

Abstract
Transcriptional regulation in Treponema pallidum subsp. pallidum is poorly understood, primarily because this organism cannot be cultivated in vitro or genetically manipulated. We have recently shown a phase variation mechanism controlling transcription initiation of Subfamily II tpr (T. pallidum repeat) genes (tprE, tprG, and tprJ), a group of virulence factor candidates. Furthermore, the same study suggested that additional mechanisms might influence the level of transcription of these tprs. The T. pallidum genome sequence has revealed a few open reading frames (ORFs) with similarity to known bacterial transcription factors (TFs), including four catabolite activator protein (CAP) homologs. In this work, sequences matching the E. coli cAMP receptor protein (CRP) binding motif were identified in silico upstream of tprE, tprG, and tprJ. Using elecrophoretic mobility shift assay (EMSA) and DNaseI footprinting assay, recombinant TP0262, a T. pallidum CRP homolog, was shown to bind specifically to amplicons obtained from the tpr promoters containing putative CRP binding motifs. Using a heterologous reporter system, binding of TP0262 to these promoters was shown to either increase (tprE and tprJ) or decrease (tprG) tpr promoter activity. This is the first characterization of a T. pallidum transcriptional modulator which influences tpr promoter activity.
Keywords: Treponema pallidum, TP0262, CRP, tpr genes, transcriptional modulation
Introduction
Treponema pallidum, the causative agent of syphilis, is an obligate human pathogen unable to survive outside of its host. It cannot be genetically manipulated and no culture system has proven effective in supporting its long-term growth in vitro (Norris & Edmondson, 1986, Cox et al., 1990). In an infected individual, the course of syphilis alternates between episodes of active clinical disease (primary, secondary, and tertiary stages) separated by periods of asymptomatic, latent infection. Primary and secondary syphilis are characterized by the appearance of typical lesions (a primary chancre, which marks the site of transmission, followed by disseminated maculopapular skin lesions in the secondary stage) containing elevated numbers of treponemes (reviewed by Radolf & Lukehart, 2006). During early syphilis, a strong humoral and cellular host immune response eliminates the great majority of treponemes, leading to spontaneous lesion resolution; nevertheless, a small number of organisms escape immune clearance and cause chronic infection (Radolf & Lukehart, 2006). Unraveling the complex biology of this spirochete has been greatly helped by analysis of the T. pallidum (Nichols strain) genome sequence (Fraser et al., 1998, Weinstock et al., 1998), which has revealed a lack of genes coding for many metabolic and biosynthetic pathways, perhaps explaining the absolute dependence of T. pallidum on its host to maintain viability and virulence. Genome analysis has also aided in the identification of putative virulence factors, such as the tpr (T. pallidum repeat) genes, whose role during infection has been actively investigated since their description.
The 12 members of the tpr gene family are homologous to the major sheath protein gene (msp) of Treponema denticola; msp is reported to encode a surface exposed protein with porin and adhesin properties (Fenno et al., 1996). Most Tpr antigens are targets of a strong humoral and cellular host immune responses during experimental syphilis, and immunization with some recombinant Tpr antigens significantly attenuates lesion development after infectious challenge (Centurion-Lara et al., 1999, Morgan et al., 2002b, Morgan et al., 2002a, Morgan et al., 2003, Leader et al., 2003, Sun et al., 2004, Giacani et al., 2005b), suggesting an important role for these antigens during infection. These genes are differentially transcribed within and among T. pallidum strains during experimental infection (Hazlett et al., 2001, Smajs et al., 2005, Giacani et al., 2007b), supporting the hypothesis of regulation of expression among these genes. Regulation of expression of virulence traits by transcription factors is a common mechanism used by bacterial pathogens to adapt and persist in their hosts; bacterial invasion, multiplication, and dissemination in an infected individual often involve temporal and spatial control of gene expression in the pathogen (Kreft & Vasquez-Boland, 2001, Abdelrahman & Belland, 2005, Seshasayee et al., 2006). Recently, the length of hypervariable homopolymeric guanosine (poly-G) tracts located upstream of Subfamily II tpr (tprE, tprG, and tprJ) transcriptional start sites (TSSs), was shown to control transcription (Giacani et al., 2007a) of these genes. This finding does not fully explains the differential expression of these genes in that there is not an absolute correlation between tpr mRNA levels and the length of the poly-G sequences seen during experimental infection (Giacani et al., 2007a). This suggests the involvement of additional factors influencing their transcription.
Using a weight matrix generated from known binding sites for the E. coli cAMP receptor protein (CRP), a member of the E. coli catabolite activator protein (CAP) family, we identified instances of the E. coli CRP binding motif upstream of the experimentally determined TSSs of the tprE, tprG, and tprJ genes.
The T. pallidum genome sequence has revealed four CAP homologs (TP0098, TP0261, TP0262, and TP0995) (Fraser et al., 1998). Except for the TroR repressor protein (Posey et al., 1999; Hazlett et al., 2003,), the CAP homologs are the only annotated proteins likely to modulate gene expression at the transcription initiation step. Among the T. pallidum CAP homologs, TP0262 shows the highest sequence homology (41%) to E. coli CRP, and conservation of the DNA and cAMP binding domains (as described in Kolb et al., 1993). Furthermore, prediction based on known crystal structures using the 3D-Jury consensus approach (http://meta.bioinfo.pl) identified TP0262 as a CRP structural homolog (data not shown). In E. coli, CRP is involved in controlling metabolic functions, but has also been reported to modulate expression of virulence factors, such as the genes encoded by the pap operon (Baga et al., 1985, Goransson et al., 1989). Because of the paucity of transcription factors in T. pallidum, the very limited metabolic capabilities of this pathogen, and the involvement of CRP in regulation of virulence genes in other systems, we began to explore the hypothesis that TP0262 modulates gene expression of important virulence factor candidates, such as the tpr genes.
Electrophoretic mobility shift assay (EMSA) and DNaseI footprinting analysis were combined to investigate recombinant TP0262's ability to specifically recognize amplicons with the predicted T. pallidum CRP binding sites upstream of these tpr genes; a heterologous reporter assay (Giacani et al., 2007a) was used to evaluate the effect in vivo of TP0262 expression on the activity of these tpr promoters. We demonstrated that TP0262 can bind these T. pallidum promoters and either up- or down-regulate their activity. This is the first characterization of a T. pallidum transcription factor that modulates transcription of tpr genes and might also function as a global regulator.
Results
TP0262 binds tpr Subfamily II promoters
Using a weight matrix generated from known binding sites for E. coli CRP (available at http://www.ccg.unam.mx/Computational_Genomics/tractorDB/) we identified instances of the E. coli CRP binding motif upstream of the experimentally determined TSSs of the tprE (−236.5), tprG (−388.5), and tprJ (−233.5) genes (Table I; Fig.1, in bold). EMSA was employed to investigate recombinant TP0262's ability to bind amplicons containing predicted T. pallidum CRP binding sites upstream of the tpr E, G, and J genes. EMSA using DNA fragments obtained from the tprE, tprG, and tprJ promoter regions, with increasing concentrations of recombinant TP0262, resulted in the shift in mobility of all fragments (Fig.2A, and B). No shift was observed when the same fragments were incubated with identical concentrations of recombinant E. coli CRP (data not shown) or in absence of cAMP (Fig.2A-C), showing that the shift induced by TP0262 depends on the presence of cAMP. Electrophoretic mobility of the amplicon containing the E. coli lac promoter, used as positive control for the assay, was appropriately retarded by recombinant E. coli CRP binding (Fig.2D).
Table I. In silico predictions and experimentally determined TpCRP binding sites.
| Gene | TP0262 binding sites predicted in silico | TP0262 binding sites determined by DNaseI footprinting | ||
|---|---|---|---|---|
| Sequence | Location1 | Sequence | Location1 | |
| tprE | TGTGGGCTCGTGCTGG | −236.5 | TGCGTGTGTGTGTGGGCTCGTGCTG | -242.5 |
| tprG | AGTGCTAGCAGTCACA | −388.5 | GTCAGTGCTAGCAGTCACATAC | -388.5 |
| tprJ | GTTTATTGCGTGCAAG | −233.5 | GTTGTCAGTGCACGAGGAGGAG | -259.5 |
location is expressed as distance (nucleotide position) from the experimentally predicted TSS (Giacani et al., 2005a). For in silico predictions, underlines indicate the predicted CRP binding motifs; for footprinting results, underlines represent the TP0262 binding sites originally identified in silico.
FIG. 1.

