the two compounds towards COX-1 and -2 were broadly similar (Holt et al., 2007). In intact cells, the compound inhibited the hydrolysis of AEA with an IC50 value of 1.2 µmol·L−1 (Holt et al., 2007). While the potency of ibu-am5 is lower than for compounds like URB597 and OL135, it is similar to the potency for N-arachidonoylserotonin (AA-5-HT; Bisogno et al., 1998; see Figure 2), a compound that is active in vivo (Maione et al., 2007, see the next section). The efficacy of ibu-am5 in pain models other than the visceral pain model have not yet been tested, but the compound can be considered a useful lead compound for the design and evaluation of dual-action FAAH/COX inhibitory agents.
Figure 3.
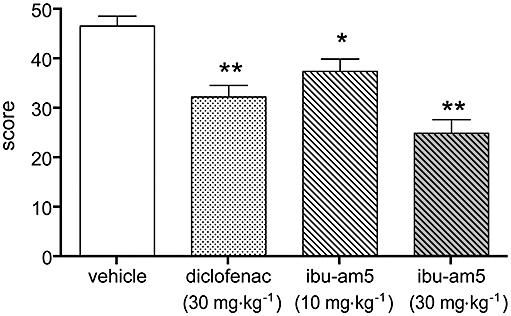
Comparison of the effects of diclofenac and N-(3-methylpyridin-2-yl)-2-(4′-isobutylphenyl)propionamide (ibu-am5) in a model of visceral pain in mice. Animals were treated subcutaneously, with the compounds 60 min prior to the instillation of 0.6% (1 mL 100 g−1, intraperitoneally) acetic acid, and the number of abdominal stretches was scored for 20 min. Data are means ± SEM, n = 8–11. *P < 0.05, **P < 0.01 vs. vehicle anova followed by Scheffe's test (A. Lichtman and V. Onnis, unpubl. data).
FAAH inhibitor/TRPV1 receptor antagonists
TRPV1 receptors are non-selective ion channels located, among other places, in sensory neurons and gate responses to painful stimuli such as heat, low pH and capsaicin, the pungent ingredient of chilli peppers. Use of both genetically modified mice and selective TRPV1 antagonists (as well as capsaicin desensitization experiments) have indicated that this receptor is a promising target for drug development (review, see Immke and Gavva, 2006), although a potential problem is the hyperthermic response elicited by antagonists (Gavva et al., 2008). AEA activates TRPV1 receptors, albeit at higher concentrations than are needed for activation of CB receptors (Zygmunt et al., 1999). However, under inflammatory conditions, such as in the presence of bradykinin and prostaglandin E2, the sensitivity of TRPV1 receptors in sensory neurons to AEA is increased (Singh Tahim et al., 2005), a finding consistent with the ability of protein kinase A and C activation to increase TRPV1 sensitivity to AEA in both heterologous expression systems and in sensory neurons (De Petrocellis et al., 2001; Ahluwalia et al., 2003). In other words, a potential antinociceptive action of an increased AEA resulting from FAAH inhibition may be offset by an increased concomitant TRPV1 receptor activation (see Dinis et al., 2004). The situation is far from simple, not the least given the propensity of TRPV1 to desensitize, and the additional involvement of both spinal and supraspinal receptors in the regulation of pain (Maione et al., 2006; Horvath et al., 2008). Nonetheless, the findings of Singh Tahim et al. (2005) and Dinis et al. (2004), together with the known antinociceptive effects of TRPV1 antagonists, suggest that a compound with FAAH inhibitory/TRPV1 receptor antagonist actions should be more efficacious than an FAAH inhibitor alone.
