

Abstract
Background and purpose:
Neuronal nicotinic acetylcholine receptors (nAChR) can modulate cell survival and memory processing. The involvement of specific nAChR subtypes in downstream signalling events has been ill defined thus far, because of a lack of subtype-selective ligands. In this study, we investigated activation and modulation of α7 nAChR-mediated phosphorylation of extracellular signal-regulated kinases (ERK1/2) in PC12 cells, using selective agonists and positive allosteric modulators.
Experimental approach:
We used undifferentiated PC12 cells endogenously expressing α7 nAChR for both biochemical and functional studies. ERK phosphorylation changes were measured by using a novel In-Cell Western procedure. α7 nAChR-mediated Ca2+ signalling was determined by using the fluorometric imaging plate reader assay.
Key results:
Robust induction of ERK phosphorylation followed exposure of PC12 cells to the selective agonist PNU-282987 in the presence of the α7 nAChR modulator PNU-120596. ERK phosphorylation was transient and was attenuated by the selective antagonist methyllycaconitine. Consistent with allosteric modulation of α7 nAChRs, PNU-120596 enhanced both the agonist potency and efficacy in activating ERK. Moreover, α7 nAChR agonists could be quantitatively differentiated based on their potency in activating ERK signalling. The rank order of potencies correlated fairly well with the corresponding binding Ki values of these α7 nAChR agonists.
Conclusions and implications:
The present work extends previous observations demonstrating the involvement of α7 nAChRs in ERK1/2 phosphorylation in PC12 cells. The In-Cell Western procedure allowed a detailed investigation of α7 nAChR function and downstream ERK signalling in response to agonist and allosteric modulators.
Keywords: PC12 cells, PNU-120596, PNU-282987, nicotine, α7 nAChR, In-Cell Western, ERK, FLIPR
Introduction
Neuronal nicotinic acetylcholine receptors (nAChRs) are pentameric ligand-gated ion channels, derived from nine α (α2–α10) and three β (β2–β4) subunits (Gotti et al., 2006). Multiple functionally distinct nAChR complexes can be assembled either as homomeric pentamers as in the case of α7 or as heteropentamers with at least two different subunits as for example, α4β2 nAChRs (Gotti et al., 2006). Among the diverse nAChRs, the role of α7 subtype in the CNS has been widely studied. In the brain, the α7 subunit is expressed at high levels in regions involved in learning and memory, particularly the hippocampus and cerebral cortex (Marks and Collins, 1982). This nAChR subtype activates and desensitizes rapidly and exhibits higher Ca2+ permeability relative to other nAChR combinations (Couturier et al., 1990). Gene knockout and antisense studies (Wehner et al., 2004; Keller et al., 2005; Curzon et al., 2006) have suggested a role for α7 nAChRs in certain cognitive and attentive tasks (Fernandes et al., 2006; Young et al., 2007). More importantly, pharmacological studies have demonstrated that augmenting α7 nAChR function is capable of ameliorating the cognitive deficits associated with neuropsychiatric and neurodegenerative diseases (Bertrand and Gopalakrishnan, 2007; Bitner et al., 2007). A range of structurally diverse α7 nAChR-selective agonists such as AR-R 17779 (Van Kampen et al., 2004), PNU-282987 (Bodnar et al., 2005), PHA-543613 (Wishka et al., 2006), SSR180711 (Pichat et al., 2006), A-582941 (Bitner et al., 2007) and ABBF (Boess et al., 2007) have demonstrated efficacy across a variety of preclinical cognition models. In addition, selective α7 nAChR agonists have demonstrated neuroprotective effects in apoptotic models in both primary neuronal cultures and PC12 cells (Li et al., 2002, Hu et al., 2007). These preclinical findings have provided support for a potential role of α7 nAChR agonists in the treatment of neurological and psychiatric disorders, including schizophrenia and Alzheimer's disease (see Levin and Rezvani, 2002; Martin et al., 2004).
It is increasingly becoming clear that functional significance of the α7 nAChR can be attributed not only to its electrogenic properties (i.e. modulation of neuronal excitability and neurotransmitter release), but also to its high Ca2+ permeability and association with biochemical signalling pathways (see Berg and Conroy, 2002; Dajas-Bailador and Wonnacott, 2004). One of the key phosphorylation cascades involved in learning and memory is the mitogen-activated protein kinase (MAPK) pathway, specifically, the phosphorylation of extracellular signal-regulated kinase (ERK). Phospho-ERK (pERK) is increased in brain regions including hippocampus following long-term memory consolidation, and pharmacological inhibition of ERK prevents long-term memory formation in rodent learning paradigms (Bitner et al., 2007). Previous studies from our laboratory have shown α7 nAChR agonism can lead to broad-spectrum efficacy in animal models at doses that enhance ERK1/2 and CREB phosphorylation and activation (Bitner et al., 2007).
In recent years, new molecules have been discovered as positive allosteric modulators (PAMs) for α7 nAChRs (Bertrand and Gopalakrishnan, 2007). At least, two different profiles of PAMs have been described thus far: type I modulators that predominantly affect the apparent peak current, agonist sensitivity and Hill coefficient, and type II modulators that cause, in addition, a modification of the desensitization profile of agonist-evoked responses. For example, compounds such as 5-HI, CCMI (Compound 6) (Ng et al., 2007) and NS-1738 (Timmermann et al., 2007) primarily increase the current amplitude of acetylcholine- or choline-evoked α7 currents and belong to the type I class of α7 PAMs. In contrast, molecules such as PNU-120596 and others (Hurst et al., 2005; Gronlien et al., 2007) exhibit the type II profile by triggering increases in both current amplitude and a distinct secondary component leading to prolongation of response to agonists. These PAMs (type II) provide additional approaches with which to examine the pharmacology of α7 nAChRs.
Several nAChR subtypes are endogenously expressed in PC12 cells, primarily α3- and α7-containing nAChRs (Blumenthal et al., 1997; Virginio et al., 2002). Previous studies have shown that nicotine activates ERK, Akt and CREB signalling in PC12 cells (Nakayama et al., 2001; 2002), and based on rank order potencies of inhibition by antagonists, a role for α3β4 nAChRs has been suggested (Nakayama et al., 2006). In contrast, other reports indicate involvement of α7 nAChRs in activating ERK signalling, under specific conditions. Ren et al. (2005) reported a modest phosphorylation of ERK in nerve growth factor-differentiated PC12 cells in response to GTS-21, a ligand that interacts with both α7 and α4β2 nAChRs. Utsugisawa et al. (2002) showed ERK phosphorylation after over-expression of α7 nAChRs in PC12 cells, independent of agonist stimulation. Most recent studies from our laboratory by using subtype-selective α7 nAChR agonists have shown dose-dependent phosphorylation of ERK and CREB in vivo in brain regions associated with cognitive processing, including cingulate cortex and hippocampus (Bitner et al., 2007). The role of α7 nAChRs in activating downstream signalling in in vitro model systems has been therefore poorly understood due to two major factors: (i) limitation of methodologies to detect signalling events triggered by relatively rapid kinetics of activation and desensitization of the α7 nAChRs; and (ii) lack of subtype-selective agonists, until recently, to investigate α7 nAChR pharmacology in native systems. In this study, we utilized PC12 cells that endogenously express α7 nAChRs and developed a novel cell-based assay to characterize the activation and modulation of α7 nAChRs and downstream phosphorylation of ERK. The present work shows that α7 nAChR function and downstream ERK signalling can be revealed by agonists in presence of PAMs, providing further support to the mechanisms by which such agents participate in cellular processes involved in learning and memory.
Methods
Test systems used
Cell culture
Rat pheochromocytoma (PC12) cells were obtained from American Type Culture Collection (ATCC, Manassas, VA). Undifferentiated PC12 cells were cultured and maintained in F-12K media supplemented with 15% horse serum, 2.5% fetal calf serum and 2 mmol·L−1 L-glutamine in poly-D lysine coated dishes at 37°C and 5% CO2. Undifferentiated PC12 cells were used in this study as they express predominantly α7 nAChR, and lower α3* expression as measured by radioligand binding. In addition, undifferentiated PC12 cells exhibited a high signal to noise ratio in the In-Cell Western (ICW) approach, compared with other cell lines tested, and thus are easier to use in high-throughput screening. Molecular target nomenclature conforms with British Journal of Pharmacology's Guide to receptors and channels (Alexander et al., 2008).
