

Abstract
Estrogen has anti-inflammatory and vasoprotective effects when administered to young women or experimental animals that appear to be converted to pro-inflammatory and vasotoxic effects in older subjects, particularly those that have been hormone free for long periods. Clinical studies have raised many important questions about the vascular effects of estrogen that cannot easily be answered in human subjects. Here we review cellular/molecular mechanisms by which estrogen modulates injury-induced inflammation, growth factor expression and oxidative stress in arteries and isolated vascular smooth muscle cells, with emphasis on the role of estrogen receptors and the Nuclear Factor-κB (NFκB) signaling pathway, as well as evidence that these protective mechanisms are lost in aging subjects.
Keywords: Estrogen, smooth muscle cells, inflammation, NFκB, vascular injury, oxidative stress
Ovarian Hormones and Cardiovascular Disease in Women
Cardiovascular disease is the leading cause of death among women in the US, and coronary heart disease (CHD) develops in women on average 10 years later than in men. This lag has been attributed, at least in part, to the protective effects of female sex hormones, particularly estrogens (defined as naturally occurring activators of estrogen receptors) before menopause (1-3). Mechanistic studies carried out in in vitro preparations and in laboratory animals have shown that both natural and synthetic estrogens have anti-inflammatory and vasoprotective effects (4-18). Further, the natural endogenous estrogen 17β-estradiol has been shown to cause rapid endothelium-independent dilation of coronary arteries of men and women, to augment endothelium-dependent relaxation of human coronary arteries ex vivo and to improve endothelial function as assessed by the brachial artery flow-mediated dilation response in postmenopausal women (19). Importantly, the latter vasoprotective effects of estrogen have been observed in the early postmenopausal years in both healthy women and those with CHD, but not in older (≥60 years) postmenopausal women, regardless of the presence or absence of CHD (19,20).
Observational studies have shown substantial benefit (∼50% reduction in CHD) of hormone therapy in women who choose to use menopausal hormones (and usually begin taking them in the perimenopausal or early postmenopausal period) (21). Randomized controlled trials of menopausal hormone therapy, which typically enroll women 10 years or longer after menopause, after many years of estrogen deprivation, have shown increases in CHD events with hormone treatment (usually conjugated equine estrogen ± a progestin) in this older (60-79 years) age group (22-24). In contrast, subgroup analyses of the Women's Health Initiative have shown that women in whom hormone therapy was initiated at a younger age (50-59 years) and earlier post menopause tended to have reduced risk of CHD and total mortality (25,26). Use of unopposed conjugated estrogen was associated with lower risk of CHD than combined estrogen + progestin (medroxyprogesterone acetate), and an ancillary study showed a statistically significant reduction in coronary artery calcium (Agatston) score in younger (50-59 years) women randomized to conjugated estrogen compared to placebo, indicating a reduced calcifiedplaque burden (27).
Possible reasons for the apparent paradox of beneficial or neutral vascular effects of menopausal hormones in young women vs detrimental effects in older women have been widely discussed in the literature (28-31). Many authors have proposed the “timing hypothesis”, which postulates that estrogen signaling pathways are altered in older women, particularly those with subclinical vascular disease, in a manner that converts anti-inflammatory/vasoprotective effects to pro-inflammatory/vasculotoxic effects (7,15,30,32,33). However, a very recent study has demonstrated potential benefit of menopausal hormones even in the setting of established atherosclerosis (34). Atherosclerotic plaques from the internal carotid arteries of postmenopausal women receiving menopausal hormones contained fewer inflammatory leukocytes, lower levels of TNF-α, activated Nuclear Factor-κB (NFκB) and matrix metalloproteinase (MMP)-9, and more collagen and inhibitor of NFκB-β (IκB-β) compared to plaques from women who had never used hormones. These findings suggest the intriguing possibility that menopausal hormones may contribute to plaque stabilization in the carotid artery by inhibiting NFκB-dependent inflammation, which is thought to be responsible for plaque rupture. The reduction in MMP-9 found in carotid plaques seems to differ from what has previously been described in coronary plaques and in the plasma of women receiving menopausal hormones (35,36). Whether these differences are related to the anatomical location of the plaques, to differences in developmental stage of the plaques, or to other, unmeasured differences between patient populations studied remains to be determined.
Thus, clinical studies have raised many important questions about the effects of estrogen on vascular inflammation that cannot easily be answered in human subjects. Insights from fundamental mechanistic studies are needed in order to delineate the cellular/molecular events that determine the response of blood vessels to inflammatory stresses and to elucidate how estrogen and other ovarian hormones interact with these processes to protect or injure blood vessels (15,30,33). These findings may pave the way to novel strategies for prevention and treatment of cardiovascular disease and other inflammatory disorders.
Estrogen Modulates Injury-induced Chemokine/Cytokine Expression and Leukocyte Infiltration
Inflammation plays an important role in the pathogenesis of many forms of vascular disease, including atherosclerosis and the response to acute vascular injury (37). Balloon injury of arteries elicits accumulation of neutrophils and monocyte/macrophages in the adventitia surrounding the injury site within hours after the insult (18) (Figure 1). The appearance of these cells is predated by expression of inflammatory mediators, including adhesion molecules and chemokines and cytokines, in acutely injured arteries (13) as well as in atherosclerotic and restenotic vessels, and is associated with activation of a variety of cell types, including adipocytes and fibroblasts, in adventitial tissues (9). Medial vascular smooth muscle cells (VSMCs) are activated early following endoluminal injury, releasing cytokines and chemokines that reach the periadventitial space to recruit leukocytes and appear to be the chief effector cells for initiation of the early inflammatory response.
Figure 1.

A simplified schematic illustration of the cellular response to endoluminal vascular injury.
