

Abstract
The highly virulent GDVII strain of Theiler's murine encephalomyelitis virus causes acute and fatal encephalomyelitis, whereas the DA strain causes mild encephalomyelitis followed by a chronic inflammatory demyelinating disease with virus persistence. The differences in the amino acid sequences of the leader protein (L) of the DA and GDVII strains are greater than those for any other viral protein. We examined the subcellular distribution of DA L and GDVII L tagged with the FLAG epitope in BHK-21 cells. Wild-type GDVII L was localized predominantly in the cytoplasm, whereas wild-type DA L showed a nucleocytoplasmic distribution. A series of the L mutant experiments demonstrated that the zinc finger domain, acidic domain, and C-terminal region of L were necessary for the nuclear accumulation of DA L. A GDVII L mutant with a deletion of the serine/threonine (S/T)-rich domain showed a nucleocytoplasmic distribution, in contrast to the predominant cytoplasmic distribution of wild-type GDVII L. A chimeric DA/GDVII L, D/G, which encodes the N region of DA L including the zinc finger domain and acidic domain, followed by the GDVII L sequence including the S/T-rich domain, was distributed exclusively throughout the cytoplasm but not in the nucleus, as observed with wild-type GDVII L. Another chimeric L, G/D (which is the converse of the D/G construct), accumulated in the nucleus as well as the cytoplasm, as was observed for wild-type DA L. The findings suggest that the differential distribution of DA L and GDVII L is determined primarily by the S/T-rich domain. The S/T-rich domain may be important for the viral activity through the regulation of the subcellular distribution of L.
Theiler's murine encephalomyelitis virus (TMEV) belongs to the genus Cardiovirus of the family Picornaviridae, and its strains are divided into two subgroups on the basis of their different biological activities. The neurovirulent strains, such as GDVII and FA, produce acute and fatal encephalomyelitis in mice. The persistent strains, such as TO, DA, BeAn, etc., induce mild and nonfatal encephalomyelitis, followed by a chronic demyelinating disease with virus persistence in the spinal cords of mice. This late demyelinating disease is thought to be an excellent experimental model for the human demyelinating disease multiple sclerosis (MS) (5, 17, 20).
The TMEV genome is a single-stranded RNA molecule and translated as a long precursor polyprotein to yield 12 viral proteins by autoproteolytic cleavage (23). Two subgroup strains of TMEV have a sequence identity of approximately 95% at the amino acid level. The amino acid sequences of the proteins encoded by the P1, P2, and P3 regions of both strains are highly conserved and show 94, 96, and 98% identity, respectively. The genome has another coding region, designated the leader (L), at the most amino-terminal location of the precursor polyprotein. The L coding region encodes 76 amino acids (aa) and shows a low sequence identity of only 85% to the above-described three regions (16, 19, 22). Therefore, L has the greatest difference in amino acid sequence among any of the viral proteins and may play an important role in subgroup-specific biological activities of TMEV. In this study, we have investigated the subcellular localization of the L proteins of GDVII and DA strains and characterized the functional domains involved in the differential distribution between DA L and GDVII L in BHK-21 cells by a series of deletion mutant and chimeric construct experiments.
MATERIALS AND METHODS
Plasmid constructs.
L is organized into the three domains: (i) the N-terminal atypical zinc finger domain, (ii) the acidic domain, and (iii) the serine/threonine (S/T)-rich domain. The amino acid sequence alignment of both strains is shown in Fig. 1A. Amino acid sequences of the zinc finger and acidic domains of both strains are identical. In contrast, the S/T-rich domain was less conserved. Five out of 13 residues are different. The C-terminal region consisting of 13 aa also contains three different amino acid residues between the strains. Wild-type L (Lwt) and mutated constructs of the DA or GDVII strain are depicted in Fig. 1B. Those cDNAs were generated by PCR amplification using pDAFL3 containing the full-length DA cDNA (24) or pGDVIIFL2 containing the full-length GDVII cDNA (10). LΔZ, LΔA, LΔS/T, and LΔC lack the zinc finger domain (aa 3 to 14), the acidic domain (aa 30 to 46), the S/T-rich domain (aa 51 to 63), and the C-terminal region (aa 64 to 76), respectively. The chimeric DA/GDVII L constructs are shown in Fig. 1C. To generate those cDNAs, DA and GDVII L cDNAs were digested with the restriction enzyme AatII (Sib Enzyme, Russia), and the fragments were religated in the following combinations. A chimeric DA/GDVII L, designated D/G, encodes the N-terminal region of DA L and the C-terminal region of GDVII L. Therefore, D/G contains the zinc finger and acidic domains of DA L, followed by the S/T-rich domain and C-terminal region of GDVII L. Another chimera, designated G/D, is the converse of the D/G construct (Fig. 1C). All of the constructs were subcloned into the EcoRI/BamHI site of the p3xFLAG-CMV14 expression plasmid (Sigma-Aldrich Biotechnology, St. Louis, MO). The FLAG epitope was tagged at the C terminus, resulting in the expression of those L constructs as FLAG-tagged fusion proteins. Lwt of both strains, D/G and G/D, also were subcloned into the p3xFLAG-CMV10 expression plasmid (Sigma-Aldrich Biotechnology, St. Louis, MO) to express the N-terminal FLAG-tagged fusion proteins. All of the mutant cDNAs were sequenced and confirmed to be identical to the expected sequences.