Measurements made
In-Cell Western blotting and quantitation
PC12 cells were plated in black-walled clear bottom 96-well Biocoat™ plates coated with poly-D-lysine (BD Biosciences, Bedford, MA) and grown for 2–3 days. Culture media was replaced with serum-free media to starve cells overnight. On the day of the assay, cell media were removed, and cells (60–80% confluent) were treated with compounds in Dulbecco's phosphate buffer saline (D-PBS) (with Ca2+, Mg2+ and 1 mg·mL−1 D-glucose). Typically, cells were treated at 37°C for 10 min with the α7 nAChR PAM followed by addition of the agonist for 5 min in a final volume of 100 µL per well, unless otherwise indicated. After treatment, D-PBS medium was discarded, and adherent cells were immediately fixed in the presence of 150 µL per well of 3.7% formaldehyde/PBS for 30–60 min at room temperature. Cells were then washed (4 × 5 min) and permeabilized with 200 µL per well of 0.1% Triton X-100/PBS. Permeabilized cells were blocked by using the Odyssey® blocking buffer (100 µL per well), and plates were rocked overnight at 4°C. Both anti-total ERK (tERK) (from rabbit) and anti-pERK (from mouse) antibodies were diluted 1:1000 and 1:500, respectively, in Odyssey® blocking buffer and added together in 50 µL per well for 2–3 h at room temperature. The plates were washed four times with 0.1% Tween 20/PBS (200 µL per well) and incubated with secondary antibodies (1:1000 dilution) in blocking buffer supplemented with 0.2% Tween for 1 h. Alexa Fluor 680-labelled goat anti-rabbit antibodies were added to recognize tERK labelling (red colour), and IRDye800-labelled donkey anti-mouse antibodies were added to recognize pERK labelling (green colour). The plates were washed four times with 0.2% Tween and 0.01% sodium dodecyl-sulfate (SDS)/PBS and scanned by using the Odyssey® infrared scanner. Well intensities were quantitated, and pERK signals were normalized to tERK signals by the Odyssey® software. Several commercially available primary antibodies against tERK and pERK were tested for their affinities and specificities (data not shown). The selected antibodies (from Sigma Aldrich, St. Louis, MO) showed the highest affinities and specificities against tERK and pERK in both ICW and standard Western blot procedures. Data were expressed as either fold increase over basal or as a percentage of maximum response.
Standard Western blotting
PC12 cells were plated in 6-well Biocoat™ plates coated with poly-D-lysine (BD Biosciences, Bedford, MA) and grown for 2–3 days. Culture medium was replaced with serum-free medium to starve cells overnight. After treatment with α7 nAChR ligands, reactions were terminated on ice, and cells were washed twice with D-PBS. Cells were then lysed and sonicated in SDS sample buffer. Samples were heated at 80°C twice for 5 min. Equal amounts were loaded on 4–12% Tris-glycine NuPage pre-cast gels and blotted to PVDF membranes, according to the manufacturer's protocol (Invitrogen, Carlsbad, CA). Immunoblots were blocked by using the Li-COR® Odyssey® blocking buffer. Immunodetection of tERK and pERK bands was carried out by using the same primary and secondary antibodies as described above for the ICW technique. Immunoblots were scanned and quantitated by using the Li-COR® Odyssey® software.
Radioligand binding
Endogenous expression of nAChR subtypes were assessed in crude membrane preparations of undifferentiated PC12 cells. α7 nAChR binding levels were determined by using the [3H]-(S,S)-2,2-dimethyl-5-(6-phenyl-pyridazin-3-yl)-5-aza-2-azonia-bicyclo[2.2.1]heptane iodide ([3H]A-585539), as described by Anderson et al. (2008). α3* expression levels were assessed by using [3H]epibatidine binding conducted in the presence of the high-affinity ligand A-585539 (100 nmol·L−1) to eliminate any interactions of [3H]epibatidine with α7 nAChRs. α4β2 nAChR binding was determined by using [3H]cytisine. [3H]epibatidine binding and [3H]cytisine binding were performed by using similar conditions as previously described (Anderson et al., 2008). Cell pellets were thawed at 4°C, washed and resuspended with a Polytron at a setting of seven in 30 volumes of BSS-Tris buffer (120 mmol·L−1 NaCl, 5 mmol·L−1 KCl, 2 mmol·L−1 CaCl2, 2 mmol·L−1 MgCl2 and 50 mmol·L−1 Tris-Cl, pH 7.4, 4°C). A total of 50–100 µg of protein, and [3H]A-585539 (0.5 nmol·L−1, 62.8 Ci·mmol−1; R46V, Abbott), [3H]epibatidine (1.4 nmol·L−1, 49 Ci·mmol−1; PerkinElmer), or [3H]cytisine (1.5 nmol·L−1, 40 Ci·mmol−1; PerkinElmer) were incubated in a final volume of 500 µL for 90 min at 4°C in duplicate. Bound radioactivity was collected on Millipore MultiScreen® harvest plates filter B (FB) pre-soaked with 0.3% polyethyleneimine by using a Packard cell harvester, washed with 2.5 mL ice-cold buffer, and radioactivity was determined by using a Packard TopCount Microplate beta counter.
Intracellular Ca2+ measurement
α7 nAChR-mediated elevation of intracellular Ca2+ levels was measured by using Ca2+-4 no-wash dye, according to manufacturer's protocol (MDS Analytical Technologies, Sunnyvale, CA). Briefly, PC12 cells were grown as a monolayer in black-walled clear bottom 96-well Biocoat™ plates coated with poly-D-lysine (BD Biosciences, Bedford, MA). Prior to the assay, the culture media was discarded and cells loaded with 100 µL of Ca2+-4 no-wash dye in NMDG buffer [10 mmol·L−1 HEPES, pH 7.4, 140 mmol·L−1 N-methyl D-glucosamine (NMDG), 5 mmol·L−1 KCl, 1 mmol·L−1 MgCl2, 10 mmol·L−1 CaCl2] for 1 h at 25°C. Because NMDG+ is poorly permeant through the α7 nAChR, the use of NMDG-containing (Na+-free) buffer enables assessment of predominantly Ca2+-triggered effects. Solutions of receptor ligands were prepared (3× for PAM and 4× for agonist) in NMDG buffer, and 50 µL were added to the cells at a delivery rate of 25 µL·s−1 to reach a final volume of 200 µL. The PAM was added 5 min before addition of the agonist. Changes in fluorescence were recorded over time at 25°C in fluorometric imaging plate reader (FLIPR) (MDS Analytical Technologies, Sunnyvale, CA) (λEX = 488 nm, λEM = 540 nm). The peak increase in fluorescence over baseline was determined and is expressed as relative fluorescence units (RFU).
Data analysis
Concentration–effect curves were fitted to the Hill equation. For graphical purposes, concentration–effect curves are typically presented as a percentage of the maximal response obtained in the control group (10 µmol·L−1 PNU-120596 for 10 min and challenged by 10 µmol·L−1 PNU-282987 for 5 min). In some cases, bottom curves were constrained to the lowest value obtained in the absence of stimulus, as indicated in figure legends. The data presented are mean ± SEM. EC50 and IC50 values were calculated from curve fits of the concentration–effect data by using the four-parameter logistic Hill equation: Y = Bottom + [(Top − Bottom)(1 + 10(LogEC50 − X).HillSlope)−1] where X is the logarithm of the ligand concentration, and Y is the response. Bottom and Top are the Y values corresponding to the top and bottom plateau values of the curve. Data were analysed by using GraphPad Prism (GraphPad Software Inc., San Diego, CA). Statistical analyses were determined by using a one-tailed Student's t-test.