tprE, tprG, tprJ promoter sequences cloned into the pGlow-TOPO vector and used for the GFP reporter assay. Underlines indicate the primers [→: sense primer(s), ←: antisense primer] used to amplify the promoter regions to obtain amplicons containing or lacking the putative TP0262 binding site. Regulatory elements are in bold. tprG and J TSSs (+1) have been experimentally demonstrated (Giacani et al., 2005a), tprE TSS (+1) is hypothetical but based on sequence homology with the other tpr genes. TP0262 bs: predicted TP0262 binding site; [GGGGGGGGGG]n indicates the hypervariable poly-G sequences upstream of the tpr promoters; +1: Transcriptional Start Site (TSS); RBS: ribosomal binding site; SC: start codon; GFP SC: green fluorescent protein start codon. The sequence Lys-Gly-Gln-Phe-Cys-Arg-Ser-Arg between the putative Tpr SCs and the GFP ORF start are vector-encoded aminoacids. Shorter amplicons were obtained from these promoter regions for EMSA and footprinting analysis using primers reported in Table II.
FIG. 2.
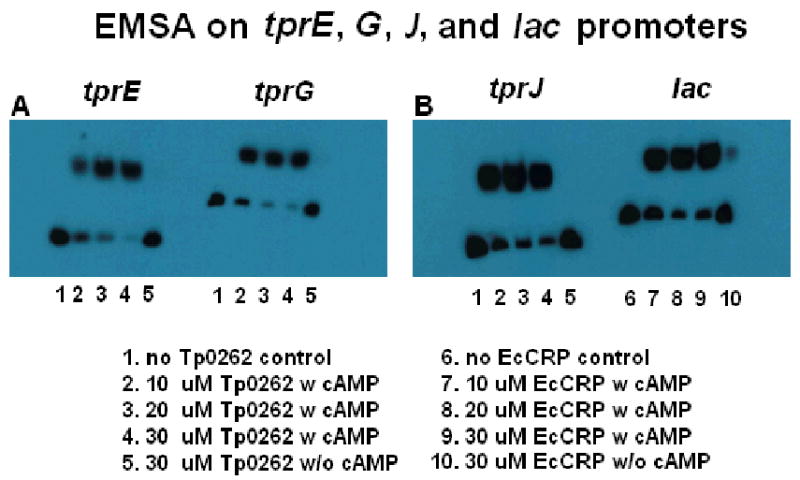
Electrophoretic mobility shift assay (EMSA) on T. pallidum tprE and tprG (Panel A), tprJ and lac (Panel B) promoters in presence of cAMP and increasing concentrations (0μM, 10 μM, 20μM, and 30 μM, respectively) of recombinant TP0262 (lanes 1-4 for the tpr genes) or E. coli CRP (lanes 6-9 for the E. coli lac promoter) proteins. TP0262 + cAMP bound the T. pallidum tpr promoters; E. coli CRP + cAMP failed to bind tpr promoters in this assay (data not shown). Recombinant E. coli CRP was shown to retard electrophoretic mobility of the lac promoter amplicon containing the TGTGA-6N-TCACA binding site. Without cAMP (lines 5 and 10), no binding was seen. 0.5 fmols of labeled probe were used in each experiment.
Identification of TP0262 binding sites on tpr Subfamily II promoters
To confirm specific promoter binding and to precisely define TP0262 binding sites, DNaseI protection assays and automated fragment analysis were performed as described by Oyamada et al. (Oyamada et al., 2007) on amplicons obtained from the tprE, tprG, tprJ upstream regions. Differences between the fluorescence intensity of the peaks between corresponding regions in protected and unprotected samples were statistically evaluated using paired Student's t-tests with significance set at p<0.05. By comparison with the size standard peaks in the same samples, protected sites were found to span the -253 to -228, -399 to -377, and -278 to -248 intervals for tprE, tprG, and tprJ, respectively, of the amplicons digested with DNaseI (Table I, right column; Fig.3A-C). The region of the lac promoter containing the TGTGA-6N-TCACA sequence, used as positive control for the footprinting assay, was protected by E. coli CRP binding from DNaseI digestion (Fig.3D).
FIG. 3.
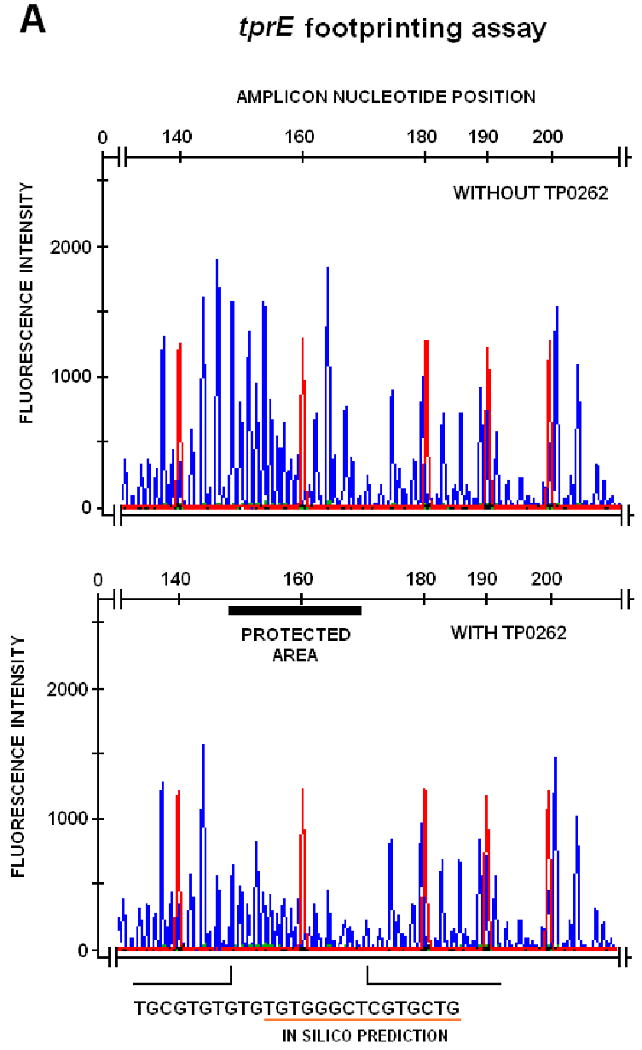
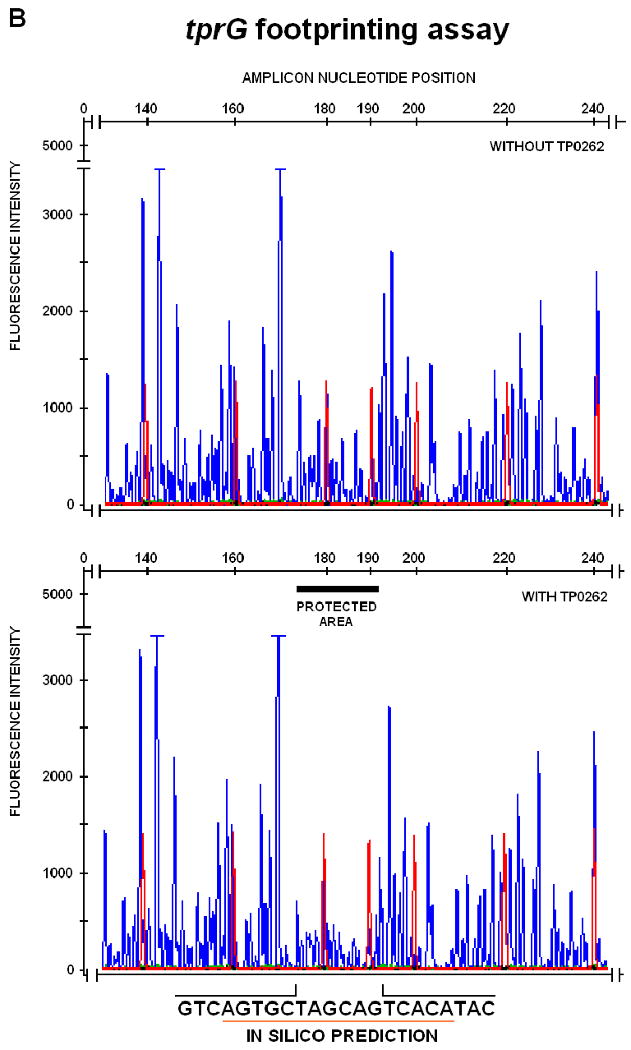

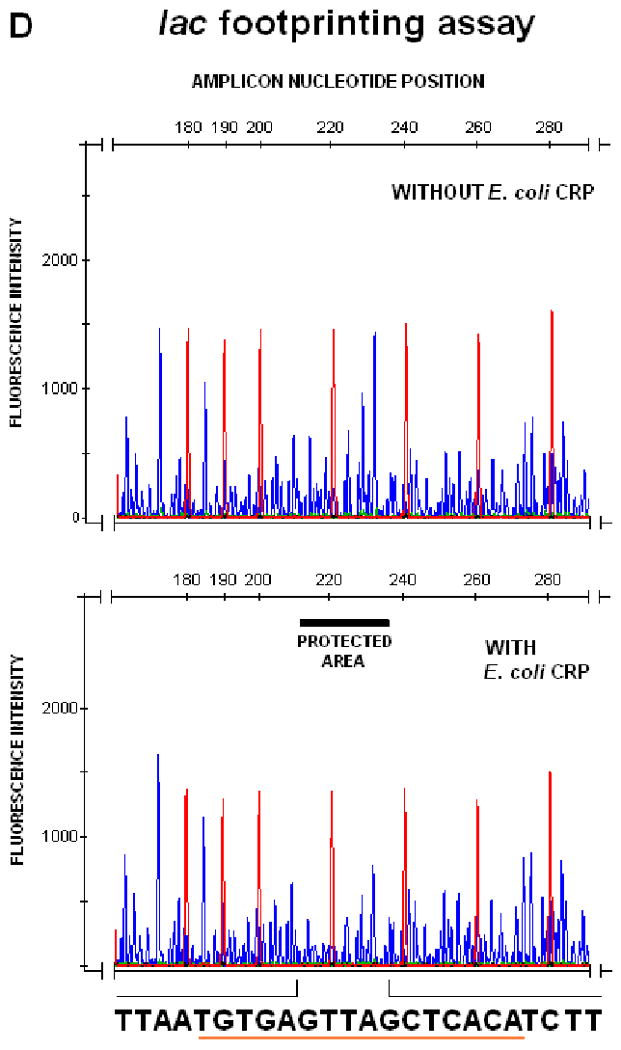
DNAseI footprinting assay on tprE (Panel A), tprG (Panel B), and tprJ (Panel C) promoters. Fluorescence intensity and peak size were evaluated using the GeneMapper 3.5 software. Signal from samples digested in presence or absence of recombinant TP0262 or E. coli CRP were statistically compared using the paired Student's t-test with significance set at p<0.05. Red peaks indicate the ROX-labeled DNA ladder, blue peaks represent fragments generated by DNaseI digestion. Sequences of the protected areas are represented at the bottom of each figure; underlined sequences represent the TP0262 binding site originally predicted in silico. In the positive control, the GTGTA-6N-TCACT sequence in the E. coli lac promoter was shown to be protected by recombinant E. coli CRP as expected (Panel D).