AA-5-HT was originally described as an FAAH inhibitor of moderate (low µmol·L−1) potency (Bisogno et al., 1998) and has been shown to increase both peripheral and central AEA levels in vivo (Capasso et al., 2005; de Lago et al., 2005). The compound also behaves in the manner expected for an FAAH inhibitor (i.e. blocked by a CB1 receptor antagonist/inverse agonist) when given i.p. in the formalin test of inflammatory pain in the mouse and with respect to its effects in stress-induced analgesia (Suplita et al., 2005; Maione et al., 2007). However, AA-5-HT is a potent (mid-nanomolar) antagonist of TRPV1 receptors expressed in HEK-293 cells (Maione et al., 2007), and this may contribute to the efficacy of the compound in models of both inflammatory (rat) and neuropathic pain (anti-allodynic effect, rat) (Maione et al., 2007). The effects of the compound in the formalin model in the rat and with respect to mechanical allodynia in the rat chronic constriction injury model are somewhat complex, because both AM251 and the TRPV1 antagonist capsazepine could antagonize its actions (Maione et al., 2007). The authors suggested that in these cases, AA-5-HT acted by increasing endocannabinoid tone at CB1 receptors and that the increased AEA levels produced a desensitization of TRPV1 receptors in sensory neurons that contributed to, but were not directly involved in, nociception (Maione et al., 2007). Several analogues of AA-5-HT have been investigated with respect to FAAH inhibition and interaction with TRPV1 receptors, but as yet none have improved on the lead compound (Ortar et al., 2007; see also Fowler et al., 2003b), although the analgesic agent arvanil may be a possible alternative lead in this respect (see Di Marzo et al., 2002).
Conclusions
In this review, a case has been made for novel analgesic compounds targeting FAAH and an additional mediator involved in pain processing. Two examples of additional targets have been presented, COX and TRPV1. These targets are not necessarily the best – indeed, in the wake of the Vioxx debacle there may be some reticence in the pharmaceutical industry to involve itself with COX – but were chosen because there are sufficient data to illustrate the concept. The report of hyperthermia associated with a TRPV1 antagonist in a clinical setting (Gavva et al., 2008) is also worrisome in this respect, although there are preclinical data to suggest that this issue is not a universal phenomenon (Bisogno et al., 1998; Lehto et al., 2008). In theory, compounds combining inhibition of FAAH with effects upon other systems known to be involved in analgesia (such as, for example, activation of opioid receptors) may be of potential interest, although to our knowledge no such compounds with such a profile have been reported in the literature. Similarly, CB2 receptor agonists may be an alternative for the endocannabinoid component of dual-action drugs, given the analgesic profile of such compounds in preclinical models (review, see Guindon and Hohmann, 2008), and that compounds interacting with both CB2 receptors and TRPV1 have been identified (Appendino et al., 2006).
A key issue, of course, is getting the balance right. Are, for example, the relative potencies of AA-5-HT towards FAAH and TRPV1, or ibu-am5 towards FAAH and COX, optimal? Isobolographic analyses of FAAH inhibitors given with either TRPV1 antagonists or NSAIDs (see e.g. Naidu and Lichtman, 2007) should shed light on relative dosages required, but in the end, the only way forward is to investigate the concept in the clinic. We hope the interest of the pharmaceutical industry in the endocannabinoid system will continue to increase, and extend to the development of designed multiple ligands (Morphy and Rankovic, 2005) of the type discussed here.
Acknowledgments
The authors thank the Swedish Research Council (Grant no. 12158, medicine; to CJF), the Research Funds of the Medical Faculty, Umeå University (to CJF) and Grant no.s R01DA15197, P01DA017259, and DA03672 (to AL) for financial support of the author's work on endocannabinoids.
Note added in proof: Our latest data with ibu-am 5 indicate that its effects in visceral pain (Figure 3 are not blocked by rimonabant or SR144528 (A. Lichtman & V. Onnis, unpublished data), suggesting that either the FAAH inhibitory component of this type of compound needs further optimation, or that its beneficial effects involve FAAH substrates other than AEA.
