

SUMMARY
Yeast Mrc1, ortholog of metazoan Claspin, is both a central component of normal DNA replication forks and a mediator of the S phase checkpoint. We report that Mrc1 interacts with Pol2, the catalytic subunit of DNA polymerase ε, essential for leading strand DNA replication and for the checkpoint. In unperturbed cells, Mrc1 interacts independently with both the N-terminal and C-terminal halves of Pol2 (Pol2N and Pol2C). Strikingly, phosphorylation of Mrc1 during the S phase checkpoint abolishes Pol2N binding but not Pol2C interaction. Mrc1 is required to stabilize Pol2 at replication forks stalled in HU. The bimodal Mrc1/Pol2 interaction may identify a novel step in regulating the S phase checkpoint response to DNA damage on the leading strand. We propose that Mrc1, which also interacts with the MCMs, may modulate coupling of polymerization and unwinding at the replication fork.
INTRODUCTION
To achieve genome stability, complicated replication machines must be sequentially assembled and precisely programmed such that S phase is appropriately integrated into the cell cycle. Assembly and programming occur during the M/G1 phases of the cell cycle. In addition, the replication machines must be protected during S phase, because progress of the replication machine along the template, while sequential, is not continuous. Replication forks stall periodically, either in difficult-to-replicate regions of the genome or due to exogenous environmental interference. How to prevent replication forks from irreversibly collapsing in S phase, when de novo assembly is no longer permitted, becomes a fascinating question. Cells must have a mechanism to stabilize the replication machinery in situ until aberrant structures can be removed and forks can restart. The simplest solution is for this mechanism to be intrinsic to the replication machine itself. The studies described here comprise a genetic and biochemical analysis of one of these components, yeast Mrc1.
Mrc1 (mediator of the replication checkpoint) plays roles in both the S phase checkpoint, a multistep response to replication stress, and at the replication fork. First, phosphorylation of Mrc1 by the “signaling” Mec1/Ddc2 kinase is required for activation of the Rad53 “effector” kinase during the S phase checkpoint (Alcasabas et al., 2001; Osborn and Elledge, 2003). Second, Mrc1, although it is not essential, is required for normal DNA replication in the absence replication stress. Mrc1 is loaded onto replication origins in every cell cycle at the same time as DNA polymerases, just after formation of the Replication Progression Complex, whose core consists of Cdc45, GINS, and the MCMs and migrates away from origins with replication forks (Alcasabas et al., 2001; Bjergbaek et al., 2005; Gambus et al., 2006; Katou et al., 2003; Szyjka et al., 2005). Mrc1 plays an intrinsic and constitutive role in replication, since in its absence all replication forks move at only half the normal rate and cells experience extensive replication fork damage in the absence of exogenous damaging agents (Alcasabas et al., 2001; Azvolinsky et al., 2006; Bjergbaek et al., 2005; Osborn and Elledge, 2003; Szyjka et al., 2005). mrc1 nulls thus exhibit a high frequency of gross chromosomal rearrangements (GCRs) and synthetic lethality with rad9 null, encoding the DNA damage checkpoint “mediator” (Bjergbaek et al., 2005; Osborn and Elledge, 2003). Several essential proteins, pol ε, Dpb11, RFC5, and Cdc7/Dbf4, like Mrc1, also perform roles in both DNA replication and in the S phase checkpoint (Araki et al., 1995; Navas et al., 1995; Sugimoto et al., 1997; Sugimoto et al., 1996). Third, Mrc1 may participate in sister chromatid cohesion and telomere capping (Tsolou and Lydall, 2007; Xu et al., 2004).
mrc1AQ, a mutant in which all of the possible Mec1 S/TQ phosphorylation sites have been changed to nonphosphorylatable AQ, is proficient in DNA replication but deficient in full Rad53 activation, and thus in the downstream global cell cycle responses of the checkpoint (Alcasabas et al., 2001; Osborn and Elledge, 2003). This separation of function mutant leads to the notion that the replication and checkpoint functions of Mrc1 are separable. The situation may be more complicated than that, however. When normal cells are transiently exposed to HU (hydroxyurea), forks stall, but the replisome, along with Mrc1, remains at the forks, which can restart when HU is removed (Bjergbaek et al., 2005; Katou et al., 2003; Szyjka et al., 2005; Tourriere et al., 2005). In an mrc1 null, however, a majority of the stalled forks fail to restart (Bjergbaek et al., 2005; Szyjka et al., 2005; Tourriere et al., 2005). This failure to restart forks correlates with an uncoupling of DNA synthesis and replisome migration (Katou et al., 2003). DNA synthesis ceases; but the replisome, including DNA polymerases α and Dpb3, Cdc45, the MCMs, and GINS, continues to translocate for thousands of base pairs without synthesizing new DNA (Bjergbaek et al., 2005; Katou et al., 2003). Mrc1 is also a component of forks paused at natural replication barriers formed by stable protein-DNA complexes, for instance in the rDNA in yeast, along with DNA polymerases α and ε, Cdc45, the MCMS, and GINS (Azvolinsky et al., 2006; Calzada et al., 2005). Thus, Mrc1 participates directly in fork stabilization during the checkpoint cascade.
One mechanism by which Mrc1 might prevent uncoupling of synthesis and unwinding and stabilize the replisome at the stalled forks is by inhibiting progression of the Cdc45/MCM helicase motor in the absence of a functioning polymerase motor. In partial support of this model, Mrc1 coimmunoprecipitates with Cdc45 (Katou et al., 2003). Mrc1 also copurifies, albeit in substoichiometric amounts, with the RPC (Gambus et al., 2006). In addition, in S. pombe, certain loss of function mutations in the Cdc45 and MCM orthologs suppress the HU sensitivity of mrc1Δ mutants (Nitani et al., 2006). However, MCM inhibition would not account for the fact that Mrc1 is a positive factor for normal DNA replication. Therefore, a more comprehensive model for Mrc1 is one in which Mrc1 is involved in molecular interactions that coordinate DNA synthesis and unwinding, i.e., polymerase and helicase, during normal DNA replication as well as during the checkpoint (Szyjka et al., 2005; Tourriere et al., 2005). In this capacity it could promote fork progression but would also be poised to inhibit fork progression during replication stress and stabilize the stalled fork.
In this work we present evidence that Mrc1 interacts not only with Cdc45 and the MCMs, but also with pol ε the primary leading strand replicative polymerase (Morrison et al., 1990; Pursell et al., 2007; Shcherbakova and Pavlov, 1996), which is also implicated in checkpoint signaling (Navas et al., 1996; Navas et al., 1995). This dual interaction supports the helicase/polymerase coupling function for Mrc1.
RESULTS
Yeast Two Hybrid Experiments Reveal a multi-site Pol2/Mrc1 Interaction
Pol ε consists of four subunits, Pol2, Dpb2, Dpb3, and Dpb4 (Kawasaki and Sugino, 2001 for review). Pol2 is the catalytic subunit (Fig. 1A). The Pol2 N-terminal half (Pol2N, residues 1–1264) encodes the polymerase and proofreading nuclease active sites. Interestingly, the remaining, noncatalytic 120 kDa Pol2 C terminus (Pol2C, residues1265–2222), comprises the only essential part of the protein. A mutant lacking Pol2N, pol2-16, is viable (Dua et al., 1999; Kesti et al., 1999). We carried out a two-hybrid screen to identify proteins that interact with Pol2C in hopes of defining its essential functions (Edwards et al., 2003). One of the genes identified, YCL061C, encodes residues 251–983 of MRC1, and full-length Mrc1 was also found to interact with Pol2C (Fig. 1B). Among other Pol2-interacting proteins (Dua et al., 2000; Edwards et al., 2003), Dpb2 appears to interact weakly with Mrc1, but DPB3, DPB4, and DPB11 do not (Fig. 1B). [Further mapping of the interaction domain within Pol2C is shown in Figure S1.] These results suggest that Mrc1 may interact directly with Pol2 and that Dpb2 may modulate that interaction.
Figure 1. Mrc1 Interacts with Pol2.