FIG. 1.

Amino acid sequences and the deletion mutants of the L proteins from DA and GDVII strains of TMEV. (A) Alignment of L sequences from DA and GDVII strains. The L sequences encoded by DA and GDVII strains are aligned. Asterisks indicate the amino acid residues that differ between the strains. TMEV L contains three domains: (i) an N-terminal atypical zinc finger domain (aa 3 to 14), (ii) an acidic domain (aa 30 to 46), and (iii) an S/T-rich domain (aa 51 to 63). (B) The deletion mutants of DA L and GDVII L. Lwt is from DA or GDVII. LΔZ, LΔA, LΔS/T, and LΔC lack the zinc finger domain, the acidic domain, the S/T-rich domain, and the C-terminal region, respectively. (C) Diagram of the chimera L. The chimera D/G consists of N-terminal DA L (aa 1 to 45) and C-terminal GDVII L (aa 46 to 76), and the chimera G/D consists of N-terminal GDVII L (aa 1 to 45) and C-terminal DA L (aa 46 to 76). (D) Diagram of Lwt and chimera L proteins subcloned into p3xFLAG-CMV10 that had been FLAG tagged at the amino terminus.
Immunoblotting.
BHK-21 cells were maintained in minimal essential medium containing 0.03% l-glutamine and 5% newborn calf serum. The expression plasmid of each construct was transfected into BHK-21 cells using Lipofectamine LTX (Invitrogen, Carlsbad, CA) according to the manufacturer's recommendations. The transfected cells were harvested at 20 h after transfection, and total cell lysates were prepared in the lysis buffer (10 mM Tris-HCl buffer [pH 8.0], 140 mM NaCl, 3 mM MgCl2, 1 mM dithiothreitol, 0.5% NP-40, and 1 mM phenylmethylsulfonyl fluoride). Cell lysates were resolved by sodium dodecyl sulfate-15% polyacrylamide gel electrophoreses and transferred to a polyvinylidene difluoride membrane (Amersham Hybond-P; GE Healthcare Ltd., United Kingdom). The membranes were incubated with anti-FLAG M2 antibody (Sigma-Aldrich, St. Louis, MO) or anti-actin AC40 antibody (Sigma-Aldrich, St. Louis, MO) at 4°C overnight, followed by incubation with horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G (IgG) (Bio-Rad, Hercules, CA) for 1 h at room temperature, and proteins were visualized using the chemiluminescence method (ECL-Plus; GE Healthcare Ltd., United Kingdom). The protein concentration was determined by the Bradford method (Bio-Rad, Hercules, CA).
Immunofluorescence microscopy.
BHK-21 cells were plated in Lab-Tek chamber slides (Nunc, Denmark) coated with 0.3 mg/ml of cell matrix type I-P (Nitta Gelatin Co., Osaka, Japan), cultured overnight, and transfected with each expression plasmid. Cells were fixed at 20 h after transfection with 4% neutralized formaldehyde (pH 7.4) for 20 min at 4°C, permeabilized with methanol for 20 min at room temperature, and then incubated with anti-FLAG M2 antibody for 2 h at room temperature. Antibody-antigen complexes were detected with Alexa Fluor 594 mouse IgG (Molecular Probes, Invitrogen Corp., Carlsbad, CA). Nuclei were stained with Hoechst 33258 (Molecular Probes, Invitrogen Corp., Carlsbad, CA). Cells were observed by a fluorescence microscope (Axiovision; Carl Zeiss). Ten to 50 transfected cells in five fields were evaluated in each experiment, and three independent experiments were performed. Protein localization was determined by scoring the ratio of the number of cells with nuclear/cytoplasmic profiles to the total number of cells. Statistical significance was tested using t tests.
Virus superinfection.
BHK-21 cells transfected with the expression plasmid for DA Lwt or GDVII Lwt were infected with DA or GDVII virus, respectively, at the indicated time after transfection at a multiplicity of infection (MOI) of 10 PFU/cell. The cells were fixed, and the distribution of L was analyzed with immunocytostaining using anti-FLAG antibody at 6 h after infection.
RESULTS
Expression of FLAG-tagged L proteins in BHK-21 cells.
Lwt and various mutant L proteins, tagged with FLAG at the C terminus and expressed in BHK-21 cells, were detected using anti-FLAG antibody by immunoblotting (Fig. 2A). The immunoblotting of chimeric D/G and G/D proteins with FLAG tagged at both the C and N termini is shown in Fig. 2B. Actin protein (a loading control) showed the predicted size at around 40 kDa, and the intensity of signals was almost constant.
FIG. 2.

