

Abstract
Enzymes are classically proposed to accelerate reactions by binding substrates within active site environments that are structurally preorganized to optimize binding interactions with reaction transition states rather than ground states. This is a remarkably formidable task considering the limited 0.1 – 1 Å scale of most substrate rearrangements. The flexibility of active site functional groups along the coordinate of substrate rearrangement, the distance scale on which enzymes can distinguish structural rearrangement, and the energetic significance of discrimination on that scale remain open questions that are fundamental to a basic physical understanding of enzyme active sites and catalysis. We bring together high resolution X-ray crystallography, 1H and 19F NMR spectroscopy, quantum mechanical calculations, and transition state analog binding measurements to test the distance scale on which non-covalent forces can constrain side chain and ligand relaxation or translation along a specific coordinate and the energetic consequences of such geometric constraints within the active site of bacterial ketosteroid isomerase (KSI). Our results strongly suggest that packing and binding interactions within the KSI active site can constrain local side chain reorientation and prevent hydrogen bond shortening by 0.1 Å or less. Further, this constraint has substantial energetic effects on ligand binding and stabilization of negative charge within the oxyanion hole. These results provide evidence that subtle geometric effects, indistinguishable in most X-ray crystallographic structures, can have significant energetic consequences and highlight the importance of using synergistic experimental approaches to dissect enzyme function.
Introduction
Enzymes and the chemical transformations they catalyze are central to all biological processes, and enzymes have evolved within the aqueous intracellular environment to accelerate reactions by greater than 20 orders of magnitude relative to the rates of the uncatalyzed reactions in water.1 Enormous progress has been made over the past fifty years in elucidating the three-dimensional structures, key catalytic groups, and chemical mechanisms of enzymes. Nonetheless, a comprehensive understanding of the physical basis for enzymatic rate enhancement remains a critical challenge of biochemistry and a key hurdle to successful rational design of novel enzymes with desired catalytic properties, motivating the need for further incisive investigations of enzyme function.2–5
Enzymology over the past decades has elucidated many of the tools utilized by enzymes to facilitate reactions, including the use of specialized functional groups distinct from water such as general acids and bases, coenzymes, metal ions, and reactive nucleophiles.6 Extensive site-directed mutagenesis studies have identified the specific groups within a protein sequence that give large rate effects upon mutation. Thousands of high-resolution enzyme structures have located such groups within active site pockets and proximal to bound substrates and transition state analogs. In many cases this information has permitted assignment of specified catalytic roles to individual groups, such as donating or abstracting a proton, hydrogen bonding to substrate atoms that develop charge in the transition state, or acting as a nucleophile to form a covalent intermediate. From these extensive advances, detailed reaction mechanisms and active site interaction maps can now be constructed for most enzymes.
Nevertheless, enzymes are more than a collection of reactive functional groups. Structural, biophysical, and computational studies have highlighted that the physical environment within an enzyme active site, defined by the complete constellation of chemical groups embedded in the folded protein that surround and solvate a reacting substrate, is very different from aqueous solution.7–13 Furthermore, these environmental differences can have important implications for catalysis. Indeed, gas-phase and solution studies have revealed that a change in solvation environment alone can alter reaction rates by >106-fold.14–16 Thus, attaining a deeper understanding of the catalytic power of enzymes will require experimental tests of the physical properties of active sites that go well beyond structural snap-shots and the rate effects from removal of reactive groups.
One of the most basic and widely-discussed properties of an enzyme active site is the orientation of its constituent backbone and side chain groups during folding to create a highly structured and idiosyncratic environment. Non-covalent contacts with neighboring active site residues position bound substrates relative to each other and relative to reactive functional groups. Indeed, Polanyi, Pauling, Haldane and others proposed early on that enzyme active sites are structurally preorganized so as to complement and preferentially bind reaction transition states rather than ground states.17–22 Experimental evidence for the transition state complementarity of active sites comes from the tight binding of inhibitors that structurally and/or electrostatically resemble expected transition states rather than ground states.21,22 Numerous structural studies have also revealed active site pockets that appear spatially organized to optimize transition state interactions, including the placement of charged residues and hydrogen bond donors that appear to anticipate the approach of complementary charges and hydrogen bond acceptors in the transition state.6,20,23–25 This widely-proposed ability of active site groups to preferentially interact with specific substrate moieties in the transition state versus the ground state rests on the changes in charge distribution and atomic positions that occur within reacting substrates along the reaction coordinate, as illustrated in the following examples.
During peptide bond hydrolysis, the carbonyl C-O bond lengthens from 1.2 Å in the ground state to 1.4 Å in the transition state as negative charge localizes on the oxygen and C-O bond order decreases. This lengthening is accompanied by a ~1.2 Å spatial translation of the carbonyl oxygen (relative to the carbonyl carbon) due to rehybridization of the carbonyl carbon from sp2 in the planar ground state to sp3 in the tetrahedral transition state (Scheme 1A).26–28 Similarly, during phosphoryl transfer reactions the non-bridging oxygen atoms translate ~0.5 Å through space as the phosphoryl group progresses from a tetrahedral ground state to a trigonal bipyramidal transition state (Scheme 1B).29,30
Scheme 1.
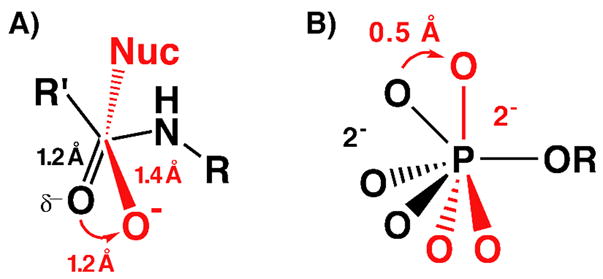
Geometric and electrostatic changes in the (A) carbonyl C-O during peptide bond hydrolysis and (B) nonbridging oxygen atoms during phosphoryl transfer reactions (nucleophile not shown) going from the ground state (black) to the transition state (red).
It was therefore reasoned that if enzymatic groups were positioned with sufficient rigidity to limit their spatial fluctuations relative to the scale of substrate rearrangement, then binding interactions between these groups and substrate atoms undergoing changes in charge localization and position during catalysis could not be optimal with both the ground state and transition state configurations of the reacting substrate.6,22 Based on these considerations, a substantial portion of enzymatic rate enhancement is classically proposed to result from binding substrates within active site environments that are structurally preorganized to optimize binding interactions with reaction transition states rather than ground states.17–20,22,31 This optimization is illustrated in Scheme 2 for proposed interactions within serine protease24,25 and protein tyrosine phosphatase29,32,33 active sites. Nonetheless, experimental evaluation of whether active site groups are positioned with sufficient rigidity to differentially interact with substrate atoms undergoing 0.1 – 1 Å spatial translations during catalysis remains a fundamental challenge, the significance and history of which we further address below.
Scheme 2.
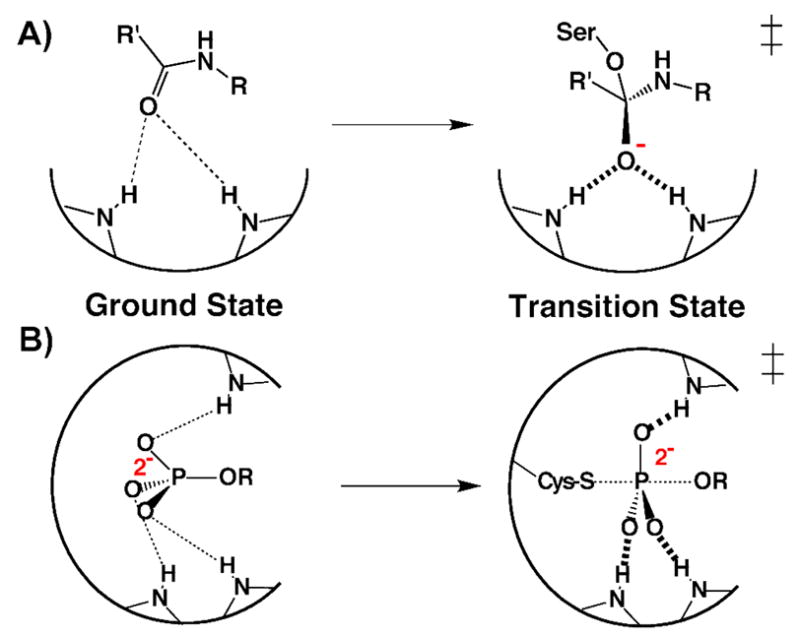
Schematic depiction of proposed optimization of transition state rather than ground state binding within the (A) serine protease and (B) protein tyrosine phosphatase active sites.
Initial proposals regarding the catalytic importance of transition state complementarity envisioned enzymes to have sufficiently rigid active sites such that large forces could be brought to bear on bound substrate molecules in the ground state, straining and deforming them along specific coordinates towards the transition state.18,19,34 Early computational modeling of the lysozyme active site, however, suggested that the forces required to significantly distort the rigid covalent bonds within substrates vastly exceeded the much softer van der Waals forces involved in positioning active site groups. Hence, it appeared physically improbable that sufficient force could be generated by non-covalent binding interactions to significantly distort substrates bound at an enzyme active site.35
Nonetheless, several lines of experimental evidence have suggested that covalent bond distortions on the scale of ~0.5 Å or smaller are possible within an enzyme active site. These studies provide evidence that groups can be positioned with sufficient rigidity to prevent plastic rearrangement of local active site architecture that would otherwise dissipate the local forces required for distortion. High and ultra-high resolution X-ray structures have revealed examples of functional groups within enzyme interiors that appear significantly distorted from canonical geometry, presumably due to non-covalent interactions with nearby residues. In several cases, these structures have been solved to sub-Å resolution with estimated coordinate uncertainties as low as 0.01 Å (unfettered by the tight geometric restraints usually needed in refinements at lower resolutions), permitting detection of subtle geometric distortions less than 0.5 Å that would be difficult to reliably detect in typical X-ray structures.36,37 These examples include distortions of sugar conformations from the chair towards the half-chair forms in 1.9 Å, 1.8 Å, and 0.94 Å resolution structures of glycosidases;38–40 bending of a Phe side chain by 0.3 Å from planarity in a 0.83 Å resolution structure of α-lytic protease;41 arching of the phenolic chromophore by 0.25 Å from planarity in a 0.82 Å structure of photoactive yellow protein;42 bending of the flavin ring by 17° in a 0.98 Å cholesterol oxidase structure;43 and deviations from planarity in peptide bonds by up to 17° in 1.0 Å, 1.1 Å, and 0.66 Å structures of cutinase, elastase, and aldose reductase, respectively.44–46 In addition, resonance Raman studies of the scissile C=O bond in bound substrates of serine proteases have detected vibrational changes indicative of 0.02 – 0.03 Å increases in the C=O bond length, suggesting that oxyanion hole groups and bound substrates are positioned with sufficient rigidity to apply force along specific trajectories and effect subtle geometric distortions.47,48
The ability of active sites to discriminate geometric differences on the 0.1 – 1 Å scale, however, is difficult to reconcile with molecular dynamics (MD) simulations and low temperature X-ray diffraction studies that have revealed that atomic positions within active sites are not static, but vary on the order of 0.1– 0.5 Å due to individual and collective atomic vibrations and librations.49–53 Such intrinsic motions may severely limit the ability of active site groups to distinguish geometric changes in reacting substrates that are of a similar magnitude. It remains possible, however, that limiting motions of active site groups along a specific trajectory may enhance rigidity along the particular coordinate needed for recognition of substrate rearrangement. Indeed, atomic displacements within covalently bonded groups are highly anisotropic due to stretching, rocking and bending motions and limits on rotational flexibility inherent to covalent bonds.51
As these examples from the literature indicate, the scale of geometric discrimination possible within an enzyme active site and the energetic importance of such discrimination remain unresolved questions that are fundamental to our most basic understanding of how enzymes work. In this work, we bring together high resolution X-ray crystallography, 1H and 19F NMR spectroscopy, quantum mechanical calculations, and transition state analog binding measurements to address two critical questions regarding the nature of geometric discrimination possible within a particular enzyme active site: (1) On what distance scale can forces within an active site constrain side chain and ligand relaxation or translation along a specified coordinate? (2) What are the energetic consequences of geometric constraints on this scale?