(A) Schematic map of the full-length POL2 gene and the zinc finger region. The full-length gene, 2222 residues, is shown at the top. Pol2N: Five conserved nuclease motifs, residues 288 and 501; seven partially conserved DNA polymerase motifs, residues 544 and 954, three conserved motifs of unknown function, residue 954 and 1183 (not indicated here); Pol2C: essential “spacer” region conserved only in the Pol2 subfamily of B family polymerases, 1180 and 2103; conserved zinc finger motif, 2103–2222(end). MutA through MutI are deletions within the zinc finger domain used for mapping the interaction subdomain in Figure S1 (Dua et al., 1998).
(B) Two hybrid assay for Mrc1/Pol2 interaction. Analyses were performed on at least three independent transformants as described previously (Edwards et al., 2003).
Meanwhile, we were surprised to find that Mrc1 interacts with Pol2N, in addition to Pol2C (Fig. 1B). Thus, there are two independent interaction sites for Mrc1 in Pol2.
We also found that mrc1AQ binds robustly to Pol2C (Fig. 1B). Since Mec1-dependent phosphorylation of Mrc1 does not appear to be essential for interaction, we conclude that Mrc1 may interact with Pol2 in normal replisome.
Pol2/Mrc1 Physical Interaction
To verify that the two hybrid results reflected physical interactions between the endogenous proteins, we studied the Mrc1/Pol2 interaction by coimmunoprecipitation. To facilitate detection of Mrc1 and Pol2 without altering the normal timing of expression or endogenous levels of the proteins, immunoprecipitation was carried out by tagging chromosomal MRC1 with 13 myc epitopes and POL2 with 3 HA (hemagglutinin) epitopes. Both tagged genes are functional (Alcasabas et al., 2001; Aparicio et al., 1997; Edwards et al., 2003). As shown in Figure 2A, lanes 1–6, untagged Mrc1 or Pol2 controls are not precipitated by anti-myc or anti-HA antibody. Tagged Mrc1-13myc and Pol2-3HA do coimmunoprecipitate, however, regardless of which species is immunoprecipitated (Fig. 2A, lanes 7–12). Comparison of the amount of protein found in the whole cell extract and immunoprecipitation supernatant, suggests that about half of the cellular Mrc1 associates with Pol2, while almost all Pol2 molecules bind to Mrc1.
Figure 2. Reciprocal Coimmunoprecipitation of Pol2 and Mrc1.

Mrc1-13myc/Pol2-3HA (strain AC2301, Table S1) is the doubly tagged strain used to test coimmunoprecipitation. Whole cell extracts (WCE) were prepared from asynchronous cells as described in Experimental Procedures. Immunoprecipitation (IP) was carried out with the antibody indicated, and proteins in the WCE, the supernatant (SN), and the IP were determined by Western blotting. WB=Western Blot. WCE and SN represent the same number of cell equivalents of protein. In control experiments, 10x excess of SN was also loaded to verify estimated precipitation efficiency.
(A) Coimmunoprecipitation. Lanes 1–6: Mrc1-13myc/Pol2 indicates strain Y1134 carrying myc-tagged Mrc1 and untagged Pol2 and serves as a control that anti-HA does not immunoprecipitate Mrc1 when Pol2 is untagged. Mrc1/Pol2-3HA, strain A1591, serves as a control that anti-myc does not immunoprecipitate Pol2-HA when Mrc1 is untagged. Lanes 7–12: As indicated in the figure, Pol2-3HA was precipitated with anti-HA or Mrc1-13myc was precipitated with anti-myc, and the amount of Pol2 and Mrc1 in the respective immunoprecipitate was determined as described in Experimental Procedures.
(B) Salt and DNase I sensitivity of coimmunoprecipitation. Lanes 1–8, wash buffers contained the amounts of NaCl indicated. Lanes 9–12. Extracts were treated with DNase I before IP. Immunoprecipitation was carried out as in A.
(C) Pol2/Mrc1 interaction as a function of cell cycle stage. Tagged strains indicated at the top of the figure were staged in the cell cycle as described in Experimental Procedures. Asynchronous cells (Asyn), cells arrested with α factor (G1), cells released from α factor-arrest into S phase (S). Immunoprecipitations were carried out with the indicated antibodies. Western blots of the precipitates are shown.
The interaction is strong enough to be observed in the absence of crosslinking and is stable at physiological salt concentrations (Fig. 2B). It is also resistant to DNaseI, and therefore is probably mediated by protein/protein and not protein/DNA interaction.
Because the cells used for these experiments were not synchronized, the fact that all of the Pol2 coimmunoprecipitated with Mrc1 suggested that Pol2 may bind to Mrc1 throughout the cell cycle. To investigate this we used cells arrested at G1 phase with α factor or cells released from α factor and allowed to progress into S phase for 60 min, as determined by flow cytometry. Pol2 and Mrc1 interacted in both G1 and S phase cells (Fig. 2C). Pol2(Dpb2) and Dpb11, a major participant in replication fork assembly and in the S phase checkpoint and Pol2(Dpb2) and GINS also interact in both the presence and absence of DNA synthesis (Masumoto et al., 2000).
To confirm the two hybrid assay result showing that Pol2N and Pol2C interacted independently with Mrc1 by coimmunoprecipitation, we first used a competition binding protocol. We overexpressed untagged Pol2N or Pol2C in an Mrc1-13myc, Pol2-3HA-containing strain. Pol2N and Pol2C each competed with Pol2, reducing the amount of Pol2 that coimmunoprecipitates with Mrc1 and increasing the amount of free Pol2 (Fig. 3A, compare lanes 1–6 with lanes 7–12). Thus, Pol2N and Pol2C can both interact with Mrc1 (see also Fig. 1B, 3D, 5, and Fig. S1B).
Figure 3. Two Independent Interactions between Pol2 and Mrc1.

(A) Competition binding assays show Pol2N and Pol2C bind Mrc1. An anti-myc IP was carried out on strain AC 2301, MRC1-myc/POL2-HA, carrying plasmid pBTM116 or pBTM116 overexpressing either Pol2C or Pol2N as potential competitors for POL2-HA binding to MRC1-myc. Lanes 1–3, no plasmid; lanes 4–6, pBTM116 vector; lanes 7–9, pBTM116 expressing Pol2C; lane 10–12, pBTM116 expressing Pol2N. Blots of IPs in were probed with anti-HA antibody in the upper row and on a separate gel with myc antibody in the lower row.
(B) Interaction of Mrc1 deletion mutants with Pol2. A schematic diagram of the MRC1 gene and the deletion mutants is shown. P’s represent likely Mec1/Ddc2 phosphorylation sites. The indicated N-terminal mrc1 deletion mutants were tagged with FLAG epitope and the C-terminal mrc1 deletion mutants were tagged with HA epitopes (see Experimental Procedures and Table S1). The POL2 gene was tagged with 13myc in strains AC2200-2204 (C-terminal mrc1 deletions) or with 3HA in strains AC2205-2209 (N-terminal mrc1 deletions) by homologous recombination as indicated. Immunoprecipitation was carried out with anti-FLAG antibody for the N-terminal Mrc1 deletions and anti-HA antibody for the C-terminal Mrc1 deletions, and immunoprecipitates were probed with anti-FLAG and anti-HA antibody as indicated. Anti-Pol2-myc or anti-Pol2-HAWestern blots of immunoprecipitates reveal efficiency of Pol2 coimmunoprecipitating with the respective mrc1 mutant proteins.
(C) S phase progression of various mrc1Δ mutants is delayed. Cells were synchronized in G1 phase with α factor and then released into the cell cycle. Samples were taken for flow cytometry analysis at the indicated times.
(D) Mrc1N binds Pol2N and Mrc1C binds Pol2C. Binding studies were carried out as in Figure 2. Strains contained the respective, chromosomally-tagged mrc1 mutant, a wild-type, untagged POL2 chromosomal gene, and HA-Pol2N or HA-Pol2C expressed from a high copy number, ADH-promoter plasmid, pACT2. The species that were immunoprecipitated are indicated on the left and coimmunoprecipitating species detected by Western blotting are indicated on the right of each panel. Rows 1 and 2: mrc1(312-1096-FLAG) carrying pACT2-HA-Pol2N; rows 3 and 4: mrc1(567-1096-FLAG) carrying pACT2-HA-Pol2C; row 5: mrc1-(752-1096-FLAG) carrying pACT2-HA-Pol2C; row 6: mrc1(312-1096-FLAG) carrying pACT2-HA-Pol2N; row 7: mrc1(567-1096-FLAG) carrying pACT2-HA-Pol2N.