Western blot analysis of L mutant and chimeric proteins. (A) Western blot analysis of DA and GDVII L mutant proteins. All of the constructs shown in Fig. 1B were subcloned into p3xFLAG-CMV14 to be expressed as FLAG-tagged fusion proteins in BHK-21 cells. The transfected cells were harvested at 20 h after transfection, and total cell lysates were prepared in the lysis buffer. The cell lysates (10 μg) were immunoblotted with anti-FLAG antibody. Lanes 1, 3, 5, 7, and 9 represent Lwt, LΔZ, LΔA, LΔS/T, and LΔC, respectively, of the DA strain. Lanes 2, 4, 6, 8, and 10 represent Lwt, LΔZ, LΔA, LΔS/T, and LΔC, respectively, of the GDVII strain. Immunoblotting for actin using the same cell lysates also is shown as a loading control. (B) Western blot analysis of DA and GDVII L chimeric proteins. The chimeric mutants shown in Fig. 1C and D were subcloned into p3xFLAG-CMV14 or p3xFLAG-CMV10, and protein expression was analyzed by Western blotting as described for panel A. Lanes 1 and 2 represent Lwt of DA, and lanes 3 and 4 represent Lwt of GDVII. Lanes 5 and 6 represent chimera D/G, and lanes 7 and 8 represent chimera G/D. Lanes 1, 3, 5, and 7 represent the proteins expressed as FLAG-tagged proteins at the C terminus, and lanes 2, 4, 6, and 8 represent the proteins expressed as FLAG-tagged proteins at the N terminus. Immunoblotting for actin using the same cell lysates also is shown as a loading control.
Each mutated protein showed nearly the predicted size at around 15 kDa, although LΔA showed a faster mobility than that of the predicted size, and ΔS/T showed different mobility between the two strains (Fig. 2A). This difference may be linked to the aberrant migration of the products from the constructs containing the highly acidic domain. In addition, the intensity of the bands of those fusion proteins was not constant, probably because of the different efficiencies of the expression of each fusion protein.
Subcellular localization of Lwt and mutants of DA L.
BHK-21 cells were transfected with the expression plasmid for Lwt and mutants of DA L, and subcellular localization was examined by indirect immunofluorescence assays. Although DA Lwt has no nuclear localization signal sequences, it was distributed not only in the cytoplasm but also in the nucleus (Fig. 3). The deletion mutant LΔS/T showed the same pattern of localization as that of Lwt in most of the cells (Fig. 3). In contrast, LΔZ, LΔA, and LΔC mutant proteins were distributed predominantly in the cytoplasm (Fig. 3). Figure 5 represents a quantitative analysis of the immunofluorescence data. Ninety percent of the cells expressing Lwt and 77% of the cells expressing LΔS/T showed nucleocytoplasmic localization. However, only 16, 35, and 4% of LΔZ, LΔA, and LΔC mutants showed nucleocytoplasmic localization, respectively (see Fig. 5). These findings suggest that the zinc finger domain, acidic domain, and C-terminal region are required for nuclear localization for DA L.
FIG. 3.

Subcellular localization of DA L and its mutants in BHK-21 cells. BHK-21 cells were transfected with the expression plasmids for DA L and its mutants (Lwt, LΔZ, LΔA, LΔS/T, and LΔC). The cells were fixed at 20 h after transfection and stained with anti-FLAG antibody for L and its mutants (anti-FLAG). Antibody-antigen complexes were detected with Alexa Fluor 594 mouse IgG. Nuclei were stained with Hoechst 33258 (Hoechst). Cells were observed by a fluorescence microscope (Axiovision; Carl Zeiss). Merge represents merged images stained with anti-FLAG and Hoechst.
FIG. 5.
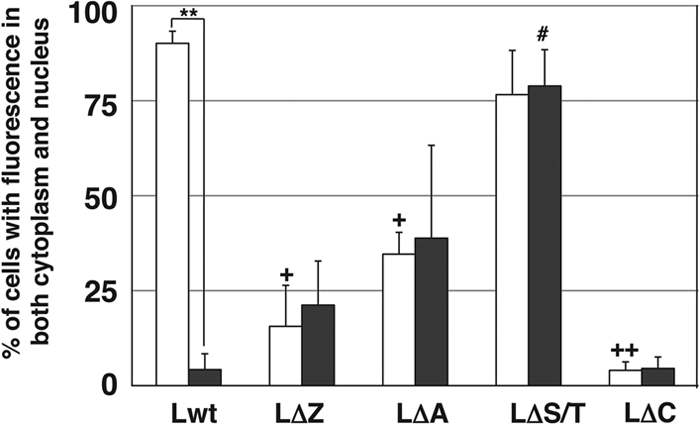
Distribution of DA and GDVII L in BHK-21 cells. Histograms show the percentages of the transfected cells showing both nuclear and cytoplasmic FLAG staining. None of the cells showed solely nuclear staining. The subcellular localization of Lwt and its mutants (LΔZ, LΔA, LΔS/T, and LΔC) was examined by fluorescence microscopy in five fields containing 10 to 50 cells positive for FLAG. Open and closed columns represent the L proteins of DA and GDVII strains, respectively. Each value represents the means ± standard errors of the means from three independent experiments. **, significant between DA Lwt and GDVII Lwt at P < 0.0001; +, significant between DA Lwt and DA LΔZ (or DA LΔA) at P < 0.005; ++, significant between DA Lwt and DA LΔC at P < 0.0001; #, significant between GDVII Lwt and GDVII LΔS/T at P < 0.005.