We have exploited the ability to observe short hydrogen bonds in the oxyanion hole of bacterial ketosteroid isomerase (KSI) spectroscopically to ask whether forces arising from non-covalent binding contacts within the active site can constrain hydrogen bonds from shortening to their preferred bond lengths. Our results strongly suggest that packing interactions within the KSI active site can constrain local side chain reorientation and prevent hydrogen bond shortening by 0.1 Å or less. Furthermore, these constraints appear to have substantial energetic consequences for ligand binding and negative charge stabilization within the oxyanion hole. This work provides evidence that subtle geometric effects indistinguishable by most X-ray structures can have substantial energetic consequences, underscoring the importance of combining structural, spectroscopic, and energetic approaches to dissect enzyme function.
Results and Discussion
KSI and the Experimental Approach
Bacterial KSI from Pseudomonas putida (pKSI) and Commamonas testosteroni (tKSI) catalyzes double-bond isomerization in a wide variety of steroid substrates via formation of a negatively charged dienolate intermediate (Scheme 3A). The intermediate is stabilized by hydrogen bonds within an active site oxyanion hole composed of Y16 (pKSI numbering) and protonated D103 and is surrounded by a dense constellation of predominantly hydrophobic side chains. Single-ring phenolate anions substituted in the meta and para positions with electron withdrawing groups have previously been shown to bind and accept two hydrogen bonds within the KSI oxyanion hole and serve as analogs of the dienolate intermediate and dienolate-like transition states (Scheme 3B).54,55 The binding of a series of phenolates of constant molecular shape but increasing oxyanion charge localization mimics the analogous charge development in the oxyanion hole during steroid isomerization. A 1.25 Å resolution X-ray structure of unsubstituted phenolate bound to pKSID40N (this mutation mimics the protonated general base, D40, present in the KSI-intermediate complex) revealed that the hydrogen bonds formed to the phenolate oxygen were substantially shorter than the vast majority of 2.8 – 3.0 Å hydrogen bonds typically observed in protein X-ray structures,56,57 with average O-O distances of 2.5 and 2.6 Å.55 Consistent with the short distances observed by X-ray crystallography, the protons bridging these two hydrogen bonds appeared as far downfield peaks (14 – 18 ppm) in NMR spectra of tKSID40N•phenolate complexes.54,55
Scheme 3.

(A)Mechanism of KSI catalyzed steroid isomerization. (B) Equilibrium binding of a substituted phenolate from water into the KSI active site. (C) Schematic depiction of a di-ortho-F-substituted phenolate bound at the KSI active site.
The hydrogen bonded protons in the oxyanion hole become progressively deshielded as the pKa and negative charge density on the oxygen of the bound phenolate is increased,58 as expected for proton migration towards the oxyanion acceptor and shortening of the hydrogen bond O•••O distance by ~0.1 Å from pKa 5.4 – 9.3.55 Shortened hydrogen bond lengths are commonly regarded as evidence for hydrogen bond strengthening,59–62 suggesting that shortened hydrogen bonds formed to phenolates of higher pKa might lead to substantially tighter binding to the KSI active site. In contrast to this expectation, the shortening of oxyanion hole hydrogen bonds upon binding phenolates of increased negative charge localization was accompanied by only a shallow increase in phenolate binding affinity (ΔΔG = −0.2 kcal/mol/pKa unit). This modest change indicated little net energetic benefit from the observed hydrogen bond changes.55 However, hydrogen bond shortening was correlated with a large favorable increase in binding enthalpy across the same series of phenolates (ΔΔH = −2.0 kcal/mol/pKa unit). This enthalpic increase is consistent with the simplest expectation for the strengthening of hydrogen bonds upon shortening59 and suggests that physical changes in oxyanion hole hydrogen bonds might nonetheless play an important role in stabilizing negative charge localization within the KSI active site.
To unify these seemingly paradoxical observations, we proposed a physical model for the KSI active site. According to this model, the progressive shortening of oxyanion hole hydrogen bonds provides a favorable enthalpic contribution to binding phenolates of increasing pKa that offsets an entropic penalty derived from localizing negative charge within an active site environment that is otherwise insufficiently preorganized and polar to stabilize localized oxyanion charge better than water. If, as our model specifies, the shortening of oxyanion hole hydrogen bonds by 0.02 Å/pKa unit results in a favorable energetic contribution to phenolate binding, then phenolate analogs that are structurally prevented from forming progressively shortened hydrogen bonds would be predicted to bind more weakly with increasing oxyanion charge (pKa) rather than slightly more strongly. In other words, ΔΔG/pKa unit for phenolate binding is predicted to be positive rather than slightly negative as seen for unhindered phenolates that allow hydrogen bond shortening.
Nevertheless, it remained unclear, given the expectation of anisotropic active site atomic motion on the 0.1 – 0.5 Å scale (see Introduction), whether groups within the KSI active site would be positioned with sufficient rigidity for non-covalent binding interactions to constrain geometric relaxation along the coordinate required for hydrogen bond shortening of ≤0.1 Å. It was additionally unclear whether geometric constraints on this distance scale would be energetically significant. The close packing of groups within the oxyanion hole observed in the pKSID40N•phenolate crystal structure suggested that the presence of ortho substituents on the phenolate ring might interfere with shortening of oxyanion hole hydrogen bonds. We hypothesized that fluoro substitution in both ortho positions of the phenolate ring (Scheme 3C) might be a sufficiently small steric perturbation to permit phenolates to bind and accept hydrogen bonds in the oxyanion hole but prevent further shortening due to close contacts with the electronegative oxygen atoms of Y16 and D103. We utilized a series of substituted phenolates containing –F groups in both ortho positions to test whether binding interactions between the phenolates and active site groups (1) can constrain oxyanion hole hydrogen bonds from shortening with increasing pKa and (2) whether interactions resulting in such a constraint significantly alter the energetics of phenolate binding.
We first determined the crystal structure of 2,6-difluorophenolate bound to pKSID40N to confirm binding and hydrogen bond formation in the oxyanion hole. We then used 1H NMR to assess the change in chemical shift of the observed hydrogen bonded protons with increasing di-ortho-fluorophenolate pKa. If hydrogen bonds were constrained from shortening in the presence of the ortho-F groups, then the chemical shift of the observed proton peaks would vary minimally with increased pKa. Alternatively, if packing within the KSI active site were insufficiently rigid to prevent plastic rearrangement of local architecture and to occlude hydrogen bond shortening, then hydrogen bond proton chemical shifts would increase with phenolate pKa, as observed previously. Finally, we assessed the change in binding affinities for a series of di-ortho-F-phenolates of increasing pKa to probe the energetic significance of any detected physical constraint on phenolate binding and hydrogen bond shortening, predicted from our model to result in weaker binding at higher pKa.
Binding of 2,6-Difluorophenolates in the KSI Oxyanion Hole
UV-VIS spectra of di-ortho-F-substituted phenols (DFPs) bound to tKSID40N confirmed binding as anionic phenolates, as observed previously for phenols substituted in only the meta or para positions (Figure S1, Supporting Information) and as expected for binding in the oxyanion hole.55 To confirm that DFPs bind in the oxyanion hole within hydrogen bonding distance to Y16 and D103, the X-ray structure of pKSID40N co-crystallized with 2,6-difluorophenolate (DFP) was determined (PDB code: 2INX, see Table S1 for complete data collection and refinement statistics). The overall KSI structure obtained at 1.5 Å resolution (Rwork = 18.5% and Rfree = 23.4%), is the same as that observed previously for pKSID40N•phenolate (PDB code: 2PZV), with an RMS deviation of 0.3 Å for backbone atoms between the two structures. The electron density map of the pKSID40N•DFP structure, contoured in Figure 1A at 1.3σ, shows well-defined densities for the modeled atomic positions within the oxyanion hole, with no indication of alternative ligand or side chain orientations.
Figure 1.

Crystal structure of DFP bound at the active site of pKSID40N. (A) Sigma-A weighted 2Fo – Fc electron density map (contoured at 1.3 σ) showing DFP (fluorine atoms in purple) bound in the oxyanion hole, positioned to receive short hydrogen bonds from Tyr16 and Asp103. (B) Superposition of the pKSID40N•DFP (carbon atoms in yellow) and pKSID40N•phenolate structures (carbon atoms in green), highlighting the short O•••O and O•••F distances to DFP. (C) Side-view of the overlay in (B), looking down the phenolate C-O axis and highlighting the different angular displacements for hydrogen bonds formed to the two ligands above and below the plane of the phenolate ring.