TP0262 binds the oligonucleotide DNA regions identified by footprinting assay
To further confirm the DNaseI protection assay results, EMSA was employed again to test recombinant TP0262's ability to recognize the binding sites experimentally identified by footprinting assay upstream of the tprE, G, and J genes. Double stranded DNA oligonucleotides (Table II) corresponding to the regions protected by TP0262 (Table I, right column; Fig.3A-D) were synthesized and used in the EMSA assay described above as competitor unlabelled DNA. In presence of competitor DNA, no shift of the labeled DNA amplicons containing the putative tprE, G, and J TP0262 binding sites was observed (Fig.4A-B, lanes 1-3). A 22 bp oligonucleotide with the E. coli CRP binding site was also shown to prevent the shift of the labeled lac promoter amplicon containing the same sequence (Fig.4B, lanes 4-6).
Table II. Primers used in this study.
| Purpose | Name1 | Sequence (5′ to 3′) | Size (bp)2 | |
|---|---|---|---|---|
| DIRECT CLONING (into pGlow-TOPO vector) | tprE-Glow-F | catggtttcggttgtaccc | 359 | |
| tprG-Glow-F | taccacgagccccatattct | 531 | ||
| tprJ-Glow-F | gtacgctgcggtcgatattt | 382 | ||
| tprE/G/J-Glow-R | tcattcccccgctcgctcct | common | ||
| t47-Sense | cgtgtggtatcaactatgg | 313 | ||
| t47-Antisense | tcaaccgtgtactcagtgc | |||
| lacPro-Glow-F | gtgagcgcaacgcaattaat | 213 | ||
| lacPro-Glow-R | tcatagctgtttcctgtgtg | |||
| CLONING into the AvrII site (cctagg, underlined) of pGlow-TOPO vector | TP0262-AvrIIF | aaacctagggccttctgcttttgctgagt | 1311 | |
| TP0262-AvrIIR | aaacctaggccaacaagggcgaatattgt | |||
| EcoliCRP-AvrIIF | aaacctaggcactgcgtcaattttcctga | 1046 | ||
| EcoliCRP-AvrIIR | aaacctagggcgtttgtcgaagtgcatag | |||
| SEQUENCING3 | T7 promoter | taatacgactcactataggg | N.A. | |
| gfp-Rev | gggtaagctttccgtatgtacg | |||
| TP0262-S | ctggagttcaatcgggagaa | N.A | ||
| TP0262-As | gggctatctcttcgatgctg | |||
| TP0262-F | gccttctgcttttgctgagt | |||
| TP0262-R | gccaacaagggcgaatattgt | |||
| EcoliCRP-S | aaacagacccgactctcgaa | N.A. | ||
| EcoliCRP-As | tgtccgggtttacctgaatc | |||
| EcoliCRP-F | gcactgcgtcaattttcctga | |||
| EcoliCRP-R | gcgtttgtcgaagtgcatag | |||
| EMSA (primers to generate tpr and lac promoter amplicons) | tprE-EMSA-F | catggtttcggttgtaccc | 116 | |
| tprE-EMSA-R | cgtaaactgatcctgcatcc | |||
| tprG-EMSA-F | taccacgagccccatattct | 151 | ||
| tprG-EMSA-R | tgacaagtcctttaaccagca | |||
| tprJ-EMSA-F | gtacgctgcggtcgatattt | 163 | ||
| tprJ-EMSA-R | gctttgcggaaatccttttt | |||
| lac-EMSA-F | gtgagcgcaacgcaattaat | 213 | ||
| lac-EMSA-R | tcatagctgtttcctgtgtg | |||
| EMSA (oligonucleotides) competitors)4 | tprE | tgcgtgtgtgtgtgggctcgtgctg | 25 | |
| tprG | gtcagtgctagcagtcacatac | 22 | ||
| tprJ | gttgtcagtgcacgaggaggag | 22 | ||
| lac | aaatgtgatctagatcacattt | 22 | ||
| FOOTPRINTING | pGLow-FAM-For | F-cggatcgggagatctaatacg5 common | ||
| tprE/J-foot-R | ccagtctagtagccccctca | 384(E); 407(J) | ||
| tprG-foot-R | ctgctcatcggctgctcta | 372 | ||
| lac-foot-R | gggtaagctttccgtatgtagc | 483 | ||
| RT-PCR6 | gfp-S | ctacctgttccatggccaac | 262 | |
| gfp-As | tgtgtccgagaatgtttcca | |||
| TP0262-S | ctggagttcaatcgggagaa | 241 | ||
| TP0262-As | gggctatctcttcgatgctg | |||
When three or more primers are grouped together, a single reverse or forward primer (indicated as “common”) was used in different amplification reactions.
The size of the amplicons relative to the tpr genes were calculated according to the genome sequence of the Nichols strain (Fraser et al., 1998), without taking into account variability within the poly-G repeats (Giacani et al., 2007a).
The first group of primers was used to sequence the promoters cloned upstream of the gfp gene in the pGlow-TOPO vector (Invitrogen); the second and third groups were used to sequence TP0262 and E. coli crp, respectively, cloned into the AvrII site of the pGlow-TOPO vector.
For simplicity only one strand of the oligonucleotide is represented.
F indicates 5′-end labeling with the fluorophor 6′-FAM.
The first primer pair was used to assess lack of DNA contamination in DNaseI-treated RNA samples; TP0262-As was used for both gene-specific reverse transcription and DNA amplification.
N.A.: Not Applicable.
FIG. 4.
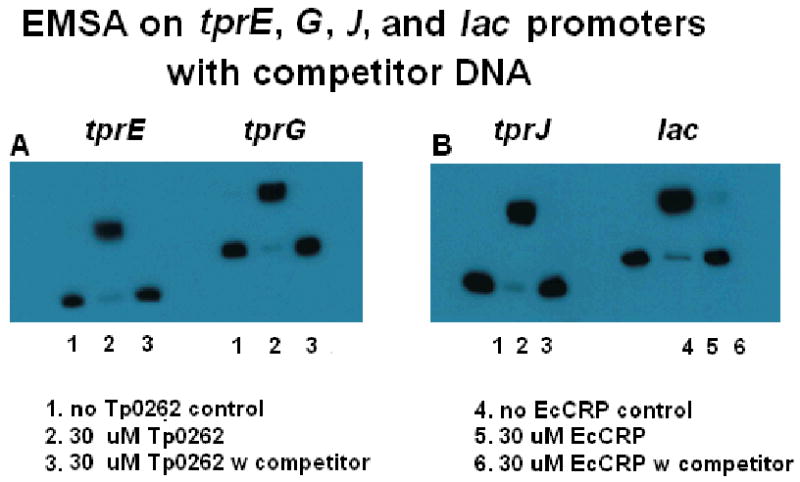
Recombinant TP0262 binds the DNA sequences identified by DNaseI footprinting assay. In presence of unlabelled competitor oligonucleotide fragments (Table II) containing the TP0262 tprE, tprG (Panel A), and J (Panel B) binding sites identified by footprinting assay, no shift of the labeled amplicons (containing the putative TP0262 binding sites) used in the previous EMSA experiments was observed. As positive control, a 22 bp unlabelled oligonucleotide (Table II) containing the E. coli CRP binding site prevented E. coli CRP from shifting the labeled lac promoter amplicon. In each experiment, the amount of the unlabelled fragments was 100 fold higher (50 fmol) than the labeled probe (0.5 fmol).