Glossary
Abbreviations:
- AA
arachidonic acid
- AA-5-HT
N-arachidonoylserotonin
- AEA
anandamide (arachidonoylethanolamide)
- 2-AG
2-arachidonoylglycerol
- AM251
N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide
- AM404
N-(4-hydroxyphenyl) arachidonylamide
- CB
cannabinoid
- COX
cyclooxygenase
- FAAH
fatty acid amide hydrolase
- HU210
(6aR)-trans-3-(1,1-dimethylheptyl)-6a,7,10,10a-tetrahydro-1-hydroxy-6,6-dimethyl-6H-dibenzo[b,d]pyran-9-methanol
- ibu-am5
N-(3-methylpyridin-2-yl)-2-(4′-isobutylphenyl)propionamide
- OL135
1-oxo-1[5-(2-pyridyl)-2-yl]-7-phenylheptane
- PEA
palmitoylethanolamide
- PG
prostaglandin
- PG-EA
prostamide (prostaglandin ethanolamide)
- PPAR
peroxisome proliferator-activated receptor
- SR144528
N-[(1S)-endo-1,3,3-trimethyl bicyclo [2.2.1] heptan-2-yl]-5-(4-chloro-3-methylphenyl)-1-(4-methylben-zyl)-pyrazole-3-carboxamide
- TRPV1
transient receptor potential vanilloid type 1
- URB597
cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester
Conflict of interest
CJF, AL and VO state no conflict of interest. PSN is now employed by BMS, Syngene International Ltd. following his postdoctoral period in the laboratory of AL.
References
- Agarwal N, Pacher P, Tegeder I, Amaya F, Constantin CE, Brenner GJ, et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat Neurosci. 2007;10:870–879. doi: 10.1038/nn1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahluwalia J, Urban L, Bevan S, Nagy I. Anandamide regulates neuropeptide release from capsaicin-sensitive primary sensory neurons by activating both the cannabinoid 1 receptor and the vanilloid receptor 1 in vitro. Eur J Neurosci. 2003;17:2611–2618. doi: 10.1046/j.1460-9568.2003.02703.x. [DOI] [PubMed] [Google Scholar]
- Appendino G, Cascio MG, Bacchiega S, Moriello AS, Minassi A, Thomas A, et al. First ‘hybrid’ ligands of vanilloid TRPV1 and cannabinoid CB2 receptors and non-polyunsaturated fatty acid-derived CB2-selective ligands. FEBS Lett. 2006;580:568–574. doi: 10.1016/j.febslet.2005.12.069. [DOI] [PubMed] [Google Scholar]
- Ashton JC, Smith PF. Cannabinoids and cardiovascular disease: the outlook for clinical treatments. Curr Vasc Pharmacol. 2007;5:175–184. doi: 10.2174/157016107781024109. [DOI] [PubMed] [Google Scholar]
- Bátkai S, Pacher P, Osei-Hyiaman D, Radaeva S, Liu J, Harvey-White J, et al. Endocannabinoids acting at cannabinoid-1 receptors regulate cardiovascular function in hypertension. Circulation. 2004;110:1996–2002. doi: 10.1161/01.CIR.0000143230.23252.D2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bátkai S, Rajesh M, Mukhopadhyay P, Haskó G, Liaudet L, Cravatt BF, et al. Decreased age-related cardiac dysfunction, myocardial nitrative stress, inflammatory gene expression, and apoptosis in mice lacking fatty acid amide hydrolase. Am J Physiol Heart Circ Physiol. 2007;293:H909–H918. doi: 10.1152/ajpheart.00373.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Melck D, De Petrocellis L, Bobrov MYu, Gretskaya NM, Bezuglov VV, et al. Arachidonoylserotonin and other novel inhibitors of fatty acid amide hydrolase. Biochem Biophys Res Commun. 1998;248:515–522. doi: 10.1006/bbrc.1998.8874. [DOI] [PubMed] [Google Scholar]