Estrogen exerts an early anti-inflammatory effect in the rat carotid injury model (4,13,18). This is reflected in an estrogen dependent sexual dimorphism in the vascular injury response, whereby neointima formation (influx of adventitial and medial cells and deposition of interstitial matrix inside the internal elastic lamella) is greater in males than in females (5,6,8,17,38). Further, treatment with a dose of estrogen that results in physiologic levels (40-60 pg/ml) of circulating hormone markedly attenuates neointima formation in gonadectomized animals of both sexes. Interestingly, coadministration of medroxyprogesterone acetate (MPA), the synthetic progestin contained in many menopausal hormone preparations and studied in the Women's Health Initiative, completely blocks the effect of estrogen on neointima formation (8). These findings are generally consistent with the observations that estrogen attenuates and either MPA or progesterone exacerbates the inflammatory response to LPS administration in the cerebral vasculature of ovariectomized rats, although when estrogen and a progestin were coadministered in this study, the anti-inflammatory effect of estrogen predominated (39).
Our laboratory has shown that systemic 17β-estradiol administration attenuates both expression of inflammatory mediators and infiltration of leukocytes into balloon-injured carotid arteries of ovariectomized rats at a very early time point post injury (13,18). 17β-estradiol had a particularly robust modulatory effect on neutrophil chemotaxis by attenuating expression of cytokine-induced neutrophil chemoattractant (CINC)-2β, a member of the cysteine-x-cysteine (CXC) chemokine family and a potent chemoattractant for neutrophils in vitro and in vivo (40) and on monocyte chemotaxis by attenuating expression of monocyte chemoattractant protein (MCP-1), a selective chemoattractant for monocytes (40).
Another mechanism that has been implicated in the anti-inflammatory/vasoprotective effects of 17β-estradiol is inhibition of expression and/or action of C-reactive protein (CRP) in injured arteries (41). We tested the hypothesis that 17β-estradiol attenuates the vascular injury response by inhibiting expression/action of CRP in a transgenic mouse model that expresses human CRP (CRPtg) in an manner that mirrors its expression in humans (41,42). Following carotid ligation injury, neointima formation was exaggerated in arteries of female CRPtg mice compared to nontransgenic (NTG) controls, whether intact or OVX, but was attenuated in both genotypes by subcutaneously administered 17β-estradiol. Human CRP protein and mRNA were expressed in the neointima of ovariectomized CRPtg mice, and expression was greatly attenuated by 17β-estradiol treatment. CRP was undetectable in other domains of injured arteries and in uninjured vessels. The findings that, as in humans and animal models of atherosclerotic disease (43), CRP is expressed in the injured/diseased vasculature and the extent of the lesion appears to correlate with the level of CRP expression provide indirect evidence that locally expressed CRP may play a functional role in the injury response. Further, these findings suggest that the modulatory effect of 17β-estradiol in the CRPtg model of acute vascular injury could be a consequence of decreased local expression of CRP in the injured artery. Ongoing studies are addressing the mechanisms by which 17β-estradiol modulates CRP gene expression and/or its downstream inflammatory actions, e.g. altered expression/activation of its IgG Fc receptors (FcγRs), which we have shown to be required for the exaggerated response to vascular injury provoked by CRP in ovariectomized CRPtg mice (44).
Estrogen Modulates Growth Factor Expression and Oxidative Stress in Injured Arteries
Estrogen modulates the acute vascular injury response and the development of other forms of vascular pathology, in part by altering the expression and/or action of various growth factors, adhesion molecules and chemokines in relevant cell types in the vessel wall (Figure 1). For example, estrogen inhibits the mitogenic effects of a number of growth factors, e.g. FGF-2 and epidermal growth factor, on VSMCs in vitro (45). The effects of estrogen on expression of growth factors and their signaling pathways are growth factor and target cell specific, e.g. estrogen appears to synergize the mitogenic effects of PDGF in VSMCs, and to enhance FGF-2 expression in a variety of cell types, including endometrial and breast cancer cell lines.
Chemoattractant/adhesion molecule expression plays a major role in vascular remodeling by directing migration of adventitial and medial cells into neointima. Our laboratory has demonstrated that expression of the chemoattractant/adhesion molecule osteopontin, which is known to be overexpressed in blood vessels in response to injury, is negatively modulated by 17β-estradiol in a dose- and ER-dependent manner in activated rat aortic SMCs in vitro (11). Immunodepletion and integrin blocking studies showed that osteopontin, via its ανβ3 integrin receptor, can direct adventitial fibroblast migration in vitro and suggested that osteopontin may be an important estrogen-sensitive mediator of adventitial activation and neointima formation in injured blood vessels (11). We observed that FGF-1 stimulates expression of osteopontin mRNA and protein in rat aortic SMCs in vitro via a signaling pathway that involves activation of FGFR-1 and Src/MEK/ERK1/2 kinases and that the pathway plays a functional role in directing adventitial fibroblast migration in vitro (46). Subsequent studies showed that 17β-estradiol dose-dependently inhibits the stimulatory effect of FGF-1 on a variety of mediators, including iNOS and the adhesion molecule periostin, as well as osteopontin. Periostin is expressed in balloon injured carotid arteries in vivo and in growth factor-treated VSMCs in vitro, and is negatively modulated by estrogen via signaling pathways distinct from that described for osteopontin (47,48).
Another important component of the vascular injury response that is modulated by estrogen involves altered production of nitric oxide (NO) due to activation or inhibition of nitric oxide synthase (NOS). The synthesis of NO in the vasculature is catalyzed by two major isoforms of NOS that are regulated by estrogen in a directionally opposite manner. NOSIII (eNOS) is constitutively expressed in endothelial cells and is up-regulated by estrogen via an ER-mediated mechanism (49). Inducible NOS (iNOS, NOSII) is not found in normal blood vessels but is highly expressed in injured arteries. Activated iNOS can produce over 1,000-fold more NO (μM range) for a longer duration than eNOS. At these concentrations, NO can be toxic to tissues via interaction with reactive oxygen species to produce powerful biologic oxidants. High levels of NO that result from expression of iNOS have been implicated in the formation of neointima following vascular injury.
Consistent with the hypothesis that oxidative stress related to iNOS activation is a major determinant of the extent of injury-induced vascular remodeling, we have demonstrated greatly attenuated neointima formation in ligated carotid arteries of mice with homozygous deletion of the iNOS gene (iNOS-/-) (50). The iNOS-/- mice responded to estrogen treatment with further attenuation of neointima formation, suggesting that the negative modulatory effect of estrogen on vascular remodeling is mediated through a variety of signaling cascades, including, but not restricted to iNOS.