TP0262 influences tpr Subfamily II promoter activity
A heterologous green fluorescent protein (GFP) reporter assay (Giacani et al., 2007a) was used to determine the effect of TP0262 on each of the tpr promoters. In this assay, tprE, tprG, and tprJ promoters (reproduced in Fig.1) with and without the region containing the putative TP0262 binding sites were first cloned into a plasmid upstream of the gfp reporter gene, and then used to transform a crp-deficient E. coli strain (G1278, Δcrp::cm). Activity of the promoters was measured as fluorescence emission in G1278 E. coli cultures expressing TP0262. G1278 cultures not expressing TP0262 were included as controls. TP0262 mRNA was also determined by qualitative RT-PCR in all E. coli transformants carrying the TP0262 gene in the reporter vector (not shown). GFP fluorescence for the tprE and tprJ promoters containing the putative CRP binding sites was found to be significantly increased by TP0262 expression with respect to the same cells in the absence of TP0262 expression (Fig.5A, B); in these constructs, omission of the regions containing the TP0262 binding site reduced the level of fluorescence regardless of TP0262 expression (Fig.5A, B), indicating a specific interaction between the putative TP0262 binding sites and TP0262 which causes an increase in promoter activity. Conversely, the activity of the tprG promoter containing the putative TP0262 binding site was found to be decreased in cells expressing TP0262 (Fig.5C). Also for the tprG promoter; no effect was observed in the absence of either TP0262 expression or the putative binding sites (Fig.5C).
FIG. 5.
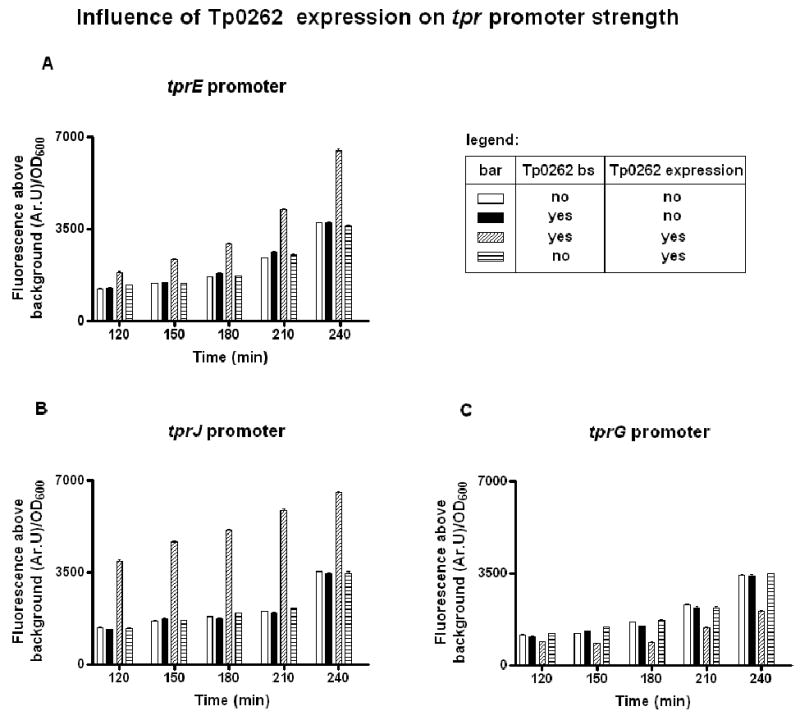
TP0262 modulates tprE, tprJ, and tprG, promoter activity (Panels A, B, and C, respectively). Graph values for the tprs are for promoter sequences containing only poly-G tracts of 8G residues within the hypervariable poly-G tract. As addressed in the Discussion paragraphs, 8G's were previously shown to be permissive for transcription, while ≥9G's were shown to drastically reduce tprE, G, and J transcription (Giacani et al., 2007a). The GFP reporter assay showed that tprE, G, and J promoters containing ≥9 G residues did not induce fluorescence above background, even in presence TP0262 expression (data not shown). Fluorescence signal induced by the E. coli lac promoter (used as positive control) was always ≥10,000 Ar.U/OD600 (data not shown). Background fluorescence induced by the negative control plasmid was subtracted from the sample-induced fluorescence signals.
Means ± SE of quadruplicate values are represented. TP0262 bs: binding site for TP0262.
Analysis of the OD600 values at the time of fluorescence measurements showed that all cultures were still within the logarithmic phase of growth at the last measurement. Fluorescence signal induced by the positive control (E. coli cells transformed with a lac promoter- gfp construct, with the vector also carrying the E. coli crp gene) was always ≥10,000 Ar.U/OD600 (data not shown). As previously reported, equal low background fluorescence was seen in cultures carrying negative control plasmid (reporter vector carrying a fragment of the tpN47 coding sequence, hence without promoter and RBS upstream of the gfp gene), and in untransformed E. coli cells, indicating absence of expression in the negative control samples (Giacani et al., 2007a).
The same heterologous reporter system was used to investigate TP0262 dependence on cAMP binding to influence promoter activity using the tprJ promoter as an example. In this experiment, the tprJ promoter-containing plasmid constructs were used to transform a crp and cya-deficient E. coli strain (G1044, Δcrp::cm::cya-, unable to synthesize cAMP). Fluorescence measurements of these cells showed that the positive regulatory effect exerted by TP0262 on the tprJ promoter (containing the region with the putative CRP binding site) was absent unless the culture media was supplemented with cAMP (Fig. 6). This further confirms the role of Tp0262 as a CRP functional homolog.
FIG. 6.
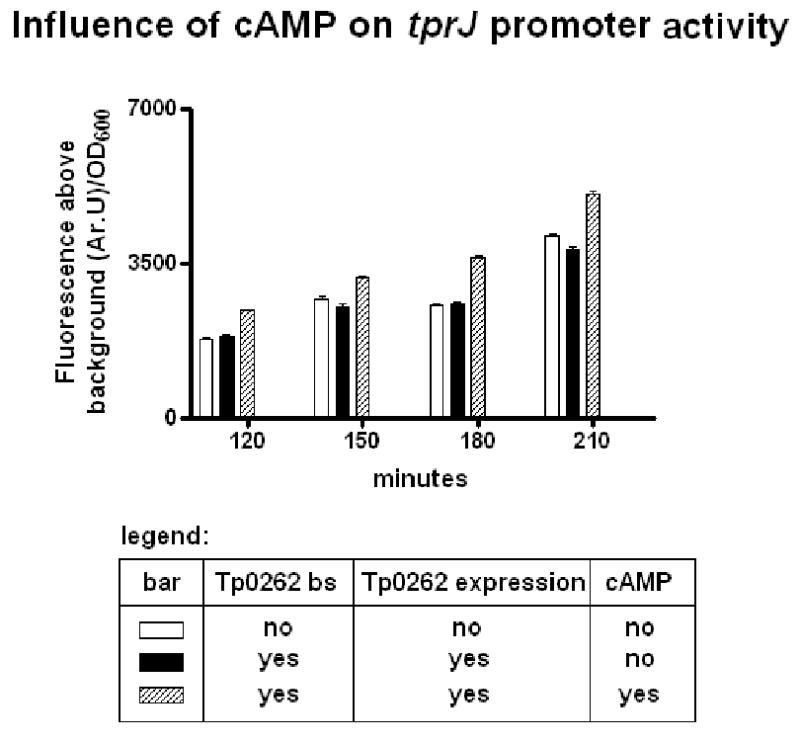
cAMP is required for TP0262 influence tprJ promoter activity. The constructs containing the tprJ promoters were used to transform a crp and cya-deficient E. coli strain (G1044, Δcrp::cm::cya-, unable to synthesize cAMP). The positive regulatory effect exerted by TP0262 on the tprJ promoter (containing the region with the putative CRP binding site) was absent in culture media not supplemented with cAMP.