- Capasso R, Matias I, Lutz B, Borrelli F, Capasso F, Marsicano G, et al. Fatty acid amide hydrolase controls mouse intestinal motility in vivo. Gastroenterology. 2005;129:941–951. doi: 10.1053/j.gastro.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Chang L, Luo L, Palmer JA, Sutton S, Wilson SJ, Barbier AJ, et al. Inhibition of fatty acid amide hydrolase produces analgesia by multiple mechanisms. Br J Pharmacol. 2006;148:102–113. doi: 10.1038/sj.bjp.0706699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocco MT, Congiu C, Onnis V, Morelli M, Cauli O. Synthesis of ibuprofen heterocyclic amides and investigation of their analgesic and toxicological properties. Eur J Med Chem. 2003;38:513–518. doi: 10.1016/s0223-5234(03)00074-6. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, et al. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci USA. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Saghatelian A, Hawkins EG, Clement AB, Bracey MH, Lichtman AH. Functional disassociation of the central and peripheral fatty acid amide signaling systems. Proc Natl Acad Sci USA. 2004;101:10821–10826. doi: 10.1073/pnas.0401292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani M, Guindon J, Lambert C, Beaulieu P. The local antinociceptive effects of paracetamol in neuropathic pain are mediated by cannabinoid receptors. Eur J Pharmacol. 2007;573:214–215. doi: 10.1016/j.ejphar.2007.07.012. [DOI] [PubMed] [Google Scholar]
- De Lago E, Gustafsson SB, Fernández-Ruiz J, Nilsson J, Jacobsson SOP, Fowler CJ. Acyl-based anandamide uptake inhibitors cause rapid toxicity to C6 glioma cells at pharmacologically relevant concentrations. J Neurochem. 2006;99:677–688. doi: 10.1111/j.1471-4159.2006.04104.x. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Harrison S, Bisogno T, Tognetto M, Brandi I, Smith GD, et al. The vanilloid receptor (VR1)-mediated effects of anandamide are potently enhanced by the cAMP-dependent protein kinase. J Neurochem. 2001;77:1660–1663. doi: 10.1046/j.1471-4159.2001.00406.x. [DOI] [PubMed] [Google Scholar]
- Deutsch DG, Chin SA. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem Pharmacol. 1993;46:791–796. doi: 10.1016/0006-2952(93)90486-g. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. Targeting the endocannabinoid system: to enhance or reduce? Nat Rev Drug Discov. 2008;7:438–455. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Griffin G, De Petrocellis L, Brandi I, Bisogno T, Williams W, et al. A structure/activity relationship study on arvanil, an endocannabinoid and vanilloid hybrid. J Pharmacol Exp Ther. 2002;300:984–991. doi: 10.1124/jpet.300.3.984. [DOI] [PubMed] [Google Scholar]
- Dinis P, Charrua A, Avelino A, Yaqoob M, Bevan S, Nagy I, et al. Anandamide-evoked activation of vanilloid receptor 1 contributes to the development of bladder hyperreflexia and nociceptive transmission to spinal dorsal horn neurons in cystitis. J Neurosci. 2004;24:11253–11263. doi: 10.1523/JNEUROSCI.2657-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CJ. The contribution of cyclooxygenase-2 to endocannabinoid metabolism and action. Br J Pharmacol. 2007;152:594–601. doi: 10.1038/sj.bjp.0707379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CJ, Tiger G, Stenström A. Ibuprofen inhibits rat brain deamidation of anandamide at pharmacologically relevant concentrations. Mode of inhibition and structure-activity relationship. J Pharmacol Exp Ther. 1997;283:729–734. [PubMed] [Google Scholar]
- Fowler CJ, Holt S, Tiger G. Acidic nonsteroidal anti-inflammatory drugs inhibit rat brain fatty acid amide hydrolase in a pH-dependent manner. J Enzyme Inhib Med Chem. 2003a;18:55–58. doi: 10.1080/1475636021000049726. [DOI] [PubMed] [Google Scholar]