Role of Estrogen Receptors (ERs) in Inflammation and Vasoprotection
Estrogen plays many roles in immunomodulation, and can be either anti- or pro-inflammatory depending on diverse factors such as the target cell type, the target organ with its specific microenvironment, the timing and concentration of estrogen administered and cell type- and microenvironment-specific variability in ER expression (51). Studies with the nonselective ER antagonist ICI 182,780 and in ER knockout mice have confirmed that the antiinflammatory/vasoprotective effect of estrogen is ER dependent (4,16,52,53). However, the ER subtype dependence of the anti-inflammatory/vasoprotective effects of estrogen and the signaling pathway(s) involved are incompletely understood. There are at least three, and possibly four distinct ERs: two ligand-activated transcription factors (ERα and ERβ), a G-protein coupled receptor (GPER, GPR30), and a putative receptor (ER-X) that has been studied mainly in brain (54). ERα and ERβ are members of the nuclear hormone receptor superfamily that are expressed in the vasculature and play a role in mediating/modulating responses to vascular injury, mainly through transcriptional regulation. GPER (GPR30) is an intracellular transmembrane ER that initiates many rapid nongenomic signaling events, including intracellular calcium mobilization and synthesis of phosphatidyl-inositol 3,4,5-triphosphate in the nucleus of many cell types (55). GPER has been identified in human internal mammary arteries and saphenous veins, but does not yet have a defined vascular function (54,56).
ERα activation has been shown to attenuate injury-induced vascular remodeling. Studies in knockout mice support ERα-mediated protective effects on vascular remodeling responses to injury (52,53), and in vitro studies have shown that ERβ also plays a protective role in injured arteries (57,58). Selective ERβ and ERα mRNA anti-sense oligomers have been used to examine the ER subtype dependence of estrogen-induced inhibition of PDGF-BB-induced p38 and p42/44 mitogen-activated protein kinase (MAPK) phosphorylation, migration, and proliferation in porcine SMCs and endothelial cells (57). The inhibitory effects of estrogen on porcine SMCs were abrogated by down-regulation of ERβ protein expression, whereas down-regulation of ERα had no effect. In contrast, down-regulation of ERα expression in porcine aortic endothelial cells inhibited estrogen-induced p38 and p42/44 MAPK activation, while down-regulation of ERβ had no effect. Thus, both ER subtypes contribute to vasoprotection in a cell type specific fashion.
Importantly, expression of ERβ is increased relative to ERα in the setting of oxidative stress, hypoxia and inflammation (59). In this situation, ERβ-mediated cross modulation of ERα can be important in regulating pathophysiologic processes, e.g. ERβ activation can inhibit ERα-stimulated IL-1 secretion.
In vivo evidence for a role for ERβ in estrogen-induced vasoprotection was provided by the observation that local delivery of an ERβ selective agonist can inhibit neointima formation induced by placement of a perivascular cuff around the femoral artery of wild type (C57/BL/65) mice (60). The ERβ-selective agonist DPN inhibited neointima formation at a dose dependent fashion when applied via a drug-eluting cuff. In contrast, the ERα-selective agonist PPT inhibited neointima formation at low but not at high concentrations. To further demonstrate the specificity of these responses, an ERα-selective antagonist, MPP (20), was used in combination with estrogen, PPT, or DPN. While the inhibitory effect of PPT on neointima formation was blocked by co-delivery of MPP, estrogen and DPN could still inhibit neointima formation. These data suggest that selective ERβ activation can inhibit neointima formation in a mouse model of restenosis.
Exciting findings from our laboratory have identified a novel mechanism by which ERβ activation may protect against injury-induced inflammation and adverse vascular remodeling: inhibition of TNF-α-induced inflammatory mediator expression in VSMCs (58). Based on published observations that TNF-α induces the rapid recruitment of leukocytes from the circulation in response to many forms of stress and is elaborated from diseased bypass grafts and atherosclerotic arteries, and our own finding that TNF-α expression is dramatically upregulated in balloon-injured rat carotid arteries and modulated by 17β-estradiol administration (13), we used TNF-α as the inflammatory stimulus. We demonstrated that TNF-α stimulated and 17β-estradiol attenuated expression of inflammatory mediators and that DPN dose dependently attenuated TNF-α-induced expression of CINC-2β, whereas PPT had no effect. The anti-inflammatory effects of DPN and 17β-estradiol were blocked by ICI-182,780. TNF-α treatment of rat aortic SMCs produced an increase in neutrophil chemotactic activity of conditioned media; treatment with 17β-estradiol, DPN and an antibody selective for CINC-2β inhibited this effect, confirming its ERβ and CINC-2β dependence. These observations are relevant to our carotid injury model, since the injured area is denuded of endothelium for several weeks and the early injury response is driven by activated SMCs and infiltrating leukocytes. Thus, modulatory effects of ERβ activation on SMC-initiated inflammatory responses likely play an important role in inhibiting early inflammatory changes in the setting of endoluminal vascular injury.
Estrogen, NFκB and Vascular Injury
In the setting of vascular injury, TNF-α activates the NFκB signaling pathway. The NFκB family of inducible transcription factors mediate the immediate-early inflammatory response (61,62). Members of this family contain an N-terminal Rel homology domain (RHD) that is important for DNA binding, dimerization, inhibitor association and nuclear localization. The most common NFκB molecule contains p65 and p50.