Figure 5. Regulation of Pol2/Mrc1 Interaction by Phosphorylation of Mrc1.

(A) Pol2 interacts with Mrc1AQ. Anti-myc IP of strain mrc1AQ-myc/POL2-HA, AC2302. IPs were probed with anti-HA antibody to detect Pol2 as in Figure 2C. The experiment was carried out simultaneously with that shown in Figure 2C.
(B) Pol2 interacts with both nonphosphorylated and phosphorylated Mrc1. Mrc1-13myc and Pol2-3HA coimmunoprecipitate in cells treated with HU. Strain AC2301, MRC1-myc/POL2-HA (lanes 1–3) or strain AC2302, mrc1AQ-myc/POL2-HA (lanes 4–6) were synchronized with α factor and released into fresh medium containing 75 mM HU for 60 min followed by immunoprecipitation with anti-HA antibody (see text for conditions compared). Mrc1-P refers to phosphorylated Mrc1.
(C) Mrc1-P does not interact with Pol2N but does interact with the Pol2-C. Pol2N and Pol2C were each tagged with HA in pACT2, and expressed in an Mrc1-13myc strain (Y1134) in the absence or presence of the indicated amounts of HU. HA-Pol2N or HA-Pol2C was IPed and Mrc1 or Mrc1-P was detected in the IP with α-myc (rows 1–7). WB HA indicates the control for Pol2N and Pol2C IP efficiencies; rows 3, 5, and 7. Rows 8 and 9 are Mrc1-myc IPs. Row 9 is control for Mrc1 IP. The various rows and lanes are described in the text.
Mrc1 contains two independent Pol2 interaction domains
The interaction of Pol2N and Pol2C with Mrc1 suggested that there might also be two Pol2 interaction sites within Mrc1. To test this prediction, a set of C-terminal and N-terminal deletions of MRC1 was generated (Fig. 3B). The deletion mutants are referred to herein by the numbers corresponding to the amino acids contained in the fragments expressed in the mutants. The MRC1 gene was precisely replaced with either C-terminal, FLAG-tagged Mrc1 fragments or N-terminal, HA-tagged Mrc1 fragments. As shown by flow cytometry (Fig. 3C), both N-terminal and C-terminal deletions cause a delay in completion of S phase. mrc1(1–219), mrc1(1–418) and mrc1(1–653) profiles resemble the slow S phase of the mrc1Δ. N-terminal deletions mrc1(312–1096), (567–1096), and (752–1096) all show a delay in entry into S phase and slow appearance of 2C DNA content. The HU sensitivity phenotypes of the mrc1 mutants are shown in Figure S2. Complete characterization of these mutants will be reported elsewhere (Komata and Shirahige, unpublished results).
The POL2 gene in each mrc1-FLAG variant was then tagged with 3HA, and the POL2 gene in each mrc1-HA variant was tagged with 13myc. Mrc1 was immunoprecipitated with either FLAG or HA antibody. The deletion mutant proteins are expressed and immunoprecipitated at equivalent levels (Fig. 3B). The anti-Pol2 Western blots of the immunoprecipitates show that Pol2 coimmunoprecipitates with all N-terminal Mrc1 fragments, although 1–219 interacts more weakly than the others (lanes 1–5). In addition, Pol2 also coimmunoprecipitates with Mrc1 C-terminal fragments, although 567–1096 shows reduced affinity. Binding of full-length Pol2 to both mrc1(1–418) and mrc1(567–1096), which don’t overlap (Fig. 3B, lanes 4 and 9, red), and to mrc1(1–219) and mrc1(318–1096) which also fail to overlap (Fig. 3B, lanes 5 and 8, blue), is consistent with there being an extended zone of Pol2 binding.
N-terminal Sequences in Mrc1 Interact with Pol2N and C-terminal Mrc1 Sequences Interact with Pol2C
To test the extended interaction domain in Mrc1, we first used a competition-binding assay. As shown in Figure 3B, lane 5, full length Pol2 interacts with mrc1(1–219). We found that expression of excess untagged Pol2N or excess full-length untagged Pol2 (Fig. S3, lanes 1–3, lanes 7–9), but not excess untagged Pol2C (lanes 4–6) competes with Pol2-myc for binding to the N-terminal mrc1(1–219)-HA fragment. Thus, Pol2N binds mrc1(1–219) and Pol2C does not.
We then showed by direct immunoprecipitation that HA-Pol2N binds mrc1(312–1096)-FLAG but not to mrc1(567–1096)-FLAG (Fig. 3D, rows 1–5). Thus, C terminal sequences beyond 567 are not required for Pol2N binding. Conversely, HA-Pol2C binds mrc1(567–1096)-FLAG and, barely detectably, even to mrc1(752–1096)-FLAG (Fig. 3D, rows 6 and 7). The stronger interaction of mrc1(567–1096) with Pol2C found here compared to interaction with full-length Pol2 in Figure 3B is likely due to the overexpression of HA-Pol2C. [We do not yet understand the cross-reacting band just below Pol2 that appears only in strains carrying the mutants shown in Figure 3D, rows 6 and 7, but Figure 3B shows that even full length Pol2 barely interacts with mrc1(752–1096)]. From these data we can conclude that the Pol2N catalytic domain binds the N-terminal, phosphorylation site-rich half of Mrc1 and that Pol2C binds the C-terminal half of Mrc1.
The Pol2/Mrc1 Interaction is Functional
To obtain evidence that Mrc1 and Pol2 interact functionally, we asked whether Mrc1 was required for the viability of various pol2 mutants or for their survival in the presence of various DNA synthesis inhibitors. pol2-16, lacking the entire N-terminal nuclease/polymerase domain of Pol2 (residues 176–1134), is both DNA replication and checkpoint proficient, although replication forks move slowly and pol2-16 fails to grow at 37°C (Dua et al., 1999; Kesti et al., 1999; Navas et al., 1995; Ohya et al., 2002). pol2-16 associates with origins of replication in S phase and is as efficient in associating with Dpb2, Dpb3, and Dpb4 subunits as Pol2 itself. Therefore, its essential function is likely in DNA replication. As tabulated in Figure 4A, MRC1 is required for the growth of pol2-16. In contrast, strain pol2-16 mrc1AQ is viable, showing that Mrc1 checkpoint mediator function is not required for pol2-16 viability. Thus, interaction of Mrc1 with Pol2C (1135–2222) may play an important role in DNA replication or some other essential process that does not require phosphorylation. pol2-16 rad9Δ is also viable.
Figure 4. Genetic interactions between POL2 and MRC1.
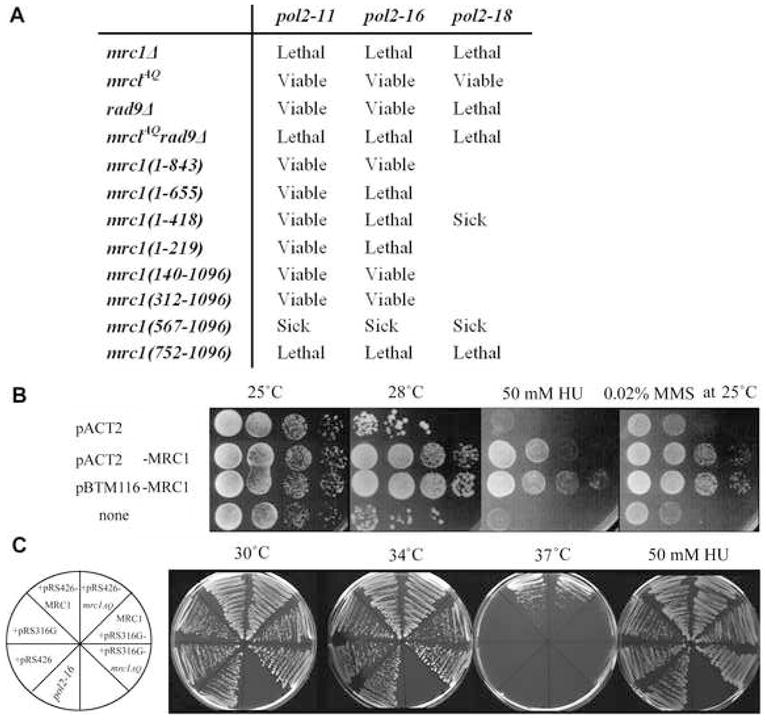
(A) Phenotypes of various pol2 mrc1 double mutants. These results were obtained using two complementary methods for creating and analyzing double mutants, as described in detail in Experimental Procedures. Strain genotypes are shown in Table S1.