The refined positions of the ring and oxygen atoms of the DFP ligand in the X-ray structure are very similar to those observed previously for the bound phenolate ligand, with both oxyanions positioned to receive short hydrogen bonds of ~2.5 and 2.6 Å from the identically oriented side chains of Tyr16 and Asp103 (Figure 1B). Thus, di-ortho-F substitution results in no changes in the observed hydrogen bond O•••O distances or side chain positions within the oxyanion hole detectable at the stated resolutions and 0.06–0.12 Å estimated coordinate error for these two structures. However, reliable detection of geometric changes of 0.1 Å or less would require a coordinate uncertainty of much less than 0.1 Å, as is only achievable in ultra-high resolution X-ray structures refined to <1 Å resolution.63
Substitution of –F for –H in the ortho positions of the phenolate ring increases the bond length from 1.05 Å (C-H) to 1.35 Å (C-F), expands the projection of the van der Waals surface along the C-F bond by 0.6 Å,64 and replaces the slightly electropositive phenyl-hydrogen with the electronegative phenyl-fluorine.65 These changes have previously been reported to subtly perturb the structure and energetics of protein interactions.66–70 The close proximity of the electronegative side chain oxygens of Y16 and D103 to the ortho positions of the phenolate ring (estimated O•••Hortho distances of 2.8 Å for Y16 and 3.1 Å for D103) might be expected to result in significant repulsive interactions upon di-ortho-F substitution. Such electrostatic repulsion has previously been reported in theoretical calculations of water-phenolate complexes between the electronegative oxygen of water and coplanar ortho-F substituents on the phenolate ring.71
While the general orientation of both ligands within the oxyanion hole is very similar, the DFP ring is rotated ~15° about its C-O axis such that the angular displacements of the hydrogen bonds formed to Tyr16 and Asp103 out the plane of the phenolate ring are increased from 15° and 47° in the case of phenolate to 34° and 58° in the case of DFP (Figure 1C). This rotation translates the ortho-F groups 0.3 Å through space from the positions they would occupy in the absence of rotation (relative to pKSID40N phenolate) and increases their distances from the Y16 and D103 oxygens by 0.1 – 0.3 Å. This rearrangement is consistent with a structural reorientation to relieve O•••F repulsive interactions. Further rotation of the phenolate ring appears to be prevented by the close packing of the A118, F86, and D40N side chains and other nearby groups around the bound DFP (Figure 2A). Indeed, quantum calculations of the energy minimized configuration of DFP receiving hydrogen bonds from Y16 and D103 (modeled as 4-Me-phenol and acetic acid, respectively) suggest that, in the absence of additional surrounding groups, the DFP ligand adopts a configuration nearly perpendicular to that observed in the pKSID40N•DFP X-ray structure (Figure 2B). This conformation presumably arises in response to electrostatic repulsion between the partial negative charges on the ortho fluorines and the nearby side chain oxygens.
Figure 2.

Structural features of KSI-bound DFP and the surrounding binding pocket. (A) Van der Waals protein surface (blue) in the oxyanion hole, showing the close packing of groups around the bound DFP (van der Waals surface shown as dots and colored by atom type: yellow, carbon; red, oxygen; purple, fluorine) and side chains of Asp103 and Tyr16 (yellow). (B) Energy minimized gas-phase structure (blue) of DFP receiving hydrogen bonds from Y16 (4-Me-phenol) and D103 (acetic acid), calculated at the B3LYP level using the 6-311++G(d,p) basis set, overlaid with the corresponding side chain and DFP orientations observed in the pKSID40N•DFP X-ray structure (yellow). (C) pKSID40N backbone colored according to the highest atomic B factor observed for each residue. The Asp103 and Tyr16 side chains are shown color-coded by their atomic B-factors. The DFP ligand is in yellow (not color-coded by B-factor).
The invariance of the Y16 and D103 side chain positions in pKSID40N•DFP relative to pKSID40N phenolate (within the ~0.1 Å coordinate uncertainty of the structures) appears to reflect local backbone rigidity and the lack of accessible conformations that can increase O•••F distances without introducing additional unfavorable contacts with neighboring groups or substantially lengthening the hydrogen bond distance with the phenolate oxyanion. Indeed, residues within the oxyanion hole, especially D103 and the dense milieu of hydrophobic groups immediately surrounding it, have the lowest atomic B-factors in the KSI structure (Figure 2C), suggesting a tightly packed environment with limited thermal motions.51 Similarly low B-factors for active site residues are observed in the pKSID40N•phenolate (Figure S2A) and pKSID40N•2-F-phenolate (Figure S2B and below) structures, in agreement with previously reported KSI structures.72 Hence, the observed orientation of DFP bound to KSI represents the conformational state with the most favorable balance of forces involving oxyanion hole hydrogen bond formation, O•••F interactions, and other packing interactions within the KSI active site.
Despite the observed rotation of the DFP ring that appears to increase O•••F distances (as discussed above), the 2.9 Å and 3.2 Å observed distances between the ortho-F atoms and oxygens of Y16 and D103 remain similar to the 3.0 Å sum of the van der Waals radii of F (1.5 Å) and O (1.5 Å).64 This close proximity suggests that repulsive interactions between the partial negative charges on these atoms have not been fully relieved. To test for the presence of significant O•••F interactions in the KSID40N•DFP complex, we acquired 19F NMR spectra of fluorophenolates bound to tKSID40N and free in solution. The 19F NMR chemical shift of a fluorine atom is dominated by the magnetic shielding conferred by its lone pair electrons and provides a sensitive probe of local interactions that perturb the distribution of these electrons and hence alter the chemical shift of the fluorine nucleus.73–75 If repulsive O•••F interactions are present in the KSID40N•DFP complex, then the 19F chemical shifts of the ortho-F atoms are expected to be substantially altered from their values in solution.
Meta and para substituted fluorophenolates bound to KSI display 19F chemical shifts within ~1 ppm of their solution values, consistent with their orientation pointing out of the active site, exposure to solvent waters, and the absence of close packing interactions with nearby residues (Figure 3A and additional fluorophenolate spectra not shown). KSI-bound DFPs, however, display 19F chemical shifts perturbed 5 to 9 ppm from their solution values, as expected for repulsive interactions between the ortho-F atoms and the nearby oxygen atoms of D103 and Y16. The presence of two peaks separated by 4.3 ppm in the 19F NMR spectrum of tKSID40N•3-Me-DFP (Figure 3B) indicates that the two ortho-F atoms experience different local environments when bound within the anisotropic KSI active site proximal to either D103 or Y16.
Figure 3.
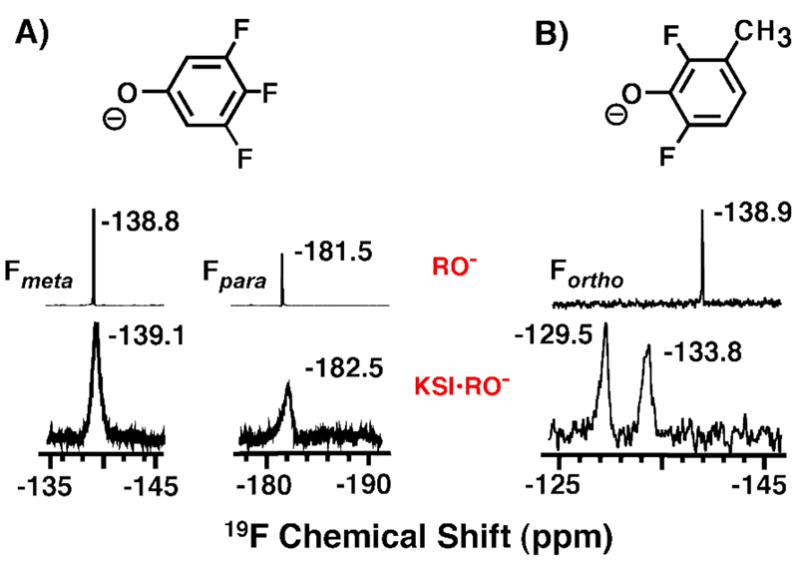
19F NMR spectra of tKSID40N•DFP complexes. (A) 19F NMR chemical shifts of the meta- and para- F atoms in KSI-bound 3,4,5-trifluorophenolate (KSI•RO−) are nearly identical to those of the free phenolate (RO-) in solution at pH 12 (from ref. 55). (B) 19F NMR chemical shifts of the ortho-F atoms in KSI-bound 3-methyl-2,6-difluorophenolate are perturbed downfield from their values when free in solution.
If there are differential interactions between the two ortho-F atoms and the distinct protein groups surrounding them and a corresponding energetic preference for accommodating the ortho-F atom of a bound phenolate nearer to Y16 or D103, then a pehnolate bearing a single ortho-F atom would be expected to bind in the KSI oxyanion hole with the fluorine preferentially oriented towards the side chain giving rise to the least unfavorable interactions. To test this possibility, the X-ray structure of pKSID40N co-crystallized with 2-F-phenol was determined (PDB code: 3CPO, data collection and refinement statistics listed in Table S2). The final structure, obtained at 1.2 Å resolution (Rwork = 16.6% and Rfree = 20.0%), indicates a phenolate binding mode nearly identical to and co-planar with pKSID40N•phenolate, without rotation about the phenolate C-O bond axis and with the single ortho-F atom positioned exclusively on the side of Y16 (Figure 4A & 4B, electron density map shown in Figure S3). This result suggests that the observed rotation in the DFP structure occurs as a consequence of the O•••F contact with the D103 carbonyl oxygen and is consistent with the O•••F contact with Y16 giving rise to a smaller repulsive interaction. Furthermore, this result predicts a smaller 19F chemical shift perturbation for the ortho F atom of DFP nearest Y16 than for the –F atom contacting D103. Indeed, the 19F spectrum of tKSID40N•2-F-4-NO2-phenol (Figure 4C) reveals a single –F peak perturbed 3.3 ppm downfield from its solution chemical shift, similar to the 5 ppm downfield shift of the less perturbed peak in the tKSID40N•3-Me-DFP spectrum (Figure 3B). This result supports assignment of the 19F peak shifted 9.4 ppm downfield and the larger O•••F repulsive interaction to the contact with the D103 carbonyl oxygen.
Figure 4.