Estrogen, via ER activation can inhibit NFκB signaling by a variety of mechanisms (Figure 2). Estrogen inhibits expression of the pro-inflammatory mediator TNF-α (A), which in turn triggers a cascade of cytokines that activate NFκB, thereby mediating a variety of chronic inflammatory diseases, including cardiovascular disease (63). Both ERα and ERβ inhibit NFκB activity in an estrogen dependent manner in many cell types, including coronary artery SMCs (64). IκB processing is a target for estrogen/ER signaling in that IκBα levels have been shown to be increased in cells that are treated with estrogen and/or overexpress ER (B), and estrogen has been shown to inhibit IκBα phosphorylation and degradation (65). Further, there is evidence that estrogen-induced stabilization of IκBα may be due to inhibition of IKK activity (C) and/or inhibition of IκBα degradation (D). Accordingly, studies in vascular cells, including rat and human VSMCs (64,66), have shown that estrogen-induced activation of ER inhibits NFκB induced transcription of chemokines/cytokines (E) and can inhibit the nuclear DNA binding activity of NFκB (F). This could occur through a direct interaction between the ER and NFκB in the nucleus and/or by ER-mediated inhibition of upstream NFκB signaling in the cytoplasm. Finally, estrogen can also block JNK induced TNF-α production (G). Our intriguing preliminary data have revealed evidence for both cytoplasmic and nuclear events in the anti-inflammatory effects of estrogen in rat aortic SMCs (67). Estrogen both enhanced synthesis of IκBα and inhibited NFκB p65 binding to promoters of pro-inflammatory genes in TNF-α treated cells, thus accelerating a negative feedback loop within the NFκB/IκBα signaling pathway, a potential novel mechanism of estrogen induced vasoprotection.
Figure 2.
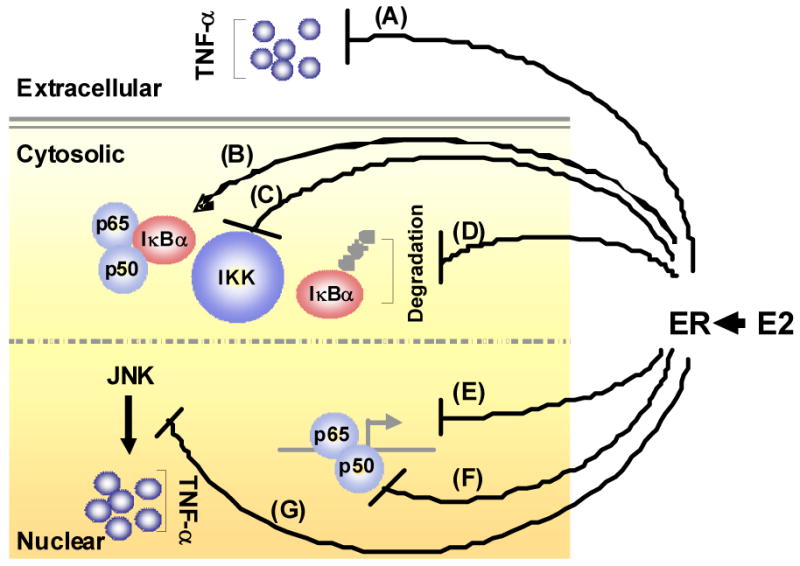
Mechanisms by which E2 treatment attenuates the pro-inflammatory (e.g. TNF-α)-stimulated IκBα/NFκB signaling pathway. E2 inhibits the expression of the pro-inflammatory mediator TNF-α (A) and increases the expression of IκBα (B). E2 can also inhibit IKK-mediated phosphorylation (C) and degradation (D) of IκBα. At the transcriptional level, E2-induced activation of estrogen receptor (ER) inhibits NFκB-induced transcription of chemokines/cytokines (E) and can inhibit the ability of NFκB to bind to DNA (F). E2 has also been shown to block JNK induced TNF-α production (G). ⊥- indicates inhibitory effects; ↑ - indicates stimulatory effects.
Aged Arteries Lose Vasoprotective and Anti-inflammatory Effects of Estrogen
In an attempt to reconcile the findings of harmful vascular effects of estrogen in older women vs. vasoprotective and anti-inflammatory effects in younger women and young female laboratory animals, we tested the hypothesis that responsiveness to estrogen is lost in balloon injured carotid arteries of aged (12 month old) ovariectomized rats (14). We observed that 17β-estradiol treatment has directionally opposite effects on neointima formation in aged (+75%) vs. young (10-12 weeks; -40%) ovariectomized rats. Further, 17β-estradiol had no effect on injury-induced increases in inflammatory mediator expression and neutrophil and monocyte infiltration in injured arteries of aged rats. ERα and ERβ expression were similar in injured carotid arteries of aged and young animals under both vehicle and 17β-estradiol treatment conditions. This is the first demonstration that 17β-estradiol stimulates, rather than attenuates, vascular injury responses in aged animals. Our findings have been confirmed by studies demonstrating that prolonged hypoestrogenicity suppresses the neuroprotective and anti-inflammatory effects of estrogen in a rodent model (68) and that estrogen signaling in the vasculature is impaired following extended periods of ovarian hormone deprivation, supporting the timing hypothesis (69).
Whether the conversion of vasoprotective/anti-inflammatory effects of estrogens to vasotoxic/pro-inflammatory effects in aging subjects is a function of prolonged hypoestrogenicity per se or is related to the aging process and/or the development of vascular disease remains an open question. There is abundant evidence that an age-related hyper-inflammatory state exists, in which circulating levels of pro-inflammatory mediators are elevated (70,71). The increased basal inflammatory activity in this environment may be further amplified by acute stimuli, e.g. stress-mediated induction of cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6 is enhanced in aged rats in vivo and in aged rat macrophages in vitro compared to young adult controls (72,73). While this increased low-grade inflammatory activity in elderly populations has variously been attributed to decreased sex steroid production, increases in amounts of hormonally-active fat tissue, and the atherosclerotic process itself (73), the underlying cellular and molecular mechanisms remain unclear.
Accelerated arterial aging has been attributed to activation of inflammatory and proteolytic pathways that result in increased collagen formation and elastin degradation and reduced VSMC number (70,71). In animal models, this age-related pathology in the larger arteries has been associated with increases in medial thickness, collagen content and collagen/elastin ratio, while numbers of medial SMC nuclei decrease. Further, the VSMCs from older rats are larger and appear to have undergone a phenotypic change toward a dedifferentiated and synthetic state. The cellular and molecular pathways that accelerate the aging process in the vasculature involve activation of NFκB, increased reactive oxygen species production, decreased nitric oxide (NO) bioavailability, and induction of matrix MMPs and TGF-β expression (71,74).