(B) Suppression of pol2-11 by MRC1 overexpression. 10-fold serial dilutions of exponentially growing cultures of pol2-11 cells transformed with plasmids expressing MRC1 (pACT2-MRC1, pBTM116-MRC1) or control pol2-11 carrying empty pACT2 or without plasmid were plated and grown at different temperatures or in the presence of HU or MMS as indicated.
(C) Suppression of pol2-16 by MRC1 Overexpression. Plates streaked from a single colony of strain pol2-16 transformed with plasmids indicated on the left (Table S2) were grown at different temperatures or in the presence of HU as indicated.
To identify the region of Mrc1 required to support pol2-16 growth, we determined which of the mrc1 C- and N-terminal deletion mutants was viable in combination with pol2-16. The results in Figure 4A show that mrc1(1–843) pol2-16 double mutants are viable but mrc1(1–218) pol2-16, mrc1(1–418) pol2-16, and mrc1(1–655) pol2-16 are inviable, suggesting that the region of Mrc1 between 655 and 843 is important for the replication function of pol2-16. Supporting this interpretation, a double mutant containing both the pol2-16 allele and mrc1(567–1096), which does interact with Pol2C, is viable, but pol2-16 mrc1(752-1096), which barely interacts with Pol2C (Fig. 3D) is inviable. Thus, the ability of Mrc1C to interact strongly with Pol2C correlates with the ability to support Pol2C-mediated roles in DNA replication in pol2-16. Therefore the Mrc1C interactions reported in Figure 3 may be functional at the replication fork in vivo.
We also tested whether the Pol2 C-terminal point mutant pol2-11 and the pol2 partial deletion mutants shown in Figure 1A required Mrc1 for viability (Fig. 4 and S4). Mutant pol2-11 carries a stop codon at residue 2196 and, unlike pol2-16, is both replication and checkpoint deficient, similar to mrc1 null mutants (Budd and Campbell, 1993; Dua et al., 1998; Frei and Gasser, 2000; Navas et al., 1995). The pol2-11 mrc1AQ double mutant is no more sensitive to the S phase checkpoint inducers HU and MMS (methyl methane sulfonate) than pol2-11 (Fig. S4). This suggests that mrc1AQ and pol2-11 are epistatic for the S phase checkpoint. However, pol2-11 mrc1Δ was found to be inviable, suggesting but not proving that Mrc1 is important for replication in the pol2-11 mutant (Fig. 4A).
Deletion of Pol2C is lethal, and therefore the role of the Mrc1N/Pol2N interaction in maintaining cell viability could not be tested in the absence of Pol2C. However, pol2-18, a temperature-sensitive mutant that retains high viability in HU, has a mutation in the N-terminal catalytic domain, Pro710Ser (Araki et al., 1992; Navas et al., 1995). The roles of Pol2C and Pol2N in vivo are partially independent, since pol2-18 and pol2-11 show partial interallelic complementation, as do pol2-18 and pol2-F, another C-terminal mutant (Dua et al., 1998; Navas et al., 1995). pol2-18 mrc1Δ is inviable, suggesting that Mrc1N/Pol2N interaction may be functional (Fig. 4A). The poor growth and lethality of pol2-18 mrc1(567-1096) and pol2-18 mrc1(752-1096) support this idea. pol2-18 mrc1AQ is viable, suggesting phosphorylation is not required to correct the pol2 defect, but that Mrc1 is providing some other function.
We also tested whether plasmids overexpressing MRC1 could suppress the replication and/or replication stress response defects, including HU and MMS sensitivity, of the pol2-16 and pol2-11 mutants. As shown in Figure 4B and 4C, respectively, overexpressing MRC1 ameliorates the temperature sensitivity and DNA damage sensitivity of pol2-11 and both MRC1 and mrc1AQ suppress pol2-16 growth and weak HU sensitivity. These studies show a functional in vivo interaction between Mrc1 and Pol2, most likely in DNA replication, since Mrc1 phosphorylation is dispensable.
Role of the Mrc1/Pol2 Interaction in the S phase Checkpoint: Effect of S/TQ Phosphorylation on Mrc1
Having provided evidence that Mrc1 and Pol2 interact during DNA replication (strictly speaking when Mrc1 is not phosphorylated) we wished to determine if their interaction played any role in the S phase checkpoint. Two hybrid assays showed that mrc1AQ interacted robustly with POL2C (Fig. 1B). Mrc1AQ was also found to coimmunoprecipitate efficiently with Pol2 (Fig. 5A), confirming that phosphorylation is not essential for the Pol2/Mrc1 interaction.
This raised the question of whether phosphorylated Mrc1 binds to Pol2 after checkpoint activation. Mrc1 becomes hyperphosphorylated in cells treated with HU, and this phosphorylation results in a Mec1/Ddc2-dependent reduced mobility on SDS gels (Fig. 5B, lanes 1–3, note Mrc1 doublet) (Osborn and Elledge, 2003). Clearly, both phosphorylated and unphosphorylated Mrc1 coimmunoprecipitate with Pol2. The non-phosphorylatable Mrc1AQ also associates with Pol2 in the presence of HU (Fig. 5B, lanes 4–6). Since phosphorylated Mrc1 can interact with Pol2, this suggests that Pol2 and Mrc1 continue to interact physically during the checkpoint cascade.
Phosphorylation of Mrc1 Abolishes Interaction with Pol2N but not with Pol2C, Suggesting a Role for Pol2N in Checkpoint Responses
Although phosphorylation of Mrc1 did not reduce binding of Mrc1 to full-length Pol2, we wished to determine if phosphorylation of Mrc1 might affect binding to either of the individual Pol2 halves. When either HA-tagged Pol2N or HA-tagged Pol2C was expressed from a high copy plasmid, pACT2, under the control of the strong ADH promoter, the respective tagged Pol2 domain coimmunoprecipitated with endogenous Mrc1-13myc (Fig. 5C, row 1, lanes 6 and 12), confirming that Pol2N and Pol2C interact independently with Mrc1. (The untagged, chromosomal POL2 gene is present to preserve viability.) Mrc1 in extracts of cells treated with 150 mM HU migrated more slowly than Mrc1 in untreated cells, presumably due to phosphorylation (Fig. 5C, row 1, and compare lanes 1–6 with 7, 8 or lanes 10–12 with 13–15). Strikingly, HA-Pol2N fails completely to interact with Mrc1-P (Fig. 5C, row 1, lane 9, and row 2, long exposure), while HA-Pol2C binds Mrc1-P avidly (Pol2C IP, row 1, lanes 13–15; Mrc1 IP, rows 8 and 9, lanes 13–15).
By reducing the levels of HU to either 100 mM HU or to 75 mM HU, we could study the interaction where only a fraction of the Mrc1 was phosphorylated, and both Mrc1 and Mrc1-P were present in the same cells. We showed clearly that HA-Pol2N binds non-phosphorylated Mrc1 (faster migrating) but not Mrc1-P, which remains in the supernatant (Fig. 5C, row 4 and row 6, lanes 7–9). Conversely, both Mrc1 and Mrc1-P species bind to HA-Pol2C in these cells (Fig. 5C, row 4 and row 6, lanes 13–15).
Therefore, phosphorylation of Mrc1 affects binding to the N- and C-terminal halves of Pol2 differentially. Mrc1-P is released from the Pol2N “catalytic” domain but remains bound to the Pol2C “structural” domain. These results suggest a role for the Mrc1N-Pol2N interaction in the checkpoint pathway.