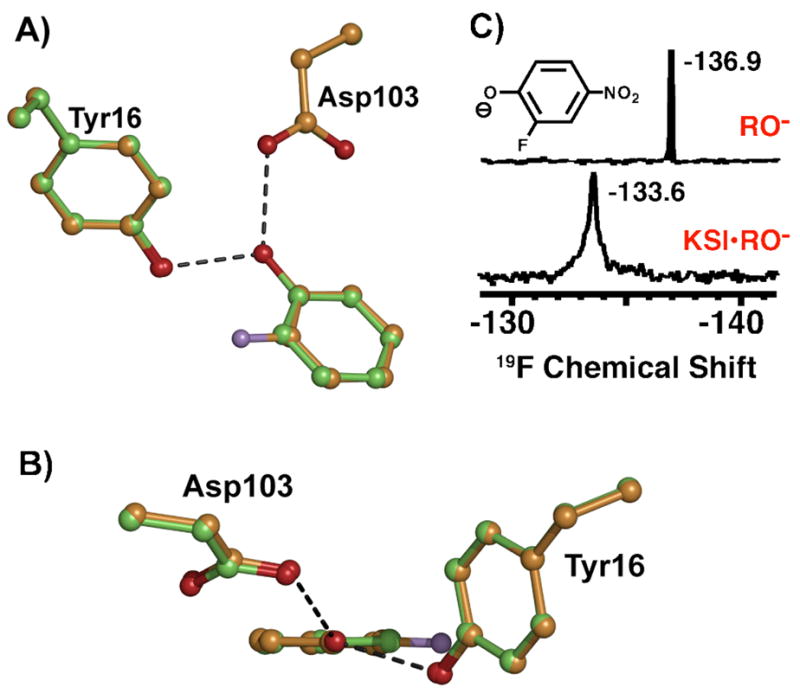
(A) Superposition of the pKSID40N•2-F-phenolate (orange) and pKSID40N•phenolate (green) structures. (B) Side-view of the overlay in (A), looking down the phenolate C-O axis. (C) 19F NMR spectra of 2-F-4-NO2-phenolate free in solution at pH 12 (RO−) and bound to tKSID40N (KSI•RO−).
As stated above, our experimental goal was to verify first that DFPs bind and accept hydrogen bonds in the KSI active site and then ask whether packing within the active site is sufficiently rigid for repulsive forces between nearby groups and the ortho-F atoms to prevent hydrogen bonded O•••O distances from shortening by 0.1 Å or less. The pKSID40N•DFP X-ray structure confirmed that DFPs bind in the KSI oxyanion hole within hydrogen bonding distance of Y16 and D103 but with interactions between the electronegative ortho-F atoms and electronegative side chain oxygens strongly suggested by 19F NMR measurements and computational modeling to be repulsive in nature. We next turned to 1H NMR to detect the hydrogen bonded protons directly and to test whether the O•••F contacts observed in the crystal structure constrain hydrogen bonds formed to the bound DFP from shortening with increasing phenolate pKa.
Hydrogen Bonds Formed to 2,6-Difluorophenolates Are Constrained from Shortening with Increasing Oxyanion Charge Localization
Structural studies of O-H•••O hydrogen bonds in small molecule complexes by low-temperature neutron diffraction have identified that the bridging proton progressively migrates towards the acceptor oxygen as the O•••O separation distance decreases and the donor O-H bond lengthens.59,76,77 Solid state and solution NMR studies of the same complexes have observed that shortened O•••O distances correlate with a more downfield chemical shift of the bridging proton.78–80 This change in proton chemical shift reflects decreased local magnetic shielding as sigma electron density within the O-H bond moves away from the bridging proton as it migrates towards the acceptor oxygen at shortened O•••O distances.81 Decreased hydrogen bond O•••O lengths, enhanced proton migration, and increased downfield chemical shift are expected upon better matching of donor and acceptor proton affinities based on extensive studies of small molecule complexes by crystallography, NMR and IR spectroscopy, and computation.59,60,82–85
Protons involved in hydrogen bonds donated by Y16 and D103 to phenolates substituted only in the meta and para positions have previously been assigned to downfield peaks detected in 1H NMR spectra of tKSID40N•phenolate complexes via H-H NOESY spectra, oxyanion hole mutations, and comparison to model compound chemical shifts.55 These hydrogen bonded proton peaks move progressively more downfield by up to 3 ppm as the phenolate pKa increases from 5.4 to 9.3 (Figure 5A & 5B, black). These changes strongly suggest increased proton migration and a steady shortening of oxyanion hole hydrogen bond O•••O distances by ~0.1 Å in response to increasing charge localization on the phenolate oxygen over the pKa range of 5.4 to 9.3. Given the apparently rigid positioning of the Y16 and D103 backbone and side chains, shortening of hydrogen bond O•••O distances by 0.1 Å is expected to predominantly reflect the combined effects of phenolate C-O bond lengthening with increased oxyanion charge localization and progressive translation of the phenolate deeper into the oxyanion hole. This observation suggests sufficient spatial freedom in the absence of ortho-substitution to permit phenolate relaxation along the coordinate required for hydrogen bond shortening. These physical changes in enzymatic hydrogen bonds upon better matching of donor and acceptor proton affinities conform to the canonical behavior of O-H•••O hydrogen bonds described above.
Figure 5.

1H NMR spectra of tKSID40N•phenolate complexes. (A) Downfield regions of spectra of non-ortho-substituted phenolates (black, from ref. 55), and DFPs (red) bound to KSI at pH 7.2. Note the spectrum for 2,4,6-F3-phenol (pKa 7.2) was acquired at pH 5.8 due to substantial overlap of the hydrogen-bonded peak with the 13 ppm enzymatic peak at pH 7.2 (see Figure S4). (B) Correlation between increasing phenolate pKa and chemical shift of the observed downfield hydrogen bonded proton peak(s). Black symbols represent the two black peaks observed in the non-ortho-substituted phenolate spectra in (A) (and additional spectra not shown; data from ref. 55), and red symbols represent the downfield peak observed in the DFP spectra in (A) (data from Table S3).
To test whether di-ortho-F-phenolates display altered hydrogen bond behavior, we acquired 1H NMR spectra for a series of DFPs ranging in pKa from 5.4 to 7.3 (Figure 5A, red). Unlike non-ortho substituted phenolates, for which the protons of both oxyanion hole hydrogen bonds appear as far downfield peaks, DFP binding gives rise to only one downfield peak >13 ppm. This observation suggests that only one of the two oxyanion hole hydrogen bonds inferred from the short O•••O distances observed in the X-ray structure involves a deshielded proton that has migrated towards the DFP oxygen. The proton bridging the second hydrogen bond may not have significantly migrated towards the DFP oxygen due to sub-optimal orbital overlap with lone pair electrons on the phenolate oxygen owing to the DFP rotation about the C-O bond axis, as described above. In the absence of significant migration, the bridging proton would be expected to have a chemical shift <11 ppm, rendering it undetectable among the very large number of proton resonances observed in the upfield region of the spectrum. It remains possible, although unlikely, that both hydrogen-bonded protons have identical, overlapping resonances for all DFPs tested. Alternatively, the second hydrogen-bonded proton could be significantly deshielded but undetectable due to broadening from rapid exchange with other groups within the active site, although both hydrogen bonded protons are observed for non-ortho phenolates and have similar peak widths. Additional H-H NOESY and low pH NMR spectra tested and confirmed assignment of the observed downfield peak to an oxyanion hole hydrogen bonded proton (Figure S4) but could not further specify whether the peak arises from the Y16 or D103 hydrogen bond. However, the continuous electron density observed in the X-ray structure between the Y16 and DFP oxygens (Figure 1A, visible up to 1.7σ), previously described for linear or nearly linear hydrogen bonds with partial covalent character,86 is consistent with the observed deshielded proton belonging to this hydrogen bond.
In contrast to the hydrogen bond behavior observed for non-ortho substituted phenolates (Figure 5A & 5B, black), the chemical shift of the downfield peak observed upon DFP binding did not increase with increasing phenolate pKa (Figure 5A & 5B, red). The minimal chemical shift variation from the average value of 13.7 ppm strongly suggests that the bridging proton position and O•••O distance within hydrogen bonds formed to DFPs change minimally with increasing phenolate pKa. Thus, forces arising from the close packing interactions and putative repulsive O•••F contacts identified earlier within the oxyanion hole appear to physically prevent hydrogen bonds formed to DFPs from shortening to the distance they would adopt in the absence of the di-ortho-F-subsititution (Scheme 4).
Scheme 4.

Physical model for phenolate binding in the KSI oxyanion hole. (A) Hydrogen bonds formed to meta-and para-substituted phenolates shorten with increasing phenolate charge localization (indicated by larger minus sign). (B) O•••F contacts constrain hydrogen bonds formed to DFPs from shortening with increasing phenolate charge localization.
To test whether this altered hydrogen bond behavior was indeed imposed by constraints within the enzyme active site and not an inherent property of all hydrogen bonds formed to di-ortho-substituted oxyanion acceptors, we characterized the hydrogen bond behavior of small molecule model complexes in solution. We measured the chemical shift of the hydrogen bonded proton formed in chloroform between a series of phenol donors and either 4-nitropyridine-N-oxide or 2,6-dichloropyridine-N-oxide87, oxyanionic acceptors of nearly equivalent pKa. The almost identical chemical shifts observed for hydrogen bonds formed to these acceptors (Figure 6) indicate that di-ortho-halo substitution per se does not perturb the length of a hydrogen bond or prevent it from shortening. These results are consistent with previous solution studies of intermolecular hydrogen bond formation between trifluoroacetic acid and substituted pyridines88 in which di-ortho- and non-ortho-substituted pyridines fell on the same trend line of 1H chemical shift versus pKa. This identical behavior between di-ortho and non-ortho-substituted hydrogen bond acceptors presumably reflects their ability to rotate in solution to avoid unfavorable interactions between the ortho substituents and the hydrogen bond donor. Thus, the apparent constraint to shortening hydrogen bonds formed to DFPs bound to KSI is a property of binding interactions within the enzymatic active site.
Figure 6.
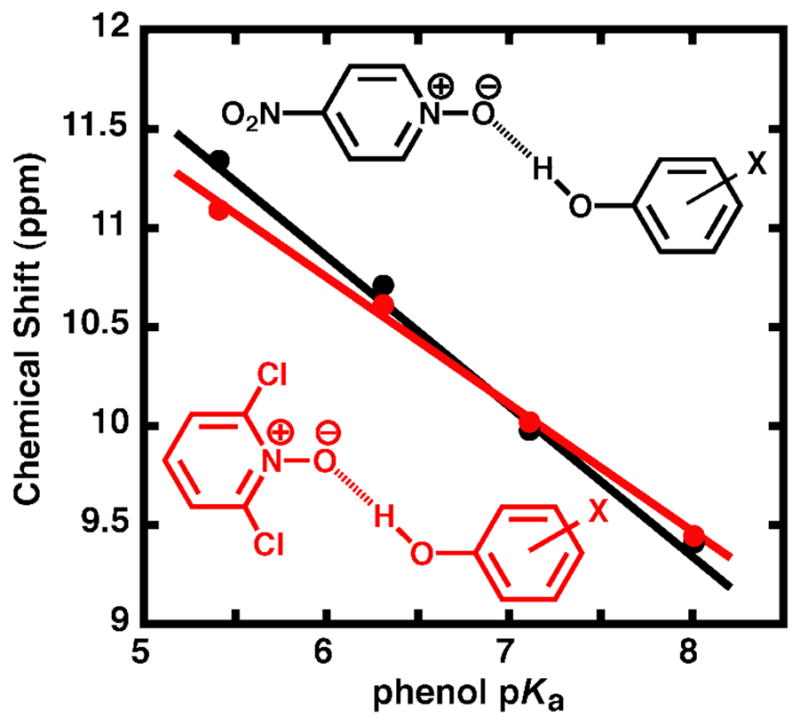
Dependence of the hydrogen bonded proton chemical shift on phenol pKa for a series of phenol donors (3,4-dinitrophenol, 3-CF3-4-nitrophenol, 4-nitrophenol, and 4-cyanophenol) hydrogen-bonded to either 4-nitropyridine-N-oxide (black, pKa = −1.7)89 or 2,6-dichloropyridine-N-oxide (red, pKa = − 2.3)90 in chloroform (data from Table S4).