Discussion
This study reports the identification of a T. pallidum CRP homolog that modulates the activity of the promoters of three tpr genes (tprE, G, and J) believed to be involved in syphilis pathogenesis. Although the characterization of elements involved in regulation of gene expression in T. pallidum is particularly challenging due to the inability to grow T. pallidum in vitro, it is plausible that transcriptional regulation plays a pivotal role in the pathogenesis of a disease that alternates periods of clinically active illness (characterized by more rapid replication of the pathogen) and periods of latent infection in which T. pallidum is rarely detected (Radolf & Lukehart, 2006). Furthermore, because T. pallidum can infect virtually every organ of the human body, modulation of gene expression in different tissues may be a mechanism to respond and adapt to these varied microenvironments.
For the tprE, G and J genes, TP0262 is not the only modulating factor, however, given the effect of the poly-G sequences (Giacani et al., 2007a) on their transcription. In several bacterial pathogens, hypervariable homopolymeric repeats located within or near promoter regions have been shown to regulate a variety of virulence factors and surface antigens through a phase variation mechanism (van der Ende et al., 1995, Saunders et al., 1998, van der Woude & Baumler, 2004); similarly, in T. pallidum, poly-G sequences of ≤8 G's located upstream of tprE, G, and J were shown to be permissive for transcription, while the presence of ≥9 G's locks these genes in an near-OFF state (Giacani et al., 2007a). Although the effect of TP0262 on tprE, G, and J promoter activity reported here was determined using promoters with 8G-repeats, the same promoters with longer poly-G tracts were also evaluated, but no fluorescence above background was ever obtained (data not shown). Thus, TP0262 likely has a fine tuning effect in expression of these genes when the length of the poly-G tract allows their transcription.
Previous studies on gene organization and transcriptional analysis of the tpr genes showed that tprG and tprJ are the first ORFs of two larger operons containing other tpr genes. tprG and tprJ precede and are co-transcribed along with the tprF and tprI genes (members of tpr Subfamily I), respectively (Giacani et al., 2005a). Transcription of both tprF and I, however, can also be driven by weak sub-operonic promoters located upstream of the tprF and tprI ORFs, respectively (Giacani et al., 2005a). Sequences homologous to the E. coli CRP binding sites were also identified upstream of both tprF and I ORFs (data not shown). The results of EMSAs and heterologous reporter assay experiments (data not shown) performed on amplicons containing these additional TP0262 binding sites showed, respectively, that 1) the DNA regions upstream of tprF and I were recognized by TP0262 when EMSA were performed; but 2) no significant difference in the level of GFP fluorescence was induced by the activity of these promoters in presence of absence of TP0262 expression when the heterologous reporter gene assay was applied to these promoters. There is no clear explanation yet for the lack of responsiveness exhibited by both tprF and tprI promoters to TP0262 expression (despite the presence of TP0262 binding sites), but it appears likely that transcription of the tprG-F and tprJ-I operons is mainly driven by the upstream tprG and J promoters. The lack of responsiveness may be also due to the binding of TP0262 under non-activating conditions or to the overriding effect of another TF that is present in the E. coli reporter system (Grainger et al., 2007, Grainger et al., 2004). It is also possible that TP0262 binding to the tprF and I promoters has no functional importance. It has recently proposed that the E. coli CRP also acts as a chromatin-shaping protein (Grainger et al., 2005). By analogy, binding of TP0262 to the tprF and tprI promoters might result in a higher hierarchy of chromosomal organization.
The E. coli CRP binding site in the lac promoter is located at nucleotide position -62.5, which allows direct interaction between the transcription factor and the carboxyl-terminal domain of the α subunits of the E.coli RNA polymerase. It is worth noting that the TP0262 binding sites identified upstream of the tprE, G, and J genes are too far away from the putative or experimentally determined TSSs to allow a similar interaction between TP0262 and the T. pallidum RNA polymerase. In these promoters, TP0262 could potentially work in concert with still unidentified transcription factors that could bind the region downstream of the TP0262 binding motifs.
Although TP0262 putative binding sites were identified by computer analyses based on the consensus sequences for the E. coli CRP binding motifs, it is interesting that neither did recombinant E. coli CRP bind the tpr DNA fragments containing the TP0262 binding sites nor was TP0262 able to bind the lac promoter or rescue lactose fermentation in crp-deficient E. coli mutants (Supplemental Information). One likely explanation is that species-specific promoter upstream and downstream regions of the binding motifs might play a role in this context. Also, evolutionary differences between these transcription modulators and their targets may be responsible for this lack of complementation between the two systems
We acknowledge the limitations of using the heterologous E. coli GFP reporter system (Giacani et al., 2007a) to study transcriptional regulation in T. pallidum. Sohaskey et al. (Sohaskey et al., 1997), for example, showed that expression driven by the ospA, ospC, flaD, and flaB promoters of Borrelia burgdorferi (the spirochetal agent of Lyme disease) varies between B. burgdorferi and E. coli when these promoters are analyzed using a transient chloramphenicol acetyltransferase (CAT) expression system. The inability to grow T. pallidum in vitro precludes transcriptional regulation studies in the bacterium itself. Nonetheless, the specific application of the E. coli GFP reporter system to the tpr promoters is supported by our previous findings that tprG and J promoters are recognized in vitro by the E. coli RNA polymerase-σ70 complex, and that the TSSs are identical in both the E. coli system and in RNA extracted directly from T. pallidum cells (Giacani et al., 2005a). A combination of tools such as this heterologous system with T. pallidum-specific in vitro transcription systems (which could be developed by reconstituting the T. pallidum RNA polymerase holoenzyme from individual recombinant peptides) will greatly enhance our ability to study transcriptional regulation for this intractable spirochete.
Ongoing genome-wide chromatin immunoprecipitation studies in our laboratories will confirm the binding specificities of TP0262 to tpr promoters and will determine the TP0262 regulon. The identification of the TP0262 regulon along with the genome-wide distribution of the T. pallidum RNA polymerase will further define the role of TP0262 as a transcriptional regulator during the pathogenesis of syphilis. In parallel, other studies will focus on the expression of the TP0485 gene that encodes for the T. pallidum adenylate cyclase. Transcription of molecules associated with stress responses, such as chaperones (dnaJ, dnaK, grpE, mopA, and mopB) and several ATP-dependent proteases (clpP, hflB, hslU, hslV) have also been shown to be regulated by catabolite repression (Gosset et al., 2004). T. pallidum possesses ORFs homologous to most of these genes, and preliminary in silico genome-wide analysis has revealed the presence of putative CRP binding sites in more than 200 upstream regions of the 1041 annotated coding sequences, including approximately 40% of the genes predicted to be associated with stress responses (data not shown). This supports involvement of TP0262 in mediating T. pallidum's ability to adapt its physiology to environmental changes in the host.
In conclusion, this work provides an important insight into the role of modulators of gene transcription in T. pallidum, a field neglected in syphilis research, and sets the stage for further studies to identify the stimuli controlling TP0262 activity, and the involvement of other factors in transcriptional modulation of these tpr genes.
Experimental Procedures
T. pallidum and E. coli strain propagation, nucleic acid extraction and preparation of competent E. coli cells
T. pallidum subsp. pallidum, Nichols strains, originally provided by James N. Miller (University of California, Los Angeles), was propagated in New Zealand white rabbits as previously reported (Lukehart et al., 1980). Briefly, rabbits were infected with 5 × 107 T. pallidum cells per testis, and treponemes were harvested at peak orchitis (day ∼10 postinfection). Collected organisms were separated from host cellular gross debris by low speed centrifugation (250 × g for 10 min at room temperature), and the supernatants were spun in a microcentrifuge for 30 min at 12,000 × g at 4°C. Bacterial pellets were resuspended in 200 μl of 1× lysis buffer (10mM Tris, pH 8.0; 0.1M EDTA; 0.5% sodium dodecyl sulfate) for DNA isolation. DNA extraction was performed as previously reported (Giacani et al., 2004) using the QIAamp DNA Mini Kit (Qiagen Inc., Chatsworth, CA). DNA samples were stored at -20°C until use in amplification reactions.
E. coli MG1655-K12 (wild type), G1278 (Δcrp::cm), and G1044 (Δcrp::cm:;cya-) strains, both derived from the K12 wild type, were kindly provided by Drs. Susan Garges, Sankar Adhya (Laboratory of Molecular Biology, NCI, NIH, Bethesda, MD), and Ding J. Jin (Gene Regulation and Chromosome Biology Laboratory, NCI, NIH, Frederick, MD) and propagated in Luria-Bertani (LB) plates supplemented with 25μg/ml chloramphenicol. DNA extraction from the MG1655-K12 strain was performed as described above for T. pallidum cells. RNA extraction to assess mRNA levels as surrogate marker for TP0262 expression in G1278 and G1044 cells transformed with TP0262-encoding plasmids used in this study was performed from cell pellets lysed in 400 μl of Ultraspec buffer (Biotecx Laboratories Inc., Houston, TX). Protocols for RNA extraction and DNaseI treatment to obtain DNA-free RNA samples were previously reported in detail (Giacani et al., 2005a). RT-PCR protocols were also reported in detail (Giacani et al., 2007b) and are only briefly described here (see GFP reporter assay). Preparation of competent G1278 and G1044 cells was performed according to Inoue et al. (Inoue H, 1990). Competent cells were transformed with approximately 300 ng of plasmid DNA. Transformants were plated on LB plates containing chloramphenicol (25μg/ml) and ampicillin (100 μg/ml) and grown overnight to be used the following day in the GFP reporter assay (see below).