Deletion of Mrc1 destabilizes Pol2 at replication forks stalled in the presence of HU
To test the idea that Mrc1 affects Pol2 at replication forks during the checkpoint, we carried out chromatin immunoprecipitation experiments using an oligonucleotide array capable of analyzing protein binding to yeast chromosome VI at 600 bp resolution (Katou et al., 2003). As shown in Fig. 6A, Pol2 is stable at replication forks after 60 minutes of treatment with HU in wild-type cells. In an mrc1Δ, however, Pol2 is clearly destabilized. It has been previously demonstrated that Tof1, which is found in a complex with Mrc1 and Csm3, is required for loading of Mrc1 onto chromatin (Katou et al., 2003). If the destabilization of Pol2 in the mrc1Δ was due to lack of Mrc1 at the replication fork, then we expected that Pol2 should also be destabilized in a tof1Δ. As shown in Fig. 6A, this is the case. We conclude that the Pol2 interaction at the fork is destabilized in the absence of Mrc1. Pol2 may have spread out on the chromatin such that it is undetectable or may have dissociated altogether; however, unlike other replication proteins previously tested (Katou et al., 2003), it is not associated with the replication fork.
Figure 6. Mrc1 stabilizes Pol2 at stalled forks and connects it with MCM complex.
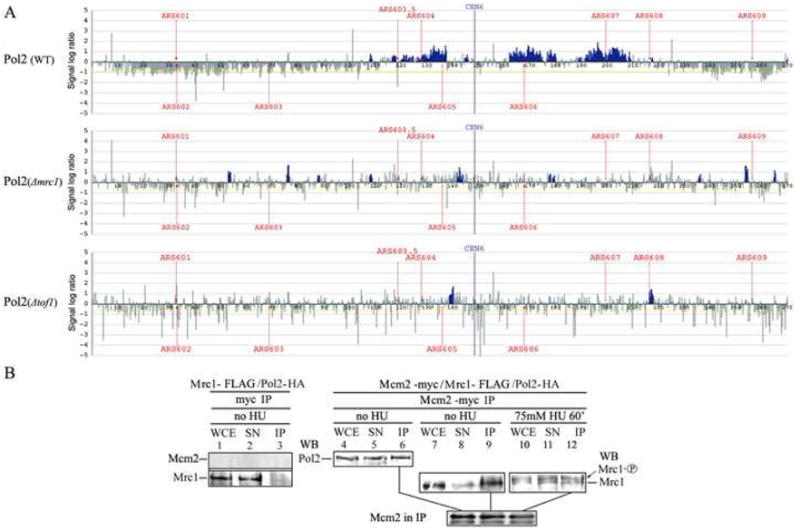
(A) Mrc1 stabilizes Pol2 at the replication fork in cells treated with HU. Wild-type, mrc1Δ or tof1Δ strains were synchronized with α factor and released from pheromone arrest in the presence of 200 mM HU. After 60 min, chromatin immunoprecipitation analysis was carried out as described previously. The blue histograms represent Pol2 enrichment. The vertical axis is enrichment on a log2 scale. The horizontal axis is length in kb.
(B) Mrc1, Pol2, and the MCMs interact in single complex. Coimmunoprecipitation of Mrc1, Pol2, and Mcm2. Mcm2-13myc/Mrc1-FLAG/Pol2-3HA triple tagged strains were used for coimmunoprecipitation (strains listed in Table S1). Whole cell extracts (WCE) were prepared from asynchronous cells (Asyn), cells arrested with α factor (G1), or cells released from α factor into S phase in the presence of HU treatment (HU). Immunoprecipitation was carried out as described in the legend to Figure 3 and Experimental Procedures. Lanes 1–3 show a control strain with untagged Mcm2. Lanes 1–3, top row, show that Mcm2 was not immunoprecipitated by the myc antibody. Lanes 1–3, bottom row, show that the Mrc1-FLAG did not immunoprecipitate with the myc antibody in the absence of myc-tagged Mcm2. Control showing that Pol2-HA does not coimmunoprecipitate with myc antibody is shown in Fig. 2A, lane 6.
Coimmunoprecipitation of Mrc1, Pol2, and Mcm2
Mrc1 coimmunoprecipitates with Cdc45 and copurifies with MCM-CDC45 (Gambus et al., 2006; Katou et al., 2003), and it has been proposed that Mrc1 couples unwinding by the MCM helicase and DNA synthesis (Szyjka et al., 2005). Our results are compatible with coupling, at least on the leading strand, occurring through Pol2, so we asked whether both Mrc1 and Pol2 coimmunoprecipitated with Mcm2. We combined Mcm2-13myc, Pol2-3HA, and Mrc1-6FLAG in the same strain, all expressed from their native promoters, and carried out a Mcm2-myc immunoprecipitate with anti-myc antibody (Fig. 6B). Both Mrc1 and Pol2 were recovered in the Mcm2-immunoprecipitate. This result makes it possible that all three proteins interact simultaneously, although the experimental design does not distinguish between the existence of a single complex with all three proteins and two different Mcm2/Mrc1 and Mcm2/Pol2 complexes. We then carried out the same experiments in HU-treated cells. Mcm2 coimmunoprecipitates with both phosphorylated and unphosphorylated Mrc1. The interaction between Mcm2 and Mrc1-P is an important and novel result showing that Mrc1 interacts with the MCM complex not only during the normal cell cycle but also during the checkpoint, as it does with Pol2 (Fig. 3 and 5).
DISCUSSION
Our results provide a new perspective on Mrc1, on pol ε and on sensing and transmission of signals in the S phase checkpoint. Our results imply at the molecular level how Mrc1 could mediate by a common mechanism, i.e., coupling between helicase and polymerase, its two apparently disparate functions, namely stimulation of replication during S phase and inhibition of replication during stress.
The Role of Mrc1 in DNA Replication
We have shown that all of the Pol2 in the cell is associated with Mrc1. This stoichiometry alone suggests that Mrc1 is very important for all of the roles played by pol ε. The suppression of pol2-11 and pol2-16 phenotypes by overexpression of Mrc1 suggests that Mrc1 either stabilizes pol ε directly or a complex containing pol ε. Since Mrc1 is not essential, it cannot be essential for wild-type pol ε at replication forks. We showed, however, that interaction between Mrc1C and Pol2C is likely important for the essential function of Pol2 and for the replication function of Mrc1, since non-phosphorylatable Mrc1 can support the viability of a number of different C-terminal pol2 mutants. Cryoelectron microscope reconstruction of the pol ε holoenzyme suggests that Pol2C sits as a tail on the globular Pol2N domain (Asturias et al., 2006 and references therein). Thus, Pol2C likely exerts its essential but non-catalytic role as a scaffold for Dpb2, Dpb3, and Dpb4, which together appear to be involved in guiding the primer/template into the Pol2N active site (Fig. 7A). The essential function for the Pol2 C-terminal scaffold in pol2-16 mutants suggests that Pol2C is also a mainstay of the entire replisome (Dua et al., 1999). Mrc1 might therefore enhance the function of the Pol2 C-terminal domain in maintaining a stable replisome.
Figure 7. Models of Functional Mrc1/Pol2 Interaction on the Leading Strand.

(A) Interaction during normal DNA replication. The shape of pol ε depicted is derived from reference (Asturias et al., 2006). The interactions between Mrc1 and Pol2 described in this study are incorporated into a rudimentary model for polymerase/helicase coupling during normal DNA replication (see text).
(B) Mrc1/Pol2 interaction on the leading strand during replication stress. Pol ε/Mrc1 senses the damage, phosphorylation of Mrc1 releases the N terminus, but there is continued interaction between Mrc1-P with the C terminus of Pol2.