While it was not possible to test DFPs with pKa values >7.3 bound to KSI, due to the strong inductive electron withdrawal of the -F substituents themselves, a nearly 2 ppm chemical shift difference is observed at pKa 7.3 for DFPs relative to non-ortho-phenolates of equivalent pKa (Figure 5B). Extensive studies of O-H•••O hydrogen bonds in small molecule complexes have identified a strong correlation between the hydrogen bond O•••O distance and the chemical shift of the bridging proton, with the chemical shift increasing from 12 to 20 ppm as the O•••O distance decreases from ~2.70 to 2.45 Å.78–80,91 Å conservative upper limit for an increase in the chemical shift of DFPs of ~0.5 ppm with increasing pKa would correspond to a hydrogen bond shortening of at most 0.02 Å. This behavior is in contrast to the ~2 ppm chemical shift increase observed for non-ortho-phenolates over the same pKa range and calculated 0.07 Å change in hydrogen bond length. Thus, the forces in and surrounding the oxyanion hole can constrain the O•••O distance of hydrogen bonds formed to bound DFPs from shortening over distances on the scale of ~0.1 Å (Scheme 4). Ab initio gas phase calculations on the formate-formic acid dimer suggest that lengthening a single O•••O hydrogen bond 0.1 Å from its optimal distance weakens the hydrogen bond by 1.2 kcal/mol.92 Does constraining KSI oxyanion hole hydrogen bonds formed to DFPs from shortening by up to 0.1 Å substantially weaken phenolate binding? We turned to direct measurement of the change in binding affinity across a series of fluorophenolates of increasing phenolate pKa to directly and quantitatively assess the energetic consequence of di-ortho-F substitution and active site constraints to hydrogen bond shortening.
Constrained Hydrogen Bonding Substantially Weakens Ligand Binding and Negative Charge Stabilization Within the KSI Oxyanion Hole
As discussed earlier, the oxyanion hole binding of a series of meta and para substituted fluorophenolates of increasing pKa and negative charge character resulted in a small increase in binding affinity (ΔΔG) by 0.2 kcal/mol per unit increase in phenolate pKa (Figure 7, open circles).55 If hydrogen bond shortening up to 0.1 Å with increasing phenolate pKa makes a substantially favorable energetic contribution to phenolate binding and negative charge stabilization (which may be nearly offset by concomitant unfavorable dipolar rearrangements within the active site, as discussed earlier),55 then the observed geometric constraints arising from di-ortho-F substitution that prevent hydrogen bond shortening would be predicted to result in weakened phenolate binding at increased pKa. Conversely, if hydrogen bond shortening by this magnitude makes a negligible energetic contribution, then the trend in ΔΔG would be similar to that previously observed with meta and para substituted fluorophenolates.
Figure 7.
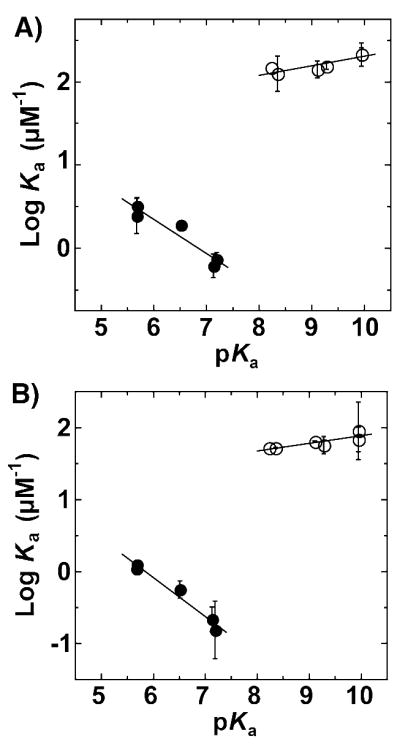
Energetics of fluorophenolate binding to KSID40N. The dependence of affinity (log Ka) on the pKa of non-ortho-fluorophenolates (open circles; from ref. 55) and di-ortho-fluorophenolates (filled circles) for binding to pKSI (A) and tKSI (B). Slopes for pKSI are 0.11 ± 0.03 and −0.41 ± 0.08 and for tKSI are 0.10 ± 0.03 and −0.54 ± 0.06. (Data from Table S5.)
Figure 7 shows the dependence of the association constant (log Ka), determined by a fluorescence competitive binding assay, on the pKa of non-ortho (open circles) or di-ortho (filled circles) fluorophenolates obtained for KSI from two different bacterial sources (Figure 7A & 7B). At a pKa of 7, interactions arising from introduction of the di-ortho-F atoms weakens phenolate binding by 3 kcal/mol relative to that expected from extrapolation for a non-ortho-fluorophenolate of identical pKa. This absolute difference in observed binding reflects the combined and convoluted energetic effects of constrained hydrogen bond shortening, O•••F repulsions, rotation of the DFP ring, and any differential solvation of the ortho F atoms within the active site relative to water. Furthermore, this affinity difference for binding to the KSI active site is in sharp contrast to solution studies of intermolecular hydrogen bond formation between 4-Br-phenol and substituted phenolates in which di-ortho-substituted phenolates fell on the same trend line of log Ka versus pKa as non-ortho-substituted phenolates.93 This identical behavior presumably reflects the ability of di-ortho-phenolates to rotate in solution to avoid unfavorable interactions between the ortho-substituents and hydrogen bond donor groups, as suggested by the quantum calculations presented earlier of the energy-minimized gas-phase structure for hydrogen bond formation to DFP (Figure 2B) and the proton chemical shift behavior of the hydrogen-bonded complexes in solution (Figure 6).
Although the absolute binding affinity (ΔG) of a DFP at any given pKa reflects many complex physical changes within the active site, the change in binding affinity per change in DFP pKa (ΔΔG) is expected to primarily report on those changes within the active site that occur in response to increasing or decreasing negative charge localization on the DFP oxygen. While the dependence of the affinity on pKa (ΔΔG) is shallow and slightly positive for the non-ortho-fluorophenolates (~0.1, ΔΔG = 0.2 kcal/mol/pKa unit), the slope is steeply negative for the di-ortho-fluorophenolates (− 0.4 to − 0.5; ΔΔG = − 0.6 to − 0.7 kcal/mol/pKa unit). This result indicates that, over the pKa ranges studied, increased charge localization on the DFP oxyanion equivalent to a 1 pKa unit increase weakens binding by ~1 kcal/mol relative to phenolates lacking di-ortho-F atoms. Thus, geometric constraints within the KSI active site that occur as a consequence of di-ortho-F substitution and constrain hydrogen bonds formed to the DFP oxygen from shortening up to 0.1 Å substantially impair the ability of the active site to stabilize increased oxyanion charge localization relative to water. This result confirms the prediction made by the KSI active site model presented earlier, in which the systematic shortening of hydrogen bonds formed to the incipient oxyanion of bound phenolates of increasing pKa makes an energetically substantial contribution to negative charge stabilization within the active site.55 Indeed, electrostatic interactions between the phenolate oxygen and preorganized dipoles present within the remainder of the active site are insufficient to off-set the observed weakening in phenolate binding when oxyanion hole hydrogen bonds are constrained from shortening.
Conclusions and Implications
Polanyi, Pauling, Haldane, Jencks, and others recognized that a fundamental difference between carrying out reactions within enzyme active sites and in aqueous solution is the ability of enzymes to exploit non-covalent binding interactions to position substrates with respect to one another and relative to reactive functional groups.17–20 Indeed, the myriad three-dimensional structures of enzyme•substrate complexes solved to date have highlighted the general complementarity between active sites and reactants.94 It has been broadly proposed that enzymes use positioning to achieve selective transition state stabilization by forming stronger binding interactions with the transition state than the ground state, based on geometric changes in the reacting substrate that allow more stabilizing contacts to form in one state than another.6,20,95–98 However, the distance scale on which enzymes can distinguish geometric rearrangement and the energetic significance of any discrimination on that scale remained unanswered questions that are fundamental to our most basic physical understanding of enzyme active sites and catalysis.
Multiple prior studies have used Glu-to-Asp mutations to test the importance of positioning reactive carboxylate groups relative to substrates. Such mutations, which remove a methylene group and perturb the positioning of the carboxylate by ~1 Å,99,100 are associated with reductions in enzyme activity as large as 104-fold.101–104 It was unclear, however, whether geometric changes less than 1 Å in magnitude, such as those occurring within reacting substrates along the reaction coordinate (Scheme 1),30 could be recognized by enzymes and whether such discriminations would be energetically important. Evidence that substantial geometric discrimination on this scale might be possible was provided by the surprisingly large reductions in catalysis observed upon oxygen to sulfur substitutions of key groups in several enzymes, highlighting the potential of the 0.3 Å longer covalent bond and larger van der Waals radius of S relative to O to disrupt the alignment and positioning of active site contacts important for catalysis.21,105–108 Nevertheless, the observed rate effects from O/S substitutions are not solely a reflection of subtle geometric perturbations but can be convoluted with additional effects due to the altered chemical properties of S relative to O.
