

Abstract
Transthyretin (TTR) amyloidogenesis inhibitors are typically composed of two aromatic rings and a linker. We have previously established optimal structures for one aromatic ring and the linker. Herein, we employ a suboptimal linker and an optimal aryl-X substructure to rank order the desirability of aryl-Z substructures–using a library of 56 N-(3,5-dibromo-4-hydroxyphenyl)benzamides. Co-consideration of amyloid inhibition potency and ex-vivo plasma TTR binding selectivity data reveal that 2,6; 2,5; 2; 3,4,5 and 3,5 aryls bearing small substituents generate the most potent and selective inhibitors, in descending order. These benzamides generally lack undesirable thyroid hormone receptor binding and COX-1 inhibition activity. Three high-resolution TTR•inhibitor crystal structures (1.31-1.35 Å) provide insight into why these inhibitors are potent and selective, enabling future structure-based design of TTR kinetic stabilizers.
Introduction
An aging-associated decline in proteostasis capacity can lead to aggregation-linked gain-of-toxic-function protein misfolding diseases such as the amyloidoses, especially when proteome maintenance is further challenged by the inheritance of mutant misfolding-prone proteins or by environmental factors.1-6 Transthyretin (TTR) is one of more than thirty human amyloidogenic proteins whose misfolding and misassembly into a variety of aggregate structures, including cross-β-sheet amyloid fibrils, appears to cause proteotoxicity.7-11 What the TTR toxic structures are and how toxicity arises are key unanswered questions.
To become amyloidogenic outside the cell, tetrameric TTR must first undergo rate-limiting dissociation, allowing the resulting monomers to partially unfold and misassemble.9 Another possibility is that TTR amyloidogenesis competes with folding and TTR tetramerization within the cellular secretory pathway, leading to intracellular proteotoxicity. Thus, proteotoxicity could have its origins both within and outside the cell and this issue remains to be resolved. Aggregation of wild-type transthyretin (WT-TTR) and the resulting proteotoxicity appears to cause senile systemic amyloidosis (SSA), a cardiac disease affecting up to ≈ 15% of the population over age 65.9, 12-14 Deposition of the V122I-TTR variant leads to familial amyloid cardiomyopathy (FAC) in up to 4% of African Americans carrying at least one V122I-TTR allele, while amyloid-associated cardiomyopathy linked to the proteotoxicity arising from other TTR variant aggregates has a lower penetrance.15, 16 Amyloidogenesis of V30M-TTR, or the aggregation of one of nearly one hundred other rarer TTR mutations, leads to familial amyloid polyneuropathy (FAP), usually presenting with peripheral neuropathy and sometimes autonomic and organ system involvement.17 The much rarer central nervous system selective amyloidoses (CNSA) result from deposition of highly destabilized TTR variants (e.g. D18G and A25T) in the brain, but not in the periphery. This is because the liver, which secretes TTR into the blood, detects these variants as misfolding prone and degrades them, unlike the choroid plexus which is a more permissive secretor of misfolding-prone variants into the brain.18-24 Without treatment, the TTR amyloidoses are fatal.
The only currently accepted therapeutic strategy to ameliorate FAP is gene therapy mediated by liver transplantation, wherein an FAP-associated mutant TTR/WT-TTR liver is replaced by a WT-TTR/WT-TTR secreting liver, eliminating the presence of amyloidogenic mutant TTR in the blood.25-27 Unfortunately, WT-TTR deposition often continues post-transplantation in the heart, leading to cardiomyopathy, consistent with the hypothesis that an age-dependent decline in proteostasis contributes to the etiology of the TTR amyloidoses.1, 28 Because liver transplantation must be performed early in the course of the disease to be effective, and owing to the shortage of livers, the expense associated with transplantation, and the requirement for life-long immune suppression, a generally applicable, oral small molecule therapeutic strategy for all the TTR-based amyloid diseases is highly desirable.9, 29, 30
Transthyretin transports the holo-retinol binding protein complex and thyroxine (T4) in the blood.9, 31-34 The four 127-amino acid β-sheet rich subunits of transthyretin create two distinct dimer-dimer interfaces. 9, 35, 36 The energetically weaker interface forms two concave thyroxine binding pockets. Small molecule binding to only one or both of the T4 sites differentially stabilizes the tetrameric ground state over the dissociative transition state, imposing kinetic stability by precluding dissociation required for amyloidogenesis.9, 35-38
Optimism that such small molecule kinetic stabilizers of TTR 24, 39-47 will be effective against the TTR amyloidoses is warranted based on human genetic evidence. T119M subunit inclusion into tetramers otherwise composed of V30M disease-associated subunits in compound heterozygotes kinetically stabilizes the tetrameric structure of TTR, preventing amyloidogenesis and ameliorating FAP.9, 37, 48-50
To increase the probability of identifying TTR kinetic stabilizers with desirable pharmacological properties, three small molecule libraries have recently been designed and synthesized to optimize the three substructures comprising a typical TTR amyloidogenesis inhibitor (Figure 1): the two aromatic rings and the linker joining them. 51, 52 In each library, one fixed substructure was purposefully chosen to be non-optimal to afford a wider range of TTR amyloidogenesis inhibitor potencies and TTR plasma binding selectivities that are together used to rank order the numerous substructures of interest. Data from the first library, focused on optimization of the aryl-X ring, revealed that thyroid hormone-like aryl substitution patterns (3,5-X2-4-OH, where X=CH3, F, Cl, Br, I) lead to potent and highly selective TTR amyloidogenesis inhibitors lacking undesirable thyroid hormone receptor and cycylooxygenase-1 (COX-1) binding activities.51 The second library, designed to reveal optimal linker substructures, demonstrates that directly linking two aryls, or tethering them through non-polar E-olefin or –CH2CH2– substructures generates the most potent and selective TTR amyloidogenesis inhibitors that also lack the undesirable thyroid hormone and COX-1 activities outlined above.52 In this study, we utilize a non-optimal amide linker and an optimal aryl-X substructure in all library members to rank order candidate aryl-Z substructures. The intention of this three part series of papers is to generate optimal substructure information that can ultimately be used to predict the structures of potent and selective TTR kinetic stabilizers that lack the thyroid hormone receptor binding capacity and COX-1 inhibition activity contraindicated for cardiomyopathy applications. These activities should increase the probability of finding a TTR kinetic stabilizer with desirable pharmacologic properties going forward.
Figure 1.

Schematic depiction of one T4 binding pocket occupied by a typical small molecule TTR aggregation inhibitor. Y represents a linker of variable chemical structure (e.g. NH, O, CH=CH, C(O)NH, etc.) joining the two aryl rings, which typically bear a combination of alkyl, carboxyl, halide, trifluoromethyl, or hydroxyl substituents (X and Z). Library 3, the focus of this manuscript, explores the SAR of the aryl-Z substructure. While the aryl-X substructure was initially envisioned to bind within the inner cavity of the thyroxine binding site, previous crystallographic data reveal that inhibitors can bind opposite to the expected orientation.34, 51, 52, 59-61 Thus, it may not be the case that the aryl-X substructure of an inhibitor will occupy the inner binding cavity as illustrated. For conceptual simplicity in terms of integrating the data from the three inhibitor substructure optimization libraries, this initial schematic has been retained.
Herein, we report a 56-member library wherein all members are composed of an optimal 3,5-dibromo-4-hydroxyphenyl aryl-X ring attached to the NH of a nonoptimal secondary amide linker connected to variable aryl-Z rings, which are the subject of this optimization study (Figure 2). Numerous aryl-Z substituents and substituent patterns dramatically increase inhibitor potency and binding selectivity in comparison to the parent N-(3,5-dibromo-4-hydroxyphenyl)benzamide inhibitor (1), while generally avoiding undesirable binding to the thyroid hormone receptor and COX-1 inhibition. High-resolution X-ray structures of three TTR•inhibitor complexes (1.31-1.35 Å) rationalizes their potency and selectivity.
Figure 2.