In addition to binding the C-terminal half of Pol2, the interaction of Mrc1 and Pol2 extends over the entire protein. Mrc1 also interacts with the polymerase/exonuclease domain, suggesting that Mrc1 may also serve as an accessory factor in polymerization by Pol2. It has been proposed that Mrc1 binds to DNA, and Mrc1 might affect the association of pol ε with the primer terminus. Our flow cytometric analysis clearly shows that deletions within the Mrc1N with Pol2 do have S phase defects. Additionally, Mrc1N might help recruit, make the primer accessible to, or stabilize another DNA polymerase, for instance one that compensates for the absence of the Pol2 polymerase in pol2-16, such as pol δ, or that participates in DNA damage repair or by-pass. pol2-16 is synthetically lethal with cdc2-1, the catalytic subunit of pol δ pol32Δ, a non-essential subunit of pol δ, and pol3-01, a mutant defective in the exonuclease proofreading domain of pol δ. A reservation in deducing that Pol2/Mrc1 interaction functions during normal DNA replication is that we have only shown that the interaction occurs in S phase and not that it occurs on chromatin. Since all of the Pol2 coimmunoprecipitates with Mrc1 in S phase, however, at least some interaction likely occurs on the DNA.
Polymerase/Helicase Coupling
Coimmunoprecipitation between Mrc1, Pol2, and MCM2 (Fig. 6) during a normal S phase and between Mrc1 and MCM2 during HU treatment, i.e., during the checkpoint, are consistent with the proposal that Mrc1 might enhance interaction between Pol2 and the replication helicase. A fraction of the cellular pol ε pool has also been shown by others to coimmunoprecipitate with Cdc45, and this is specific to pol ε since pol α does not associate with Cdc45 (Zou and Stillman, 2000). These results provide molecular evidence for the proposal that Mrc1 may be involved in coupling polymerase and helicase function, originally proposed based on observed functional uncoupling of DNA synthesis and unwinding in mrc1 null strains in the presence of replication stress (Szyjka et al., 2005). GINS, a third component of the RPC, is almost certainly also involved in coupling, and the association of Mrc1 with the RPC also indirectly supports a role for Mrc1 in coupling. A model for Mrc1/Pol2/MCM coupling is shown in Figure 7A. A parallel coupling model has been proposed for the lagging strand in which Mcm10 and And-1/Ctf4 have been proposed to link pol α and the MCM helicase (Zhu et al., 2007).
At the prokaryotic replication fork, polymerase/helicase coupling is both required for maximum elongation rates and also appears to be at the root of a replisome in which the polymerases (are programmed to?) transiently dissociate from the primer/template and yet remain associated with the moving fork through interaction with the other proteins at the fork. This “dynamic processivity” is accomplished by direct interaction of the polymerase with the helicase in phage T7, indirect polymerase/helicase interaction mediated through the clamp-loader component of the pol III holoenzyme in E. coli, and possibly by interaction with the clamp subunits in phage T4. It is easy to envision how this transient mode of interaction of the polymerases with the primer terminus but processive interaction with the fork provides for release and rapid recycling of polymerases to new primers on the lagging strand as well as for rapid exchange to “reserve” polymerases when damage to the template or to the engaged polymerase or barriers in the template are encountered. This also provides a new way to view the coordination of leading and lagging strands (Lovett, 2007). Indirect evidence for replicative DNA polymerase interchangeability in eukaryotes (between an actively synthesizing polymerase and a “reserve” polymerase) is provided by the pol2-16 mutant, in which a second polymerase, presumably pol δ, can fulfill the role of the missing polymerase of pol ε on the leading strand (Dua et al., 1999).
S Phase Checkpoint Function of the Mrc1/Pol2 Interaction
Mrc1 was first described as a mediator between the signaling and effector kinases, Mec1 and Rad53, and as such regulates the global checkpoint responses, such as slowing of the cell cycle, induction of transcription, and inhibition of late origin firing. Our results suggest Mrc1 might also be involved in sensing and additional signaling mechanisms. Association with Pol2N places Mrc1N near the polymerase active site, where it might sense replication fork blocks generating a checkpoint signal (Fig. 7B). When Mrc1N is phosphorylated, presumably by Mec1, in response to replication stress (HU addition), Mrc1N dissociates from Pol2N (Fig. 5). One possible consequence of this dissociation might be slowing of the replication fork. In this case, Mrc1 would itself be an effector of the S phase checkpoint. Release could also allow Mrc1-P to interact with another protein, perhaps an MCM component or Cdc45, at the replication fork, inhibiting fork movement.
Another consequence of the release of the phosphorylated Mrc1 N terminus might be recruitment, stabilization, and/or activation of Rad53, in turn activating the downstream checkpoint events. Alternatively, the release of Mrc1N might cause a conformational change in Pol2C that leads to activation of the checkpoint through the C-terminal checkpoint domain. If so, then Pol2N may constitute the elusive suppressor of checkpoint activation postulated to operate during normal, unimpeded DNA replication. In sum, the results suggest a molecular mechanism by which DNA damage on the leading strand can be sensed and the signal amplified through activation of downstream kinases.
Concomitant with sensing and signaling through its bimodal N-terminal interactions with Pol2N, Mrc1 might be involved in stabilizing the fork through its continued interaction with Pol2C during the checkpoint, in preparation for replication restart. Stabilization of the replication forks is often considered the most important function of Mec1 (Pasero et al., 2003 for review). Mrc1C might be acting to anchor Pol2C to the helicase and to the primer/template junction during the checkpoint. In the complete absence of Mrc1, both Pol2N and Pol2C may have weakened interaction with primer/template. Unwinding can still occur but synthesis ceases (Nedelcheva et al., 2005).
A critical finding is that Pol2, which is associated with HU-stalled replication forks in wild-type cells, is destabilized at stalled forks in mutants lacking Mrc1 or in mutants where Mrc1 fails to associate with DNA (Fig. 6A). In this sense Pol2 is exceptional, since in previous studies most replication proteins tested were shown to be uncoupled from the site of DNA synthesis but remained colocalized on the chromatin at a distal site (Katou et al., 2003). Unlike Pol2, Dpb3, a subunit of pol ε, remains with the replisome in the mrc1 Δ mutant in the presence of HU (Katou et al., 2003). This discrepancy could be explained by the fact that Dpb3 and Dpb4, histone fold-containing proteins encoded by non-essential genes (Iida and Araki, 2004), form a subcomplex independent of Pol2/Dpb2 (Dua et al., 2000; Dua et al., 2002). The Dpb3/Dpb4 complex may be stabilized by interactions with fork components or chromatin components other than Mrc1. Dpb3 and Dpb4 may also be components of chromatin remodeling complexes (Iida and Araki, 2004). Pol2 association with ARS607 is also significantly reduced in mrc1Δ compared to wild-type after 60 minutes of HU treatment in an independent study (Bjergbaek et al., 2005).
The Pol2/Mrc1 interactions described here may be conserved in metazoans. Metazoan Claspin is a functional Mrc1 ortholog. An evolutionary comparison of Mrc1/Claspin structure, as well as a summary of the interaction data presented in this paper, are shown in Figure S5, emphasizing that although the functions are conserved, the overall organization of the proteins differ. Similar to our observations in yeast, pol ε is associated with Claspin immunoprecipitated from aphidicolin-treated Xenopus egg extract chromatin eluates (Lee et al., 2005). Furthermore, hyperunwinding that involves uncoupling of the polymerase and the helicase has been observed in Xenopus in the presence of aphidicolin, a polymerase inhibitor (Byun et al., 2005; Walter and Newport, 2000).
EXPERIMENTAL PROCEDURES
Yeast Strains and Plasmids
Strains, plasmids, and oligonucleotides used in this study are listed in Tables S1-S3.
Analysis of Double Mutants
Viability of double mutants carrying various MRC1 alleles and either pol2-11, pol2-16, or pol2-18 alleles was determined by two methods. First, spores from standard crosses and tetrad dissection were incubated and genotyped at 23°C. Any combination failing to generate viable double mutants was deemed synthetically lethal. At least 12 tetrads were dissected for each double mutant, and usually more were dissected. Second, POL2/pol2 and MRC1/mrc1 heterozygous diploids carrying plasmid encoded MRC1-URA3 (cloned in pRS416) were generated for each pol2 and mrc1 mutant. mrc1 pol2 double mutants were identified after tetrad dissection and then tested for viability in the absence of MRC1-URA3 by plating on 5-FOA.