Subcellular localization of Lwt and mutants of GDVII L.
We next examined the subcellular distribution of Lwt and mutants of GDVII L. In contrast to the nucleocytoplasmic distribution of DA Lwt, GDVII Lwt was localized predominantly in the cytoplasm (Fig. 4). LΔZ, LΔA, and LΔC mutants showed the same cytoplasmic distribution pattern as Lwt (Fig. 4). On the other hand, LΔS/T was distributed in both the nucleus and the cytoplasm (Fig. 4). Figure 5 represents a quantitative analysis of the immunofluorescence data. Seventy-nine percent of the cells expressing LΔS/T showed nucleocytoplasmic localization. In contrast, less than 5% of Lwt and LΔC showed nucleocytoplasmic localization. Only 21 and 39% of cells expressing LΔZ and LΔA, respectively, showed nucleocytoplasmic localization (Fig. 5). GDVII L was localized in the cytoplasm, and none of the deletion mutants changed its localization, except ΔS/T, which altered the localization to nuclear.
FIG. 4.

Subcellular localization of GDVII L and its mutants in BHK-21 cells. BHK-21 cells were transfected with the expression plasmids for GDVII L and its mutants (Lwt, LΔZ, LΔA, LΔS/T, and LΔC). The cells were fixed at 20 h after transfection and stained with anti-FLAG antibody for L and its mutants (anti-FLAG). Antibody-antigen complexes were detected with Alexa Fluor 594 mouse IgG. Nuclei were stained with Hoechst 33258 (Hoechst). Cells were observed by a fluorescence microscope (Axiovision; Carl Zeiss). Merge represents merged images stained with anti-FLAG and Hoechst.
These finding suggested that the S/T-rich domain is necessary for the cytoplasmic accumulation of GDVII L. The zinc binding domain and the acidic domain also may contribute to helping L remain in the cytoplasm, although the values showed no significant difference.
Subcellular localization of the chimeric L from DA and GDVII strains.
To further analyze the differential distribution of DA and GDVII L, we generated chimeric constructions and examined their subcellular distribution. The chimeric D/G containing the S/T-rich domain of GDVII L was distributed exclusively throughout the cytoplasm in the same pattern as that of GDVII Lwt. The chimeric G/D containing the S/T-rich domain of DA L was distributed not only in the cytoplasm but also in the nucleus. The pattern was the same as that of DA Lwt (Fig. 6A). Quantitative analysis demonstrated that 96.9% of the cells expressing the chimeric G/D showed nucleocytoplasmic localization, compared to 9.9% of chimeric D/G (Fig. 6B). These findings suggest that the C-terminal one-third sequence of DA L (aa 46 to 76) is important for the nuclear accumulation of TMEV L. The region of aa 46 to 76 encodes the S/T-rich domain and the C-terminal region, as shown in Fig. 1A. The data regarding the distribution of DA LΔC and GDVII LΔC (Fig. 3, 4, and 5) suggest that the C-terminal region is required for the nuclear localization of L. The S/T-rich domain of GDVII is considered to be involved in a shift in the distribution to the cytoplasm, because ΔS/T was the only GDVII mutant to show a localization different from that of GDVII L (Fig. 4 and 5). Therefore, it is suggested that the S/T-rich domain of GDVII L is the key sequence to distribute L exclusively in the cytoplasm. Previously, we reported that the amino acid at position 57 of L, which belongs to the S/T-rich domain, regulated subgroup-specific virus growth on BHK-21 cells (26). We investigated the effect of residue 57 on L protein distribution by using the mutant DALPro, in which the residue 57 serine was mutated to proline (as in GDVII L), and GDVIILSer, in which the residue 57 proline was mutated to serine (as in DA L). Quantitative analysis demonstrated that 79.5% ± 7.2% of the cells expressing DALPro showed nucleocytoplasmic localization, while 17.0% ± 7.4% of the cells expressed GDVIILSer. DALPro was not significantly different from DA Lwt, although its level of expression was a little lower. GDVIILSer was not significantly different from GDVII Lwt either, although its level of expression was a little higher. These findings suggested that residue 57 was not the only residue to decide the distribution of L.
FIG. 6.