How these processes interfere with ER signaling is an unanswered question, but provocative preliminary observations from our own laboratory have identified a novel molecular mechanism that may contribute to the hyper-inflammatory environment and possibly promote a pro-inflammatory effect of estrogens in the aging vasculature (75-77). We are currently testing the novel hypothesis that inflammation-induced injury in the aging vasculature is related to excessive accumulation and impaired dynamic O-GlcNAcylation of critical proteins in the IκBα/NFκB signaling pathway. We postulate that this differential O-GlcNAcylation plays a mechanistic role in the age-related loss of estrogen-induced vasoprotection.
Acknowledgments
This work was supported, in part, by National Heart, Lung, and Blood Institute grants HL07457, HL64614, HL75211, HL087980 (Oparil S); HL080017, HL044195 (Chen YF) and by American Heart Association Greater Southeast Affiliate grant, 0425455B, 0765398B (Xing D).
References
- 1.Barton M, Meyer MR, Haas E. Hormone replacement therapy and atherosclerosis in postmenopausal women: does aging limit therapeutic benefits? Arterioscler Thromb Vasc Biol. 2007;27:1669–1672. doi: 10.1161/ATVBAHA.106.130260. [DOI] [PubMed] [Google Scholar]
- 2.Kannel WB, Wilson PW. Risk factors that attenuate the female coronary disease advantage. Arch Intern Med. 1995;155:57–61. [PubMed] [Google Scholar]
- 3.Ouyang P, Michos ED, Karas RH. Hormone replacement therapy and the cardiovascular system. J Amer Coll Cardiol. 2006;47:1741–1753. doi: 10.1016/j.jacc.2005.10.076. [DOI] [PubMed] [Google Scholar]
- 4.Bakir S, Mori T, Durand J, Chen YF, Thompson JA, Oparil S. Estrogen-induced vasoprotection is estrogen receptor dependent: evidence from the balloon-injured rat carotid artery model. Circulation. 2000;101:2342–2344. doi: 10.1161/01.cir.101.20.2342. [DOI] [PubMed] [Google Scholar]
- 5.Chen SJ, Li H, Durand J, Oparil S, Chen YF. Estrogen reduces myointimal proliferation after balloon injury of rat carotid artery. Circulation. 1996;93:577–584. doi: 10.1161/01.cir.93.3.577. [DOI] [PubMed] [Google Scholar]
- 6.Chen YF, Oparil S. Effects of sex steroids in vascular injury. In: Levin ER, Nadler JL, editors. Endocrinology of Cardiovascular Function. Kluwer Academic Publishers; Boston/Dordrecht/London: 1998. pp. 45–49. [Google Scholar]
- 7.Clarkson TB, Appt SE. Controversies about HRT-lessons from monkey models. Maturitas. 2005;51:64–74. doi: 10.1016/j.maturitas.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 8.Levine RL, Chen SJ, Durand J, Chen YF, Oparil S. Medroxyprogesterone attenuates estrogen-mediated inhibition of neointima formation after balloon injury of the rat carotid artery. Circulation. 1996;94:2221–2227. doi: 10.1161/01.cir.94.9.2221. [DOI] [PubMed] [Google Scholar]
- 9.Li G, Chen SJ, Oparil S, Chen YF, Thompson JA. Direct in vivo evidence demonstrating neointimal migration of adventitial fibroblasts after balloon injury of rat carotid arteries. Circulation. 2000;101:1362–1365. doi: 10.1161/01.cir.101.12.1362. [DOI] [PubMed] [Google Scholar]
- 10.Li G, Chen YF, Greene GL, Oparil S, Thompson JA. Estrogen inhibits vascular smooth muscle cell-dependent adventitial fibroblast migration in vitro. Circulation. 1999;100:1639–1645. doi: 10.1161/01.cir.100.15.1639. [DOI] [PubMed] [Google Scholar]
- 11.Li G, Chen YF, Kelpke SS, Oparil S, Thompson JA. Estrogen attenuates integrin-β3-dependent adventitial fibroblast migration after inhibition of osteopontin production in vascular smooth muscle cells. Circulation. 2000;101:2949–2955. doi: 10.1161/01.cir.101.25.2949. [DOI] [PubMed] [Google Scholar]
- 12.Mendelsohn ME, Karas RH. HRT and the young at heart. N Engl J Med. 2007;356:2639–2641. doi: 10.1056/NEJMe078072. [DOI] [PubMed] [Google Scholar]
- 13.Miller AP, Feng W, Xing D, Weathington NM, Blalock JE, Chen YF, Oparil S. Estrogen modulates inflammatory mediator expression and neutrophil chemotaxis in injured arteries. Circulation. 2004;110:1664–1669. doi: 10.1161/01.CIR.0000142050.19488.C7. [DOI] [PubMed] [Google Scholar]
- 14.Miller AP, Xing D, Feng W, Fintel M, Chen YF, Oparil S. Aged rats lose vasoprotective and anti-inflammatory effects of estrogen in injured arteries. Menopause. 2007;14:251–260. doi: 10.1097/01.gme.0000235366.39726.f6. [DOI] [PubMed] [Google Scholar]
- 15.Miller VM, Shuster LT, Hayes SN. Controversy of hormone treatment and cardiovascular function: Need for strengthened collaborations between preclinical and clinical scientists. Curr Opin Investig Drugs. 2003;4:1220–1232. [PubMed] [Google Scholar]
- 16.Mori T, Durand J, Chen Y, Thompson JA, Bakir S, Oparil S. Effects of short-term estrogen treatment on the neointimal response to balloon injury of rat carotid artery. Am J Cardiol. 2000;85:1276–1279. doi: 10.1016/s0002-9149(00)00748-7. [DOI] [PubMed] [Google Scholar]
- 17.Oparil S, Chen SJ, Chen YF, Durand JN, Allen L, Thompson JA. Estrogen attenuates the adventitial contribution to neointima formation in injured rat carotid arteries. Cardiovasc Res. 1999;44:608–614. doi: 10.1016/s0008-6363(99)00240-0. [DOI] [PubMed] [Google Scholar]