Construction of Mrc1 Deletion Mutants
PCR products of the Mrc1 promoter region (300 bp upstream of the MRC1 gene) were obtained using specific primers, as indicated in Table S3. The PCR product was digested with BglII and PacI and ligated into BglII/PacI digested pFA6a-His3MX6-PGAL thus replacing PGAL1 with the Mrc1 promoter. Mrc1 N-terminal deletion mutants were generated using this plasmid and Mrc1 FLAG strain which was deleted 200 bp upstream of the MRC1 gene with URA3 marker. Mrc1 C terminal deletions were constructed using the primers indicated in the Mrc1-HA strain.
Cell Synchronization
Strains were arrested in G1 phase by supplementing the media of exponential cultures with 10 μg/ml αfactor. Synchronized cells were released from G1 into fresh YPD and growth continued for 60 min, after which all cells showed S phase DNA content by flow cytometry (Reis and Campbell, 2007).
Immunoprecipitation Assays
Coimmunoprecipitation analysis of wild-type or mutant Mrc1 with Pol2 was performed using strains coexpressing a 13myc-tagged version of Mrc1 and 3HA tagged version of Pol2. Other tags employed for specific experiments are described in the legends to the figures. Samples (2.0×109 cells) were collected by centrifugation and the extract was prepared as previously described. The cells were crushed by glass beads in 180 μl lysis buffer [45 mM Hepes-KOH pH 7.2, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 0.2% NP40] containing protease inhibitors [1% Protease Inhibitor Cocktail (Sigma) and 1 mM PMSF] and cleared by centrifugation (WCE). WCE total protein (6 mg) was incubated with 6 μg of antibody indicated in the figures for 3h at 4°C. anti-myc antibody was monoclonal 9E10 and anti-HA antibody was 12CA5. anti-FLAG antibody was from Sigma Aldrich. Precipitation was carried out with pre-washed protein G beads (GE Healthcare) for 3h at 4°C. The supernatant was recovered and washed 4x with lysis buffer (or with increasing NaCl concentrations where indicated). To degrade chromosomal DNA in cell extracts, DNase I (Worthington) was added to a final concentration of 200 units per ml and incubated for 1–2h at 4 °C before IP. DNA degradation was confirmed by agarose gel electrophoresis and EtBr staining.
Antibody-bound fractions were suspended in a minimal amount of SDS loading buffer (62.5 mM Tris–HCl pH 6.8, 2% sodium lauryl sulfate, 10% glycerol, 5% 2-mercaptethanol) and half of the sample was analyzed by Western blotting using an ECL detection reagent (Amersham). WCE and SN (immunoprecipitate supernatant), both corresponding to approximately 100 μg total protein, were also analyzed.
Competition binding assays were carried out using the same strains as standard coimmunoprecipitations, except that strains contained plasmids (pBTM116) overexpressing untagged Pol2, Pol2N, or Pol2C. To determine binding of Pol2N and Pol2C to Mrc1 directly, Mrc1 tagged strains were transformed with pACT2 plasmids overexpressing HA-tagged Pol2, Pol2N, and Pol2C. Pol2C (residues 1265–2222) carrying a series of 10 amino acid deletions in the C-terminal 120 amino acids as well as the pol2-11 mutant were cloned into pACT2, which overexpresses HA tagged versions of the mutant proteins (Dua et al., 2000; Dua et al., 1999; Dua et al., 1998). These constructs were introduced into an Mrc1-13myc strain. We then carried out myc IPs to detect interaction between Mrc1 and pol2-HA mutant proteins.
(ChIP)-chip Assays
The oligonucleotide arrays of chromosome VI with 300 nucleotide resolution, chromatin immunoprecipitation procedures, internal controls, and strains used have been described (Katou et al., 2003). Strains contained Pol2-3HA.
Data in this paper can be obtained from GEO (http://www.ncbi.nlm.nih.gov/geo) with accession number GSE12138.
Supplementary Material
Acknowledgments
We thank Stephen Elledge and Oscar Aparicio for strains. This work is dedicated to a gifted teacher, Ernest Russ. This work was supported by MEXT Japan, Grant-in-Aid for Scientific Research on Priority Areas, “Chromosome Cycle” to K.S. and M.K., GCOE program from MEXT Japan to Y.K., a CIRM fellowship to HL and NIH GM25508 and NIH GM087666 to JLC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJH, Boussett K, Furuya K, Diffley JFX, Carr AM, Elledge SJ. Mrc1 transduces signals of DNA replication stress to activiate Rad53. Nature Cell Biol. 2001;3:958–965. doi: 10.1038/ncb1101-958. [DOI] [PubMed] [Google Scholar]
- Aparicio OM, Weinstein DM, Bell S. Components and dynamics of DNA replication complexes in S. cerevisiae: Redistribution of MCM proteins and Cdc45p during S phase. Cell. 1997;91:59–69. doi: 10.1016/s0092-8674(01)80009-x. [DOI] [PubMed] [Google Scholar]
- Araki H, Leem SH, Amornrat P, Sugino A. Dpb11, which interacts with DNA polymerase II(ε) in Saccharomyces cerevisiae, has a dual role in S-phase progression and at a cell cycle checkpoint. Proc Natl Acad Sci USA. 1995;92:11791–11795. doi: 10.1073/pnas.92.25.11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki H, Ropp PA, Johnson AL, Johnston LH, Morrison A, Sugino A. DNA polymerase II, the probable homolog of mammalian DNA polymerase ε, replicates chromosomal DNA in the yeast Saccharomyces cerevisiae. EMBO J. 1992;11:733–740. doi: 10.1002/j.1460-2075.1992.tb05106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asturias FJ, Cheung IK, Sabouri N, Chilkova O, Wepplo D, Johansson E. Structure of Saccharomyces cerevisiae DNA polymerase epsilon by cryo-electron microscopy. Nat Struct Mol Biol. 2006;13:35–43. doi: 10.1038/nsmb1040. [DOI] [PubMed] [Google Scholar]
- Azvolinsky A, Dunaway S, Torres JZ, Bessler JB, Zakian VA. The S. cerevisiae Rrm3p DNA helicase moves with the replication fork and affects replication of all yeast chromosomes. Genes Dev. 2006;20:3104–3116. doi: 10.1101/gad.1478906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjergbaek L, Cobb JA, Tsai-Pflugfelder M, Gasser SM. Mechanistically distinct roles for Sgs1p in checkpoint activation and replication fork maintenance. Embo J. 2005;24:405–417. doi: 10.1038/sj.emboj.7600511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd ME, Campbell JL. DNA polymerases δ and ε are required for chromosomal replication in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:496–505. doi: 10.1128/mcb.13.1.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzada A, Hodgson B, Kanemaki M, Bueno A, Labib K. Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev. 2005;19:1905–1919. doi: 10.1101/gad.337205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dua R, Edwards S, Levy DL, Campbell JL. Subunit interactions within the Saccharomyces cerevisiae DNA polymerase epsilon (pol ε) complex- demonstration of a dimeric pol ε. J Biol Chem. 2000;275:28816–28825. doi: 10.1074/jbc.M002376200. [DOI] [PubMed] [Google Scholar]
- Dua R, Levy D, Campbell JL. Analysis of the essential functions of the C-terminal protein/protein interaction domain of Saccharomyces cerevisiae pol ε and its unexpected ability to support growth in the absence of the DNA polymerase domain. J Biol Chem. 1999;274:22283–22288. doi: 10.1074/jbc.274.32.22283. [DOI] [PubMed] [Google Scholar]
- Dua R, Levy DL, Campbell JL. Role of the putative zinc finger domain of Saccharomyces cerevisiae DNA polymerase ε in DNA replication and the S/M checkpoint pathway. J Biol Chem. 1998;273:30046–30055. doi: 10.1074/jbc.273.45.30046. [DOI] [PubMed] [Google Scholar]
- Dua R, Levy DL, Li CX, Snow PM, Campbell JL. In vivo reconstitution of Saccharomyces cerevisiae DNA polymerase ε in insect cells: Purification and characterization. J Biol Chem. 2002;277:7889–7896. doi: 10.1074/jbc.M108546200. [DOI] [PubMed] [Google Scholar]