Amplification and cloning
All primers in Table II were designed using the Primer3 software (http://fokker.wi.mit.edu/primer3/input.htm). All PCR amplifications were performed in 50 μl reactions containing 200 μM each dNTP, 20 mM Tris-HCl (pH 8.4), 1.5 mM MgCl2, 50 mM KCl, 400 nM of each primer, and 1.0 U of Taq DNA Polymerase (Promega, Madison, WI) with approximately 100 ng of DNA template in each reaction. Cycling conditions were denaturation for 5 min at 95°C, followed by 1 min at 95°C, annealing for 1 min at 60°C and extension for 1 min at 72°C for a total of 45 cycles. Final extension was 10 min at 72°C. Amplicons for the tprE, tprG, and tprJ promoters, as well as for the E. coli lac promoter (used as positive control in the GFP reporter assay) were directly cloned into the pGlow-TOPO vector (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions and for each promoter, insert-containing plasmids were extracted using the Qiagen Plasmid Mini Kit (Qiagen) and sequenced in both strands with the dye terminator kit (Applied Biosystems, Foster City, CA) to ensure the correct orientation of the insert with respect to the gfp gene and the absence of amplification errors. Because the poly-G repeats of varying length located upstream of the tprE, tprG, and tprJ TSSs (Giacani et al., 2005a, Giacani et al., 2007a) influence promoter strength (Giacani et al., 2007a), sequencing was also used to evaluate the number of G residues contained in these poly-G tracts. The T. pallidum TP0262 and E. coli crp genes were amplified and cloned into the AvrII restriction site of the pGlow-TOPO vector to enable the crp-deficient G1287 and G1044 E. coli strains to express these genes when required in the GFP reporter assay. Standard cloning procedures (Maniatis et al., 1989) were adopted to insert the TP0262 and E. coli crp genes into the pGlow-TOPO vector in the opposite orientation with respect to the gfp coding sequence.
Recombinant peptides
To obtain recombinant 6X-His-tagged TP0262 and E coli CRP proteins for the EMSA and footprinting assays, amplicons with the two ORFs (primers in Table II) were first cloned into the TOPO-TA cloning vector (Invitrogen), checked for sequence accuracy, excised and inserted into the pQE30 expression vector (Qiagen) between the KpnI and HindIII restriction sites. Recombinant proteins were expressed in M15 (pREP4) cells (Qiagen) according to the manufacturer's guidelines. Briefly cultures were induced with 1.0 mM IPTG when OD600 reached 0.4-0.6 Absorbance Units and harvested after 4 hour incubation at 37°C. Cell pellets were resuspended in 1X lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 1 mg/ml lysozyme, pH 8.0) with EDTA-free protease inhibitor cocktail tablets (Complete; Roche, Basel, Switzerland), subjected to 12 rounds of sonication in ice for a total of 60 seconds, and centrifuged at 15,000 rpm for 30 min. Supernatants were filtered through a 0.45 μm filter prior to purification by nickel affinity chromatography. Wash and elution buffers were identical to the lysis buffer except for the increased concentration of imidazole (20 and 250 mM, respectively) and for the absence of lysozyme and protease inhibitors. Elution products were tested for size and purity by SDS-PAGE and quantified using a Bicinchoninic Acid Assay kit (Pierce, Rockford, IL). More than 95% of the protein was found in the expected size of the rTP0262 (∼26 kDa). Protein reactivity with anti 6xHis antibody (Sigma, St. Louis, MI) was tested according to the antibody manufacturer's instructions. Purified proteins were dialyzed against phosphate buffered saline (PBS)-10% glycerol and stored in aliquots at -20°C until use.
EMSA
Amplicons of the upstream regions of tprE, tprG, and tprJ containing the putative TP0262 binding sites were obtained using the primers listed in Table II; primer pairs were designed in order to obtain short amplicons (116-163 bp, Table II) with the putative TP0262 binding site equally spaced from the 3′ and 5′ ends. Amplicons were gel-purified and concentrations measured spectrophotometrically using a ND-1000 instrument (NanoDrop Technologies, Wilmington, DE). Fragments were labeled with the Biotin 3′ End DNA Labeling Kit, suitable for labeling ds DNA with 3′ overhangs (Pierce). Labeling efficiency was determined following the manufacturer's protocol. Labeled fragments were then purified using the QIAquick PCR purification kit (Qiagen) and stored at -20°C until use. Twenty microliter reactions containing 0.5 fmol of DNA and increasing concentrations of purified recombinant TP0262 and E. coli CRP (10 μM, 20 μM, and 30 μM) in reaction buffer [10 mM Tris, 50 mM KCl, 10 mM DTT, pH7.5, 5 mM MgCl2, 0.1 mM EDTA, 50 ng/μl Poly(dI·dC), 0.05% NP-40, and 2.5 % glycerol] were incubated at room temperature for 20 min. cAMP (Sigma) was added to all reactions to the final concentration of 0.1 mM, except to investigate binding activity in absence of cAMP. After incubation, reactions were mixed with 5μl of 5× DNA loading buffer and separated by electrophoresis in 4% polyacrylamide gels in 0.5X TBE buffer supplemented with 0.1 mM cAMP at 30 V/gel for 45 min. Gels were transferred to nylon membranes in a Mini Trans-Blot Cell (Bio-Rad, Hercules, CA) in 0.5X TBE at 200 mA for 30 min. After UV-crosslinking DNA to membranes for 10 minutes on a transilluminator equipped with 312 nm bulbs, biotin-labeled DNA was detected by using the LightShift Chemiluminescent EMSA Kit (Pierce) following the manufacturer's instruction. The streptavidin-HRP conjugate was used at 1:2000 dilution. As a positive control, the lac promoter of E. coli was also amplified (primers in Table II), labeled and analyzed for electrophoretic mobility retardation in presence of recombinant E. coli CRP, with and without cAMP.
The same protocol was applied when unlabelled competitor oligonucleotides were used investigate TP0262 interaction with the sequences experimentally identified by DNaseI footprinting assay (protocol below). In these experiments, the final concentration of the competitor DNA was approximately ∼100 fold higher (50 fmol) than the labeled probe.
DNaseI footprinting assay
Footprinting analysis was performed using a modified protocol published by Oyamada et al. (Oyamada et al., 2007). Probes containing the tprE, tprG, and tprJ putative TP0262 binding sites, and a control probe with the E. coli lac promoter containing the E. coli CRP binding site were obtained by PCR amplification from a plasmid template in which these regions were previously cloned. Amplification was carried out using the conditions described above and primers listed in Table II. A common sense primer, which is labeled with the fluorescent dye 6-FAM, anneals to the plasmid (pGlow-TOPO) and can be used on multiple templates in combination with a specific antisense primer (Table II). Amplification reaction products were run in 1% agarose gels and amplicons were gel-purified to eliminate excess primers. Concentration was determined using a ND-1000 spectrophotometer (NanoDrop Technologies). Approximately 2 pmol of DNA per fragment were digested with 3 U of DNaseI (Promega) for 1 min at room temperature in 100 μl of binding buffer (25 mM Tris-HCL, pH 8.0; 50 mM KCl, 6.25 mM MgCl2; 0.5 mM EDTA; 10% glycerol, and 1 mM DTT) in presence of 1 mM cAMP and 25 pmol of recombinant TP0262 (tprE, tprG, and tprJ) or recombinant E. coli CRP (lac promoter). Reactions were stopped by adding 90 μl of stop solution (200 mM NaCl, 30 mM EDTA, 1% SDS, and 100 μg/ml yeast DNA), phenol-chloroform purified, precipitated with 3 volumes of 100% ethanol, and washed with 70% ethanol. Samples were then vacuum dried and resuspended in 17 μl Hi-Di formamide (Applied Biosystems). Digestions were then mixed with 0.5 μl of ROX-labeled DNA ladder (MapMarker400, Bioventures, Murfreesboro, TN) and run in an ABI3100 Genetic Analyzer (Applied Biosystems). After fragment separation, fluorescence intensity and peak areas were analyzed with the GeneMapper 3.5 software (Applied Biosystems). The identification of the DNA regions protected by TP0262 or E. coli CRP binding was determined by comparison of both fluorescence intensity and peak areas between samples digested in presence or absence of recombinant TP0262 or E. coli CRP using the paired Student's t-test with significance set at p<0.05.