- 18.Xing D, Miller A, Novak L, Chen YF, Oparil S. Estradiol and progestins differentially modulate leukocyte infiltration after vascular injury. Circulation. 2004;109:234–241. doi: 10.1161/01.CIR.0000105700.95607.49. [DOI] [PubMed] [Google Scholar]
- 19.Sherwood A, Bower JK, McFetridge-Durdle J, Blumenthal JA, Newby LK, Hinderliter AL. Age moderates the short-term effects of transdermal 17beta-estradiol on endothelium-dependent vascular function in postmenopausal women. Arterioscler Thromb Vasc Biol. 2007;27:1782–1787. doi: 10.1161/ATVBAHA.107.145383. [DOI] [PubMed] [Google Scholar]
- 20.Harrington WR, Sheng S, Barnett DH, Petz LN, Katzenellenbogen JA, Katzenellenbogen BS. Activities of estrogen receptor alpha- and beta-selective ligands at diverse estrogen responsive gene sites mediating transactivation or transrepression. Mol Cell Endocrinol. 2003;206:13–22. doi: 10.1016/s0303-7207(03)00255-7. [DOI] [PubMed] [Google Scholar]
- 21.Grodstein F, Manson JE, Colditz GA, Willett WC, Speizer FE, Stampfer MJ. A prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Ann Intern Med. 2000;133:933–941. doi: 10.7326/0003-4819-133-12-200012190-00008. [DOI] [PubMed] [Google Scholar]
- 22.Anderson GL, Limacher M, Assaf AR, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women's Health Initiative randomized controlled trial. JAMA. 2004;291:1701–1712. doi: 10.1001/jama.291.14.1701. [DOI] [PubMed] [Google Scholar]
- 23.Grady D, Herrington D, Bittner V, Blumenthal R, Davidson M, Hlatky M, Hsia J, Hulley S, Herd A, Khan S, Newby LK, Waters D, Vittinghoff E, Wenger N HERS Research Group. Cardiovascular disease outcomes during 6.8 years of hormone therapy: Heart and Estrogen/progestin Replacement Study follow-up (HERS II) JAMA. 2002;288:49–57. doi: 10.1001/jama.288.1.49. [DOI] [PubMed] [Google Scholar]
- 24.Manson JE, Hsia J, Johnson KC, Rossouw JE, Assaf AR, Lasser NL, Trevisan M, Black HR, Heckbert SR, Detrano R, Strickland OL, Wong ND, Crouse JR, Stein E, Cushman M Women's Health Initiative Investigators. Estrogen plus progestin and the risk of coronary heart disease. N Engl J Med. 2003;349:523–534. doi: 10.1056/NEJMoa030808. [DOI] [PubMed] [Google Scholar]
- 25.Hsia J, Langer RD, Manson JE, Kuller L, Johnson KC, Hendrix SL, Pettinger M, Heckbert SR, Greep N, Crawford S, Eaton CB, Kostis JB, Caralis P, Prentice R Women's Health Initiative Investigators. Conjugated equine estrogens and coronary heart disease: the Women's Health Initiative. Arch Intern Med. 2006;166:357–365. doi: 10.1001/archinte.166.3.357. [DOI] [PubMed] [Google Scholar]
- 26.Rossouw JE, Prentice RL, Manson JE, Wu L, Barad D, Barnabei VM, Ko M, LaCroix AZ, Margolis KL, Stefanick ML. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA. 2007;297:1465–1477. doi: 10.1001/jama.297.13.1465. [DOI] [PubMed] [Google Scholar]
- 27.Manson JE, Allison MA, Rossouw JE, Carr JJ, Langer RD, Hsia J, Kuller LH, Cochrane BB, Hunt JR, Ludlam SE, Pettinger MB, Gass M, Margolis KL, Nathan L, Ockene JK, Prentice RL, Robbins J, Stefanick ML WHI and WHI-CACS Investigators. Estrogen therapy and coronary-artery calcification. N Engl J Med. 2007;356:2591–2602. doi: 10.1056/NEJMoa071513. [DOI] [PubMed] [Google Scholar]
- 28.Dubey RK, Imthurn B, Barton M, Jackson EK. Vascular consequences of menopause and hormone therapy: importance of timing of treatment and type of estrogen. Cardiovasc Res. 2005;66:295–306. doi: 10.1016/j.cardiores.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 29.Manson JE, Bassuk SS, Harman SM, Brinton EA, Cedars MI, Lobo R, Merriam GR, Miller VM, Naftolin F, Santoro N. Postmenopausal hormone therapy: new questions and the case for new clinical trials. Menopause. 2006;13:139–147. doi: 10.1097/01.gme.0000177906.94515.ff. [DOI] [PubMed] [Google Scholar]
- 30.Phillips LS, Langer RD. Postmenopausal hormone therapy: critical reappraisal and a unified hypothesis. Fertil Steril. 2005;83:558–566. doi: 10.1016/j.fertnstert.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 31.Turgeon JL, McDonnell DP, Martin KA, Wise PM. Hormone therapy: physiological complexity belies therapeutic simplicity. Science. 2004;304:1269–1273. doi: 10.1126/science.1096725. [DOI] [PubMed] [Google Scholar]
- 32.Miller AP, Chen YF, Xing D, Feng S, Oparil S. Hormone replacement therapy and inflammation: Interaction in cardiovascular disease. Hypertension. 2003;42:657–663. doi: 10.1161/01.HYP.0000085560.02979.0C. [DOI] [PubMed] [Google Scholar]
- 33.Miller VM, Clarkson TB, Harman SM, Brinton EA, Cedars M, Lobo R, Manson JE, Merriam GR, Naftolin F, Santoro N. Women, hormones, and clinical trials: a beginning, not an end. J Appl Physiol. 2005;99:381–383. doi: 10.1152/japplphysiol.00248.2005. [DOI] [PubMed] [Google Scholar]
- 34.Marfella R, Di Filippo C, Portoghese M, Ferraraccio F, Crescenzi B, Siniscalchi M, Barbieri M, Bologna C, Rizzo MR, Rossi F, D'Amico M, Paolisso G. Proteasome activity as a target of hormone replacement therapy-dependent plaque stabilization in postmenopausal women. Hypertension. 2008;51:1135–1141. doi: 10.1161/HYPERTENSIONAHA.107.105239. [DOI] [PubMed] [Google Scholar]