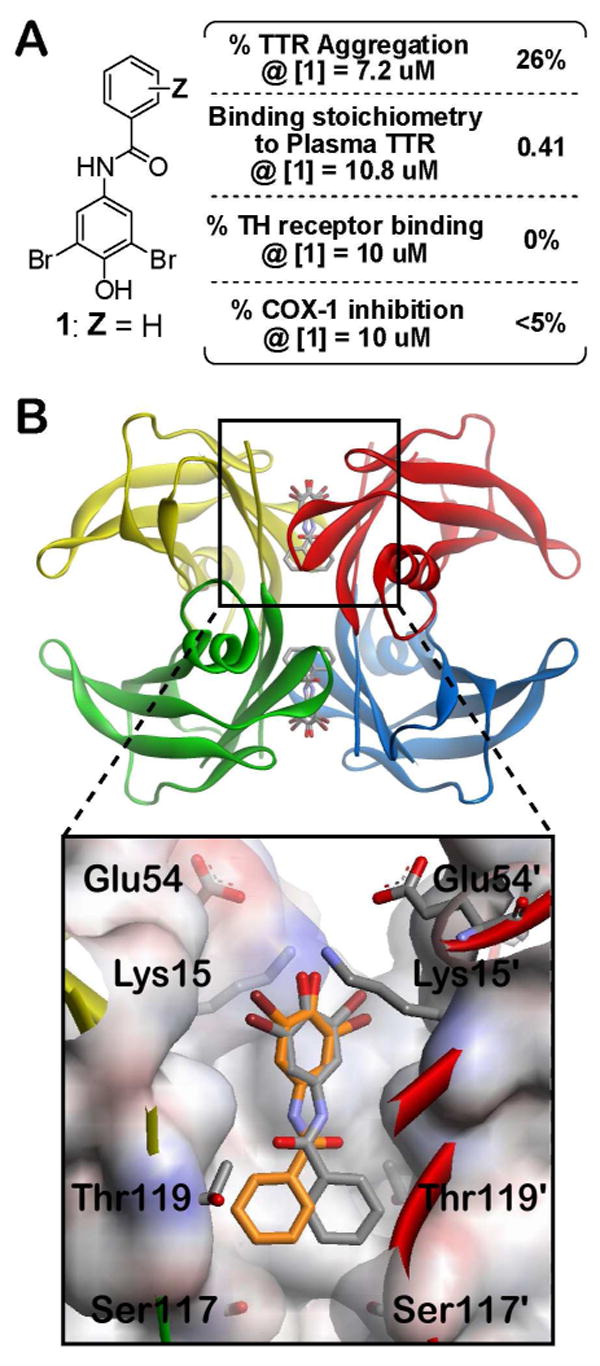
Selection of an appropriate molecular scaffold for structure-guided drug design to identify favorable aryl-Z substituents and substitution patterns to evaluate in this study. A. N-(3,5-dibromo-4-hydroxyphenyl)benzamide (1) was previously found to be a moderately potent and selective inhibitor of TTR amyloidogenesis, allowing 26% aggregation of WT-TTR in vitro (7.2 μM inhibitor, 3.6 μM TTR, pH 4.4, 37°C, 72 h) and displaying a stoichiometry of 0.41 equivalents bound to TTR in human blood plasma ex vivo.52 Importantly, compound 1 does not target the thyroid hormone (TH) receptor or inhibit COX-1 activity. B. Molecular modeling was performed using the previously determined TTR•(1)2 crystal structure (PDB accession code 3CN4), in which compound 1 binds in the reverse orientation with its 3,5-dibromo-4-hydroxyphenyl substructure occupying the outer binding site cavity, projecting the aryl-Z ring under evaluation into the halogen binding pockets of the inner cavity.52 Individual TTR monomers in the three-dimensional ribbon diagram have been color coded red, yellow, green, and blue for differentiation. The zoomed image of one site shows 1 bound in its two symmetry-related binding modes (orange and grey). The protein surface is shown colored by electrostatic potential, with the front portion cut away to allow easier visualization into the pocket.
Results
Design and synthesis of the aryl-Z optimization library
In the recently published linker optimization component (part 2) of this three-part study, N-(3,5-dibromo-4-hydroxyphenyl)benzamide (1) was established to be a moderately potent and selective transthyretin amyloidogenesis inhibitor (Figure 2A), 52 lacking contraindicated thyroid hormone receptor binding or COX-1 inhibition properties. The amide linker is isostructural with the most potent 2-arylbenzoxazole and E-olefin linkers, but is nonoptimal because its polarity is poorly matched with the hydrophobic properties of TTR subsite that is proximal to the linker. Thus the bisarylamide library described within to optimize the aryl-Z ring should prove to be highly relevant, especially considering that all library members are made up of an optimized aryl-X ring identified in the first paper in this series.
The TTR•(1)2 crystal structure (PDB accession code 3CN4) solved previously,52 enables structure-based design principles to be used to identify favorable aryl-Z substituents and substitution patterns, thus enhancing the probability for success. Compound 1 binds such that the 3,5-Br2-4-hydroxyphenyl substructure (having a calculated pKa of 6.5) occupies the outer thyroxine binding site (true of all low pKa phenols structurally characterized to date) where the phenolate can interact with the Lys-15 and 15′ side chain ε-NH3+ groups (Figure 2B).51, 52 This orientation projects the aryl-Z ring into the inner cavity, where its substituents can occupy the symmetric hydrophobic depressions referred to as halogen binding pockets 3 and 3′(HBP-3 and 3′). Upon slight repositioning of the aryl-Z ring, it appears that ortho and meta substituents could interact favorably with HBP-3 and 3′. In addition, polar meta or para substituents (e.g. amino groups or high pKa phenols) could enhance binding affinity through hydrogen bonding with the Ser-117/117′ hydroxyls.
Certain aryl-Z substructures, such as low pKa phenols, could change the binding orientation, such that the 3,5-Br2-4-hydroxyphenyl substructure common to all library members now occupies the inner binding cavity. If this were to occur, modeling suggests that the same aryl-Z substructures bearing ortho and meta alkyl and halide substituents could interact favorably with the hydrophobic HBP-1 and 1′, while meta or para carboxyl, amino or phenolic substituents could make electrostatic interactions with the Lys-15 and 15′ ε-NH3+ groups or the Glu-54 and 54′ carboxylate groups.
Using structure-based principles as a rough guideline, a library of 56 bisarylamides was synthesized to evaluate 10 different aryl-Z substituents (a-j) in 8 distinct substitution patterns (2-9). Co-consideration of amyloid inhibition and ex vivo plasma TTR binding selectivity data using a simple equation allows us to rank order the aryl-Z substructures from most desirable to least desirable using an efficacy score (Figure 3; see below).
Figure 3.
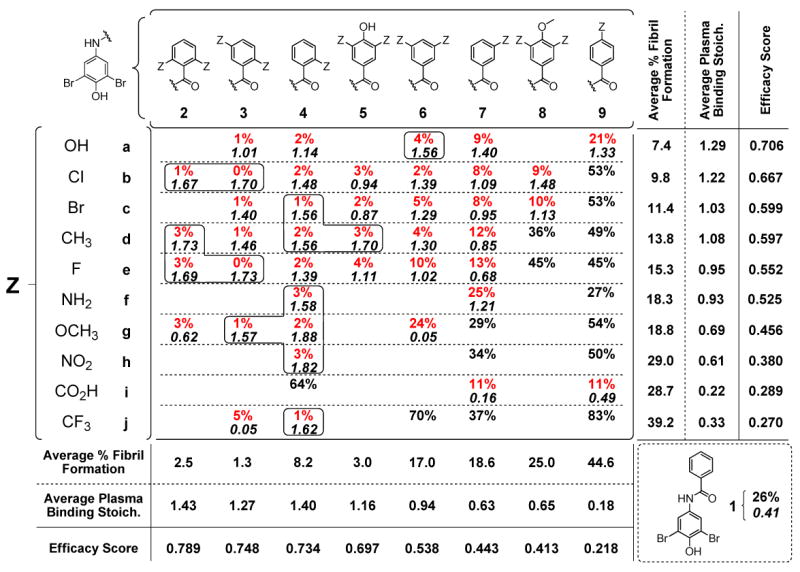
Inhibition of TTR aggregation and stoichiometry of inhibitor bound to TTR in human blood plasma. Percent (%) values represent the extent of in vitro WT-TTR fibril formation in the presence of inhibitor (7.2 μM inhibitor, 3.6 μM TTR, pH 4.4, 37°C, 72 h) relative to aggregation in the absence of inhibitor (100%), with the best values shown in red (<25% aggregation; errors are typically less than ±5 percentage points). The average stoichiometries of the most potent aggregation inhibitors bound to TTR in human blood plasma ex vivo are shown in italics (10.8 μM inhibitor, 1.8-5.4 μM TTR; theoretical maximum binding stoichiometry = 2). Those exhibiting exceptional binding selectivity to TTR are boxed (errors are typically less than ±0.1).
Synthesis of these bisarylamides was accomplished primarily via coupling of a substituted benzoyl chloride (phthalic anhydride in the case of 4i) with 4-amino-2,6-dibromophenol (Scheme 1) to form the amide bond. The amide bond coupling reactions occurred rapidly in THF and, after 1 hour of mixing, the resultant bisarylamides were precipitated by dilution with water. In many cases, simple filtration and rinsing of the solid with saturated sodium bicarbonate (no basic rinse in the case of 4i) resulted in compounds of acceptable purity for biophysical evaluation (>95% purity as determined by RP-HPLC and 1H-NMR). Compounds not meeting this criterion were further purified by column chromatography over silica. Several compounds required further substituent conversions (i.e. anisole or ester deprotection or NO2 reduction to NH2), accomplished using standard procedures (refer to Scheme 1 and the Experimental and Supporting Information sections). In some cases, synthesis of the appropriate benzoyl chloride building block was required, accomplished by reacting the commercially available benzoic acids with thionyl chloride at elevated temperatures, followed by concentration in vacuo.
Scheme 1.