- Fowler CJ, Tiger G, López-Rodríguez ML, Viso A, Ortega-Gutiérrez S, Ramos JA. Inhibition of fatty acid amidohydrolase, the enzyme responsible for the metabolism of the endocannabinoid anandamide, by analogues of arachidonoyl-serotonin. J Enzyme Inhib Med Chem. 2003b;18:225–231. doi: 10.1080/1475636031000080216. [DOI] [PubMed] [Google Scholar]
- Gavva NR, Treanor JJ, Garami A, Fang L, Surapaneni S, Akrami A, et al. Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain. 2008;136:202–210. doi: 10.1016/j.pain.2008.01.024. [DOI] [PubMed] [Google Scholar]
- Gühring H, Hamza M, Sergejeva M, Ates M, Kotalla CE, Ledent C, et al. A role for endocannabinoids in indomethacin-induced spinal antinociception. Eur J Pharmacol. 2002;454:153–163. doi: 10.1016/s0014-2999(02)02485-8. [DOI] [PubMed] [Google Scholar]
- Guindon J, Hohmann AG. Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br J Pharmacol. 2008;153:319–334. doi: 10.1038/sj.bjp.0707531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, De Léan A, Beaulieu P. Local interactions between anandamide, an endocannabinoid, and ibuprofen, a nonsteroidal anti-inflammatory drug, in acute and inflammatory pain. Pain. 2006a;121:85–93. doi: 10.1016/j.pain.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Guindon J, LoVerme J, De Léan A, Piomelli D, Beaulieu P. Synergistic antinociceptive effects of anandamide, an endocannabinoid, and nonsteroidal anti-inflammatory drugs in peripheral tissue: a role for endogenous fatty-acid ethanolamides? Eur J Pharmacol. 2006b;550:68–77. doi: 10.1016/j.ejphar.2006.08.045. [DOI] [PubMed] [Google Scholar]
- Guindon J, Desroches J, Beaulieu P. The antinociceptive effects of intraplantar injections of 2-arachidonoyl glycerol are mediated by cannabinoid CB2 receptors. Br J Pharmacol. 2007;150:693–701. doi: 10.1038/sj.bjp.0706990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häbler C. Über den K- und Ca-gehalt von eiter und exsudaten und seine beziehungen zum entzündungsschmerz. Klin Wochenschr. 1929;8:1569–1572. [Google Scholar]
- Haller VL, Cichewicz DL, Welch SP. Non-cannabinoid CB1, non-cannabinoid CB2 antinociceptice effects of several novel compounds in the PPQ stretch test in mice. Eur J Pharmacol. 2006;546:60–68. doi: 10.1016/j.ejphar.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Ho W-SV, Randall MD. Endothelium-dependent metabolism by endocannabinoid hydrolases and cyclooxygenases limits vasorelaxation to anandamide and 2-arachidonoylglycerol. Br J Pharmacol. 2007;150:641–651. doi: 10.1038/sj.bjp.0707141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Högestätt ED, Jönsson BAG, Ermund A, Andersson DA, Björk H, Alexander JP, et al. Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid amide hydrolase-dependent arachidonic acid conjugation in the nervous system. J Biol Chem. 2005;280:31405–31412. doi: 10.1074/jbc.M501489200. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, et al. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- Holt S, Fowler CJ. Anandamide metabolism by fatty acid amide hydrolase in intact C6 glioma cells. Increased sensitivity to inhibition by ibuprofen and flurbiprofen upon reduction of extra- but not intracellular pH. Naunyn Schmiedebergs Arch Pharmacol. 2003;367:237–244. doi: 10.1007/s00210-002-0686-z. [DOI] [PubMed] [Google Scholar]