GFP reporter assay
PCR products for the tprE, tprG, and tprJ promoters were obtained from T. pallidum DNA using primers (Table II) specifically designed for expression of a GFP fusion protein using the pGlow-TOPO system (Invitrogen). Amplicons were gel purified using the QIAquick gel extraction kit (Qiagen) and cloned into the pGlow-TOPO vector according to manufacturer's instructions. For each tpr promoter, the amplicon included the predicted TP0262 binding sites, the predicted tpr gene RBS (-AGGAG-), and the GTG start codon in frame with the gfp coding sequence in the vector (Fig. 1A-C). The start codons of these tpr genes have not yet been experimentally identified, and, as previously reported (Giacani et al., 2007a), we selected RBSs and start codons for the pGlow-TOPO constructs separated 8 to 12 nucleotides from each other in order to provide E. coli with the optimal translational spacing. To facilitate recognition of the tpr start codons (GTG) by E. coli, the GTG triplet was mutated to an ATG in all clones (Fig.1A-C) by site directed mutagenesis. Expression of GFP from these constructs resulted in the addition of nine extra amino acids to the actual GFP peptide, encoded by the tpr start codon and eight additional, vector-encoded aminoacids (Fig.1A-C). In total, four different constructs were obtained for both tprE and J promoters (with poly-G repeats 8-11 G nucleotides long), and three constructs for tprG (with 8-10 G's) (Fig.1). Identical tpr promoter-pGlow TOPO plasmids with the same poly-G variants but without the DNA region containing the putative CRP binding sites were already available from previous studies (Giacani et al., 2007a). As mentioned above, when expression of TP0262 in the G1278 E. coli cells was required, the DNA fragment containing the TP0262 ORF and ∼150 bp of the 5′- and 3′- flanking regions was cloned into the AvrII site of the pGlow-TOPO vector, in opposite orientation to the gfp coding sequence. A construct containing both the lac promoter and the operator upstream of the gfp gene was used as a positive control; because transcription of the lac promoter does not occur in a crp-deficient E. coli strain, the E. coli crp gene was also cloned into the vector (in the AvrII site, in opposite orientation to the gfp gene). As a negative control, to determine background fluorescence, a ∼300 bp fragment of the tpN47 (TP0574) coding sequence with no RBS and no promoter activity was inserted out-of-frame with the gfp coding sequence into the pGlow-TOPO vector. All constructs were sequenced in both directions to verify correct orientation, continuity in the reading frame, and absence of mutations, as well as to ensure that the length of the poly-G repeats did not change after amplification and propagation in E. coli. Constructs were subsequently used to transform G1278 E. coli cells. For GFP fluorescence measurements, cells were inoculated from a Petri dish into 4 ml of LB medium at room temperature containing 100 μg/ml ampicillin and 25 μg/ml chloramphenicol, and grown at 37°C for 4 hrs. Cultures with optical densities (OD600) of ∼ 0.5 Absorbance units (AU) were used for fluorescence measurements in quadruplicate every 30 min until cultures reached OD600 of ∼2 AU. Briefly, 400 μl of culture were spun for 4 minutes at 12,000 × g and resuspended in an equal volume of phosphate buffered saline (PBS); cells were then divided in four wells (100 μl/well) of a black OptiPlate-96F (Perkin Elmer, Boston MA) for top fluorescence reading. Excitation and emission wavelength were 405 and 505 nm, respectively, and readings were performed in a Fusion Universal Microplate Analyzer (Perkin Elmer). Before each fluorescence readings, OD600 of the cultures was recorded again for normalization of the fluorescence measurements (expressed in Arbitrary Units, Ar.U) to the optical density of the culture. Background values obtained from each experiment (using E. coli cells transformed with the tpN47-pGlow TOPO vector) were subtracted from the sample values. Differences between levels of fluorescence were compared using Student's t-test, with significance set at p<0.05.
To confirm TP0262 expression in G1278 cells carrying the TP0262 gene in the pGLow-TOPO vector, we used RT-PCR analysis to determine TP0262 mRNA levels as a surrogate marker. 400 μl of each E. coli culture was spun immediately after fluorescence measurements and cells resuspended in an equal volume of Ultraspec buffer (Biotecx Laboratories) for RNA isolation. Extraction was performed according to the manufacturer's guidelines and followed by RNA treatment with DNaseI (Turbo-DNase, Ambion, Austin, TX) according to the provided protocol. DNase-treated RNA was tested for residual plasmid DNA contamination by qualitative amplification using gfp -specific primers (gfp-S and gfp-As primers, Table II) and the amplification conditions described above. DNA-free RNA was stored in aliquots at -80°C until use. Reverse transcription of total RNA was performed using the Superscript II First Strand Synthesis Kit (Invitrogen) with the TP0262-As primer (Table II) according to the provided protocol. All cDNA samples were then tested for TP0262 specific message using qualitative conventional PCR with the TP0262-S and -As primers (Table II) and the amplification conditions described above.
Identical procedures were applied when G1044 E. coli cells were used to evaluate TP0262 dependency on cAMP (co-effector) to exert its modulatory effect on the tprJ promoter. In this experiment, cAMP effect was evaluated by supplementing the growth medium with a final concentration of 0.75 mM cAMP (Sigma), and comparing fluorescence measurements of cultures grown in absence or presence of cAMP.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. Susan Garges (National Institutes of Health) for her valuable advice and suggestions; to Drs. Susan Garges, Sankar Adhya, and Ding J. Jin (National Institutes of Health) for providing the E. coli strains; Nan Li (University of Washington) for help with the in silico identification of the TP0262 binding sites; to Drs. Julio Cabrera, Dale Lewis (National Institutes of Health), Mofang Liu (Shanghai Institutes for Biological Sciences, China) and Elisabetta Marotti (University of Ancona, Italy) for their invaluable suggestions; Bruce Godfrey (Comparative Genomics Center at the University of Washington) for assistance with the footprinting experiments, and to Miko Robertson for manuscript preparation. This work was supported by NIH grants AI42143 and AI63940 (to SAL and ACL), and GM076222 (to MT).