General synthesis of the bisarylamide inhibitors under evaluation. a) Ar-COCl, THF (20-95%); b) Ar-CO2H, SOCl2, heat, concentrate, then 4-amino-2,6-dibromophenol, THF (9-76%); c) Phthalic anhydride, THF (4i, 66%); d) BBr3, CH2Cl2 (19-87%); e) LiOH•H2O, THF, MeOH, H2O (7h, 70%; 9h, 64%); f) Sn, HCl/AcOH (4f, 93%; 7f, 41%; 9f, 86%).
Small molecule-mediated inhibition of WT-TTR amyloidogenesis
The ability of the bisarylamide library members to inhibit WT-TTR aggregation was evaluated using the acid-mediated aggregation assay used previously to identify optimal aryl-X and linker-Y substructures.51, 52 Briefly, physiologically relevant concentrations of WT-TTR (3.6 μM) were incubated with a candidate inhibitor (7.2 μM) prior to subjecting the solution to partial denaturation conditions that enable ≈ 90% of WT-TTR to aggregate in the absence of an inhibitor after 72 h (pH 4.4, 37 °C). The extent of TTR aggregation (Figure 3) was monitored by measuring sample turbidity in the presence of the test compound, expressed as a percentage of TTR aggregation observed in inhibitor-free samples (assigned to be 100% fibril formation). Of the 56 new compounds evaluated, 41 display increased inhibitor potencies relative to the parent compound 1 (<25% fibril formation, values in red). Twenty eight of the benzamides completely inhibit TTR aggregation (<5% fibril formation) within the 72 h time frame of the assay.
Binding selectivity of amyloidogenesis inhibitors to TTR in human blood plasma
The 41 compounds displaying increased inhibitor potencies relative to 1 (i.e. <25% fibril formation) were further evaluated for their ability to bind selectively to TTR in blood plasma using an ex vivo TTR plasma binding selectivity assay.51-53 Briefly, the candidate inhibitor (10.8 μM) is incubated in human blood plasma in the dark at 37°C for 24 h. Transthyretin, with any bound inhibitor, is then captured by a resin-conjugated anti-TTR antibody and any unbound material is washed away (including weakly or nonspecifically bound inhibitors). The captured TTR•(inhibitor)n complex is then dissociated from the antibody under alkaline conditions and TTR and inhibitor stoichiometry is quantified by RP-HPLC. Results represent the average stoichiometry of inhibitor bound to TTR in blood plasma (Figure 3, lower black italicized values), the maximum value being 2 owing to the presence of the two thyroxine binding sites in each tetramer. Of the 41 inhibitors evaluated, 31 display average binding stoichiometries exceeding 1 equivalent bound per TTR tetramer, 14 of which are exceptionally selective displaying >1.5 equivalents bound (boxed values). Six bisarylamides display modest yet likely sufficient binding stoichiometries (0.5-1.0 equivalents bound), while the remaining 4 exhibit minimal binding selectivity to TTR (<0.5 equivalents).
Evaluating the potent TTR amyloidogenesis inhibitors for COX-1 enzymatic inhibition and binding to the thyroid hormone nuclear receptor
The 41 most potent TTR aggregation inhibitors were further evaluated for their ability to inhibit COX-1 enzymatic activity and for their binding to the thyroid hormone nuclear receptor. These analyses were contracted out to the Cerep laboratories in Redmond, WA, USA (refer to the Experimental section for a detailed description of the assay protocols).51, 52, 54, 55 For the COX-1 inhibition analyses, results represent the % inhibition of arachidonic acid conversion to PGE2 due to competitive binding of test compound (10μM) to COX-1 (Figure 4, lower, black values). Of the 41 compounds evaluated, all but four display <5% inhibition of COX-1 activity: compounds 4e, 4h, 7i, and 8c display slight to modest (10-33%) COX-1 inhibition. For the thyroid hormone receptor binding analyses, the % displacement of [125I]-labeled triiodothyronine (T3, the primary thyroid hormone) was determined from competitive binding of test compound (10μM) to the thyroid hormone receptor (Figure 4, red, italicized values). Of the 41 compounds evaluated, five display >10% inhibition of T3 binding to the thyroid hormone nuclear receptor (3a, 4a, 5e, 6e, and 7a), the maximum activity observed being 28% for compound 6e.
Figure 4.

Thyroid hormone receptor T3 displacement and COX-1 inhibition for the most potent aggregation inhibitors (those displaying <25% aggregation). The extent of the test compounds (10μM) competitively binding to the thyroid hormone receptor and displacing T3 are shown in red italics (errors are typically less than ±2 percentage points). COX-1 inhibition results are shown below in black, with values representing the % inhibition by the test compounds (10μM) of COX-1-mediated conversion of arachidonic acid to prostaglandin-E2 (errors are typically less than ±6 percentage points).
X-ray crystallographic analysis of WT-TTR bound to inhibitors 2d, 3b, and 5d
Crystal structures of compounds 2d, 3b, and 5d in complex with WT-TTR were determined to 1.35 Å, 1.31 Å and 1.35 Å resolutions, respectively (Table 1). In all three structures, the observed electron density allowed unambiguous placement of the kinetic stabilizer. The 3,5-Br2-4-hydroxyphenyl substructure bound in the outer thyroid hormone binding pocket as was the case for the parent TTR•(1)2 co-crystal structure (Figure 5).52 As anticipated, the bromine substituents extend into HBP-1 and 1′, while the putative phenolate makes electrostatic interactions with the Lys-15 and 15′ ε-NH3+ groups. In all cases, at least one of the aryl-Z substituents of 2d, 3b, and 5d positioned in the inner binding cavity project their methyl and chloro substituents into HBP-3 and 3′.
Table 1.
Data collection and refinement statistics for the crystal structures of WT-TTR in complex with inhibitors 2d, 3b, and 5d.
| WT-TTR/2d | WT-TTR/3b | WT-TTR/5d | |
|---|---|---|---|
| Data Collection | |||
| Beamline | SSRL 9-2 | SSRL 9-2 | SSRL 9-2 |
| Wavelength (Å) | 0.9795 | 0.9795 | 0.9795 |
| Resolution (Å) | 1.35 (1.35-1.40)a | 1.31 (1.31-1.36) | 1.35 (1.35-1.36) |
| Space group | P21212 | P21212 | P21212 |
| a, b, c (Å) | 42.65,84.62,64.94 | 42.50,85.00, 64.74 | 42.58,85.34,64.28 |
| No. of molecules in a.u. | 2 | 2 | 2 |
| No. of observations | 346,443 (22,334)a | 353,372 (16,464) | 358,657 (16,884) |
| No. of unique reflections | 51,708 (4,558)a | 54,365 (3,829) | 55,178 (4,221) |
| Redundancy | 6.7 (64.9)a | 6.5 (4.3) | 6.5 (4.0) |
| Completeness (%) | 98.4 (88.1)a | 95.1 (68.0) | 96.4 (75.1) |
| Rsym (%)b | 3.7 (50.1)a | 3.4 (45.2) | 4.0 (33.1) |
| Average I/σ | 38.3 (2.3)a | 39.1 (2.3) | 36.6 (3.6) |
| Refinement statistics | |||
| Resolution (Å) | 64.96-1.35 | 64.68-1.31 | 64.28-1.31 |
| No. of reflections (working set) | 49,039 (3,100)a | 51,574 (2,573) | 52,337 (2,930) |
| No. of reflections (test set) | 2,629 (173)a | 2,744 (142) | 2,794 (156) |
| Rcryst (%)a,c | 17.3 a,c | 17.7 | 17.7 |
| Rfree (%)a,d | 18.8 a,d | 20.4 | 20.0 |
| No. of protein/ligand/water atoms | 891,884/38/215 | 891,884/38/208 | 891,884/44/209 |
| Average B-values | |||
| Protein | 16.6 | 15.2 | 14.9 |
| Ligand | 19.0 | 19.3 | 19.4 |
| Wilson B-value | 20.8 | 18.4 | 17.5 |
| Ramachandran plot | |||
| Most favored (%) | 92.1 | 92.1 | 92.1 |
| Additionally allowed (%) | 7.9 | 7.9 | 7.9 |
| Generously allowed (%) | 0 | 0 | 0 |
| Disallowed (%) | 0 | 0 | 0 |
| R.M.S. deviations | |||
| Bond lengths (Å) | 0.018 | 0.018 | 0.019 |
| Angles (°) | 1.73 | 1.78 | 1.84 |
Numbers in parentheses are for highest resolution shell of data.
Rsym = Σhkl | I-<I> | Σhkl I
Rcryst = Σhkl | Fo-Fc | Σhkl Fo
Rfree is the same as Rcryst, but for 5% of data excluded from the refinement.
Figure 5.