Subcellular localization of chimeric L proteins of DA and GDVII strains. (A) Subcellular localization of chimera D/G and G/D. BHK-21 cells were transfected with the expression plasmids for FLAG-tagged chimera D/G or G/D at the C terminus. The cells were fixed 20 h after transfection and stained with anti-FLAG antibody for L and its mutants (anti-FLAG). Antibody-antigen complexes were detected with Alexa Fluor 594-conjugated anti-mouse IgG. Nuclei were stained with Hoechst 33258 (Hoechst). Cells were observed by a fluorescence microscope (Axiovision; Carl Zeiss). Merge represents merged images stained with anti-FLAG and Hoechst. (B) Distributions of the chimeras D/G and G/D. Histograms show the percentages of the transfected cells showing both nuclear and cytoplasmic FLAG staining. None of the cells showed solely nuclear staining. The localization was observed by fluorescence microscopy in five fields containing 10 to 50 cells positive for FLAG expression. Closed and open columns showed the results for the proteins FLAG tagged at the C and N termini, respectively. Each value represents the means ± standard errors of the means from three independent experiments. *, significant between DA and GDVII or D/G tagged at the C terminus at P < 0.0001; +, significant between DA and GDVII or D/G tagged at the N terminus at P < 0.0001.
Localization of FLAG-tagged L at different positions.
To determine whether the position of the FLAG tagging affected L localization, DA Lwt, GDVII Lwt, D/G, and G/D were subcloned into plasmid p3xFLAG-CMV10 to express N-terminally tagged proteins (Fig. 1D). The quantitative analysis of localization is shown in Fig. 6B. In any case, N-terminally tagged protein and C-terminally tagged protein did not show any difference in terms of L distribution.
Time course of subcellular localization of L after transfection.
To investigate whether the subcellular distribution of L alters during the expression course after transfection, we fixed cells at 8, 12, 16, 20, and 24 h after transfection with plasmids expressing FLAG-tagged DA Lwt and GDVII Lwt fused at both the N and C termini (Fig. 7). All of the proteins already were expressed at 8 h after transfection. Subcellular distributions of L were not altered significantly during the time course of expression. The constructs of DA Lwt that were FLAG tagged at the N and C termini showed the same nucleocytoplasmic localization throughout the time examined. GDVII L that had been FLAG tagged at the C terminus was localized in the cytoplasm, while GDVII L that had been FLAG tagged at the N terminus showed a relatively higher nucleocytoplasmic distribution ratio at 8 and 12 h and then showed the same cytoplasmic distribution at 16, 20, and 24 h as that of GDVII L tagged at the C terminus. Because L protein expression is toxic and induces apoptosis, we could not examine the distribution at longer than 24 h after transfection.
FIG. 7.
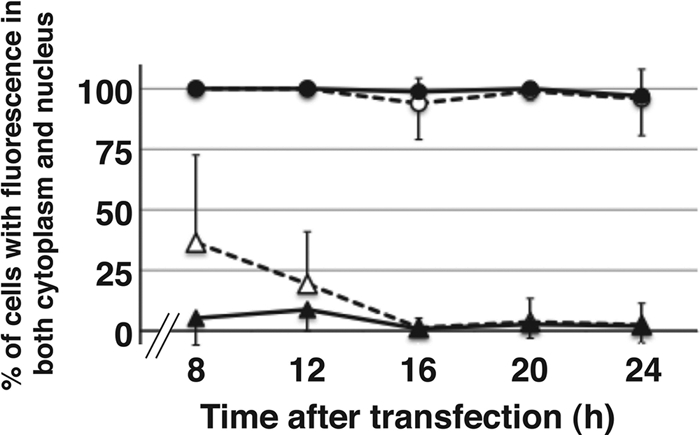
Time course of subcellular localization after transfection. Transitions of subcellular localizations were graphed as percentages of the cells transfected with DA (circle) or GDVII (triangle), showing both nuclear and cytoplasmic FLAG staining. None of the cells showed solely nuclear staining. Localization was observed by fluorescence microscopy in five fields containing 10 to 50 cells positive for FLAG expression. Closed and open symbols show the results of the expression of FLAG-tagged proteins at the C and N termini.
Virus superinfection.
Because no antibodies recognizing neither DA L or GDVII L were available for immunocytostaining, we could not show the distributions of L derived from virus infection directly. We chose an alternative approach to study whether the localization of FLAG-tagged L is affected by virus superinfection. The cells expressing either DA L or GDVII L were infected with DA or GDVII virus, respectively, at an MOI of 10 PFU/cell at 4 and 7 h after transfection. We analyzed the distribution of FLAG-tagged L at 6 h after viral infection. At longer times of more than 6 h after infection, it was difficult to analyze the distribution because of the apoptotic condensation of nuclei and/or the cytopathic effect. The quantitative analysis of localization is shown in Fig. 8. Neither DA L nor GDVII L had any effect on cellular distribution by virus infection, suggesting that the distribution of L expressed by authentic TMEV infection is the same as that of FLAG-tagged L expressed by transfection in BHK-21 cells.
FIG. 8.