- 35.Lewandowski KC, Komorowski J, Mikhalidis DP, Bienkiewicz M, Tan BK, O'Callaghan CJ, Lewinski A, Prelevic G, Randeva HS. Effects of hormone replacement therapy type and route of administration on plasma matrix metalloproteinases and their tissue inhibitors in postmenopausal women. J Clin Endocrinol Metab. 2006;91:3123–3130. doi: 10.1210/jc.2005-2789. [DOI] [PubMed] [Google Scholar]
- 36.Sowers MR, Randolph J, Jr, Jannausch M, Lasley B, Jackson E, McConnell D. Levels of sex steroid and cardiovascular disease measures in premenopausal and hormone-treated women at midlife: implications for the “timing hypothesis”. Arch Intern Med. 2008;168:2146–2153. doi: 10.1001/archinte.168.19.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 38.Oparil S, Levine RL, Chen SJ, Durand J, Chen YF. Sexually dimorphic response of the balloon-injured rat carotid artery to hormone treatment. Circulation. 1997;95:1301–1307. doi: 10.1161/01.cir.95.5.1301. [DOI] [PubMed] [Google Scholar]
- 39.Sunday L, Tran MM, Krause DN, Duckles SP. Estrogen and progestagens differentially modulate vascular proinflammatory factors. Am J Physiol. 2006;291:E261–E267. doi: 10.1152/ajpendo.00550.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luster AD. Chemokines: chemotactic cytokines that mediate inflammation. N Eng J Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 41.Wang D, Oparil S, Chen YF, McCrory MA, Feng W, Szalai AJ. Estrogen treatment abrogates neointima formation in human C-reactive protein transgenic mice. Arterioscler Thromb Vasc Biol. 2005;25:2094–2099. doi: 10.1161/01.ATV.0000179602.85797.3f. [DOI] [PubMed] [Google Scholar]
- 42.Szalai AJ, McCrory MA. Varied biologic functions of C-reactive protein: lessons learned from transgenic mice. Immunol Res. 2002;26:279–287. doi: 10.1385/IR:26:1-3:279. [DOI] [PubMed] [Google Scholar]
- 43.Paul A, Ko KW, Li L, Yechoor V, McCrory MA, Szalai AJ, Chan L. C-reactive protein accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109:647–655. doi: 10.1161/01.CIR.0000114526.50618.24. [DOI] [PubMed] [Google Scholar]
- 44.Xing D, Hage FG, Chen YF, McCrory MA, Feng W, Skibinski GA, Majid-Hassan E, Oparil S, Szalai AJ. Exaggerated neointima formation in human C-reactive protein transgenic mice is IgG Fc receptor type I (FcγRI) dependent. Am J Pathol. 2008;172:22–30. doi: 10.2353/ajpath.2008.070154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suzuki A, Mizuno K, Ino Y, Okada M, Kikkawa F, Mitzutani S, Tomoda Y. Effects of 17β-estradiol and progesterone on growth-factor-induced proliferation and migration in human female aortic smooth muscle cells in vitro. Cardiovasc Res. 1996;31:516–523. [PubMed] [Google Scholar]
- 46.Li G, Oparil S, Kelpke SS, Chen YF, Thompson JA. Fibroblast growth factor receptor-1 signaling induces osteopontin expression and vascular smooth muscle cell-dependent adventitial fibroblast migration in vitro. Circulation. 2002;106:854–859. doi: 10.1161/01.cir.0000024113.26985.cc. [DOI] [PubMed] [Google Scholar]
- 47.Li G, Wang D, Chen YF, Oparil S. Vascular injury induces expression of periostin: a novel vascular extracellular matrix protein via the PI3-kinase-MAP kinase pathway. (Abstract) J Am Coll Cardiol. 2003;41:34A. [Google Scholar]
- 48.Li P, Oparil S, Chen YF. Hypoxia-responsive growth factors upregulate periostin and osteopontin expression via distinct signaling pathways in rat pulmonary arterial smooth muscle cells. J Appl Physiol. 2004;97:1550–1558. doi: 10.1152/japplphysiol.01311.2003. [DOI] [PubMed] [Google Scholar]
- 49.Hayashi T, Yamada K, Esaki T, Kuzuya M, Satake S, Ishikawa T, Hidaka H, Iguchi A. Estrogen increases endothelial nitric oxide by a receptor mediated system. Biochem Biophys Res Commun. 1995;214:847–855. doi: 10.1006/bbrc.1995.2364. [DOI] [PubMed] [Google Scholar]
- 50.Tolbert T, Thompson JA, Bouchard P, Oparil S. Estrogen-induced vasoprotection is independent of inducible nitric oxide synthase expression: Evidence from the mouse carotid artery ligation model. Circulation. 2001;104:2740–2745. doi: 10.1161/hc4701.099581. [DOI] [PubMed] [Google Scholar]
- 51.Straub RH. The complex role of estrogens in inflammation. Endocr Rev. 2007;28:521–574. doi: 10.1210/er.2007-0001. [DOI] [PubMed] [Google Scholar]
- 52.Brouchet L, Krust A, Dupont S, Chambon P, Bayard F, Arnal JF. Estradiol accelerates reendothelialization in mouse carotid artery through estrogen receptor-alpha but not estrogen receptor-beta. Circulation. 2001;103:423–428. doi: 10.1161/01.cir.103.3.423. [DOI] [PubMed] [Google Scholar]
- 53.Karas RH, Hodgin JB, Kwoun M, Krege JH, Aronovitz M, Mackey W, Gustafsson JA, Korach KS, Smithies O, Mendelsohn ME. Estrogen inhibits the vascular injury response in estrogen receptor beta-deficient mice. Proc Natl Acad Sci USA. 1999;96:15133–15136. doi: 10.1073/pnas.96.26.15133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller VM, Duckles SP. Vascular actions of estrogens: functional implications. Pharmacol Rev. 2008;60:210–241. doi: 10.1124/pr.107.08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 56.Haas E, Meyer MR, Schurr U, Bhattacharya I, Minotti R, Nguyen HH, Heigl A, Lachat M, Genoni M, Barton M. Differential effects of 17β-estradiol on function and expression of estrogen receptor α, estrogen receptor β, and GPR30 in arteries and veins of patients with atherosclerosis. Hypertension. 2007;49:1358–1363. doi: 10.1161/HYPERTENSIONAHA.107.089995. [DOI] [PubMed] [Google Scholar]