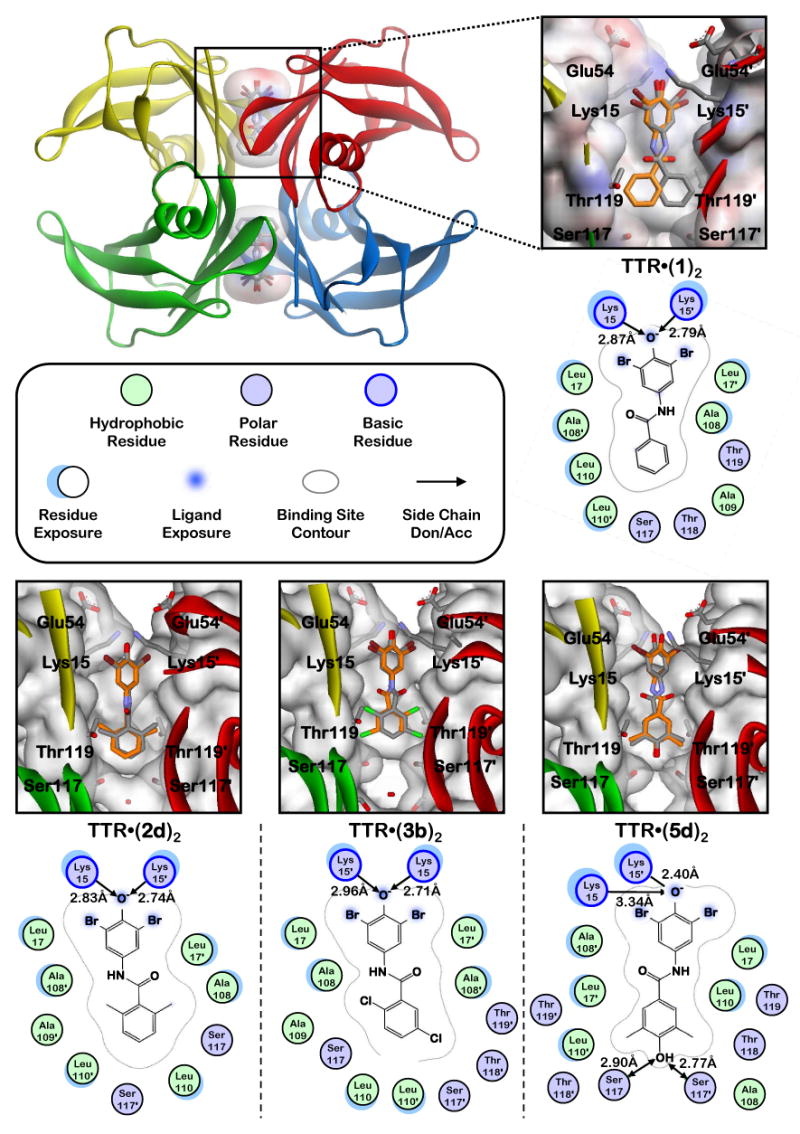
X-ray crystallography structures and schematic representations of key features of homotetrameric WT-TTR co-crystallized with inhibitors 1, 2d, 3b, and 5d (PDB accession codes 3CN4,52 3ESN, 3ESO, and 3ESP, respectively). The zoomed images show the respective inhibitors bound in their two symmetry-related binding modes (orange and grey), with the protein surface shown colored by electrostatic potential (n.b., the front portion has been cut away to allow easier visualization into the pocket). Schematic representations of each bound inhibitor are presented as two-dimensional topology diagrams (generated using MOE (2006.08), Chemical Computing Group, Montreal, Canada). Inhibitors are shown in only one of their two symmetry-related binding modes for clarity. Important hydrogen bonding and electrostatic interactions between protein side chains and the ligands are indicated by arrows, with distances shown in Å. A graphical legend for interpretation of key binding site characteristics is displayed below the TTR tetramer ribbon diagram. Ligand exposure indicates specific portions of the ligand structures that are solvent accessible (i.e. not completely buried within the binding pocket). Residue exposure indicates those amino acids whose side chains and/or peptide backbones are partially solvent accessible.
In the 2d and 3b structures, an ordered water molecule is located between the Ser-117 hydroxyls of each T4 binding site, which is not observed in the TTR•(1)2 structure. In the TTR•(5d)2 structure, this ordered water molecule is absent and instead the 4-phenolic substituent creates a hydrogen bonding network that bridges the Ser-117 and 117′ hydroxyls on adjacent TTR subunits. While additional ordered water molecules have been modeled in elsewhere, these do not make significant interactions with any of the bound inhibitors.
Taking the positioning of the parent ligand in the TTR•(1)2 co-crystal structure as a reference, compound 2d appears to bind ∼0.4 Å deeper within the pocket, while compounds 3b and 5d bind ∼0.3 Å and ∼1.4 Å shallower, respectively. The non-planarity of the aryl-X and aryl-Z rings observed in the TTR•(1)2 co-crystal structure was also observed for these new kinetic stabilizers (46-51° for 2d, 41-44° for 3b, and 43-46° for 5d, compared to 42-45° for 1).
Discussion
Optimal aryl-Z substructures for creating potent TTR amyloidogenesis inhibitors that also exhibit high TTR binding selectivity in blood
To use the previously employed substructure ranking calculation52 that co-considers inhibition efficacy and plasma TTR binding selectivity, we first averaged the percent fibril formation (% F.F.ave) and plasma TTR binding stoichiometry (P.S.ave) values across the columns reflecting different substitution patterns and across the rows reflecting the influence of a given substituent in several contexts. Compounds that were not tested in the plasma selectivity assay because they allowed >25% aggregation of TTR were assigned plasma stoichiometry values of 0 for averaging purposes, as experience demonstrates that poor aggregation inhibitors typically display little, if any, binding to TTR in blood plasma. To obtain an “Efficacy Score” for each linker substructure, the % F.F.ave and P.S.ave values were then input into equation 1.
| Eq. 1 |
Subtraction of the % F.F.ave values from 100% converts the data into % inhibition. The P.S.ave values are adjusted by adding 1 in order to be able to differentiate inhibitors with plasma binding stoichiometries of 0 from each other based on % inhibition data. Division by 300% restricts the efficacy scores to a range between 0 and 1, the latter being the best score. Co-consideration of efficacy and binding selectivity using equation 1 affords clear rank-orderings of the different aryl-Z substituents and substitution patterns (Figure 3). Nearly all of the 2,6; 2,5; 2; 3,4,5; and 3,5 substitution patterns all afforded excellent TTR amyloidogenesis inhibitors that exhibited selective binding to TTR in plasma ex vivo in descending order. The hydroxy, Cl, Br, CH3, F and NH2 substituents generally afforded excellent TTR amyloidogenesis inhibitors that also exhibited selective TTR binding in plasma in descending order. Strictly analogous ranking of aryl-X substituents and substitution patterns as well as optimal linkers now allows highly efficacious and selective inhibitor structures to be predicted.
Generally the most potent and selective TTR amyloidogenesis inhibitors interact minimally with COX-1 enzyme and thyroid hormone receptor
Since many of the potent TTR amyloidogenesis inhibitors in this aryl-Z optimization library bear resemblance to some common non-steroidal anti-inflammatory drugs (NSAIDs), their ability to inhibit the COX-1 enzyme was evaluated. COX-1 binding is undesirable, especially for familial amyloid cardiomyopathy patients, as NSAID activity can further compromise renal blood flow.56 Of the 41 compounds evaluated, four display minimal COX-1 inhibition (4e, 4h, 7i, and 8c).
Since these compounds all contain the thyroid hormone-like 3,5-Br2-4-OH aryl-X ring, we evaluated the 41 most potent kinetic stabilizers for their ability to bind to the thyroid hormone nuclear receptor. Five compounds, the majority bearing OH or F substitutents in distinct substitution patterns displace >10% of T3 from the thyroid hormone receptor (3a, 4a, 5e, 6e, and 7a). The random distribution of substitution patterns suggests that this undesirable characteristic could be prevented by employing different linkers and by minimizing the use of the 3,5-Br2-4-OH aryl-X ring.52 It might be prudent to pursue future inhibitors bearing hydroxyl and fluoro substituents in meta positions with caution.
Structural characterization of potent and selective TTR amyloidogenesis inhibitors with optimal aryl-Z substitution patterns
Structures of kinetic stabilizers 2d, 3b, and 5d in complex with WT-TTR (Figure 5) support the hypothesis that low pKa phenols, including 3,5-Br2-4-hydroxyphenyl substructures, bind in the outer cavity of the thyroxine binding site where their phenolates interact with the Lys-15 and 15′ side chain ε-NH3+ groups, whereas high pKa phenols like 3,5-(CH3)2-4-hydroxyphenyl prefer to occupy the inner binding cavity, H-bonding with the Ser-117 and 117′ hydroxyl groups.51, 52 That 5d is one of the most potent and selective TTR amyloidogenesis inhibitors discovered to date is not surprising as it incorporates both preferred subsite binding aryls. Based on previous structural data51, 52 and that obtained herein, it is likely that the remaining compounds bind with their 3,5-Br2-4-OH aryl ring occupying the outer binding cavity.
Even though the aryl-Z substituents under evaluation are fairly diverse, every one is capable of increasing inhibitor potency and selectivity if displayed in the proper substitution pattern. Bisarylamides bearing ortho aryl-Z substituents are generally more potent than their counterparts bearing meta and especially para substituents, almost independent of the nature of the substituent. Consistent with previously published TTR•(inhibitor)2 X-ray co-crystal structures, the three structures determined in this study reveal that the aryl-X and aryl-Z rings are oriented out-of-plane with respect to one another.9, 34, 38, 57-62 With ortho substituents, unbound inhibitors may have a steric predisposition to the non-planar bound conformations, thereby displaying more favorable binding energetics.