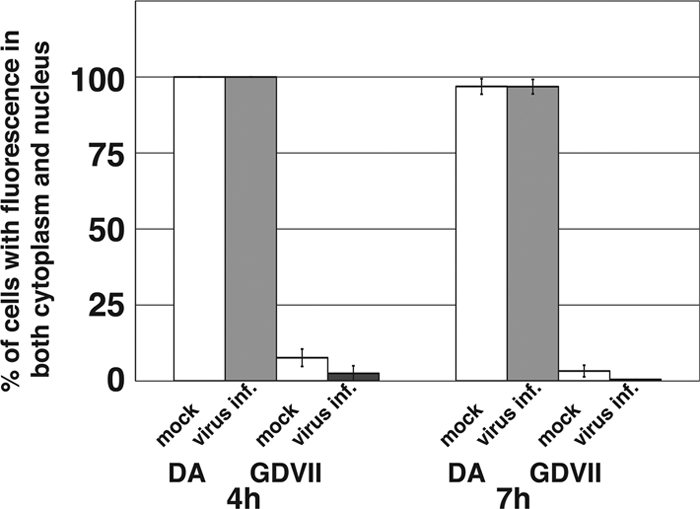
Virus superinfection. Histograms show the percentages of the cells showing both nuclear and cytoplasmic FLAG staining. The cells expressing either DA L or GDVII L FLAG tagged at the C terminus were mock infected (inf.) or were infected with DA (gray bars) or GDVII (black bars) at an MOI of 10 PFU/cell at 4 and 7 h after transfection. The localization was observed by fluorescence microscopy in five fields containing 10 to 50 cells positive for FLAG expression.
DISCUSSION
TMEV has been studied as an animal model for MS (5, 17, 20). Although various studies using recombinant viruses between GDVII and DA (or BeAn) strains demonstrated that capsid proteins, especially VP1 and VP2, are important for virus persistence and demyelination (14, 23), the precise localization of the region responsible for those biological activities remains to be clarified.
L and L* are nonstructural proteins, and those coding regions are located at the 5′ end of the polyprotein coding regions. L* is synthesized only in DA subgroup strains and is not incorporated into virions (18). Studies have demonstrated that L* is required for virus growth in macrophages in vitro (13, 27) and also is important for virus persistence and demyelination in vivo (7, 11). The importance of L* for virus persistence and demyelination is studied using the DA strain with a mutation from AUG to ACG at the L* initiation codon. However, the finding still is controversial, since the mutation from AUG to ACG at the L* initiation codon in another molecular clone of the same virus strain has a weak influence on persistence (28, 29). L is a 76-aa-long protein (19) and also is reported to be crucial for neurovirulence (6) and in virion assembly (3). L contains a zinc binding motif (8) that inhibits gamma/beta inteferon (IFN-α/β) production early after viral infection, leading to the persistence of DA subgroup strains (30). It also has been suggested that the trafficking of IFN regulatory factor 3 (IRF-3) from the cytoplasm to the nucleus is perturbed by L, leading to the inhibiting of IFN-α/β production (9). Therefore, these studies suggest that L works as a multifunctional regulator for TMEV biological activities. In this report, we examined the subcellular localization of L of both subgroup strains in BHK-21 cells using several L constructs.
Surprisingly, the distribution of DA L is totally different from that of GDVII L in BHK-21 cells. GDVII L was localized predominantly in the cytoplasm, while DA L showed nucleocytoplasmic distribution. A series of deletion mutant experiments further demonstrated that the zinc finger domain, acidic domain, and C-terminal region were necessary for the nuclear accumulation of DA L. We also found that the GDVII L mutant deleted of the S/T-rich domain showed nuclear accumulation. The S/T-rich domain of L may regulate the nuclear-targeting function of other regions. Our findings using the chimeric proteins further support this hypothesis. The chimeric protein containing the S/T-rich domain of GDVII L was distributed exclusively throughout the cytoplasm in the same fashion as GDVII Lwt. Accumulation within the nuclei of the chimeric protein containing the S/T-rich domain of DA L was observed to have the same localization pattern as wild-type DA L. Amino acid sequences of the zinc finger domain of L are completely identical between DA and GDVII strains, while the S/T-rich domains are less conserved and 5 out of 13 amino acid residues are different. Since the replacement of residue 57 showed no effects on the localization of L, the change of one amino acid is not enough to affect the subcellular localization of L. In addition, the S/T-rich domain of GDVII L contains a potential phosphorylation site. Recently, Hato et al. have shown that the phosphorylation at threonine 47 in L of mengovirus was necessary for viral virulence and the inhibition of IFN-α/β (12). The phosphorylation status of the S/T-rich domain could be influenced by the cellular or environmental conditions. The mechanism(s) for the different distributions of the two L proteins still remains to be clarified. One possibility is that the two L proteins are small enough to enter into the nucleus by diffusion, and only GDVII L can be exported from the nucleus by some mechanism(s) through the S/T-rich domain. In that case, each domain except the S/T-rich one may play a role in retaining a normal L structure.
Although essential infectious steps of picornaviruses have been believed to take place mainly in the cytoplasm, virus-specific proteins are reported to be detected in the nucleus (1, 2, 4). The infection by TMEV considerably disturbed the nucleocytoplasmic trafficking of cellular proteins. L can promote the redistribution of a nuclear protein to the cytoplasm. L also can promote the redistribution of a cytoplasmic protein to the nucleus, i.e., IRF-3 (9). As a result, L inhibits the production of IFN-α4 and IFN-β at the transcriptional level. This leads to the dysregulation of the whole defense system, fostering the persistence of TMEV.