References
- Abdelrahman YM, Belland RJ. The chlamydial developmental cycle. FEMS Microbiol Rev. 2005;29:949–959. doi: 10.1016/j.femsre.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Baga M, Goransson M, Normark S, Uhlin BE. Transcriptional activation of a pap pilus virulence operon from uropathogenic Escherichia coli. Embo J. 1985;4:3887–3893. doi: 10.1002/j.1460-2075.1985.tb04162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centurion-Lara A, Castro C, Barrett L, Cameron C, Mostowfi M, Van Voorhis WC, Lukehart SA. Treponema pallidum major sheath protein homologue Tpr K is a target of opsonic antibody and the protective immune response. J Exp Med. 1999;189:647–656. doi: 10.1084/jem.189.4.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DL, Riley B, Chang P, Sayahtaheri S, Tassell S, Hevelone J. Effects of molecular oxygen, oxidation-reduction potential, and antioxidants upon in vitro replication of Treponema pallidum subsp. pallidum. Appl Environ Microbiol. 1990;56:3063–3072. doi: 10.1128/aem.56.10.3063-3072.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenno JC, Muller KH, McBride BC. Sequence analysis, expression, and binding activity of recombinant major outer sheath protein (Msp) of Treponema denticola. J Bacteriol. 1996;178:2489–2497. doi: 10.1128/jb.178.9.2489-2497.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CM, Norris SJ, Weinstock GM, White O, Sutton GG, Dodson R, Gwinn M, Hickey EK, Clayton R, Ketchum KA, Sodergren E, Hardham JM, McLeod MP, Salzberg S, Peterson J, Khalak H, Richardson D, Howell JK, Chidambaram M, Utterback T, McDonald L, Artiach P, Bowman C, Cotton MD, Venter JC. Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science. 1998;281:375–388. doi: 10.1126/science.281.5375.375. [DOI] [PubMed] [Google Scholar]
- Giacani L, Hevner K, Centurion-Lara A. Gene organization and transcriptional analysis of the tprJ, tprI, tprG and tprF loci in the Nichols and Sea 81-4 Treponema pallidum isolates. J Bacteriol. 2005a;187:6084–6093. doi: 10.1128/JB.187.17.6084-6093.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacani L, Lukehart S, Centurion-Lara A. Length of guanosine homopolymeric repeats modulates promoter activity of Subfamily II tpr genes of Treponema pallidum ssp. pallidum. FEMS Immunol Med Microbiol. 2007a;51:289–301. doi: 10.1111/j.1574-695X.2007.00303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacani L, Molini B, Godornes C, Barrett L, Van Voorhis WC, Centurion-Lara A, Lukehart SA. Quantitative analysis of tpr gene expression in Treponema pallidum isolates: differences among isolates and correlation with T-cell responsiveness in experimental syphilis. Infect Immun. 2007b;75:104–112. doi: 10.1128/IAI.01124-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacani L, Sambri V, Marangoni A, Cavrini F, Storni E, Donati M, Corona S, Lanzarini P, Cevenini R. Immunological evaluation and cellular location analysis of the TprI antigen of Treponema pallidum subsp. pallidum. Infect Immun. 2005b;73:3817–3822. doi: 10.1128/IAI.73.6.3817-3822.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacani L, Sun ES, Hevner K, Molini BJ, Van Voorhis WC, Lukehart SA, Centurion-Lara A. Tpr homologs in Treponema paraluiscuniculi Cuniculi A strain. Infect Immun. 2004;72:6561–6576. doi: 10.1128/IAI.72.11.6561-6576.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goransson M, Forsman P, Nilsson P, Uhlin BE. Upstream activating sequences that are shared by two divergently transcribed operons mediate cAMP-CRP regulation of pilus-adhesin in Escherichia coli. Mol Microbiol. 1989;3:1557–1565. doi: 10.1111/j.1365-2958.1989.tb00141.x. [DOI] [PubMed] [Google Scholar]
- Gosset G, Zhang Z, Nayyar S, Cuevas WA, Saier MH., Jr Transcriptome analysis of Crp-dependent catabolite control of gene expression in Escherichia coli. J Bacteriol. 2004;186:3516–3524. doi: 10.1128/JB.186.11.3516-3524.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger DC, Aiba H, Hurd D, Browning DF, Busby SJ. Transcription factor distribution in Escherichia coli: studies with FNR protein. Nucleic Acids Res. 2007;35:269–278. doi: 10.1093/nar/gkl1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger DC, Hurd D, Harrison M, Holdstock J, Busby SJ. Studies of the distribution of Escherichia coli cAMP receptor protein and RNA polymerase along the E. coli chromosome. Proc Natl Acad Sci U S A. 2005;102:17693–17698. doi: 10.1073/pnas.0506687102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger DC, Overton TW, Reppas N, Wade JT, Tamai E, Hobman JL, Constantinidou C, Struhl K, Church G, Busby SJ. Genomic studies with Escherichia coli MelR protein: applications of chromatin immunoprecipitation and microarrays. J Bacteriol. 2004;186:6938–6943. doi: 10.1128/JB.186.20.6938-6943.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazlett KR, Rusnak F, Kehres DG, Bearden SW, La Vake CJ, La Vake ME, Maguire ME, Perry RD, Radolf JD. The Treponema pallidum tro operon encodes a multiple metal transporter, a zinc-dependent transcriptional repressor, and a semi-autonomously expressed phosphoglycerate mutase. J Biol Chem. 2003;278:20687–20694. doi: 10.1074/jbc.M300781200. [DOI] [PubMed] [Google Scholar]
- Hazlett KRO, Sellati TJ, Nguyen TT, Cox DL, Clawson ML, Caimano MJ, Radolf JD. The TprK protein of Treponema pallidum is periplasmic and is not a target of opsonic antibody or protective immunity. J Exp Med. 2001;193:1015–1026. doi: 10.1084/jem.193.9.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, N H, Okayama H. High efficiency transformation of Escherichia coli with plasmids. Gene. 1990;96:23–28. doi: 10.1016/0378-1119(90)90336-p. [DOI] [PubMed] [Google Scholar]
- Kolb A, Busby S, Buc H, Garges S, Adhya S. Transcriptional regulation by cAMP and its receptor protein. Annu Rev Biochem. 1993;62:749–795. doi: 10.1146/annurev.bi.62.070193.003533. [DOI] [PubMed] [Google Scholar]
- Kreft J, Vazquez-Boland JA. Regulation of virulence genes in Listeria. Int J Med Microbiol. 2001;291:145–157. doi: 10.1078/1438-4221-00111. [DOI] [PubMed] [Google Scholar]
- Leader BT, Hevner K, Molini BJ, Barrett LK, Van Voorhis WC, Lukehart SA. Antibody responses elicited against the Treponema pallidum repeat proteins differ during infection with different isolates of Treponema pallidum subsp. pallidum. Infect Immun. 2003;71:6054–6057. doi: 10.1128/IAI.71.10.6054-6057.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukehart SA, Baker-Zander SA, Sell S. Characterization of lymphocyte responsiveness in early experimental syphilis. I. In vitro response to mitogens and Treponema pallidum antigens. J Immunol. 1980;124:454–460. [PubMed] [Google Scholar]
- Maniatis T, Sambrook J, Fritsch EF. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- Morgan CA, Lukehart SA, Van Voorhis WC. Immunization with the N-terminal portion of Treponema pallidum repeat protein K attenuates syphilitic lesion development in the rabbit model. Infect Immun. 2002a;70:6811–6816. doi: 10.1128/IAI.70.12.6811-6816.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan CA, Lukehart SA, Van Voorhis WC. Protection against syphilis correlates with specificity of antibodies to the variable regions of Treponema pallidum repeat protein K. Infect Immun. 2003;71:5605–5612. doi: 10.1128/IAI.71.10.5605-5612.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan CA, Molini BJ, Lukehart SA, Van Voorhis WC. Segregation of B and T cell epitopes of Treponema pallidum repeat protein K to variable and conserved regions during experimental syphilis infection. J Immunol. 2002b;169:952–957. doi: 10.4049/jimmunol.169.2.952. [DOI] [PubMed] [Google Scholar]
- Norris SJ, Edmondson DG. Factors affecting the multiplication and subculture of Treponema pallidum subsp. pallidum in a tissue culture system. Infect Immun. 1986;53:534–539. doi: 10.1128/iai.53.3.534-539.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyamada T, Yokoyama K, Morinaga M, Suzuki M, Makino K. Expression of Escherichia coli DcuS-R two-component regulatory system is regulated by the secondary internal promoter which is activated by CRP-cAMP. J Microbiol. 2007;45:234–240. [PubMed] [Google Scholar]
- Posey JE, Hardham JM, Norris SJ, Gherardini FC. Characterization of a manganese-dependent regulatory protein, TroR, from Treponema pallidum. Proc Natl Acad Sci U S A. 1999;96:10887–10892. doi: 10.1073/pnas.96.19.10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radolf JD, Lukehart SA. Pathogenic Treponema: Molecular and Cellular Biology. Norfolk, England: Caister Academic Press; 2006. [Google Scholar]
- Saunders NJ, Peden JF, Hood DW, Moxon ER. Simple sequence repeats in the Helicobacter pylori genome. Mol Microbiol. 1998;27:1091–1098. doi: 10.1046/j.1365-2958.1998.00768.x. [DOI] [PubMed] [Google Scholar]
- Seshasayee AS, Bertone P, Fraser GM, Luscombe NM. Transcriptional regulatory networks in bacteria: from input signals to output responses. Curr Opin Microbiol. 2006;9:511–519. doi: 10.1016/j.mib.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Smajs D, McKevitt M, Howell JK, Norris SJ, Cai WW, Palzkill T, Weinstock GM. Transcriptome of Treponema pallidum: Gene Expression Profile during Experimental Rabbit Infection. J Bacteriol. 2005;187:1866–1874. doi: 10.1128/JB.187.5.1866-1874.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohaskey CD, Arnold C, Barbour AG. Analysis of promoters in Borrelia burgdorferi by use of a transiently expressed reporter gene. J Bacteriol. 1997;179:6837–6842. doi: 10.1128/jb.179.21.6837-6842.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun ES, Molini BJ, Barrett LK, Centurion-Lara A, Lukehart SA, Van Voorhis WC. Subfamily I Treponema pallidum repeat protein family: sequence variation and immunity. Microbes Infect. 2004;6:725–737. doi: 10.1016/j.micinf.2004.04.001. [DOI] [PubMed] [Google Scholar]
- van der Ende A, Hopman CT, Zaat S, Essink BB, Berkhout B, Dankert J. Variable expression of class I outer membrane protein in Neisseria meningitidis is caused by variation in the spacing between the -10 and -35 regions of the promoter. J Bacteriol. 1995;177:2475–2480. doi: 10.1128/jb.177.9.2475-2480.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Woude MW, Baumler AJ. Phase and antigenic variation in bacteria. Clin Microbiol Rev. 2004;17:581–611. doi: 10.1128/CMR.17.3.581-611.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock GM, Hardham JM, McLeod MP, Sodergren EJ, Norris SJ. The genome of Treponema pallidum: new light on the agent of syphilis. FEMS Microb Rev. 1998;22:323–332. doi: 10.1111/j.1574-6976.1998.tb00373.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.