Para-substitution, reflected in the 8 and 9 series, generally exhibits decreased potency and selectivity, likely due to the inability of these aryl-Z rings to engage the HBPs and because of steric clashing with the Ser-117/117′ side chains when positioned in the inner binding cavity. The inability of many of these substituents to hydrogen bond with the Ser-117/117′ hydroxyls in the inner binding cavity or the Lys-15/15′ ε-NH3+ and/or Glu-54/54′ carboxyl groups when positioned in the outer binding cavity could also reduce ligand binding affinity and thus inhibitor potency. This hypothesis is consistent with the observation that hydrogen bonding capable substituents such as phenols (9a and 5b-e) and carboxyls (9i) are well tolerated in this position.
It is clear that alkyl and halide substituents on the aryl-Z ring produce the most potent and selective inhibitors, likely owing to their hydrophobic and charge transfer interactions with the halogen binding pockets, as described in more detail in the first study and elsewhere.51, 63, 64 The hydroxyl substituent on the aryl-Z ring also contributes to high potency and selectivity over a range of substitution patterns, likely owing in large part to its ability to hydrogen bond.
Concluding Remarks
The systematic optimization of the three substructures comprising a typical transthyretin kinetic stabilizer (i.e. the two aromatic rings and the linker joining them) has enabled the rank ordering of substructures that can be combined to produce potent and selective inhibitors. Because most of these TTR kinetic stabilizers are devoid of thyroid hormone receptor binding and COX-1 inhibition activity, it is likely that kinetic stabilizers derived from combinations of the best aryl and linker substructures will be as well, mitigating toxicity in the envisioned long term use of these drugs.51, 52
The high-resolution X-ray crystallographic data obtained from this three part series supports the hypothesis that thyroid-hormone-like, 3,5-X2-4-hydroxyphenyl aryl substructures (X = halide) prefer to bind in the outer binding site when combined with most other substituted aromatics including phenols exhibiting high pKas.51, 52 These structural insights enable us to move beyond simply screening collections of compounds to now being able to confidently employ structure-based drug design for the development of diverse, potent, and highly selective transthyretin amyloidogenesis inhibitors. Use of the structural data, along with the rankings of the three substructural elements enabled by this three-part series, is currently motivating the synthesis of inhibitors that should be exceptionally potent and selective, and thus potential clinical candidates. Upon the identification of clinical candidates, it will be highly desirable to evaluate their cellular and organismal toxicity.
Experimental
General Synthetic Methods
Unless otherwise stated, all chemicals were purchased from commercial suppliers and used without further purification. Reaction progress was monitored by thin-layer chromatography on silica gel 60 F254 coated glass plates (EM Sciences). Flash chromatography was performed using 230-400 mesh silica gel 60 (EM Sciences). NMR spectra were recorded on a Bruker 500 MHz spectrometer. 1H-NMR spectra were calibrated to the d6-DMSO solvent peak at 2.49 ppm. Reverse phase high performance liquid chromatography (RP-HPLC) was performed using a Waters 600 E multi-solvent delivery system employing a Waters 486 tunable absorbance detector and 717 autosampler. Samples were chromatographically separated using a ThermoHypersil-Keystone Betabasic-18 column (model 71503-034630, 150 Å pore size, 3 μm particle size), eluting with a H2O:CH3CN gradient solvent system. Linear gradients were run from either 100:0, 80:20, or 60:40 A:B to 0:100 A:B (A = 95:5 H2O:CH3CN, 0.25% trifluoroacetic acid (TFA); B = 5:95 H2O:CH3CN, 0.25% TFA). Final compound purities were additionally evaluated under distinct RP-HPLC conditions by chromatographically separating samples using a Vydac-C4 column (model 214TP5415, 300 Å pore size, 5 μm particle size), eluting with a H2O:MeOH gradient solvent system. Linear gradients were run from 100:0 to 0:100 C:D (C = 99.75% H2O, 0.25% TFA; D = 100% MeOH). Representative synthetic procedures are presented below, while experimental details for compounds synthesized analogously are presented in the Supporting Information.
Representative amide coupling procedures: Method A
4-Amino-2,6-dibromophenol (∼0.3-0.7 mmol, 1.0 eq.) was mixed with the respective substituted benzoyl chloride (∼0.3-0.7 mmol, ∼1-1.2 eq.) in THF (∼0.5-2.0 mL) at ambient temperature. After ∼1 h the reaction mixtures were diluted with H2O (∼20 mL), sonicated, and the resulting precipitates were filtered, rinsed with H2O, collected, sonicated with sat. NaHCO3 (∼10 mL), filtered, rinsed with H2O, and collected. When necessary, compounds were further purified by flash chromatography over silica, employing a hexanes:EtOAc elution system. All compounds were characterized by 1H-NMR and RP-HPLC and were >95% in purity. See the Supporting Information for specific synthetic procedures and characterization data.
Representative amide coupling procedures: Method B
Substituted benzoic acids (∼0.3-0.7 mmol, ∼1-1.2 eq.) were stirred in SOCl2 (∼2.0 mL) at ∼70-80°C. After ∼1 h the reactions were concentrated, then the resulting acid chlorides were mixed with 4-amino-2,6-dibromophenol (∼0.3-0.7 mmol, 1.0 eq.) in THF (∼0.5-2.0 mL) at ambient temperature. After ∼1 h the reaction mixtures were diluted with H2O (∼20 mL), sonicated, and the resulting precipitates were filtered, rinsed with H2O, collected, sonicated with sat. NaHCO3 (∼10 mL), filtered, rinsed with H2O, and collected. When necessary, compounds were further purified by flash chromatography over silica, employing a hexanes:EtOAc elution system. All compounds were characterized by 1H-NMR and RP-HPLC and were >95% in purity. See the Supporting Information for specific synthetic procedures and characterization data.
Representative anisole deprotection procedures
Boron tribromide (0.5-3.5 mmol of 1 M BBr3 in hexanes, 5-10 eq.) was added to a stirring mixture of the respective anisole (0.1-0.35 mmol, 1 eq.) in anhydrous CH2Cl2 (5-10 mL) and the reaction was stirred at room temperature under an argon atmosphere. After 18 h the reaction was quenched with MeOH (5 mL), extracted into EtOAc (50 mL), and washed with 1 N HCl (25 mL), sat. NaHCO3 (25 mL), and brine (25 mL). The organics were then dried over Na2SO4, filtered, and concentrated. Compounds were purified by flash chromatography over silica, employing a hexanes:EtOAc elution system. All compounds were characterized by 1H-NMR and RP-HPLC and were >95% in purity. See the Supporting Information for specific synthetic procedures and characterization data.
Representative methyl ester hydrolysis procedures
LiOH•H2O (0.5-0.85 mmol, ∼4 eq.) was added to a stirring mixture of the respective methyl ester (0.13-0.2 mmol, 1 eq.) in H2O/MeOH/THF (0.5/0.5/1.5 mL) and the reaction was stirred at room temperature. After 18 h the reaction was acidified with 1 N HCl and extracted into EtOAc (50 mL), washed with H2O (2×25 mL) and brine (25 mL), dried over Na2SO4, filtered, and concentrated. Compounds were characterized by 1H-NMR and RP-HPLC and were >95% in purity. See the Supporting Information for specific synthetic procedures and characterization data.
Representative NO2 to NH2 reduction procedures
Tin powder (0.6-2.2 mmol, 4.0-4.7 eq.) was added to a stirring mixture of the respective nitro compound (0.13-0.55 mmol, 1 eq.) in an HCl/AcOH mixture (0.2/2.0 mL). After 18 h the reaction was diluted with H2O (25 mL), neutralized with NaHCO3, extracted into EtOAc (50 mL) and the organics were washed with H2O (25 mL) and brine (25 mL), dried over Na2SO4, filtered, and concentrated. Compounds were purified by flash chromatography over silica, employing a hexanes:EtOAc elution system. All compounds were characterized by 1H-NMR and RP-HPLC and were >95% in purity. See the Supporting Information for specific synthetic procedures and characterization data.
Evaluating small molecule-mediated inhibition of WT-TTR amyloidogenesis
WT-TTR was purified from an E. coli expression system as described previously.65 To a 495 μL aliquot of 0.4 mg/mL WT-TTR (7.2 μM, 10 mM phosphate, pH 7.0, 100 mM KCl, 1 mM EDTA) in a disposable cuvette was added 5.0 μL of the desired small molecule in DMSO (1.44 mM) and the sample was vortexed briefly. After incubating for 30 min at ambient temperature, the pH of the sample was lowered to 4.4 by addition of 500 μL of acidic buffer (100 mM acetate, pH 4.2, 100 mM KCl, 1 mM EDTA) and the solution was briefly vortexed and incubated in the dark for 72 h at 37°C (final protein and inhibitor concentrations were 3.6 and 7.2 μM, respectively). The sample was then vortexed to evenly distribute any precipitate, and the turbidity was measured at 400 nm on a Hewlett Packard model 8453 UV-Vis Spectrophotometer. The extent of WT-TTR aggregation (% fibril formation, f.f.; % inhibition = 100 - %f.f.) was determined by comparing the sample turbidity in the presence of small molecule as a percentage of control WT-TTR sample incubated with 5.0 μL of pure DMSO (representing 100% aggregation, 0% inhibition). All samples were performed in at least quintuplicate, with average values obtained presented in Figure 3 (errors are typically less than ±5 percentage points).