Previously, it was shown that the L protein of a neurovirulent TMEV strain could functionally replace that of the DA strain in vitro and in vivo (21) using chimeric viruses. They used L929 cells for in vitro experiments. In fact, we found that GDVII L as well as DA L was localized not only in the nucleus but also in the cytoplasm in L929 cells (data not shown). It is possible that DA L and GDVII L play different roles in some types of cells, including BHK-21 cells. We also examined the distribution of L in RAW264.7 cells, a mouse macrophage cell line, and found the same L distribution as that of BHK-21 cells in both strains (data not shown). It is reported that macrophages are the principal cells for the viral persistence of the DA strain during central nervous system infection (15, 25). One speculation is that DA L localizes in both the cytoplasm and nucleus in macrophages and inhibits IFN transcription via IRF-3, leading to virus persistence, whereas GDVII L localizes exclusively in the cytoplasm and cannot persist in macrophages because of the production of IFN.
Since TMEV L is reported to be toxic to cells when expressed alone (9), we fixed cells at 20 h after the transfection of L. Therefore, this is the first report to investigate the behavior of L in cells using various L constructs, not viruses. DA L may affect cellular factors by translocating itself into the nucleus.
Here, we report different subcellular localizations of DA L and GDVII L in BHK-21 cells. The differences in the behaviors of DA L and GDVII L depend on the S/T-rich domain. The S/T-rich domain of TMEV may play an important role in regulating TMEV biological activities. It remains to be clarified which cellular molecules interact with L. This may lead to the elucidation of the mechanism(s) of TMEV persistence and TMEV-induced demyelinating disease.
Acknowledgments
This work was supported in part by the Health and Labor Sciences Research Grant of Intractable Diseases (Neuroimmunological Diseases) from the Ministry of Health, Labor, and Welfare of Japan and a Grant for Collaborative Research from Kanazawa Medical University (C2007-5, C2008-2)
Footnotes
Published ahead of print on 22 April 2009.
REFERENCES
- 1.Aminev, A. G., S. P. Amineva, and A. C. Palmenberg. 2003. Encephalomyocarditis viral protein 2A localizes to nucleoli and inhibits cap-dependent mRNA translation. Virus Res. 9545-57. [DOI] [PubMed] [Google Scholar]
- 2.Aminev, A. G., S. P. Amineva, and A. C. Palmenberg. 2003. Encephalomyocarditis virus (EMCV) proteins 2A and 3BCD localize to nuclei and inhibit cellular mRNA transcription but not rRNA transcription. Virus Res. 9559-73. [DOI] [PubMed] [Google Scholar]
- 3.Badshah, C., M. A. Calenoff, and K. Rundell. 2000. The leader polypeptide of Theiler's murine encephalomyelitis virus is required for the assembly of virions in mouse L cells. J. Virol. 74875-882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bienz, K., D. Egger, Y. Rasser, and W. Bossart. 1982. Accumulation of poliovirus proteins in the host cell nucleus. Intervirology 18189-196. [DOI] [PubMed] [Google Scholar]
- 5.Brahic, M., J. F. Bureau, and T. Michiels. 2005. The genetics of the persistent infection and demyelinating disease caused by Theiler's virus. Annu. Rev. Microbiol. 59279-298. [DOI] [PubMed] [Google Scholar]
- 6.Calenoff, M. A., C. S. Badshah, M. C. Dal Canro, H. L. Lipton, and M. K. Rundell. 1995. The leader polypeptide of Theiler's virus is essential for neurovirulence but not for virus growth in BHK cells. J. Virol. 695544-5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, H. H., W. P. Kong, L. Zhang, P. L. Ward, and R. P. Roos. 1995. A picornaviral protein synthesized out of frame with the polyprotein plays a key role in a virus-induced immune-mediated demyelinating disease. Nat. Med. 1927-931. [DOI] [PubMed] [Google Scholar]
- 8.Chen, H. H., W. P. Kong, and R. P. Roos. 1995. The leader peptide of Theiler's murine encephalomyelitis virus is a zinc-binding protein. J. Virol. 698076-8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delhaye, S., V. van Pesch, and T. Michiels. 2004. The leader protein of Theiler's virus interferes with nucleocytoplasmic trafficking of cellular proteins. J. Virol. 784357-4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fu, J., S Stein, L. Rosenstein, T. Bodwell, M. Routbort, B. L. Semler, and R. P. Roos. 1990. Neurovirulence determinants of genetically engineered Theiler viruses. Proc. Natl. Acad. Sci. USA 874125-4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghadge, G. D., L. Ma, S. Sato, J. Kim, and R. P. Roos. 1998. A protein critical for a Theiler's virus-induced immune system-mediated demyelinating disease has a cell type-specific antiapoptotic effect and a key role in virus persistence. J. Virol. 728605-8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hato, S. V., C. Ricour, B. M. Schulte, K. H. W. Lanke, M. de Bruijni, J. Zoll, W. J. G. Melchers, T. Michiels, and J. M. van Kuppeveld. 2007. The mengovirus leader protein blocks interferon-α/β gene transcription and inhibits activation of interferon regulatory factor 3. Cell. Microbiol. 92921-2930. [DOI] [PubMed] [Google Scholar]