Drugs, chemicals and other materials
Polyclonal rabbit anti-ERK1/2 and monoclonal mouse anti-pERK1/2 were purchased from Sigma-Aldrich (St. Louis, MO). Alexa Fluor 680-labelled goat anti-rabbit antibodies were obtained from Molecular Probes (Eugene, OR). IRDye 800CW-labelled donkey anti-mouse antibodies were purchased from Rockland (Gilbertsville, PA). The infrared Odyssey® scanner and Odyssey® blocking buffer were obtained from Li-COR Inc. (Lincoln, NE). Cell culture reagents were obtained from Invitrogen (Carlsbad, CA). The α7 nAChR agonist, N-[(3R)-1-Azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride (PNU-282987; Bodnar et al., 2005) and the corresponding PAM, 1-(5-chloro-2,4-dimethoxy-phenyl)-3-(5-methyl-isoxazol-3-yl)-urea (PNU-120596; Hurst et al., 2005) are commercially available from Tocris Bioscience (Ellisville, MO); the selective antagonist methyllycaconitine (MLA) was from Sigma-Aldrich (St. Louis, MO). Other α7 nAChR ligands used in the present study were synthesized at Abbott. These include (2.4)-dimethoxybenzylidene anabaseine dihydrochloride (GTS-21; Briggs et al., 1997), 1,4-diazabicyclo[3.2.2]nonane-4-carboxylic acid, 4-bromophenyl ester (SSR180711; Biton et al., 2007), 4-(5-phenyl-[1,3,4]oxadiazol-2-yl)-1,4-diaza-bicyclo[3.2.2]nonane (A-803401, also known as NS6784) and 2-methyl-5-(6-phenyl-pyridazin-3-yl)-octahydro-pyrrolo[3,4-c]pyrrole dihydrochloride (A-582941; Bitner et al., 2007). Other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) or Tocris Bioscience (Ellisville, MO) unless otherwise indicated.
Results
Endogenous expression of nAChR subtypes in undifferentiated PC12 cells
Expression of α7-, α3- and α4-containing nAChRs in membrane preparations from undifferentiated PC12 cells were assessed by binding of [3H]A-585539, [3H]epibatidine and [3H]cytisine respectively, as described in Methods. Expression levels of α7 nAChRs were estimated at 134 ± 15 fmol·mg−1 protein (n = 3), while only weak expression of α3-containing nAChRs were found (10 ± 2 fmol·mg−1 protein, n = 4). No detectable binding to α4-containing nAChRs was observed in undifferentiated PC12 cells.
Phosphorylation of ERK via α7 nAChR
Prior to evaluation of the anti-tERK and anti-pERK antibodies by the ICW procedure, we characterized these antibodies using standard Western blot procedures. Undifferentiated PC12 cells were treated with an α7 nAChR agonist with or without α7 nAChR PAM, and cell lysate samples were resolved on SDS-PAGE, followed by immunoblotting, as described in Methods. Western blots were then scanned by using the Li-COR® Odyssey® scanner to enable dual near-infrared detection of tERK (red) and pERK (green). Figure 1A shows that the anti-tERK antibodies were highly selective for ERK1 (≈42 kDa) and ERK2 (≈40 kDa) proteins, under these conditions. Similarly, anti-pERK antibodies recognized only bands of pERK1 and pERK2 proteins (Figure 1A). Treatment of cells with the α7 nAChR agonist PNU-282987 (10 µmol·L−1) or the PAM (PNU-120596; 10 µmol·L−1) alone did not affect the basal phosphorylation levels of ERK. However, when cells were treated with both PNU-120596 and PNU-282987, the levels of phosphorylation of ERK1/2 were dramatically increased (Figure 1A,B). The ratio of pERK/tERK was increased 4–5-fold, compared with the control levels. This effect was attenuated by the selective antagonist MLA (100 nmol·L−1), consistent with an α7 nAChR-mediated effect.
Figure 1.
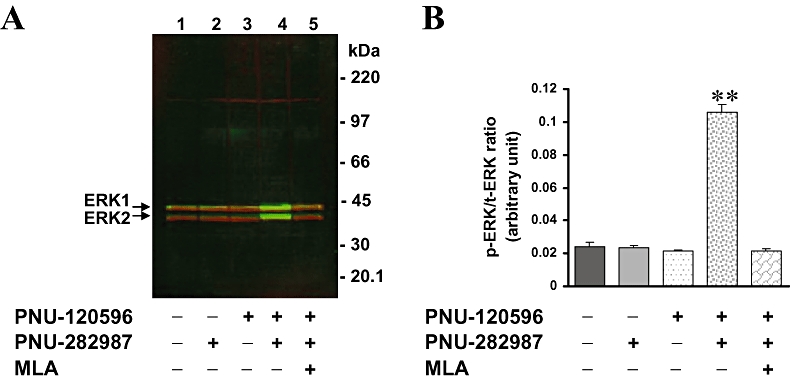
α7 nAChR-mediated ERK phosphorylation in undifferentiated PC12 cells (A) Immunoblotting of tERK1/2 (red) and pERK1/2 (green) as visualized by the dual near-infrared detection capabilities of Li-COR® Odyssey® scanner. PC12 cells were untreated (lane 1) or treated (lane 2), with agonist (10 µmol·L−1 PNU-282987) for 5 min; (lane 3), PAM (10 µmol·L−1 PNU-120596) for 10 min; (lane 4), PAM for 10 min followed by agonist for 5 min; (lane 5), 100 nmol·L−1 MLA for 2 min followed by PAM for 10 min and agonist for 5 min. (B) Intensities of pERK1/2 bands were quantitated and normalized to those of tERK1/2 by using the Li-COR® Odyssey® software. Data are mean ± SEM; n = 4. **P < 0.01, compared with basal control values. ERK, extracellular signal-regulated kinase; MLA, methyllycaconitine; nAChR, nicotinic acetylcholine receptor; PAM, positive allosteric modulator; pERK, phospho-ERK; tERK, total ERK.
Characterization of α7 nAChR-mediated ERK activation using the ICW procedure
We next optimized immunodetection conditions using whole PC12 cells cultured in 96-well plate format. After treatment with α7 nAChR ligands, both anti-tERK and anti-pERK antibodies were added to fixed cells, followed by incubation with corresponding secondary antibodies, as described in Methods. The 96-well plates were then scanned by using the Odyssey® scanner, and both tERK (red) and pERK (green) signals were visualized and quantitated by the Odyssey® software. Increasing concentrations of the PAM (PNU-120596) alone up to 100 µmol·L−1 had no effect on ERK phosphorylation (Figure 2A,B). However, the PAM did induce a concentration-dependent stimulation of ERK (EC50 = 3.1 µmol·L−1; 95% CI 2.26–4.11 µmol·L−1), in the presence of the agonist PNU-282987 (1 µmol·L−1; Figure 2A,B). The increase in ERK phosphorylation levels obtained by using the ICW procedure (4–6-fold) was comparable to that obtained by standard Western technique (Figures 1B and 2B). Moreover, phosphorylation of ERK in response to PNU-120596 and PNU-282987 stimulation was attenuated in a concentration-dependent manner by the antagonist MLA (IC50 = 2.75 nmol·L−1; 95% CI 1.54–4.92 nmol·L−1) (Figure 3A,B). MLA did not significantly affect basal ERK phosphorylation. These data validated the ICW procedure and demonstrated that ERK phosphorylation was evoked by α7 nAChR activation and required the presence of both α7 nAChR agonist and PAM under these experimental conditions.
Figure 2.
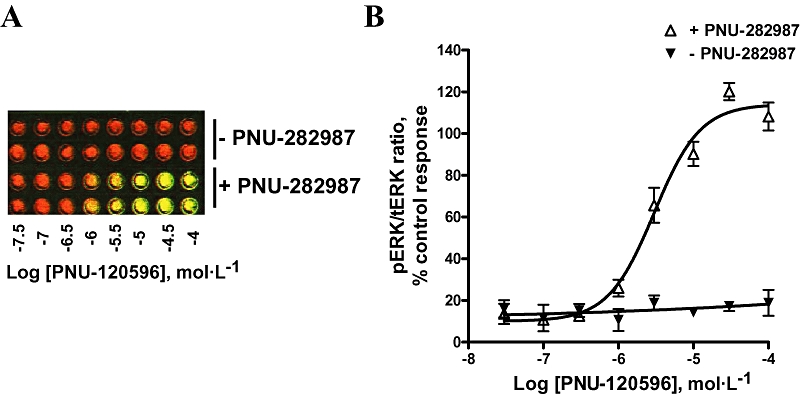
In-Cell Western of ERK signals in 96-well plate format. Immunodetection of ERK was optimized by using whole PC12 cells in 96-well plate format, as described in Methods. (A) Representative image of cell wells as visualized by the Li-COR® Odyssey® scanner of tERK (red) and pERK (green) immunoreactive signals. Well columns represent duplicates of concentration–response effects of the PAM (PNU-120596) alone (10 min) or in the presence of agonist (1 µmol·L−1 PNU-282987) (5 min). (B) Graph plots of normalized pERK signals in response to activation of α7 nAChR by PNU-120596 in the absence or in the presence of PNU-282987. Data are mean ± SEM; n = 3 in duplicate. ERK, extracellular signal-regulated kinase; nAChR, nicotinic acetylcholine receptor; PAM, positive allosteric modulator; pERK, phospho-ERK; tERK, total ERK.