- Holt S, Nilsson J, Omeir R, Tiger G, Fowler CJ. Effects of pH on the inhibition of fatty acid amidohydrolase by ibuprofen. Br J Pharmacol. 2001;133:513–520. doi: 10.1038/sj.bjp.0704113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt S, Paylor B, Boldrup L, Alajakku K, Vandevoorde S, Sundström A, et al. Inhibition of fatty acid amide hydrolase, a key endocannabinoid metabolizing enzyme, by analogues of ibuprofen and indomethacin. Eur J Pharmacol. 2007;565:26–36. doi: 10.1016/j.ejphar.2007.02.051. [DOI] [PubMed] [Google Scholar]
- Horvath G, Kekesi G, Nagy E, Benedek G. The role of TRPV1 receptors in the antinociceptive effect of anandamide at spinal level. Pain. 2008;134:277–284. doi: 10.1016/j.pain.2007.04.032. [DOI] [PubMed] [Google Scholar]
- Hu SS-J, Bradshaw HB, Chen JS-C, Tan B, Walker JM. Prostaglandin E2 glycerol ester, an endogenous COX-2 metabolite of 2-arachidonoylglycerol, induces hyperalgesia and modulates NFκB activity. Br J Pharmacol. 2008;153:1538–1549. doi: 10.1038/bjp.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immke DC, Gavva NR. The TRPV1 receptor and nociception. Semin Cell Dev Biol. 2006;17:582–591. doi: 10.1016/j.semcdb.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Jayamanne A, Greenwood R, Mitchell VA, Aslan S, Piomelli D, Vaughan CW. Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br J Pharmacol. 2006;147:281–288. doi: 10.1038/sj.bjp.0706510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri MD, Richardson D, Kendall DA, Barrett DA, Chapman V. Analgesic effects of fatty acid amide hydrolase inhibition in a rat model of neuropathic pain. J Neurosci. 2006;26:13318–13327. doi: 10.1523/JNEUROSCI.3326-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kage KL, Richardson PL, Traphagen L, Severin J, Pereda-Lopez A, Lubben T, et al. A high throughput fluorescent assay for measuring the activity of fatty acid amide hydrolase. J Neurosci Methods. 2007;161:47–54. doi: 10.1016/j.jneumeth.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nature Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- La Rana G, Russo R, Campolongo P, Bortolato M, Mangieri RA, Cuomo V, et al. Modulation of neuropathic and inflammatory pain by the endocannabinoid transport inhibitor AM404 [N-(4-hydroxyphenyl)-eicosa-5,8,11,14-tetraenamide. J Pharmacol Exp Ther. 2006;317:1365–1371. doi: 10.1124/jpet.105.100792. [DOI] [PubMed] [Google Scholar]
- de Lago E, Petrosino S, Valenti M, Morera E, Ortega-Gutierrez S, Fernandez-Ruiz J, et al. Effect of repeated systemic administration of selective inhibitors of endocannabinoid inactivation on rat brain endocannabinoid levels. Biochem Pharmacol. 2005;70:446–452. doi: 10.1016/j.bcp.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Lang W, Qin C, Lin S, Khanolkar AD, Goutopoulos A, Fan P, et al. Substrate specificity and stereoselectivity of rat brain microsomal anandamide amidohydrolase. J Med Chem. 1999;42:896–902. doi: 10.1021/jm980461j. [DOI] [PubMed] [Google Scholar]
- Lehto S, Tamir R, Deng H, Klionsky L, Kuang R, Le A, et al. Antihyperalgesic effects of AMG8562, a novel vanilloid receptor TRPV1 modulator that does not cause hyperthermia in rats. J Pharmacol Exp Ther. 2008;326:218–229. doi: 10.1124/jpet.107.132233. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Shelton CC, Advani T, Cravatt BF. Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor-mediated phenotypic hypoalgesia. Pain. 2004a;109:319–327. doi: 10.1016/j.pain.2004.01.022. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Leung D, Shelton CC, Saghatelian A, Hardouin C, Boger DL, et al. Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J Pharmacol Exp Ther. 2004b;311:441–448. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]