Evaluating the binding selectivity of amyloidogenesis inhibitors to TTR in human blood plasma
The procedure for the antibody capture method to evaluate the stoichiometry of inhibitor bound to TTR in human blood plasma has been described in detail elsewhere.53 Briefly, 7.5 μL of a 1.44 mM DMSO solution of test compound was incubated with human blood plasma (1.0 mL) in a 2 mL microfuge tube at 37°C for 24 h on a rocker (30 rpm). Then, 125 μL of a 1:1 v:v slurry of unfunctionalized sepharose resin in TSA (10 mM Tris, pH 8.0, 140 mM NaCl) was added and the mixture was incubated for another hour at 4°C on a rocker (18 rpm). The sample was then centrifuged and the supernatant divided into 2 aliquots of 400 μL each, which were each added to 200 μL of a 1:1 v:v slurry of sepharose resin conjugated to an anti-TTR antibody in TSA, and the sample was incubated again at 4°C for 20 min on a rocker (18 rpm). The sample was then centrifuged, the supernatant removed, and the TTR bound resin was washed three times (10 min each wash) with 1 mL TSA/0.05% saponin, then twice (10 min each wash) with 1 mL TSA at 4°C on a rocker (18 rpm). After centrifugation and removal of the final supernatant, dissociation of the TTR and bound test compound from the resin-bound antibody was achieved through addition of 155 μL of aqueous triethylamine (100 mM, pH∼11.5) and rocking (18 rpm) at 4°C for 30 min. The suspension was then centrifuged and 145 μL of the supernatant containing both TTR and test compound was removed, neutralized with 0.84 mL of glacial acetic acid (to prevent peak bleeding during HPLC analysis), and then analyzed by RP-HPLC to determine the stoichiometry of small molecule bound to TTR (the test compound-TTR complex dissociates and the small molecule and protein are chromatographically separable under the HPLC buffer conditions). Quantification of test compound and TTR is achieved by comparing the integrated peak areas to standard curves: the ratio of the amount of test compound to TTR yields the binding stoichiometry, of which a theoretical maximum value of 2 is possible owing to the two T4 binding sites per TTR tetramer. Analyses were performed in at least duplicates of duplicates from at least two different blood plasma samples (i.e. at least 4 analyses), with average values obtained presented in Figure 3 (errors are typically less than ±0.1).
Evaluating the inhibition of COX-1 enzymatic activity by the potent TTR amyloidosis inhibitors
The evaluation of the most potent TTR aggregation inhibitors (displaying <25% fibril formation) at inhibiting COX-1 activity was contracted out to the Cerep laboratories in Redmond, WA, USA. Compound analyses were performed using assay catalog reference #777-1hr, which uses procedures developed by Glaser et al.54 A brief experimental protocol as provided by Cerep is outlined below. The enzyme (∼2 μg) is pre-incubated in the absence (water control) or presence of test compound (10.0 μM) for 20 min at 22°C in 250 μL of buffer (100 mM Tris-HCl, pH 8, 2 mM phenol, 1 μM hematine). Arachidonic acid (4 μM) is then added to initiate the reaction (no arachidonic acid added for basal control measurements). After incubation at 22°C for 10 min, the reaction is quenched by addition of 2 M HCl and 1 M Tris-HCl (pH 7.8) and cooling at 4°C. Prostaglandin-E2 (PGE2) quantification is performed using an EIA detection kit with measurements made using a microplate reader. Values of analyses performed in singleton are presented in Figure 4 (black values), which represent the % inhibition of arachidonic acid conversion to prostaglandin-E2 due to competitive binding of test compound to COX-1 (previous studies typically show representative errors in the data of less than ±6 percentage points).51, 52 Control analyses are performed analogously with the standard inhibitory reference compound diclofenac tested at several concentrations to obtain an inhibition curve from which its IC50 is calculated (12 nM).
Evaluating the binding of the potent TTR amyloidosis inhibitors to the thyroid hormone nuclear receptor
The evaluation of the most potent TTR aggregation inhibitors (displaying <25% fibril formation) at binding to the thyroid hormone receptor was contracted out to the Cerep laboratories in Redmond, WA, USA. Compound analyses were performed using assay catalog reference #855, which uses procedures developed by Inoue et al.55 The experimental protocol as provided by Cerep is briefly outlined below. Liver membrane homogenates (100 μg protein) are incubated for 18 h at 4°C with 0.1 nM 125I-labeled triiodothyronine ([125I]T3, the primary thyroid hormone) in the absence or presence of test compound (10.0 μM) in 500 μL of buffer (20 mM Tris-HCl, pH 7.6, 50 mM NaCl, 2 mM EDTA, 10% glycerol, and 5 mM β-mercaptoethanol). The samples are then vacuum filtered through glass fiber filters (GF/B, Packard), rinsed several times with ice-cold buffer (50 mM Tris-HCl and 150 mM NaCl), and the filters are dried and counted for radioactivity in a scintillation counter (Topcount, Packard) using a scintillation cocktail (Microscint 0, Packard). Non-specific binding, determined in the presence of 1 μM T3, is subtracted from the [125I]T3 binding results. Values of analyses performed in singleton are presented in Figure 4 (red italicized values), which represent the % displacement of 125I-labeled triiodothyronine (T3, the primary thyroid hormone) due to competitive binding of test compound to the thyroid hormone receptor (previous studies typically show representative errors in the data of less than ±2 percentage points).51, 52 Control analyses are performed analogously with T3 tested at several concentrations to obtain a competition curve from which its IC50 is calculated (0.38 nM).
X-ray crystallographic analysis of WT-TTR bound to inhibitors 2d, 3b, and 5d
WT-TTR was purified from an E. coli expression system as described previously.65 The protein was concentrated to 4 mg/mL in 10 mM NaPi, 100 mM KCl, at pH 7.6 and co-crystallized at room temperature with various inhibitors at a 5 molar excess using the vapor-diffusion sitting drop method. Crystals were grown from 1.395 M sodium citrate, 3.5% v/v glycerol at pH 5.5. The crystals were cryo-protected with inhibitor-free 1.395 M sodium citrate, 10% v/v glycerol at pH 5.5. Data were collected on beam line 9-2 at the Stanford Synchrotron Radiation Laboratory (SSRL) at a wavelength of 0.9795 Å. Data sets were integrated and scaled using HKL2000.66 The crystals were indexed in orthorhombic space group P21212 with two subunits per asymmetric unit (see Table 1 for full data collection and refinement statistics). The three crystal structures were determined by molecular replacement using the 2FBR67 coordinates in the program Phaser.68 Further model building and refinement were completed using Coot69 and Refmac.70 For all structures, hydrogens were added during refinement and anisotropic B values calculated. Final models were validated using the JCSG quality control server (http://jcsgsrv2/QC) incorporating Molprobity,71 ADIT (http://rcsb-deposit.rutgers.edu/validate) WHATIF,72 Resolve,73 and Procheck.74 Full data collection and refinement statistics are presented in Table 1.
Supplementary Material
Supporting Information Available: Detailed synthesis and characterization data on additional compounds prepared analogously to the general synthetic procedures presented in the Experimental section, tabulation of compound purity as determined by C4 and C18 RP-HPLC, and calculated phenolic pKa values. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We are grateful for the financial support of the NIH DK 46335 (J.W.K), CA58896 and AI42266 (I.A.W), as well as the Skaggs Institute for Chemical Biology and the Lita Annenberg Hazen Foundation. Assistance with the thyroid hormone receptor and COX-I binding assays by the Cerep laboratories (Redmond, WA, USA and France) and Richard Labaudiniere of FoldRx Pharmaceuticals (Boston, MA, USA), as well as the technical expertise of Ted Foss, M. T. Dendle, and Mike Saure, are also greatly appreciated. X-ray diffraction data were collected at beam line 9-2 at the Stanford Synchrotron Radiation Laboratory (SSRL), a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. The authors would also like to thank Drs. Xiaoping Dai, Andre Schiefner, and Xiaojin Xu in the Wilson laboratory for assistance with data collection.
Abbreviations
- TTR
transthyretin
- COX-1
cyclooxygenase-1
- WT
wild type
- HBP
halogen binding pocket
- PGE2
prostaglandin E2
- T4
thyroxine
- T3
triiodothyronine
- NSAID
non-steroidal anti-inflammatory drug
Footnotes
Protein Data Bank Accession Codes. Atomic coordinates have been deposited in the RCSB Protein Data Bank (www.pdb.org) and are available under accession codes 3ESN (WT-TTR in complex with 2d), 3ESO (WT-TTR in complex with 3b), and 3ESP (WT-TTR in complex with 5d).