Figure 3.
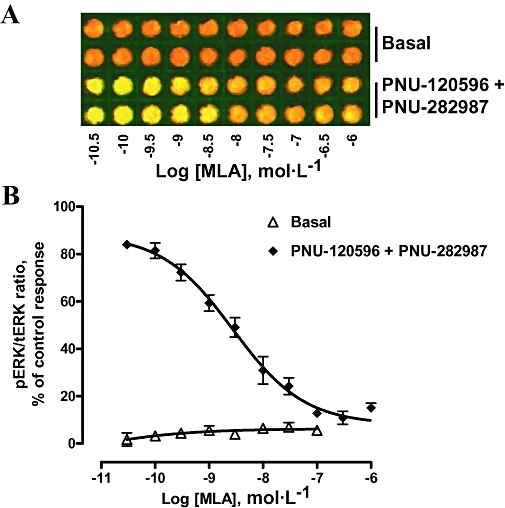
Blockade of α7 nAChR-mediated ERK phosphorylation by the α7 nAChR antagonist, MLA. (A) Concentration-dependent effect of MLA alone or in the presence of PNU-120596 (10 µmol·L−1) and PNU-282987 (10 µmol·L−1). (B) Plots of pERK signals showed that MLA blocked α7 nAChR-mediated ERK phosphorylation in a concentration-dependent manner (IC50 = 2.75 nmol·L−1; 95% CI 1.54–4.92 nmol·L−1). Data are mean ± SEM; n = 3 in duplicate. ERK, extracellular signal-regulated kinase; MLA, methyllycaconitine; nAChR, nicotinic acetylcholine receptor; pERK, phospho-ERK; tERK, total ERK.
Transient phosphorylation of ERK in response to α7 nAChR activation
Figure 4A shows the time course for agonist-induced ERK phosphorylation in the continuous presence of the PAM, PNU-120596, as described in Methods. pERK reached a maximal level within 3–7 min of adding the agonist PNU-282987. The levels of ERK phosphorylation declined when cells were treated with the agonist PNU-282987 for longer time periods (15 or 30 min) (Figure 4A), indicating the transient nature of the response. In addition, ERK phosphorylation did not completely return to the basal levels after prolonged exposure (up to 30 min) to α7 nAChR agonist and PAM.
Figure 4.
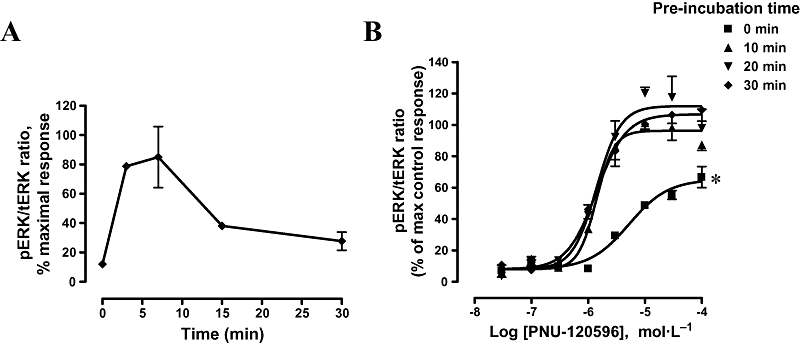
Time course of agonist and PAM on α7 nAChR-mediated ERK phosphorylation. (A) PC12 cells were treated with PAM (10 µmol·L−1 PNU-120596) for 10 min and challenged with agonist (10 µmol·L−1 PNU-282987) for different incubation times: 3, 7, 15 and 30 min. pERK signals were quantitated and pERK/tERK ratio were normalized to maximal response. Data are mean ± SEM; n = 2 in duplicate. (B) PC12 cells were pretreated with increasing concentrations of PNU-120596 for varying time intervals (0, 10, 20 and 30 min) before challenge with 10 µmol·L−1 PNU-282987 for 5 min. pERK signals were normalized and plotted as % of control response (10 min pre-incubation with PAM). Concentration–effect curves were fitted to non-linear regression curves with curve bottoms constrained to the basal value obtained in the absence of agonist. Curve parameters are shown in Table 1. Data are mean ± SEM; n = 3–5 in duplicate. *P < 0.05, compared with control response (10 min pre-incubation with PAM). ERK, extracellular signal-regulated kinase; nAChR, nicotinic acetylcholine receptor; PAM, positive allosteric modulator; pERK, phospho-ERK; tERK, total ERK.
We next investigated the time course of PAM effects on ERK phosphorylation while keeping incubation time with the agonist (PNU-282987) constant at 5 min (Figure 4B). Pre-incubation with PAM for 10 min (control) increased ERK phosphorylation with an EC50 value of 1.39 µmol·L−1 (95% CI 1.11–174 µmol·L−1). Longer pre-incubation times did not significantly enhance the potency or the efficacy of the PAM in phosphorylating ERK. EC50 values were 1.41 µmol·L−1 and maximal efficacy of 112% and 107% of control response, after 20 and 30 min pre-incubation with PAM respectively (Figure 4B, Table 1). However, when PNU-120596 and PNU-282987 were added simultaneously, maximal efficacy was reduced to 65% and the EC50 value increased to 5.23 µmol·L−1 (Figure 4B, Table 1). Additionally, a similar reduction in ERK response was observed when cells were first pretreated with the agonist PNU-282987 before addition of the PAM, compared with that observed after simultaneous addition of agonist and PAM to the cells (data not shown).
Table 1.
Effects of varying exposure times on potency and efficacy of PNU-120596
| Pre-incubation time (min) |
Potency and efficacy of PNU-120596 |
||
|---|---|---|---|
| EC50 (µmol·L−1) | Hill slope | % Efficacy | |
| 0 | 5.23 (3.33–8.22) | 1.26 ± 0.28 | *65 ± 4 |
| 10 | 1.39 (1.11–1.74) | 2.73 ± 0.67 | 96 ± 3 |
| 20 | 1.41 (1.03–1.93) | 2.06 ± 0.55 | 112 ± 6 |
| 30 | 1.41 (1.12–1.78) | 1.67 ± 0.27 | 107 ± 4 |
Concentration–response parameters were determined by non-linear fitting of the Hill equation to the values shown in Figure 4B. Data are shown as mean (95% confidence interval) for EC50 and mean ± SEM for Hill coefficient and maximal response relative to 10 min pre-incubation time with PNU-120596.
n = 3–5 independent determinations in duplicate.
P < 0.05, compared with the 10 min control.
PNU-120596 allosterically modulates agonist activation of ERK
To further elucidate agonist–PAM interactions, concentration–response curves for PAM (or agonist) were generated in the presence of varying concentrations of agonist (or PAM). Increasing concentrations of the PAM (PNU-120596) induced a modest leftward shift in the potency of the agonist [1.6-fold decrease in EC50 value along with an increase in the Hill slope value (from 2.04 ± 1.10 to 2.60 ± 0.53) as well as an increase in the maximal efficacy (Figure 5A)]. Maximal ERK response gradually increased in a concentration-dependent manner from 80% to 121% (Figure 5A). These data are consistent with an allosteric modulation by PNU-120596 of the PNU-282987-induced, α7 nAChR-mediated, phosphorylation of ERK. However, different concentrations of the agonist had minimal effects on the concentration–response curves for the PAM (Figure 5B). EC50 values for the PAM were about 2.9–4.2 µmol·L−1 in the presence of different concentrations of agonist (PNU-282987) (Figure 5B). In addition, increasing concentrations (>100 nmol·L−1) of PNU-282987 did not affect the maximal ERK response, nor the Hill slope values. Lower concentrations of PNU-282987 showed minimal to no response of ERK phosphorylation (Figure 5B). These results collectively support the allosteric interaction between the modulatory and agonist sites on the α7 nAChRs.
Figure 5.