- Edwards SE, Li CX, Levy DL, Brown J, Snow PM, Campbell JL. Saccharomyces cervisiae DNA polymerase epsilon and polymerase sigma interact physically and functionally, suggesting a role for polymerase epsilon in sister chromatid cohesion. Mol Cell Biol. 2003;23:2733–2748. doi: 10.1128/MCB.23.8.2733-2748.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei C, Gasser SM. The yeast Sgs1p helicase acts upstream of Rad53p in the DNA replication checkpoint and colocalizes with Rad53p in S-phase-specific foci. Genes Dev. 2000;14:81–96. [PMC free article] [PubMed] [Google Scholar]
- Gambus A, Jones RC, Sanchez-Diaz A, Kanemaki M, van Deursen F, Edmondson RD, Labib K. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat Cell Biol. 2006;8:358–366. doi: 10.1038/ncb1382. [DOI] [PubMed] [Google Scholar]
- Iida T, Araki H. Noncompetitive Counteractions of DNA Polymerase {varepsilon} and ISW2/yCHRAC for Epigenetic Inheritance of Telomere Position Effect in Saccharomyces cerevisiae. Mol Cell Biol. 2004;24:217–227. doi: 10.1128/MCB.24.1.217-227.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katou Y, Kanoh Y, Bando M, Noguchi H, Tanaka H, Ashikari T, Sugimoto K, Shirahige K. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature. 2003;424:1078–1083. doi: 10.1038/nature01900. [DOI] [PubMed] [Google Scholar]
- Kawasaki YY, Sugino AA. Yeast replicative DNA polymerases and their role at the replication fork. Molecules and cells. 2001;12:277–285. [PubMed] [Google Scholar]
- Kesti T, Flick K, Keranen S, Syvaoja JE, Wittenberg C. DNA polymerase epsilon catalytic domains are dispensable for DNA replication, DNA repair, and cell viability. Mol Cell. 1999;3:679–685. doi: 10.1016/s1097-2765(00)80361-5. [DOI] [PubMed] [Google Scholar]
- Lee J, Gold DA, Shevchenko A, Shevchenko A, Dunphy WG. Roles of replication fork-interacting and Chk1-activating domains from Claspin in a DNA replication checkpoint response. Mol Biol Cell. 2005;16:5269–5282. doi: 10.1091/mbc.E05-07-0671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett ST. Polymerase switching in DNA replication. Mol Cell. 2007;27:523–526. doi: 10.1016/j.molcel.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Masumoto H, Sugino A, Araki H. Dpb11 controls the association between DNA polymerases alpha and varepsilon and the autonomously replicating sequence region of budding yeast. Mol Cell Biol. 2000;20:2809–2817. doi: 10.1128/mcb.20.8.2809-2817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison A, Araki H, Clark AB, Hamatake RK, Sugino A. A third essential DNA polymerase in S. cerevisiae. Cell. 1990;62:1143–1151. doi: 10.1016/0092-8674(90)90391-q. [DOI] [PubMed] [Google Scholar]
- Navas TA, Sanchez Y, Elledge SJ. RAD9 and DNA polymerase epsilon form parallel sensory branches for transducing the DNA damage checkpoint signal in Saccharomyces cerevisiae. Genes Dev. 1996;10:2632–2643. doi: 10.1101/gad.10.20.2632. [DOI] [PubMed] [Google Scholar]
- Navas TA, Zhou Z, Elledge SJ. DNA polymerase epsilon links the DNA replication machinery to the S phase checkpoint. Cell. 1995;80:29–39. doi: 10.1016/0092-8674(95)90448-4. [DOI] [PubMed] [Google Scholar]
- Nedelcheva MN, Roguev A, Dolapchiev LB, Shevchenko A, Taskov HB, Shevchenko A, Stewart AF, Stoynov SS. Uncoupling of unwinding from DNA synthesis implies regulation of MCM helicase by Tof1/Mrc1/Csm3 checkpoint complex. J Mol Biol. 2005;347:509–521. doi: 10.1016/j.jmb.2005.01.041. [DOI] [PubMed] [Google Scholar]
- Nitani N, Nakamura K, Nakagawa C, Masukata H, Nakagawa T. Regulation of DNA replication machinery by Mrc1 in fission yeast. Genetics. 2006;174:155–165. doi: 10.1534/genetics.106.060053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohya T, Kawasaki Y, Hiraga S, Kanbara S, Nakajo K, Nakashima N, Suzuki A, Sugino A. The DNA polymerase domain of pol(epsilon) is required for rapid, efficient, and highly accurate chromosomal DNA replication, telomere length maintenance, and normal cell senescence in Saccharomyces cerevisiae. J Biol Chem. 2002;277:28099–28108. doi: 10.1074/jbc.M111573200. [DOI] [PubMed] [Google Scholar]
- Osborn AJ, Elledge SJ. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 2003;17:1755–1767. doi: 10.1101/gad.1098303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasero PP, Shimada KK, Duncker BPBP. Multiple roles of replication forks in S phase checkpoints: sensors, effectors and targets. Cell cycle. 2003;2:568–572. [PubMed] [Google Scholar]
- Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–130. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis CC, Campbell JL. Contribution of Trf4/5 and the Nuclear Exosome to Genome Stability Through Regulation of Histone mRNA Levels in Saccharomyces cerevisiae. Genetics. 2007;175:993–1010. doi: 10.1534/genetics.106.065987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova PV, Pavlov YI. 3′-5′ exonucleases of DNA polymerases e and d correct base analog induced DNA replication errors on opposite DNA strands in Saccharomyces cerevisiae. Genetics. 1996;142:717–726. doi: 10.1093/genetics/142.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto K, Ando S, Shimomura T, Matsumoto K. Rfc5, a replication factor C component, is required for regulation of Rad53 protein kinase in the yeast checkpoint pathway. Mol Cell Biol. 1997;17:5905–5914. doi: 10.1128/mcb.17.10.5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto K, Shimomura T, Hashimoto K, Araki H, Sugino A, Matsumoto K. RFC5, a small subunit of replication factor C complex, couples DNA replication and mitosis in budding yeast. Proc Natl Acad Sci USA. 1996;93:7048–7052. doi: 10.1073/pnas.93.14.7048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szyjka SJ, Viggiani CJ, Aparicio OM. Mrc1 is required for normal progression of replication forks throughout chromatin in S. cerevisiae. Mol Cell. 2005;19:691–697. doi: 10.1016/j.molcel.2005.06.037. [DOI] [PubMed] [Google Scholar]
- Tourriere H, Versini G, Cordon-Preciado V, Alabert C, Pasero P. Mrc1 and Tof1 promote replication fork progression and recovery independently of Rad53. Mol Cell. 2005;19:699–706. doi: 10.1016/j.molcel.2005.07.028. [DOI] [PubMed] [Google Scholar]
- Tsolou AA, Lydall DD. Mrc1 protects uncapped budding yeast telomeres from exonuclease EXO1. DNA repair. 2007;6:1607–1617. doi: 10.1016/j.dnarep.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, Newport J. Initiation of eukaryotic DNA replication: origin unwinding and sequential chromatin association of Cdc45, RPA, and DNA polymerase alpha. Mol Cell. 2000;5:617–627. doi: 10.1016/s1097-2765(00)80241-5. [DOI] [PubMed] [Google Scholar]
- Xu H, Boone C, Klein HL. Mrc1 is required for sister chromatid cohesion to aid in recombination repair of spontaneous damage. Mol Cell Biol. 2004;24:7082–7090. doi: 10.1128/MCB.24.16.7082-7090.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Ukomadu C, Jha S, Senga T, Dhar SK, Wohlschlegel JA, Nutt LK, Kornbluth S, Dutta A. Mcm10 and And-1/CTF4 recruit DNA polymerase {alpha} to chromatin for initiation of DNA replication. Genes Dev. 2007;21:2288–2299. doi: 10.1101/gad.1585607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Stillman B. Assembly of a complex containing Cdc45p, replication protein A, and Mcm2p at replication origins controlled by S-phase cyclin-dependent kinases and Cdc7p-Dbf4p kinase. Mol Cell Biol. 2000;20:3086–3096. doi: 10.1128/mcb.20.9.3086-3096.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.