- LoVerme J, Russo R, La Rana G, Fu J, Farthing J, Mattace-Raso G, et al. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor. J Pharmacol Exp Ther. 2006;319:1051–1061. doi: 10.1124/jpet.106.111385. [DOI] [PubMed] [Google Scholar]
- McCarberg BH, Barkin RL. The future of cannabinoids as analgesic agents: a pharmacologic, pharmacokinetic, and pharmacodynamic overview. Am J Ther. 2007;14:475–483. doi: 10.1097/MJT.0b013e3180a5e581. [DOI] [PubMed] [Google Scholar]
- Maione S, Bisogno T, de Novellis V, Palazzo E, Cristino L, Valenti M, et al. Elevation of endocannabinoid levels in the ventrolateral periaqueductal grey through inhibition of fatty acid amide hydrolase affects descending nociceptive pathways via both cannabinoid receptor type 1 and transient receptor potential vanilloid type-1 receptors. J Pharmacol Exp Ther. 2006;316:969–982. doi: 10.1124/jpet.105.093286. [DOI] [PubMed] [Google Scholar]
- Maione S, De Petrocellis L, de Novellis V, Moriello AS, Petrosino S, Palazzo E, et al. Analgesic actions of N-arachidonoyl-serotonin, a fatty acid amide hydrolase inhibitor with antagonistic activity at vanilloid TRPV1 receptors. Br J Pharmacol. 2007;150:766–781. doi: 10.1038/sj.bjp.0707145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem. 2005;48:6523–6543. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- Naidu PS, Lichtman AH. Synergistic antinociceptive effects of URB597 and diclofenac in a mouse visceral pain model. 17th Annual Symposium on the Cannabinoids, Burlington, Vermont, International Cannabinoid Research Society, 2007, #172. Available online at http://cannabinoidsociety.org/SYMPOSIUM.2007/2007.ICRS.Program.and.Abstracts.pdf.
- Ortar G, Cascio MG, De Petrocellis L, Morera E, Rossi F, Schiano-Moriello A, et al. New N-arachidonoylserotonin analogues with potential ‘dual’ mechanism of action against pain. J Med Chem. 2007;50:6554–6569. doi: 10.1021/jm070678q. [DOI] [PubMed] [Google Scholar]
- Ottani A, Leone S, Sandrini M, Ferrari A, Bertolini A. The analgesic activity of paracetamol is prevented by the blockade of cannabinoid CB1 receptors. Eur J Pharmacol. 2006;531:280–281. doi: 10.1016/j.ejphar.2005.12.015. [DOI] [PubMed] [Google Scholar]
- Pacher P, Bátkai S, Osei-Hyiaman D, Offertáler L, Liu J, Harvey-White J, et al. Hemodynamic profile, responsiveness to anandamide, and baroreflex sensitivity of mice lacking fatty acid amide hydrolase. Am J Physiol Heart Circ Physiol. 2005;289:H533–H541. doi: 10.1152/ajpheart.00107.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Bátkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paylor B, Holt S, Fowler CJ. The potency of the fatty acid amide hydrolase inhibitor URB597 is dependent upon the assay pH. Pharmacol Res. 2006;54:481–485. doi: 10.1016/j.phrs.2006.07.006. Corrigendum published in Pharmacol Res55: 80 (2007. [DOI] [PubMed] [Google Scholar]
- Ray WA, Chung CP, Stein CM, Smalley WE, Hall K, Arbogast PG, et al. Risk of peptic ulcer hospitalizations in users of NSAIDs with gastroprotective cotherapy versus coxibs. Gastroenterology. 2007;133:790–798. doi: 10.1053/j.gastro.2007.06.058. [DOI] [PubMed] [Google Scholar]
- Reynolds JR. On the therapeutical uses and toxic effects of canabis indica. Lancet. 1890;135:637–638. [Google Scholar]
- Russo R, Loverme J, La Rana G, Compton TR, Parrott J, Duranti A, et al. The fatty acid amide hydrolase inhibitor URB597 (cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester) reduces neuropathic pain after oral administration in mice. J Pharmacol Exp Ther. 2007;322:236–242. doi: 10.1124/jpet.107.119941. [DOI] [PubMed] [Google Scholar]