We have reported herein a selective test of the limits of structural precision and geometrical discrimination possible within the active site of a particular enzyme, bacterial ketosteroid isomerase. The results provide strong evidence that forces arising from non-covalent packing and binding interactions within this active site can constrain the positioning of a bound phenolate ligand and contacting side chains with a precision of ±0.1 Å or better. These constraints are sufficient to prevent relaxation of side chain and ligand positions along the specific coordinate required for shortening of oxyanion hole hydrogen bond O•••O distances by ~0.1 Å upon increased charge localization. The structural rigidity within the KSI active site that enables geometric discrimination on this scale appears to derive from tight packing of hydrophobic groups around D103 and Y16 that limits the rotational rearrangement and thermal mobility of these groups, coupled with structural constraints in the substrate binding pocket. While the amplitude of zero-point bond vibrations, which represent the minimum temperature independent mobility of covalently bonded atoms, are on the same scale as the ~0.1 Å geometric constraints observed in KSI, vibrational modes are highly anisotropic and rotational modes can be highly constrained.51,52 More limited atomic motions on scales less than 0.1 Å within the oxyanion hole along specific trajectories orthogonal to allowed motional modes may enhance rigidity along the coordinate needed to constrain hydrogen bond shortening by up to 0.1 Å. Local flexibility may be further dampened by simultaneous motions of neighboring atoms towards each other that result in unfavorable van der Waals overlap, thereby limiting the amplitude of motions in a particular direction. Such motions are sometimes referred to as “anticorrelated”,109 and similar effects for the KSI active site have been reported based on MD simulations.110
Geometric constraints in KSI that prevent side chain and ligand relaxation by ~0.1 Å have substantial energetic consequences. Indeed, increased charge localization on the phenolate oxygen equivalent to a 1 pKa unit increase weakens binding by ~1 kcal/mol relative to phenolates lacking di-ortho-F atoms. This energetic difference over the limited pKa range tested suggests that over a much larger pKa range of 10–15 units, such as that traversed going from the ground state to transition state during steroid isomerization, the presence or absence of geometric constraints akin to those resulting in this system from di-ortho-F substitution could lead to differential stabilization energies and binding affinities of several kcal/mol.
While the specific physical interactions underlying the geometric constraints described in this study are distinct from interactions during catalysis by KSI, they are conceptually related to the types of geometric discriminations that have been proposed to make significant contributions to selective transition state stabilization in KSI,55 serine proteases and other oxyanion hole containing hydrolases,24–26,111,112 and phosphoryl transfer enzymes.29,32,33,113 In all of these cases, the rigid positioning of active site residues has been proposed to constrain favorable interactions from forming in the ground state but permit them to form in the transition state once geometric constraints have been relieved due to changes in the hybridization of substrate atoms.
In the case of KSI, structural features of the oxyanion hole suggest that hydrogen bond formation to the reacting substrate is geometrically optimal in the transition state but not in the ground state. During steroid isomerization, the hybridization of the substrate oxygen changes from a planar sp2 carbonyl to a tetrahedral sp3 dienolate (Scheme 3A), altering the spatial distribution of its lone pair electrons. This reorientation of atomic orbitals about the substrate oxygen alters its geometric preference for accepting hydrogen bonds. Whereas the ground state carbonyl preferentially accepts hydrogen bonds within the sp2 orbital plane of the carbonyl group, optimal orbital overlap and hydrogen bond formation to the dienolate intermediate occurs above and below the plane of the steroid and C-O bond. However, the D103 and Y16 side chains appear consistently positioned above and below the plane of the steroid and C-O bond in previously published structures with bound transition state analogs55,114 (Figure 1C) and are identically oriented in structures of unliganded KSI.115 This invariance of D103 and Y16 orientations is consistent with tight packing of the surrounding hydrophobic residues and rigid constraints on side chain positioning. Empirically-derived and quantum mechanical potential energy functions for hydrogen bond formation to oxygen acceptors suggest that significant energetic discrimination may be possible from positioning hydrogen bond donors within the KSI oxyanion hole for optimal interaction with the transition state rather than the ground state.116,117
One of the most prominent and widely-discussed examples of an enzyme that achieves a remarkable degree of rate enhancement is OMP decarboxylase. This enzyme appears to exclusively use non-covalent binding interactions within its active site, and its 1017-fold rate enhancement ranks among the largest measured to date for any enzyme.118 However, despite numerous X-ray structures, extensive computational modeling, and a tremendous breadth of mutagenesis and other mechanistic dissections of this enzyme, substantial uncertainty and controversy remain regarding the physical origin of the prolific rate enhancement achieved by this enzyme.97,119–123 It seems improbable that a single catalytic mechanism provides the entirety of the observed rate enhancement, for this or any other enzyme. Nonetheless, we suggest that geometric discrimination of substrate rearrangements along the reaction coordinate, such as differential interactions between K93 and the oxygens of the substrate’s reactive carboxylate group due to changes in position, hybridization, and orbital geometry going from the ground state to the transition state, may account for some of the prodigious yet puzzling selective transition state stabilization achieved by this enzyme.
Favorable features of KSI permitted a selective test of the scale of geometrical discrimination and accompanying energetic consequences possible in this particular enzyme active site. Multi-faceted structural, spectroscopic, computational, and energetic tests of enzyme function, similar to the approaches taken herein, will be necessary to test the generality of our conclusions, to broaden our understanding of geometric effects in other enzymatic systems, and ultimately to garner a full understanding of the catalytic power of enzymes.
Methods
Materials
All reagents were of the highest purity commercially available (≥97%). Substituted phenols and pyridine-N-oxides were purchased from Sigma-Aldrich, Oakwood Products, and Acros Organics. All buffers were prepared with reagent grade chemicals or better.
KSI Expression and Purification
KSI was expressed and purified as previously described.55 Final purity was >99% as estimated from a Coomassie-stained SDS-PAGE gel, and protein concentration was determined using the calculated molar extinction coefficient in 6 M guanidium chloride.124
Absorbance Spectra of KSI•2,6-difluorophenolate Complexes
Absorbance spectra of substituted 2,6-F2-phenolates were acquired in a microcuvette with a Uvikon 9310 absorbance spectrophotometer. Spectra of 50 μM phenols in 10 mM HCl (pH 2), and 10 mM NaOH (pH 12) were compared with the spectrum of the same concentration phenol in 40 mM KPi buffer, pH 7.2, in the presence of 300 μM tKSID40N (phenol >95% bound, data not shown) after subtraction of the free enzyme spectrum.
KSI X-ray Crystallography
pKSID40N•2,6-difluorophenolate and pKSID40N•2-fluorophenolate co-crystals in space group C2221 were obtained using the hanging drop vapor diffusion method by mixing 2 μl of pKSID40N at 25 mg/ml and 2 μl of reservoir solution (1.4 M ammonium sulfate, 6.5 % (v/v) 2-propanol, and 2–3 mM 2,6-difluorophenol or 2-fluorophenol at pH 7.0). Cube-shaped crystals measuring ~0.4 mm × 0.4 mm × 0.3 mm appeared after one week of incubation at room temperature. Cryo-protection was achieved by first soaking the crystals in a solution of mother liquor diluted 1:1 with 2.9 M sodium malonate (pH 7.0), then transferring the crystals directly into the 2.9 M sodium malonate solution. Crystals were shock-cooled by immersion into liquid nitrogen. Diffraction data from single crystals maintained at 100 K were collected for pKSID40N•2,6-difluorophenolate at beamline 14-BMC of the Advanced Photon Source (Argonne National Laboratory, Argonne, Illinois, USA) and for pKSID40N•2-fluorophenolate at beamline BL9-1 of the Stanford Synchrotron Radiation Laboratory (Stanford Linear Accelerator Center, Stanford, CA, USA). Data were integrated and scaled using the HKL2000 program package.125 An initial model of the protein was obtained by the molecular replacement method with the program PHASER,126 using the coordinates of a previously reported structure of pKSID40N (PDB code: 2PZV). Refinement and manual rebuilding were carried out using the programs REFMAC5127 and COOT,128 respectively. Model quality was assessed using the program PROCHECK [5] and the Ramachandran plot showed no residues outside the allowed regions. Data and model statistics are included as Table S1 (pKSID40N•2,6-difluorophenolate) and Table S2 (pKSID40N•2-fluorophenolate) in Supporting Information. Structural coordinates have been deposited in the RCSB Protein Data Bank (http://www.rcsb.org/pdb) under accession codes 2INX (pKSID40N•2,6-difluorophenolate) and 3CPO (pKSID40N•2-fluorophenolate).
KSI NMR Spectroscopy
1H NMR spectra of KSI were acquired at the Stanford Magnetic Resonance Laboratory (SMRL) on an 800-MHz Varian UNITYINOVA spectrometer running VNMR v6.1C and equipped with a 5-mm, triple resonance, gradient 1H(13C/15N) probe. NMR samples consisted of 0.5–2.0 mM tKSID40N and 0.5–5.0 mM substituted phenol, in 40 mM KPi buffer (pH 7.2), 1 mM EDTA, 2 mM DTT, and 10% (v/v) DMSO-d6 (which served as the deuterium lock solvent and prevented freezing at subzero temperatures) in 5 mM Shigemi symmetrical microtubes at −3.0 ± 0.5 °C. One-dimensional proton spectra were acquired using the 1331 binomial pulse sequence129 to suppress the water signal, with a spectral width of 30 ppm (carrier frequency set on the water resonance) and an excitation maximum of 14–17 ppm. Data were collected with 32,000 points and a 1.9 s recycle delay for 512–5120 scans and processed using a 10-Hz line broadening, with a baseline correction applied over the peaks of interest. Chemical shifts were referenced internally to the water resonance (5.1 ppm at −3 °C) and externally to a sample of sodium-3-trimethylsilylpropionate-2,2,3,3-d4 (0 ppm) in the same buffer conditions and were reproducible to ±0.1 ppm in independent samples.130 As previously described, spectra were consistent with slow exchange on the NMR timescale.55
Two dimensional 1H-1H NOESY spectra were acquired using the SS-NOESY pulse sequence to minimize saturation of protons exchanging with bulk water.131 Data were acquired over 23.8 ppm spectral widths of 2048 data points with a 50 ms mixing time, 1.55 second recycle delay, and 64 scans per t1 increment over 256 increments. Spectra were processed using shifted, squared sine bell windows functions in both dimensions and one-fold zero filling in the t1 dimension and viewed with the program SPARKY.132
One dimensional 19F spectra of tKSID40N-bound F-phenolates were acquired on a 500-MHz Varian UNITYINOVA NMR (operating at 470.23-MHz) running VNMR v6.1c and equipped with a 5 mm PFG switchable probe operating at ambient temperature (20 °C). 10% (v/v) D2O served as the lock solvent, and 19F chemical shifts were referenced to an external standard of trifluoroacetic acid (−76.1 ppm relative to CFCl3). Spectra for 20 mM free phenols were obtained (12 scans) in 10 mM NaOH (pH 12), condition in which each phenol is present as the ionized phenolate. 200–4000 scans were obtained for each tKSID40N-bound phenol under conditions in which the phenol was >95% bound (40 mM KPi, 1 mM EDTA, 2 mM DTT, 1.5 mM enzyme, 1 mM phenol, data not shown) and processed using a 10 Hz line broadening with a baseline correction applied over the observed peaks.