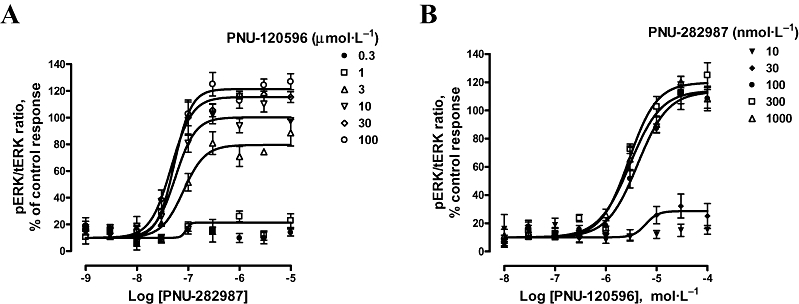
Concentration–effect curves of α7 nAChR ligand-induced ERK phosphorylation. Incubation times for PAM (10 min) and agonist (5 min) were selected in order to achieve maximal ERK phosphorylation. (A) Concentration–response curves of agonist (PNU-282987)-induced ERK phosphorylation were generated in the presence of various concentrations of PAM (PNU-120596). EC50 values of PNU-282987 varied from 47 to 80 nmol·L−1 (Hill slope nH = 2.04–2.60) while the percentage of efficacy increased from 80% to 120%, after exposure to 3 and 100 µmol·L−1 respectively. No pERK changes were observed in the presence of PNU-120596 < 0.3 µmol·L−1, and the corresponding curves were not plotted for the sake of clarity. Data are mean ± SEM; n = 3–5 in duplicate. (B) Conversely, concentration–response curves of PAM (PNU-120596)-induced ERK phosphorylation were generated in the presence of various concentrations of agonist (PNU-282987). EC50 values of PNU-120596 ranged from 2.85 to 4.22 µmol·L−1 in the presence of >100 nmol·L−1 PNU-282987. Under these conditions, no increase in percentage of efficacy of the PAM was observed (Hill slope nH = 1.41–1.58). Curves corresponding to PNU-282987<10 nmol·L−1 showed no pERK changes and have not been shown. Curve bottoms were constrained to basal value (10) obtained in the absence of stimulus. Data are mean ± SEM; n = 3–5 in duplicate. ERK, extracellular signal-regulated kinase; nAChR, nicotinic acetylcholine receptor; PAM, positive allosteric modulator; pERK, phospho-ERK; tERK, total ERK.
Pharmacology of α7 nAChR agonists in phosphorylating ERK
We next investigated the modulation of ERK phosphorylation in the presence of nicotine and other structurally different nAChR agonists, including GTS-21, A-803401 (NS6784), PNU-282987, A-582941 and SSR180711A. High concentration of nicotine (100 µmol·L−1) and other α7 nAChR ligands (10 µmol·L−1), given alone, did not induce any ERK phosphorylation (data not shown). In the presence of 10 µmol·L−1 PNU-120596, however, all agonists tested exhibited a concentration-dependent increase in pERK (Figure 6A,B). Interestingly, the ICW procedure differentiated between agonists based on their potency in activating ERK signalling with the following rank order of EC50 values: A-803401 (NS6784) (8.2 nmol·L−1) < PNU-282987 (18.3 nmol·L−1) < SSR180711A (54 nmol·L−1) < A-582941 (180 nmol·L−1) < GTS-21 (198 nmol·L−1) < nicotine (1121 nmol·L−1) (Figure 6, Table 2). This rank order showed a good correlation (r2 = 0.88) with agonist Ki values for displacement of radioligand binding to α7 nAChRs (Briggs et al., 2007; Anderson et al., 2008) (Figure 6C). This provides further support for the specific interaction with α7 nAChRs to induce ERK phosphorylation.
Figure 6.
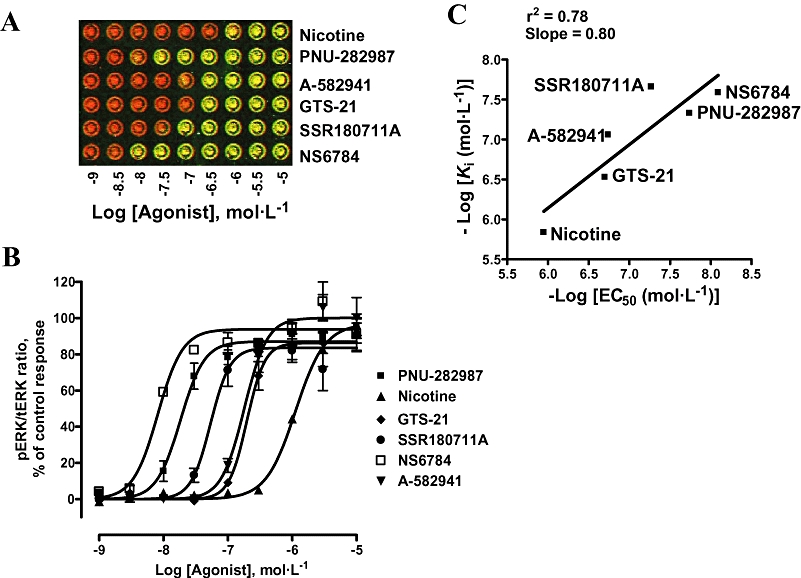
Characterization of α7 nAChR agonists using the ICW procedure. Concentration–response curves of different agonists were generated in the presence of 10 µmol·L−1 PNU-120596. (A) Representative image of plate wells showing different degrees of ERK phosphorylation in response to various α7 nAChR agonists. (B) pERK signals were quantitated and plotted as a % of the response to PNU-282987. Curve bottoms were constrained to basal value (0) obtained in the absence of agonist. Data are mean ± SEM; n = 2–5 in duplicate. (C) Correlation between EC50 values of α7 nAChR agonists generated in the present ICW procedure and affinity Ki values reported recently in the [3H]MLA binding in rat brain (in the absence of α7 nAChR PAM) (Briggs et al., 2007; Anderson et al., 2008). Ki value for SSR180711A to rat α7 nAChR was reported in Biton et al. (2007). ERK, extracellular signal-regulated kinase; ICW, In-Cell Western; MLA, methyllycaconitine; nAChR, nicotinic acetylcholine receptor; PAM, positive allosteric modulator; pERK, phospho-ERK; tERK, total ERK.
Table 2.
Characterization of agonists in extracellular signal-regulated kinase phosphorylation, mediated by activation of α7 nicotinic acetylcholine receptors
| EC50 (nmol·L−1) | Hill slope | |
|---|---|---|
| Nicotine | 1121 (937–1340) | 1.90 ± 0.29 |
| GTS-21 | 198 (165–237) | 3.14 ± 0.53 |
| NS6784 | 8.2 (6.3–10.7) | 2.26 ± 0.65 |
| PNU-282987 | 18.3 (14.8–22.7) | 2.35 ± 0.43 |
| A-582941 | 180 (144–225) | 2.46 ± 0.44 |
| SSR180711A | 54 (39.5–74) | 2.86 ± 0.68 |
Concentration–response parameters were determined by non-linear fitting of the Hill equation to the values shown in Figure 6B. Data are shown as mean (95% confidence interval) for EC50 and mean ± SEM for Hill coefficient. n = 2–5 independent determinations in duplicate.
α7 nAChR-mediated intracellular Ca2+ increases in PC12 cells
We further examined the effect of activation of α7 nAChRs to evoke intracellular Ca2+ changes, using the FLIPR assay, as described in Methods. As shown in Figure 7A, the agonist (10 µmol·L−1 PNU-282987) alone did not evoke an increase in intracellular Ca2+, but did induce a robust response in the presence of the PAM (PNU-120596; 10 µmol·L−1) (Figure 7A,B). Likewise, nicotine (100 µmol·L−1) alone evoked minimal intracellular Ca2+ increase, but triggered a more robust increase in the presence of PNU-120596 (Figure 7A,B).
Figure 7.