References
- 1.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Q, Powers ET, Nieva J, Huff ME, Dendle MA, Bieschke J, Glabe CG, Eschenmoser A, Wentworth P, Jr, Lerner RA, Kelly JW. Metabolite-initiated protein misfolding may trigger Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:4752–4757. doi: 10.1073/pnas.0400924101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 4.Mu TW, Fowler DM, Kelly JW. Partial restoration of mutant enzyme homeostasis in three distinct lysosomal storage disease cell lines by altering calcium homeostasis. PLoS Biol. 2008;6:e26. doi: 10.1371/journal.pbio.0060026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morley JF, Brignull HR, Weyers JJ, Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002;99:10417–10422. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–1474. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- 7.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 8.Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 9.Johnson SM, Wiseman RL, Sekijima Y, Green NS, Adamski-Werner SL, Kelly JW. Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: a focus on the transthyretin amyloidoses. Acc Chem Res. 2005;38:911–921. doi: 10.1021/ar020073i. [DOI] [PubMed] [Google Scholar]
- 10.Cohen FE, Kelly JW. Therapeutic approaches to protein-misfolding diseases. Nature. 2003;426:905–909. doi: 10.1038/nature02265. [DOI] [PubMed] [Google Scholar]
- 11.Kelly JW, Colon W, Lai Z, Lashuel HA, McCulloch J, McCutchen SL, Miroy GJ, Peterson SA. Transthyretin quaternary and tertiary structural changes facilitate misassembly into amyloid. Adv Protein Chem. 1997;50:161–181. doi: 10.1016/s0065-3233(08)60321-6. [DOI] [PubMed] [Google Scholar]
- 12.Westermark P, Bergstrom J, Solomon A, Murphy C, Sletten K. Transthyretin-derived senile systemic amyloidosis: clinicopathologic and structural considerations. Amyloid. 2003;10 1:48–54. [PubMed] [Google Scholar]
- 13.Westermark P, Sletten K, Johansson B, Cornwell GG., 3rd Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci USA. 1990;87:2843–2845. doi: 10.1073/pnas.87.7.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buxbaum JN, Tagoe CE. The genetics of the amyloidoses. Annu Rev Med. 2000;51:543–569. doi: 10.1146/annurev.med.51.1.543. [DOI] [PubMed] [Google Scholar]
- 15.Jiang X, Buxbaum JN, Kelly JW. The V122I cardiomyopathy variant of transthyretin increases the velocity of rate-limiting tetramer dissociation, resulting in accelerated amyloidosis. Proc Natl Acad Sci USA. 2001;98:14943–14948. doi: 10.1073/pnas.261419998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacobson DR, Pastore RD, Yaghoubian R, Kane I, Gallo G, Buck FS, Buxbaum JN. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336:466–473. doi: 10.1056/NEJM199702133360703. [DOI] [PubMed] [Google Scholar]
- 17.Plante-Bordeneuve V, Said G. Transthyretin related familial amyloid polyneuropathy. Curr Opin Neurol. 2000;13:569–573. doi: 10.1097/00019052-200010000-00011. [DOI] [PubMed] [Google Scholar]
- 18.Gambetti P, Russo C. Human brain amyloidoses. Nephrol Dial Transplant. 1998;13 7:33–40. doi: 10.1093/ndt/13.suppl_7.33. [DOI] [PubMed] [Google Scholar]
- 19.Sekijima Y, Hammarstrom P, Matsumura M, Shimizu Y, Iwata M, Tokuda T, Ikeda S, Kelly JW. Energetic characteristics of the new transthyretin variant A25T may explain its atypical central nervous system pathology. Lab Invest. 2003;83:409–417. doi: 10.1097/01.lab.0000059937.11023.1f. [DOI] [PubMed] [Google Scholar]
- 20.Hammarstrom P, Sekijima Y, White JT, Wiseman RL, Lim A, Costello CE, Altland K, Garzuly F, Budka H, Kelly JW. D18G transthyretin is monomeric, aggregation prone, and not detectable in plasma and cerebrospinal fluid: a prescription for central nervous system amyloidosis? Biochemistry. 2003;42:6656–6663. doi: 10.1021/bi027319b. [DOI] [PubMed] [Google Scholar]
- 21.Sekijima Y, Wiseman RL, Matteson J, Hammarstrom P, Miller SR, Sawkar AR, Balch WE, Kelly JW. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121:73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 22.Hurshman Babbes AR, Powers ET, Kelly JW. Quantification of the thermodynamically linked quaternary and tertiary structural stabilities of transthyretin and its disease-associated variants: the relationship between stability and amyloidosis. Biochemistry. 2008;47:6969–6984. doi: 10.1021/bi800636q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiseman RL, Powers ET, Buxbaum JN, Kelly JW, Balch WE. An adaptable standard for protein export from the endoplasmic reticulum. Cell. 2007;131:809–821. doi: 10.1016/j.cell.2007.10.025. [DOI] [PubMed] [Google Scholar]
- 24.Hammarstrom P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc Natl Acad Sci U S A. 2002;99:16427–16432. doi: 10.1073/pnas.202495199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holmgren G, Ericzon BG, Groth CG, Steen L, Suhr O, Andersen O, Wallin BG, Seymour A, Richardson S, Hawkins PN. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet. 1993;341:1113–1116. doi: 10.1016/0140-6736(93)93127-m. [DOI] [PubMed] [Google Scholar]
- 26.Tan SY, Pepys MB, Hawkins PN. Treatment of amyloidosis. Am J Kidney Dis. 1995;26:267–285. doi: 10.1016/0272-6386(95)90647-9. [DOI] [PubMed] [Google Scholar]
- 27.Suhr OB, Herlenius G, Friman S, Ericzon BG. Liver transplantation for hereditary transthyretin amyloidosis. Liver Transpl. 2000;6:263–276. doi: 10.1053/lv.2000.6145. [DOI] [PubMed] [Google Scholar]
- 28.Olofsson BO, Backman C, Karp K, Suhr OB. Progression of cardiomyopathy after liver transplantation in patients with familial amyloidotic polyneuropathy, Portuguese type. Transplantation. 2002;73:745–751. doi: 10.1097/00007890-200203150-00015. [DOI] [PubMed] [Google Scholar]
- 29.Tojo K, Sekijima Y, Kelly JW, Ikeda S. Diflunisal stabilizes familial amyloid polyneuropathy-associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neurosci Res. 2006;56:441–449. doi: 10.1016/j.neures.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 30.Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13:236–249. doi: 10.1080/13506120600960882. [DOI] [PubMed] [Google Scholar]
- 31.Blake CC, Geisow MJ, Oatley SJ, Rerat B, Rerat C. Structure of prealbumin: secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A. J Mol Biol. 1978;121:339–356. doi: 10.1016/0022-2836(78)90368-6. [DOI] [PubMed] [Google Scholar]
- 32.Hornberg A, Eneqvist T, Olofsson A, Lundgren E, Sauer-Eriksson AE. A comparative analysis of 23 structures of the amyloidogenic protein transthyretin. J Mol Biol. 2000;302:649–669. doi: 10.1006/jmbi.2000.4078. [DOI] [PubMed] [Google Scholar]
- 33.Monaco HL, Rizzi M, Coda A. Structure of a complex of two plasma proteins: transthyretin and retinol-binding protein. Science. 1995;268:1039–1041. doi: 10.1126/science.7754382. [DOI] [PubMed] [Google Scholar]
- 34.Klabunde T, Petrassi HM, Oza VB, Raman P, Kelly JW, Sacchettini JC. Rational design of potent human transthyretin amyloid disease inhibitors. Nat Struct Biol. 2000;7:312–321. doi: 10.1038/74082. [DOI] [PubMed] [Google Scholar]
- 35.Foss TR, Kelker MS, Wiseman RL, Wilson IA, Kelly JW. Kinetic stabilization of the native state by protein engineering: Implications for inhibition of transthyretin amyloidogenesis. J Mol Biol. 2005;347:841–854. doi: 10.1016/j.jmb.2005.01.050. [DOI] [PubMed] [Google Scholar]
- 36.Foss TR, Wiseman RL, Kelly JW. The pathway by which the tetrameric protein transthyretin dissociates. Biochemistry. 2005;44:15525–15533. doi: 10.1021/bi051608t. [DOI] [PubMed] [Google Scholar]
- 37.Hammarstrom P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299:713–716. doi: 10.1126/science.1079589. [DOI] [PubMed] [Google Scholar]
- 38.Wiseman RL, Johnson SM, Kelker MS, Foss T, Wilson IA, Kelly JW. Kinetic stabilization of an oligomeric protein by a single ligand binding event. J Am Chem Soc. 2005;127:5540–5551. doi: 10.1021/ja042929f. [DOI] [PubMed] [Google Scholar]