- Schmid PC, Zuzarte-Augustin ML, Schmid HHO. Properties of rat liver N-acylethanolamine amidohydrolase. J Biol Chem. 1985;260:14145–14149. [PubMed] [Google Scholar]
- Singh Tahim A, Sántha P, Nagy I. Inflammatory mediators convert anandamide into a potent activator of the vanilloid type 1 transient receptor potential receptor in nociceptive primary sensory neurons. Neuroscience. 2005;136:539–548. doi: 10.1016/j.neuroscience.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Sit SY, Conway C, Bertekap R, Xie K, Bourin C, Burris K, et al. Novel inhibitors of fatty acid amide hydrolase. Bioorg Med Chem Lett. 2007;17:3287–3291. doi: 10.1016/j.bmcl.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Suplita RL, II, Farthing JN, Gutierrez T, Hohmann AG. Inhibition of fatty-acid amide hydrolase enhances cannabinoid stress-induced analgesia: sites of action in the dorsolateral periaqueductal gray and rostral ventromedial medulla. Neuropharmacology. 2005;49:1201–1209. doi: 10.1016/j.neuropharm.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Timmons A, Seirstad M, Apodaca R, Epperson M, Pippel D, Brown S, et al. Novel ketooxazole based inhibitors of fatty acide amide hydrolase (FAAH) Bioorg Med Chem Lett. 2008;18:2109–2113. doi: 10.1016/j.bmcl.2008.01.091. [DOI] [PubMed] [Google Scholar]
- Turner EH, Matthews AM, Linardatos E, Tell RA, Rosenthal R. Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med. 2008;358:252–260. doi: 10.1056/NEJMsa065779. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM, Christoph T, Kögel B, Schiene K, Hennies H-H, Englberger W, et al. )-(1R,2R)-3-(3-dimethylamino-1-ethyl-2-methyl-propyl)-phenol hydrochloride (tapentadol HCl): a novel µ-opioid receptor agonist/norepinephrine reuptake inhibitor with broad-spectrum analgesic properties. J Pharmacol Exp Ther. 2007;323:265–276. doi: 10.1124/jpet.107.126052. [DOI] [PubMed] [Google Scholar]
- Wahn H, Wolf J, Kram F, Frantz S, Wagner JA. The endocannabinoid arachidonoyl ethanolamide (anandamide) increases pulmonary arterial pressure via cyclooxygenase-2 products in isolated rabbit lungs. Am J Physiol Heart. 2005;289:2491–2496. doi: 10.1152/ajpheart.00718.2005. [DOI] [PubMed] [Google Scholar]
- Walker JM, Huang SM, Strangman NM, Tsou K, Sañudo-Peña MC. Pain modulation by release of the endogenous cannabinoid anandamide. Proc Natl Acad Sci USA. 1999;96:12198–12203. doi: 10.1073/pnas.96.21.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White WB. Cardiovascular effects of the cyclooxygenase inhibitors. Hypertension. 2007;49:408–418. doi: 10.1161/01.HYP.0000258106.74139.25. [DOI] [PubMed] [Google Scholar]
- Zeilhofer HU, Brune K. Analgesic strategies beyond the inhibition of cyclooxygenases. Trends Pharmacol Sci. 2006;27:467–474. doi: 10.1016/j.tips.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Zias J, Stark H, Sellgman J, Levy R, Werker E, Breuer A, et al. Early medical use of cannabis. Nature. 1993;363:215. doi: 10.1038/363215a0. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H-h, Sørgård M, Di Marzo V, et al. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Chuang H-h, Movahed P, Julius D, Högestätt ED. The anandamide transport inhibitor AM404 activates vanilloid receptors. Eur J Pharmacol. 2000;396:39–42. doi: 10.1016/s0014-2999(00)00207-7. [DOI] [PubMed] [Google Scholar]