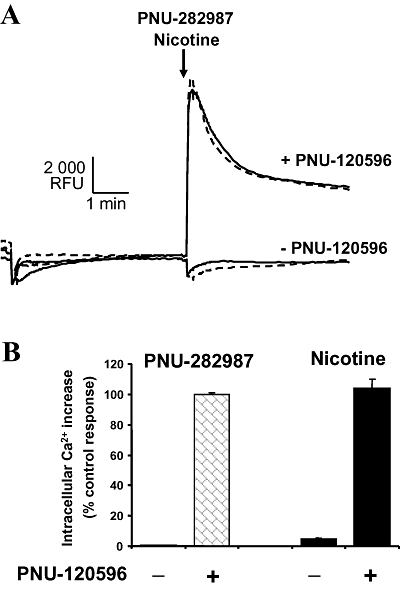
Activation of α7 nicotinic acetylcholine receptor-mediated intracellular Ca2+ increase in PC12 cells. PC12 cells were treated with 10 µmol·L−1 PNU-282987 or 100 µmol·L−1 -(-)nicotine alone or in the presence of 10 µmol·L−1 PNU-120596. PNU-120596 (or vehicle) was applied 5 min before addition of agonist. (A) Representative fluorometric imaging plate reader trace showing time course of intracellular Ca2+ changes in response to PNU-282987 (dashed line) or nicotine (solid line) added at the arrow. Ca2+ increases were measured as relative fluorescence units (RFU). (B) Ca2+ peak responses were quantitated and expressed as a % of control response (PNU-282987). Data are mean ± SEM; n = 4 in duplicate.
Discussion
In the present study we investigated the pharmacology of α7 nAChR-mediated ERK activation using selective α7 nAChR ligands. We demonstrated that α7 nAChR, endogenously expressed in undifferentiated PC12 cells, activated downstream ERK signalling. Consistent with allosteric modulation, PNU-120596 affected both potency and efficacy of α7 nAChR agonist in activating ERK signalling. The ICW procedure enabled us to provide a detailed pharmacology of α7 nAChR-mediated ERK activation and provided a quantitative analysis for potencies of α7 nAChR ligands in activating this pathway. The present work indicates that allosteric potentiation of α7 nAChRs could be translated into downstream cellular responses and extends our understanding of the molecular signalling of this nAChR subtype.
The nAChRs are involved in a wide variety of cognitive functions and implicated in pathological conditions, associated with neurodegenerative diseases such as Alzheimer's and Parkinson's diseases (Dani et al., 2001). The neuroprotective role of nicotine has been extensively studied in several cell and neuronal models (Dajas-Bailador and Wonnacott, 2004). Nicotine has been shown to activate ERK/MAPK and CREB signalling in neurons, including hippocampal neurons (Dajas-Bailador et al., 2002; Hu et al., 2002) as well as in cell lines such as SH-SY5Y and PC12 cells (Nakayama et al., 2001; Dajas-Bailador et al., 2002). These cells endogenously express several nAChR subunits, which can be activated by nicotine, including α3-, α7- and β2-containing nAChR. The downstream signalling of α7 nAChRs has therefore been not well defined until recently, because of a lack of subtype-selective ligands. In the present work, we showed that selective potentiation and activation of α7 nAChRs induced a robust activation of ERK1/2 signalling (≈4–6-fold increase in ERK phosphorylation) in undifferentiated PC12 cells. Moreover, this activation required the presence of a PAM in order to elicit a substantial ERK phosphorylation. The present study provides a basis for understanding the pharmacology and further exploring the biochemical pathways associated with these rapidly desensitized channels.
PNU-120596 is an example of a prototypical type II PAM that both increases agonist-evoked peak currents and prolongs the evoked response in the continued presence of the agonist (Hurst et al., 2005; Gronlien et al., 2007). In the present study, we showed that PNU-120596 dramatically enhanced agonist-evoked ERK phosphorylation, in a time- and concentration-dependent manner. PNU-120596 affected both the potency and efficacy of the agonist in inducing ERK phosphorylation. Our data are consistent with the allosteric effects of PNU-120596 reported in electrophysiological studies. Interestingly, in vivo studies have shown that an α7 nAChR agonist, given alone, can trigger enhancement of ERK1/2 and CREB phosphorylation in specific regions of the brain (Bitner et al., 2007). Thus, the lack of ERK activation in response to agonist alone in in vitro model systems such PC12 cells could be due to the rapid desensitization of α7 nAChRs. When applied to Xenopus oocytes or cultured hippocampal neurons, PNU-282987 evoked a rapidly desensitizing (milliseconds) inward whole-cell current that was concentration-dependent and blocked by the antagonist MLA (Bodnar et al., 2005; Gronlien et al., 2007). Considering the time scale of our in vitro assay conditions (where α7 nAChRs would typically activate and desensitize upon agonist addition), inclusion of a PAM enables amplification of responses and provides a robust measurable signal. The fact that agonists that are active in the in vitro ERK assay (as for example, A-582941) can evoked ERK phosphorylation when given alone in vivo as measured by immunohistochemical techniques, suggest that this in vitro assay has a degree of physiological relevance. It is also likely that in vivo efficacy and kinetics of α7 nAChRs may be regulated by other endogenous modulators.
The ICW procedure showed a high-throughput advantage over the conventional Western technique and enabled us to investigate in detail the modulation of α7 nAChRs and their signalling cascade via phosphorylation of ERK. This procedure presents a great advantage in investigating α7 nAChR signalling, in particular, and that of other receptors or channels in general. This provides a complementary approach to other high-throughput assays used in drug screening, such as binding or FLIPR assays. The dual near-infrared detection capabilities of the Li-COR® Odyssey® scanner enabled a simultaneous determination of both pERK and tERK levels. α7 nAChR ligands could be quantitatively differentiated based on their potency and efficacy in activating ERK signalling. Although the ICW procedure has been adopted to investigate downstream signalling of G-protein coupled receptors such as dopamine D2 and D3 receptors (Wong, 2004), the present study is the one of the first to examine the signalling pathways mediated by ligand-gated ion channels, by utilizing this procedure.
As reported previously, PC12 cells provide an excellent cellular model to investigate different signalling pathways involved in proliferation, differentiation or neuroprotection and are responsive to most growth factors, neurotrophins and hormones (Vaudry et al., 2002). In addition, PC12 cells are particularly attractive to address nAChR pharmacology as they endogenously express several nAChR subunits. They have been shown to express α3, α5, α7, β2, β3 and β4 at functional, mRNA or protein levels (Boulter et al., 1987; 1990; Rogers et al., 1992; Henderson et al., 1994). The role of specific nAChR subtypes in the neuroprotective effects of nicotine and induction of ERK/MAPK signalling has been unclear, because of the lack of subtype-specific agonists and antagonists. Both α3- and α7-containing nAChRs have been reported to mediate nicotine effects in activating and phosphorylating ERK/MAPK pathway in PC12 cells (Nakayama et al., 2001; Utsugisawa et al., 2002; Nakayama et al., 2006). Using subtype-selective ligands, the present work implicates α7 nAChRs in the activation of ERK signalling. Interestingly, we showed that 100 µmol·L−1 nicotine, which activates both α7- and α3-containing nAChRs, did not elicit an increase in ERK phosphorylation itself, consistent with the low expression of α3-containing nAChRs in these cells. Lower concentrations of nicotine were also used and showed no ERK response (data not shown). However, nicotine induced a dramatic increase in ERK phosphorylation after pre-incubation with the PAM (PNU-120596), suggesting that the α7 nAChR-mediated component of nicotine's action, as measured by ERK signalling, can be amplified in the presence of a PAM.
Similarly, in the absence of PNU-120596, nicotine (up to 100 µmol·L−1; data not shown) did not induce Ca2+ flux, but did induce a robust Ca2+ response in the presence of PNU-120596. Our present data indicate that undifferentiated PC12 cells do not express enough functional α3-containing nAChR to elicit a noticeable response in both Ca2+ flux and ERK assays. Determination of specific binding to α3* and α7 nAChRs in undifferentiated PC12 cells showed relatively low expression levels of α3*, compared with that of α7 nAChRs. The expression ratio between α3* and α7 nAChRs obtained in the present study differed substantially from previous reports where α3* expression was equal to or greater than α7 nAChR expression in PC12 cells (Blumenthal et al., 1997; Nakayama et al., 2001; Dickinson et al., 2007). In addition, Dickinson et al. (2007) have shown that increases in intracellular Ca2+ levels in response to α7 nAChR activation were affected by modulators of intracellular Ca2+ stores and were independent from voltage-operated Ca2+ channel activation. Taken together, our present work indicates that the α7 nAChR-induced intracellular Ca2+ increase could be responsible for downstream ERK phosphorylation. Although the present work demonstrates the implication of α7 nAChRs in ERK activation, it does not exclude the role of α3* in nicotine-induced neuroprotection and ERK phosphorylation in PC12 cells (Nakayama et al., 2001; 2006). The difference observed between our work and others could be due to the difference in PC12 cells used for the studies. It is known that PC12 cells are influenced by a number of factors, including their source, the number of growth passages and the culture media and conditions. Particularly, expression levels of endogenous nAChRs are drastically affected by such factors (Blumenthal et al., 1997). Nonetheless, this issue may be overcome with the availability of subtype-specific ligands for nAChRs.