- 39.Green NS, Foss TR, Kelly JW. Genistein, a natural product from soy, is a potent inhibitor of transthyretin amyloidosis. Proc Natl Acad Sci U S A. 2005;102:14545–14550. doi: 10.1073/pnas.0501609102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petrassi HM, Johnson SM, Purkey H, Chiang KP, Walkup T, Jiang X, Powers ET, Kelly JW. Potent and selective structure-based dibenzofuran inhibitors of transthyretin amyloidogenesis: Kinetic stabilization of the native state. J Am Chem Soc. 2005;127:6662–6671. doi: 10.1021/ja044351f. [DOI] [PubMed] [Google Scholar]
- 41.Razavi H, Powers ET, Purkey HE, Adamski-Werner SL, Chiang KP, Dendle MT, Kelly JW. Design, synthesis, and evaluation of oxazole transthyretin amyloidogenesis inhibitors. Bioorg Med Chem Lett. 2005;15:1075–1078. doi: 10.1016/j.bmcl.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 42.Miller SR, Sekijima Y, Kelly JW. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab Invest. 2004;84:545–552. doi: 10.1038/labinvest.3700059. [DOI] [PubMed] [Google Scholar]
- 43.Oza VB, Petrassi HM, Purkey HE, Kelly JW. Synthesis and evaluation of anthranilic acid-based transthyretin amyloid fibril inhibitors. Bioorg Med Chem Lett. 1999;9:1–6. doi: 10.1016/s0960-894x(98)00696-9. [DOI] [PubMed] [Google Scholar]
- 44.Baures PW, Oza VB, Peterson SA, Kelly JW. Synthesis and evaluation of inhibitors of transthyretin amyloid formation based on the non-steroidal anti-inflammatory drug, flufenamic acid. Bioorg Med Chem. 1999;7:1339–1347. doi: 10.1016/s0968-0896(99)00066-8. [DOI] [PubMed] [Google Scholar]
- 45.Peterson SA, Klabunde T, Lashuel HA, Purkey H, Sacchettini JC, Kelly JW. Inhibiting transthyretin conformational changes that lead to amyloid fibril formation. Proc Natl Acad Sci USA. 1998;95:12956–12960. doi: 10.1073/pnas.95.22.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baures PW, Peterson SA, Kelly JW. Discovering transthyretin amyloid fibril inhibitors by limited screening. Bioorg Med Chem. 1998;6:1389–1401. doi: 10.1016/s0968-0896(98)00130-8. [DOI] [PubMed] [Google Scholar]
- 47.Miroy GJ, Lai Z, Lashuel HA, Peterson SA, Strang C, Kelly JW. Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc Natl Acad Sci USA. 1996;93:15051–15056. doi: 10.1073/pnas.93.26.15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hammarstrom P, Schneider F, Kelly JW. Trans-suppression of misfolding in an amyloid disease. Science. 2001;293:2459–2462. doi: 10.1126/science.1062245. [DOI] [PubMed] [Google Scholar]
- 49.Coelho T, Carvalho M, Saraiva MJ, Alves I, Almeida MR, Costa PP. A strikingly benign evolution of FAP in an individual found to be a compound heterozygote for two TTR mutations: TTR Met 30 and TTR Met 119. J Rheumatol. 1993;20:179. [Google Scholar]
- 50.Coelho T, Chorao R, Sausa A, Alves I, Torres MF, Saraiva MJ. Compound heterozygotes of transthyretin Met30 and transthyretin Met119 are protected from the devastating effects of familial amyloid polyneuropathy. Neuromusc Disord. 1996;6:27. [Google Scholar]
- 51.Johnson SM, Connelly S, Wilson IA, Kelly JW. Biochemical and structural evaluation of highly selective 2-arylbenzoxazole-based transthyretin amyloidogenesis inhibitors. J Med Chem. 2008;51:260–270. doi: 10.1021/jm0708735. [DOI] [PubMed] [Google Scholar]
- 52.Johnson SM, Connelly S, Wilson IA, Kelly JW. Toward Optimization of the Linker Substructure Common to Transthyretin Amyloidogenesis Inhibitors Using Biochemical and Structural Studies. J Med Chem. 2008 doi: 10.1021/jm800435s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Purkey HE, Dorrell MI, Kelly JW. Evaluating the binding selectivity of transthyretin amyloid fibril inhibitors in blood plasma. Proc Natl Acad Sci USA. 2001;98:5566–5571. doi: 10.1073/pnas.091431798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Glaser K, Sung ML, O'Neill K, Belfast M, Hartman D, Carlson R, Kreft A, Kubrak D, Hsiao CL, Weichman B. Etodolac selectively inhibits human prostaglandin G/H synthase 2 (PGHS-2) versus human PGHS-1. Eur J Pharmacol. 1995;281:107–111. doi: 10.1016/0014-2999(95)00302-2. [DOI] [PubMed] [Google Scholar]
- 55.Inoue A, Yamakawa J, Yukioka M, Morisawa S. Filter-binding assay procedure for thyroid hormone receptors. Anal Biochem. 1983;134:176–183. doi: 10.1016/0003-2697(83)90280-4. [DOI] [PubMed] [Google Scholar]
- 56.Epstein M. Non-steroidal anti-inflammatory drugs and the continuum of renal dysfunction. J Hypertens Suppl. 2002;20:S17–23. [PubMed] [Google Scholar]
- 57.Johnson SM, Petrassi HM, Palaninathan SK, Mohamedmohaideen NN, Purkey H, Nichols C, Chiang KP, Walkup T, Sacchettini JC, Sharpless KB, Kelly JW. Bisaryloxime ethers as potent inhibitors of transthyretin amyloid fibril formation. J Med Chem. 2005;48:1576–1587. doi: 10.1021/jm049274d. [DOI] [PubMed] [Google Scholar]
- 58.Oza VB, Smith C, Raman P, Koepf EK, Lashuel HA, Petrassi HM, Chiang KP, Powers ET, Sachettinni J, Kelly JW. Synthesis, structure, and activity of diclofenac analogues as transthyretin amyloid fibril formation inhibitors. J Med Chem. 2002;45:321–332. doi: 10.1021/jm010257n. [DOI] [PubMed] [Google Scholar]
- 59.Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. Diflunisal analogues stabilize the native state of transthyretin. Potent inhibition of amyloidogenesis. J Med Chem. 2004;47:355–374. doi: 10.1021/jm030347n. [DOI] [PubMed] [Google Scholar]
- 60.Purkey HE, Palaninathan SK, Kent KC, Smith C, Safe SH, Sacchettini JC, Kelly JW. Hydroxylated polychlorinated biphenyls selectively bind transthyretin in blood and inhibit amyloidogenesis: rationalizing rodent PCB toxicity. Chem Biol. 2004;11:1719–1728. doi: 10.1016/j.chembiol.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 61.Razavi H, Palaninathan SK, Powers ET, Wiseman RL, Purkey HE, Mohamedmohaideen NN, Deechongkit S, Chiang KP, Dendle MT, Sacchettini JC, Kelly JW. Benzoxazoles as transthyretin amyloid fibril inhibitors: synthesis, evaluation, and mechanism of action. Angew Chem Int Ed Engl. 2003;42:2758–2761. doi: 10.1002/anie.200351179. [DOI] [PubMed] [Google Scholar]
- 62.Petrassi HM, Klabunde T, Sacchettini JC, Kelly JW. Structure-based design of N-phenyl phenoxazine transthyretin amyloid fibril inhibitors. J Am Chem Soc. 2000;122:2178–2192. [Google Scholar]
- 63.Andrea TA, Cavalieri RR, Goldfine ID, Jorgensen EC. Binding of thyroid hormones and analogues to the human plasma protein prealbumin. Biochemistry. 1980;19:55–63. doi: 10.1021/bi00542a009. [DOI] [PubMed] [Google Scholar]
- 64.Auffinger P, Hays FA, Westhof E, Ho PS. Halogen bonds in biological molecules. Proc Natl Acad Sci U S A. 2004;101:16789–16794. doi: 10.1073/pnas.0407607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lashuel HA, Wurth C, Woo L, Kelly JW. The most pathogenic transthyretin variant, L55P, forms amyloid fibrils under acidic conditions and protofilaments under physiological conditions. Biochemistry. 1999;38:13560–13573. doi: 10.1021/bi991021c. [DOI] [PubMed] [Google Scholar]
- 66.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 67.Green NS, Palaninathan SK, Sacchettini JC, Kelly JW. Synthesis and characterization of potent bivalent amyloidosis inhibitors that bind prior to transthyretin tetramerization. J Am Chem Soc. 2003;125:13404–13414. doi: 10.1021/ja030294z. [DOI] [PubMed] [Google Scholar]
- 68.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 69.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 70.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 71.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 72.Vriend G. WHAT IF: a molecular modeling and drug design program. J Mol Graph. 1990;8:52–56. 29. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 73.Terwilliger TC. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr D Biol Crystallogr. 2003;59:38–44. doi: 10.1107/S0907444902018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Laskowski R, MacArthur M, Moss D, Thornton J. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Detailed synthesis and characterization data on additional compounds prepared analogously to the general synthetic procedures presented in the Experimental section, tabulation of compound purity as determined by C4 and C18 RP-HPLC, and calculated phenolic pKa values. This material is available free of charge via the Internet at http://pubs.acs.org.