1H NMR of Small Molecule Hydrogen Bonded Complexes
1H NMR spectra were acquired at −45 °C on a 400 MHz Varian Mercury NMR equipped with a 5 mm PFG auto-switchable 4-nucleus (13C/31P-1H/19F) probe and running VNMR v6.1c. Samples of 1:1 hydrogen-bonded complexes (50 mM) were prepared in CDCl3 (with 0.05% TMS for referencing) and contained either 4-NO2-pyridine-N-oxide or 2,6-Cl2-pyridine-N-oxide as the hydrogen bond acceptor and 3,4-(NO2)2-phenol, 3-CF3-4-NO2-phenol, 4-NO2-phenol, or 4-CN-phenol as the hydrogen bond donor. Spectra were taken (25–100 scans) for each hydrogen bonded complex and the data were processed using a line-broadening of 5 Hz.
Determination of Phenolate Affinities for KSI
Fluorophenolate affinities for KSI were determined using a fluorescence competition binding assay described previously.55 Briefly, the KSI affinity of EqA488-1, the KSI reaction intermediate analog equilenin conjugated to Alexa Fluor 488, was determined by titration with enzyme using a FluoroLog-3 spectroflourometer to monitor quenching of the Alexa Fluor fluorescence upon binding to KSI (excitation at 480 nm, emission at 515 nm, bandpass of 10 nm and 14.7 nm, respectively, and 0.1 nM EqA488-1 in a 45 μL microcuvette). Enzyme concentration (typically 0.1 – 100 nM) was varied above and below the Kd for the ligand (~1 nM for EqA488-1). After correcting for background fluorescence, the observed EqA488-1 fluorescence as a function of enzyme concentration was fit to a quadratic binding isotherm using nonlinear regression analysis to determine the EqA488-1 affinity for KSI, as previously described.55 The EqA488-1 Kd of 1.0 nM determined by this method was the same as previously reported.55
To determine the observed KSI affinities for the series of 2,6-F2-phenol ligands (with varying fluorine substitution at the other ring positions) by competition, EqA488-1 was displaced from the active site of KSI by the addition of the substituted 2,6-F2-phenol to a mixture of the enzyme (with concentration 1 to 4-fold above the EqA488-1 Kd) and EqA488-1 (0.1 nM). The observed fluorescence as a function of phenol concentration was fit to a quadratic binding isotherm and the KSI affinity of the 2,6-F2-phenol ligand under assay conditions as well as the pH-independent KSI affinity of the substituted 2,6-F2-phenolate were determined as previously described.55 Error bars are the average deviation of 2–6 independent assays.
Quantum Mechanical Calculations
Energy minimization and geometry optimization of the isolated 2,6-F2-phenolate ligand (DFP) receiving hydrogen bonds from the Y16 and protonated D103 side chains were performed with the B3LYP density functional method and the 6-311++G(d,p) basis set using the Gaussian 03 software package.133 Initial side chain and ligand coordinates were obtained from the pKSID40N•phenolate X-ray structure (PDB code: 2PVZ). The side chains were each truncated at their respective Cβ atoms by converting the methylene group into a methyl group, thereby modeling Y16 as 4-Me-phenol and D103 as acetic acid, and –F atoms were added to both ortho positions of the phenolate ligand. Side chain positions were fixed with respect to each other while the DFP position and orientation were permitted to vary during all calculations.
Acknowledgments
We thank Corey Liu and Steve Lynch for assistance with NMR experiments; Tzanko Doukov for help with crystallographic data collection at SSRL; Dave Agard, Steve Boxer, Aaron Fafarman, Pehr Harbury, Chaitan Khosla, Judith Klinman, Jonathan Lassila, and Jason Schwans for helpful discussions; and members of the Herschlag lab for comments on the manuscript. This work was funded by an NSF grant to DH, DR, and GAP (MCB-0641393). PAS and DAK were supported in part by HHMI Predoctoral Fellowships. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357. Use of the Stanford Magnetic Resonance Laboratory was supported in part by the Stanford University School of Medicine. Use of the Stanford Synchrotron Radiation Laboratory was supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
Footnotes
Supporting Information Available: Complete reference 133, UV absorbance spectra for tKSID40N•DFP complexes (Figure S1), Atomic B-factors for the pKSID40N•phenolate (Figure S2A) and pKSID40N•2-F-phenolate (Figure S2B) structures, electron density map of pKSID40N•2-F-phenolate oxyanion hole (Figure S3), 1H-1H NOESY and low pH NMR spectra for tKSID40N•phenolate complexes (Figure S4), data collection and crystallographic refinement statistics for the pKSID40N•2,6-F2-phenolate (Table S1) and pKSID40N•2-F-phenolate (Table S2) X-ray structures, 1H NMR chemical shifts of downfield peaks in spectra of tKSID40N•DFP complexes (Table S3), 1H NMR chemical shifts of hydrogen-bonded proton observed for complexes between substituted phenols and 4-nitropyridine-N-oxide or 2,6-dichloropyridine-N-oxide (Table S4), and binding affinities of DFPs to KSID40N determined by fluorescence (Table S5). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lad C, Williams NH, Wolfenden R. Proc Natl Acad Sci. 2003;100:5607–5610. doi: 10.1073/pnas.0631607100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bolon DN, Voigt CA, Mayo SL. Curr Opin Chem Biol. 2002;6:125–129. doi: 10.1016/s1367-5931(02)00303-4. [DOI] [PubMed] [Google Scholar]
- 3.Jiang L, Althoff EA, Clemente FR, Doyle L, Rothlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas CF, 3rd, Hilvert D, Houk KN, Stoddard BL, Baker D. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaplan J, DeGrado WF. Proc Natl Acad Sci. 2004;101:11566–11570. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lippow SM, Tidor B. Curr Opin Biotechnol. 2007;18:305–311. doi: 10.1016/j.copbio.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fersht AR. Structure and Mechanism in Protein Science. 2. W.H. Freeman and Company; New York: 1999. [Google Scholar]
- 7.Cohen BE, McAnaney TB, Park ES, Jan YN, Boxer SG, Jan LY. Science. 2002;296:1700–1703. doi: 10.1126/science.1069346. [DOI] [PubMed] [Google Scholar]
- 8.Warshel A, Aqvist J, Creighton S. Proc Natl Acad Sci. 1989;86:5820–5824. doi: 10.1073/pnas.86.15.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sigala PA, Fafarman AT, Bogard PE, Boxer SG, Herschlag D. J Am Chem Soc. 2007;129:12104–12105. doi: 10.1021/ja075605a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Macgregor RB, Weber G. Nature. 1986;319:70–73. doi: 10.1038/319070a0. [DOI] [PubMed] [Google Scholar]
- 11.Perutz MF. Science. 1978;201:1187–1191. doi: 10.1126/science.694508. [DOI] [PubMed] [Google Scholar]
- 12.Mertz EL, Krishtalik LI. Proc Natl Acad Sci. 2000;97:2081–2086. doi: 10.1073/pnas.050316997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simonson T, Archontis G, Karplus M. J Phys Chem B. 1999;103:6142–6156. [Google Scholar]
- 14.Tanaka K, Mackay GI, Payzant JD, Bohme DK. Can J Chem. 1976;54:1643–1659. [Google Scholar]
- 15.Crosby J, Stone R, Lienhard GE. J Am Chem Soc. 1970;92:2891. doi: 10.1021/ja00712a048. [DOI] [PubMed] [Google Scholar]
- 16.Chabinyc ML, Craig SL, Regan CK, Brauman JI. Science. 1998;279:1882–1886. doi: 10.1126/science.279.5358.1882. [DOI] [PubMed] [Google Scholar]
- 17.Polanyi M. Z Elektrochem. 1921;27:142–150. [Google Scholar]
- 18.Pauling L. Chem Eng News. 1946;24:1375–1377. [Google Scholar]
- 19.Haldane JBS. Enzymes. Longmans Green; London: 1930. [Google Scholar]
- 20.Jencks WP. Catalysis in Chemistry and Enzymology. 2. Dover; New York: 1987. [Google Scholar]
- 21.Wolfenden R. Acc Chem Res. 1972;5:10–18. [Google Scholar]
- 22.Lienhard GE. Science. 1973;180:149–154. doi: 10.1126/science.180.4082.149. [DOI] [PubMed] [Google Scholar]
- 23.Blake CC, Johnson LN, Mair GA, North AC, Phillips DC, Sarma VR. Proc R Soc Lond B Biol Sci. 1967;167:378–388. doi: 10.1098/rspb.1967.0035. [DOI] [PubMed] [Google Scholar]
- 24.Robertus JD, Kraut J, Alden RA, Birktoft JJ. Biochemistry. 1972;11:4293–4303. doi: 10.1021/bi00773a016. [DOI] [PubMed] [Google Scholar]
- 25.Fersht AR, Blow DM, Fastrez J. Biochemistry. 1973;12:2035–2041. doi: 10.1021/bi00735a002. [DOI] [PubMed] [Google Scholar]
- 26.Fersht AR. Proc R Soc Lond B Biol Sci. 1974;187:397–407. doi: 10.1098/rspb.1974.0084. [DOI] [PubMed] [Google Scholar]
- 27.Wilmouth RC, Edman K, Neutze R, Wright PA, Clifton IJ, Schneider TR, Schofield CJ, Hajdu J. Nat Struct Biol. 2001;8:689–694. doi: 10.1038/90401. [DOI] [PubMed] [Google Scholar]
- 28.Katona G, Wilmouth RC, Wright PA, Berglund GI, Hajdu J, Neutze R, Schofield CJ. J Biol Chem. 2002;277:21962–21970. doi: 10.1074/jbc.M200676200. [DOI] [PubMed] [Google Scholar]