- 57.Geraldes P, Sirois MG, Tanguay JF. Specific contribution of estrogen receptors on mitogen-activated protein kinase pathways and vascular cell activation. Circ Res. 2003;93:399–405. doi: 10.1161/01.RES.0000088640.18462.42. [DOI] [PubMed] [Google Scholar]
- 58.Xing D, Feng W, Miller AP, Weathington NM, Chen YF, Novak L, Blalock E, Oparil S. Estrogen modulates TNF-α-induced inflammation in rat aortic smooth muscle cells through estrogen receptor-β activation. Am J Physiol. 2007;292:H2607–H2612. doi: 10.1152/ajpheart.01107.2006. [DOI] [PubMed] [Google Scholar]
- 59.Rider V, Li X, Peterson G, Dawson J, Kimler BF, Abdou NI. Differential expression of estrogen receptors in women with systemic lupus erythematosus. J Rheumatol. 2006;33:1093–1101. [PubMed] [Google Scholar]
- 60.Krom YD, Pires NM, Jukema JW, de Vries MR, Frants RR, Havekes LM, van Dijk KW, Quax PH. Inhibition of neointima formation by local delivery of estrogen receptor alpha and beta specific agonists. Cardiovasc Res. 2007;73:217–226. doi: 10.1016/j.cardiores.2006.10.024. [DOI] [PubMed] [Google Scholar]
- 61.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 62.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IκB-NFκB signaling module: temporal control and selective gene activation. Science. 2002;298:1241–1245. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 63.Arenas IA, Armstrong SJ, Xu Y, Davidge ST. Chronic tumor necrosis factor-alpha inhibition enhances NO modulation of vascular function in estrogen-deficient rats. Hypertension. 2005;46:76–81. doi: 10.1161/01.HYP.0000168925.98963.ef. [DOI] [PubMed] [Google Scholar]
- 64.Speir E, Yu ZX, Takeda K, Ferrans VJ, Cannon RO., 3rd Antioxidant effect of estrogen on cytomegalovirus-induced gene expression in coronary artery smooth muscle cells. Circulation. 2000;102:2990–2996. doi: 10.1161/01.cir.102.24.2990. [DOI] [PubMed] [Google Scholar]
- 65.Kalaitzidis D, Gilmore TD. Transcription factor cross-talk: the estrogen receptor and NF-kappaB. Trends Endocrinol Metab. 2005;16:46–52. doi: 10.1016/j.tem.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 66.Sharma RV, Gurjar MV, Bhalla RC. Selected contribution: estrogen receptor-alpha gene transfer inhibits proliferation and NF-kappaB activation in VSM cells from female rats. J Appl Physiol. 2001;91:2400–2406. doi: 10.1152/jappl.2001.91.5.2400. [DOI] [PubMed] [Google Scholar]
- 67.Xing D, Nozell S, Chen YF, Feng W, Oparil S. Estrogen enhances IκBα synthesis and inhibits NFκB p65 binding to the promoters of inflammatory genes in rat aortic smooth muscle cells. (Abstract) EB. 2009 in press. [Google Scholar]
- 68.Suzuki S, Brown CM, Dela Cruz CD, Yang E, Bridwell DA, Wise PM. Timing of estrogen therapy after ovariectomy dictates the efficacy of its neuroprotective and antiinflammatory actions. Proc Natl Acad Sci USA. 2007;104:6013–6018. doi: 10.1073/pnas.0610394104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pinna C, Cignarella A, Sanvito P, Pelosi V, Bolego C. Prolonged ovarian hormone deprivation impairs the protective vascular actions of estrogen receptor alpha agonists. Hypertension. 2008;51:1210–1217. doi: 10.1161/HYPERTENSIONAHA.107.106807. [DOI] [PubMed] [Google Scholar]
- 70.Lakatta EG. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises – Part III: Cellular and molecular clues to heart and arterial aging. Circulation. 2003;107:490–497. doi: 10.1161/01.cir.0000048894.99865.02. [DOI] [PubMed] [Google Scholar]
- 71.Najjar SS, Scuteri A, Lakatta EG. Arterial aging: Is it an immutable cardiovascular risk factor? Hypertension. 2005;46:454–462. doi: 10.1161/01.HYP.0000177474.06749.98. [DOI] [PubMed] [Google Scholar]
- 72.Tang Y, DiPietro L, Feng Y, Wang X. Increased TNF-alpha and PGI(2), but not NO release from macrophages in 18-month-old rats. Mech Ageing Dev. 2000;114:79–88. doi: 10.1016/s0047-6374(00)00090-7. [DOI] [PubMed] [Google Scholar]
- 73.Kovacs EJ. Aging, traumatic injury, and estrogen treatment. Exp Gerontol. 2005;40:549–555. doi: 10.1016/j.exger.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 74.Kregel KC, Zhang HJ. An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. Am J Physiol. 2007;292:R18–R36. doi: 10.1152/ajpregu.00327.2006. [DOI] [PubMed] [Google Scholar]
- 75.Miller AP, Robertson C, Xing D, Li P, Chen YF, Chatham JC, Oparil S. Aging-related accumulation of O-GlcNacylated proteins in the cardiovascular system. J Invest Med. 2007;55:S279. [Google Scholar]
- 76.Fülöp N, Feng W, Xing D, He K, Not LG, Brocks CA, Marchase RB, Miller AP, Chatham JC. Aging leads to increased levels of protein O-linked N-acetylglucosamine in heart, aorta, brain and skeletal muscle in Brown-Norway rats. Biogerontology. 2008;9:139–151. doi: 10.1007/s10522-007-9123-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Laczy B, Hill BG, Wang K, Paterson AJ, White CR, Xing D, Chen YF, Darley-Usmar V, Oparil S, Chatham JC. Protein O-GlcNAcylation: A new signaling paradigm for the cardiovascular system. Am J Physiol. 2008 Nov 21; doi: 10.1152/ajpheart.01056.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]