Other α7 nAChR agonists used in the present study showed similar effects on ERK phosphorylation and required the presence of the PAM, PNU-120596, to achieve substantial ERK phosphorylation. Interestingly, the potency of PNU-120596 was independent from that of the agonist used (data not shown), while in the presence of PNU-120596, a range of α7 nAChR agonists could be differentiated based on their potency in activating ERK signalling. Comparison of the EC50 values of agonists (in presence of PNU-120596) showed a good correlation with their binding Ki values and demonstrates that the pharmacology of ERK activation was consistent with interaction with α7 nAChRs. In addition, here we show that the PAM provides an excellent tool to elucidate the subtype-selective signalling pathways of this highly diversified nAChR family. Taken together, our present results implicate α7 nAChRs in the ERK signalling and associated pathways relevant to cognitive and neuroprotective processes.
Glossary
Abbreviations:
- ERK
extracellular signal-regulated kinase
- FLIPR
fluorometric imaging plate reader
- ICW
In-Cell Western
- MLA
methyllycaconitine
- nAChR
nicotinic acetylcholine receptor
- PAM
positive allosteric modulator
Conflict of interest
This study was supported by Abbott Laboratories.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153(Suppl.)(2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DJ, Bunnelle WH, Surber BW, Du J, Surowy CS, Tribollet E, et al. 3H]A-585539[(1S,4S)-2,2-dimethyl-5-(6-phenylpyridazin-3-yl)-5-aza-2-azoniabicyclo[2.2.1]heptane], a novel high-affinity alpha7 neuronal nicotinic receptor agonist: radioligand binding characterization to rat and human brain. J Pharmacol Exp Ther. 2008;324:179–187. doi: 10.1124/jpet.107.130062. [DOI] [PubMed] [Google Scholar]
- Berg DK, Conroy WG. Nicotinic alpha7 receptors: synaptic options and downstream signaling in neurons. J Neurobiol. 2002;53:512–523. doi: 10.1002/neu.10116. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Gopalakrishnan M. Allosteric modulation of nicotinic acetylcholine receptors. Biochem Pharmacol. 2007;74:1155–1163. doi: 10.1016/j.bcp.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Bitner RS, Bunnelle WH, Anderson DJ, Briggs CA, Buccafusco J, Curzon P, et al. Broad-spectrum efficacy across cognitive domains by alpha7 nicotinic acetylcholine receptor agonism correlates with activation of ERK1/2 and CREB phosphorylation pathways. J Neurosci. 2007;27:10578–10587. doi: 10.1523/JNEUROSCI.2444-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biton B, Bergis OE, Galli F, Nedelec A, Lochead AW, Jegham S, et al. SSR180711, a novel selective alpha7 nicotinic receptor partial agonist: (1) binding and functional profile. Neuropsychopharmacology. 2007;32:1–16. doi: 10.1038/sj.npp.1301189. [DOI] [PubMed] [Google Scholar]
- Blumenthal EM, Conroy WG, Romano SJ, Kassner PD, Berg DK. Detection of functional nicotinic receptors blocked by alpha-bungarotoxin on PC12 cells and dependence of their expression on post-translational events. J Neurosci. 1997;16:6094–6104. doi: 10.1523/JNEUROSCI.17-16-06094.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar AL, Cortes-Burgos LA, Cook KK, Dinh DM, Groppi VE, Hajos M, et al. Discovery and structure-activity relationship of quinuclidine benzamides as agonists of alpha7 nicotinic acetylcholine receptors. J Med Chem. 2005;48:905–908. doi: 10.1021/jm049363q. [DOI] [PubMed] [Google Scholar]
- Boess FG, De Vry J, Erb C, Flessner T, Hendrix M, Luithle J, et al. The novel alpha7 nicotinic acetylcholine receptor agonist N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-7-[2-(methoxy)phenyl]-1-benzofuran-2-carboxamide improves working and recognition memory in rodents. J Pharmacol Exp Ther. 2007;321:716–725. doi: 10.1124/jpet.106.118976. [DOI] [PubMed] [Google Scholar]
- Boulter J, Connolly J, Deneris E, Goldman D, Heinemann S, Patrick J. Functional expression of two neuronal nicotinic acetylcholine receptors from cDNA clones identifies a gene family. Proc Natl Acad Sci USA. 1987;84:7763–7767. doi: 10.1073/pnas.84.21.7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter J, O'Shea-Greenfield A, Duvoisin RM, Connolly JG, Wada E, Jensen A, et al. Alpha 3, alpha 5, and beta 4: three members of the rat neuronal nicotinic acetylcholine receptor-related gene family form a gene cluster. J Biol Chem. 1990;265:4472–4482. [PubMed] [Google Scholar]
- Briggs CA, Anderson DJ, Brioni JD, Buccafusco JJ, Buckley MJ, Campbell JE, et al. Functional characterization of the novel neuronal nicotinic acetylcholine receptor ligand GTS-21 in vitro and in vivo. Pharmacol Biochem Behav. 1997;57:231–241. doi: 10.1016/s0091-3057(96)00354-1. [DOI] [PubMed] [Google Scholar]
- Briggs CA, Gronlien JH, Timmermann DB, Kerr P, Curzon P, Damsgaard J, et al. Role of channel activation in alpha7 nAChR-mediated cognitive enhancement. Biochem Pharmacol. 2007;74:25–26. Satellite Meeting Abstracts. [Google Scholar]
- Couturier S, Bertrand D, Matter JM, Hernandez MC, Bertrand S, Millar N, et al. A neuronal nicotinic acetylcholine receptor subunit (alpha7) is developmentally regulated and forms a homo-oligomeric channel blocked by alpha-BTX. Neuron. 1990;6:847–856. doi: 10.1016/0896-6273(90)90344-f. [DOI] [PubMed] [Google Scholar]
- Curzon P, Anderson DJ, Nikkel AL, Fox GB, Gopalakrishnan M, Decker MW, et al. Antisense knockdown of the rat alpha7 nicotinic acetylcholine receptor produces spatial memory impairment. Neurosci Lett. 2006;410:15–19. doi: 10.1016/j.neulet.2006.09.061. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador F, Wonnacott S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci. 2004;25:317–324. doi: 10.1016/j.tips.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador FA, Soliakov L, Wonnacott S. Nicotine activates the extracellular signal-regulated kinase 1/2 via the alpha7 nicotinic acetylcholine receptor and protein kinase A, in SH-SY5Y cells and hippocampal neurones. J Neurochem. 2002;80:520–530. doi: 10.1046/j.0022-3042.2001.00725.x. [DOI] [PubMed] [Google Scholar]
- Dani JA, Ji D, Zhou FM. Synaptic plasticity and nicotine addiction. Neuron. 2001;3:349–352. doi: 10.1016/s0896-6273(01)00379-8. [DOI] [PubMed] [Google Scholar]
- Dickinson JA, Hanrott KE, Mok MH, Kew JN, Wonnacott S. Differential coupling of alpha7 and non-alpha7 nicotinic acetylcholine receptors to calcium-induced calcium release and voltage-operated calcium channels in PC12 cells. J Neurochem. 2007;100:1089–1096. doi: 10.1111/j.1471-4159.2006.04273.x. [DOI] [PubMed] [Google Scholar]
- Fernandes C, Hoyle E, Dempster E, Schalkwyk LC, Collier DA. Performance deficit of alpha7 nicotinic receptor knockout mice in a delayed matching-to-place task suggests a mild impairment of working/episodic-like memory. Genes Brain Behav. 2006;5:433–440. doi: 10.1111/j.1601-183X.2005.00176.x. [DOI] [PubMed] [Google Scholar]
- Gotti C, Zoli M, Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci. 2006;27:482–491. doi: 10.1016/j.tips.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Gronlien HJ, Hakerud M, Ween H, Thorin-Hagene K, Briggs CA, Gopalakrishnan M, et al. Distinct profiles of {alpha}7 nAChR positive allosteric modulation revealed by structurally diverse chemotypes. Mol Pharmacol. 2007;72:715–724. doi: 10.1124/mol.107.035410. [DOI] [PubMed] [Google Scholar]