- 13.Himeda, T., Y. Ohara, K. Asakura, Y. Kontani, M. Murakami, H. Suzuki, and M. Sawada. 2005. A lentiviral expression system demonstrates that L* protein of Theiler's murine encephalomyelitis virus (TMEV) is essential for virus growth in a murine macrophage-like cell line. Virus Res. 10823-28. [DOI] [PubMed] [Google Scholar]
- 14.Lipton, H. L., and M. L. Jelachich. 1997. Molecular pathogenesis of Theiler's murine encephalomyelitis virus-induced demyelinating disease in mice. Intervirology 40143-152. [DOI] [PubMed] [Google Scholar]
- 15.Lipton, H. L., G. Twaddle, and M. L. Jelachich. 1995. The predominant virus antigen burden is present in macrophages in Theiler's murine encephalomyelitis virus-induced demyelinating disease. J. Virol. 692523-2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michiels, T., N. Jarousse, and M. Brahic. 1995. Analysis of the leader and capsid coding regions of persistent and neurovirulent strains of Theiler's virus. Virology 214550-558. [DOI] [PubMed] [Google Scholar]
- 17.Obuchi, M., and Y. Ohara. 1998. Theiler's murine encephalomyelitis virus and mechanisms of its persistence. Neuropathology 1813-18. [Google Scholar]
- 18.Obuchi, M., T. Odagiri, K. Asakura, and Y. Ohara. 2001. Association of L* protein of Theiler's murine encephalomyelitis virus with microtubules in infected cells. Virology 28995-102. [DOI] [PubMed] [Google Scholar]
- 19.Ohara, Y., S. Stein, J. Fu, L. Stillman, L. Klaman, and R. P. Roos. 1988. Molecular cloning and sequence determination of DA strain of Theiler's murine encephalomyelitis viruses. Virology 164245-255. [DOI] [PubMed] [Google Scholar]
- 20.Oleszak, E. L., J. R. Chang, H. Friedman, C. D. Katsetos, and C. D. Platsoucas. 2004. Theiler's virus infection: a model for multiple sclerosis. Clin. Microbiol. Rev. 17174-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paul, S., and T. Michiels. 2006. Cardiovirus leader proteins are functionally interchangeable and have evolved to adapt to virus replication fitness. J. Gen. Virol. 871237-1246. [DOI] [PubMed] [Google Scholar]
- 22.Pevear, D. C., J. Borkowski, M. Calenoff, C. K. Oh, B. Ostrowski, and H. L. Lipton. 1988. Insights into Theiler's virus neurovirulence based on a genomic comparison of the neurovirulent GDVII and less virulent BeAn strains. Virology 1651-12. [DOI] [PubMed] [Google Scholar]
- 23.Roos, R. P. 2002. Pathogenesis of Theiler's murine encephalomyelitis virus-induced disease. p. 427-436. In B. L. Semler, and E. Wimmer (ed.), Molecular biology of picornaviruses, ASM Press, Washington, DC.
- 24.Roos, R. P., S. Stein, Y. Ohara, J. Fu, and B. L. Semler. 1989. Infectious cDNA clones of the DA strain of Theiler's murine encephalomyelitis virus. J. Virol. 635492-5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rossi, C. P., M. Delcroix, I. Huitinga, A. McAllister, N. van Rooijen, E. Claassen, and M. Brahic. 1997. Role of macrophages during Theiler's virus infection. J. Virol. 713336-3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takano-Maruyama, M., Y. Ohara, K. Asakura, and T. Okuwa. 2006. Theiler's murine encephalomyelitis virus leader protein amino acid residue 57 regulates subgroup-specific virus growth on BHK-21 cells. J. Virol. 8012025-12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takata, H., M. Obuchi, J. Yamamoto, T. Odagiri, R. P. Roos, H. Iizuka, and Y. Ohara. 1998. L* protein of the DA strain of Theiler's murine encephalomyelitis virus is important for virus growth in a murine macrophage-like cell line. J. Virol. 724950-4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Eyll, O., and T. Michiels. 2000. Influence of the Theiler's virus L* protein on macrophage infection, viral persistence, and neurovirulence. J. Virol. 749071-9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Eyll, O., and T. Michiels. 2002. Non-AUG-initiated internal translation of the L* protein of Theiler's virus and importance of this protein for viral persistence. J. Virol. 7610665-10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Pesch, V., O. van Eyll, and T. Michiels. 2001. The leader protein of Theiler's virus inhibits immediate-early alpha/beta interferon production. J. Virol. 757811-7817. [DOI] [PMC free article] [PubMed] [Google Scholar]