- Henderson LP, Gdovin MJ, Liu C, Gardner PD, Maue RA. Nerve growth factor increases nicotinic ACh receptor gene expression and current density in wild-type and protein kinase A-deficient PC12 cells. J Neurosci. 1994;14:1153–1163. doi: 10.1523/JNEUROSCI.14-03-01153.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Liu QS, Chang KT, Berg DK. Nicotinic regulation of CREB activation in hippocampal neurons by glutamatergic and nonglutamatergic pathways. Mol Cell Neurosci. 2002;21:616–625. doi: 10.1006/mcne.2002.1202. [DOI] [PubMed] [Google Scholar]
- Hu M, Schurdak ME, Puttfarcken PS, El Kouhen R, Gopalakrishnan M, Li J. High content screen microscopy analysis of A beta 1-42-induced neurite outgrowth reduction in rat primary cortical neurons: neuroprotective effects of alpha7 neuronal nicotinic acetylcholine receptor ligands. Brain Res. 2007;1151:227–235. doi: 10.1016/j.brainres.2007.03.051. [DOI] [PubMed] [Google Scholar]
- Hurst RS, Hajos M, Raggenbass M, Wall TM, Higdon NR, Lawson JA, et al. A novel positive allosteric modulator of the alpha7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization. J Neurosci. 2005;25:4396–4405. doi: 10.1523/JNEUROSCI.5269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JJ, Keller AB, Bowers BJ, Wehner JM. Performance of alpha7 nicotinic receptor null mutants is impaired in appetitive learning measured in a signaled nose poke task. Behav Brain Res. 2005;162:143–152. doi: 10.1016/j.bbr.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Levin ED, Rezvani AH. Nicotinic treatment for cognitive dysfunction. Curr Drug Targets CNS Neurol Disord. 2002;4:423–431. doi: 10.2174/1568007023339102. [DOI] [PubMed] [Google Scholar]
- Li Y, Meyer EM, Walker DW, Millard WJ, He YJ, King MA. Alpha7 nicotinic receptor activation inhibits ethanol-induced mitochondrial dysfunction, cytochrome c release and neurotoxicity in primary rat hippocampal neuronal cultures. J Neurochem. 2002;4:853–858. doi: 10.1046/j.1471-4159.2002.00891.x. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Collins AC. Characterization of nicotine binding in mouse brain and comparison with the binding of alpha-bungarotoxin and quinuclidinyl benzilate. Mol Pharmacol. 1982;3:554–564. [PubMed] [Google Scholar]
- Martin LF, Kem WR, Freedman R. Alpha-7 nicotinic receptor agonists: potential new candidates for the treatment of schizophrenia. Psychopharmacology (Berl) 2004;1:54–64. doi: 10.1007/s00213-003-1750-1. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Numakawa T, Ikeuchi T, Hatanaka H. Nicotine-induced phosphorylation of extracellular signal-regulated protein kinase and CREB in PC12h cells. J Neurochem. 2001;79:489–498. doi: 10.1046/j.1471-4159.2001.00602.x. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Numakawa T, Ikeuchi T. Nicotine-induced phosphorylation of Akt through epidermal growth factor receptor and Src in PC12h cells. J Neurochem. 2002;83:1372–1379. doi: 10.1046/j.1471-4159.2002.01248.x. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Shimoke K, Isosaki M, Satoh H, Yoshizumi M, Ikeuchi T. Subtypes of neuronal nicotinic acetylcholine receptors involved in nicotine-induced phosphorylation of extracellular signal-regulated protein kinase in PC12h cells. Neurosci Lett. 2006;392:101–104. doi: 10.1016/j.neulet.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Ng HJ, Whittemore ER, Tran MB, Hogenkamp DJ, Broide RS, Johnstone TB, et al. Nootropic alpha7 nicotinic receptor allosteric modulator derived from GABAA receptor modulators. Proc Natl Acad Sci USA. 2007;104:8059–8064. doi: 10.1073/pnas.0701321104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichat P, Bergis OE, Terranova JP, Urani A, Duarte C, Santucci V, et al. SSR180711, a novel selective alpha7 nicotinic receptor partial agonist: (II) efficacy in experimental models predictive of activity against cognitive symptoms of schizophrenia. Neuropsychopharmacology. 2006;32:17–34. doi: 10.1038/sj.npp.1301188. [DOI] [PubMed] [Google Scholar]
- Ren K, Puig V, Papke RL, Itoh Y, Hughes JA, Meyer EM. Multiple calcium channels and kinases mediate alpha7 nicotinic receptor neuroprotection in PC12 cells. J Neurochem. 2005;94:926–933. doi: 10.1111/j.1471-4159.2005.03223.x. [DOI] [PubMed] [Google Scholar]
- Rogers SW, Mandelzys A, Deneris ES, Cooper E, Heinemann S. The expression of nicotinic acetylcholine receptors by PC12 cells treated with NGF. J Neurosci. 1992;12:4611–4623. doi: 10.1523/JNEUROSCI.12-12-04611.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermann DB, Grønlien JH, Kohlhaas KL, EØ N, Dam E, Jørgensen TD, et al. An allosteric modulator of the alpha7 nicotinic acetylcholine receptor possessing cognition-enhancing properties in vivo. J Pharmacol Exp Ther. 2007;323:294–307. doi: 10.1124/jpet.107.120436. [DOI] [PubMed] [Google Scholar]
- Utsugisawa K, Nagane Y, Obara D, Tohgi H. Overexpression of alpha7 nicotinic acetylcholine receptor prevents G1-arrest and DNA fragmentation in PC12 cells after hypoxia. J Neurochem. 2002;81:497–505. doi: 10.1046/j.1471-4159.2002.00823.x. [DOI] [PubMed] [Google Scholar]
- Van Kampen M, Selbach K, Schneider R, Schiegel E, Boess F, Schreiber R. AR-R 17779 improves social recognition in rats by activation of nicotinic alpha7 receptors. Psychopharmacology (Berl) 2004;172:375–383. doi: 10.1007/s00213-003-1668-7. [DOI] [PubMed] [Google Scholar]
- Vaudry D, Stork PJ, Lazarovici P, Eiden LE. Signaling pathways for PC12 cell differentiation: making the right connections. Science. 2002;296:1648–1649. doi: 10.1126/science.1071552. [DOI] [PubMed] [Google Scholar]
- Virginio C, Giacometti A, Aldegheri L, Rimland JM, Terstappen GC. Pharmacological properties of rat alpha7 nicotinic receptors expressed in native and recombinant cell systems. Eur J Pharmacol. 2002;445:153–161. doi: 10.1016/s0014-2999(02)01750-8. [DOI] [PubMed] [Google Scholar]
- Wehner JM, Keller JJ, Keller AB, Picciotto MR, Paylor R, Booker TK, et al. Role of neuronal nicotinic receptors in the effects of nicotine and ethanol on contextual fear conditioning. Neuroscience. 2004;129:11–24. doi: 10.1016/j.neuroscience.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Wishka DG, Walker DP, Yates KM, Reitz SC, Jia S, Myers JK, et al. Discovery of N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]furo[2,3-c]pyridine-5-carboxamide, an agonist of the alpha7 nicotinic acetylcholine receptor, for the potential treatment of cognitive deficits in schizophrenia: synthesis and structure-activity relationship. J Med Chem. 2006;49:4425–4436. doi: 10.1021/jm0602413. [DOI] [PubMed] [Google Scholar]
- Wong SK. A 384-well cell-based phospho-ERK assay for dopamine D2 and D3 receptors. Anal Biochem. 2004;333:265–272. doi: 10.1016/j.ab.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Young JW, Crawford N, Kelly JS, Kerr LE, Marston HM, Spratt C, et al. Impaired attention is central to the cognitive deficits observed in alpha7 deficient mice. Eur Neuropsychopharmacol. 2007;17:145–155. doi: 10.1016/j.euroneuro.2006.03.008. [DOI] [PubMed] [Google Scholar]