- 29.Alhambra C, Wu L, Zhang ZY, Gao J. J Am Chem Soc. 1998;120:3858–3866. [Google Scholar]
- 30.Schramm VL, Shi WX. Curr Opin Str Biol. 2001;11:657–665. doi: 10.1016/s0959-440x(01)00269-x. [DOI] [PubMed] [Google Scholar]
- 31.Wolfenden R. Annu Rev Biophys Bioeng. 1976;5:271–306. doi: 10.1146/annurev.bb.05.060176.001415. [DOI] [PubMed] [Google Scholar]
- 32.Zhang ZY, Wang Y, Wu L, Fauman EB, Stuckey JA, Schubert HL, Saper MA, Dixon JE. Biochemistry. 1994;33:15266–15270. doi: 10.1021/bi00255a007. [DOI] [PubMed] [Google Scholar]
- 33.Zhang M, Zhou M, Van Etten RL, Stauffacher CV. Biochemistry. 1997;36:15–23. doi: 10.1021/bi961804n. [DOI] [PubMed] [Google Scholar]
- 34.Lumry R. Ann N Y Acad Sci. 1974;227:46–73. doi: 10.1111/j.1749-6632.1974.tb14373.x. [DOI] [PubMed] [Google Scholar]
- 35.Levitt M. In: Peptides, polypeptides, and proteins. Blout ER, Bovey FA, Goodman M, Lotan N, editors. Wiley; New York: 1974. pp. 99–113. [Google Scholar]
- 36.Vrielink A, Sampson N. Curr Opin Struct Biol. 2003;13:709–715. doi: 10.1016/j.sbi.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 37.Cachau RE, Podjarny AD. J Mol Recognit. 2005;18:196–202. doi: 10.1002/jmr.738. [DOI] [PubMed] [Google Scholar]
- 38.Kuroki R, Weaver LH, Matthews BW. Science. 1993;262:2030–2033. doi: 10.1126/science.8266098. [DOI] [PubMed] [Google Scholar]
- 39.Sidhu G, Withers SG, Nguyen NT, McIntosh LP, Ziser L, Brayer GD. Biochemistry. 1999;38:5346–5354. doi: 10.1021/bi982946f. [DOI] [PubMed] [Google Scholar]
- 40.Guerin DMA, Lascombe MB, Costabel M, Souchon H, Lamzin V, Beguin P, Alzari PM. J Molec Biol. 2002;316:1061–1069. doi: 10.1006/jmbi.2001.5404. [DOI] [PubMed] [Google Scholar]
- 41.Fuhrmann CN, Kelch BA, Ota N, Agard DA. J Mol Biol. 2004;338:999–1013. doi: 10.1016/j.jmb.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 42.Getzoff ED, Gutwin KN, Genick UK. Nat Struct Biol. 2003;10:663–668. doi: 10.1038/nsb958. [DOI] [PubMed] [Google Scholar]
- 43.Lyubimov AY, Heard K, Tang H, Sampson NS, Vrielink A. Protein Sci. 2007;16:2647–2656. doi: 10.1110/ps.073168207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Longhi S, Czjzek M, Lamzin V, Nicolas A, Cambillau C. J Mol Biol. 1997;268:779–799. doi: 10.1006/jmbi.1997.1000. [DOI] [PubMed] [Google Scholar]
- 45.Wurtele M, Hahn M, Hilpert K, Hohne W. Acta Crystallogr, Sect D: Biol Crystallogr. 2000;56:520–523. doi: 10.1107/s0907444900000299. [DOI] [PubMed] [Google Scholar]
- 46.Howard EI, Sanishvili R, Cachau RE, Mitschler A, Chevrier B, Barth P, Lamour V, Van Zandt M, Sibley E, Bon C, Moras D, Schneider TR, Joachimiak A, Podjarny A. Proteins: Struct Funct Bioinf. 2004;55:792–804. doi: 10.1002/prot.20015. [DOI] [PubMed] [Google Scholar]
- 47.Tonge PJ, Carey PR. Biochemistry. 1992;31:9122–9125. doi: 10.1021/bi00153a002. [DOI] [PubMed] [Google Scholar]
- 48.Whiting AK, Peticolas WL. Biochemistry. 1994;33:552–561. doi: 10.1021/bi00168a021. [DOI] [PubMed] [Google Scholar]
- 49.Frauenfelder H, Petsko GA, Tsernoglou D. Nature. 1979;280:558–563. doi: 10.1038/280558a0. [DOI] [PubMed] [Google Scholar]
- 50.Artymiuk PJ, Blake CC, Grace DE, Oatley SJ, Phillips DC, Sternberg MJ. Nature. 1979;280:563–568. doi: 10.1038/280563a0. [DOI] [PubMed] [Google Scholar]
- 51.Ringe D, Petsko GA. Methods Enzymol. 1986;131:389–433. doi: 10.1016/0076-6879(86)31050-4. [DOI] [PubMed] [Google Scholar]
- 52.Karplus M, McCammon JA. Annu Rev Biochem. 1983;52:263–300. doi: 10.1146/annurev.bi.52.070183.001403. [DOI] [PubMed] [Google Scholar]
- 53.Ladner JE, Wladkowski BD, Svensson LA, Sjolin L, Gilliland GL. Acta Crystallogr, Sect D: Biol Crystallogr. 1997;53:290–301. doi: 10.1107/S090744499601582X. [DOI] [PubMed] [Google Scholar]
- 54.Petrounia IP, Pollack RM. Biochemistry. 1998;37:700–705. doi: 10.1021/bi972262s. [DOI] [PubMed] [Google Scholar]
- 55.Kraut DA, Sigala PA, Pybus B, Liu CW, Ringe D, Petsko GA, Herschlag D. PLoS Biol. 2006;4:e99. doi: 10.1371/journal.pbio.0040099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anderson S, Crossoon S, Moffat K. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:1008–1016. doi: 10.1107/S090744490400616X. [DOI] [PubMed] [Google Scholar]
- 57.Fuhrmann CN, Daugherty MD, Agard DA. J Am Chem Soc. 2006;128:9086–9102. doi: 10.1021/ja057721o. [DOI] [PubMed] [Google Scholar]
- 58.The correlation between phenolate pKa and the amount of negative charge localized on the phenolate oxygen is supported by quantum calculations (Gross, K. C., Seybold, P. G. Int. J. of Quant. Chem. 2001, 85, 569–579 and Gross, K. C., Seybold, P. G., Hadad, C. M. Int. J. Quant. Chem. 2002, 90, 445–458) and the 0.01 Å decrease in C-O bond length per unit decrease in phenolate pKa observed in phenolate X-ray structures in the Cambridge Database (unpublished analysis), as expected for increased C-O bond order from withdrawal of electron density from the phenolate oxygen into the ring and substituent groups.
- 59.Hibbert F, Emsley J. Adv Phys Org Chem. 1990;26:255–379. [Google Scholar]
- 60.Kumar GA, McAllister MA. J Org Chem. 1998;63:6968–6972. doi: 10.1021/jo980759h. [DOI] [PubMed] [Google Scholar]
- 61.Cleland WW, Kreevoy MM. Science. 1994;264:1887. doi: 10.1126/science.8009219. [DOI] [PubMed] [Google Scholar]
- 62.Frey PA, Whitt SA, Tobin JB. Science. 1994;264:1927. doi: 10.1126/science.7661899. [DOI] [PubMed] [Google Scholar]
- 63.Ladd MFC, Palmer RA. Structure Determination by X-Ray Crystallography. Kluwer Academic/Plenum Publishers; New York: 2003. [Google Scholar]
- 64.Bondi A. J Phys Chem. 1964;68:441–451. [Google Scholar]
- 65.Auffinger P, Hays FA, Westhof E, Ho PS. Proc Natl Acad Sci. 2004;101:16789–16794. doi: 10.1073/pnas.0407607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xiao G, Parsons JF, Tesh K, Armstrong RN, Gilliland GL. J Mol Biol. 1998;281:323–339. doi: 10.1006/jmbi.1998.1935. [DOI] [PubMed] [Google Scholar]
- 67.Pal PP, Bae JH, Azim MK, Hess P, Friedrich R, Huber R, Moroder L, Budisa N. Biochemistry. 2005;44:3663–3672. doi: 10.1021/bi0484825. [DOI] [PubMed] [Google Scholar]
- 68.Hammond SJ, Birdsall B, Feeney J, Searle MS, Roberts GC, Cheung HT. Biochemistry. 1987;26:8585–8590. doi: 10.1021/bi00400a014. [DOI] [PubMed] [Google Scholar]
- 69.Phillips RS, VonTersch RL, Secundo F. Eur J Biochem. 1997;244:658–663. doi: 10.1111/j.1432-1033.1997.00658.x. [DOI] [PubMed] [Google Scholar]
- 70.Park YW, Luo JK, Schultz PG, Kirsch JF. Biochemistry. 1997;36:10517–10525. doi: 10.1021/bi970298e. [DOI] [PubMed] [Google Scholar]
- 71.Andrea TA, Dietrich SW, Murray WJ, Kollman PA, Jorgensen EC, Rothenberg S. J Med Chem. 1979;22:221–232. doi: 10.1021/jm00189a002. [DOI] [PubMed] [Google Scholar]
- 72.Oh KS, Cha SS, Kim DH, Cho HS, Ha NC, Choi G, Lee JY, Tarakeshwar P, Son HS, Choi KY, Oh BH, Kim KS. Biochemistry. 2000;39:13891–13896. doi: 10.1021/bi001629h. [DOI] [PubMed] [Google Scholar]
- 73.Gerig JT. Methods Enzymol. 1989;177:3–23. doi: 10.1016/0076-6879(89)77003-8. [DOI] [PubMed] [Google Scholar]
- 74.Danielson MA, Falke JJ. Annu Rev Biophys Biomol Struct. 1996;25:163–195. doi: 10.1146/annurev.bb.25.060196.001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lau EY, Gerig JT. J Am Chem Soc. 2000;122:4408–4417. [Google Scholar]
- 76.Steiner T, Saenger W. Acta Crystallogr, Sect B: Struct Sci. 1994;50:348–357. [Google Scholar]
- 77.Gilli P, Bertolasi V, Ferretti V, Gilli G. J Am Chem Soc. 1994;116:909–915. [Google Scholar]
- 78.Jeffrey GA, Yeon Y. Acta Crystallogr, Sect B: Struct Sci. 1986;42:410–413. [Google Scholar]
- 79.Berglund B, Vaughan RW. J Chem Phys. 1980;73:2037–2043. [Google Scholar]
- 80.McDermott A, Rydenour CF. In: Encyclopaedia of Nuclear Magnetic Resonance. Grant DM, Harris RK, editors. Wiley; Hoboken: 1996. pp. 3820–3824. [Google Scholar]
- 81.Rohlfing CM, Allen LC, Ditchfield R. J Chem Phys. 1983;79:4958–4966. [Google Scholar]
- 82.Gilli P, Bertolasi V, Pretto L, Ferretti V, Gilli G. J Am Chem Soc. 2004;126:3845–3855. doi: 10.1021/ja030213z. [DOI] [PubMed] [Google Scholar]
- 83.Zundel G. Adv Chem Phys. 2000;111:1–217. [Google Scholar]
- 84.Novak A. Structure and Bonding. Vol. 18. Springer-Verlag; New York: 1974. pp. 177–216. [Google Scholar]
- 85.Brycki B, Brzezinski B, Zundel G, Keil T. Mag Res Chem. 1992;30:507–510. [Google Scholar]
- 86.Stevens ED, Lehmann MS, Coppens P. J Am Chem Soc. 1977;99:2829–2831. [Google Scholar]
- 87.Pyridine-N-oxides were used as oxyanionic hydrogen bond acceptors instead of phenolates since they did not require the presence of a counter-ion. The dichloro compound was used instead of the difluoro variant due to the commercial availability of the former.
- 88.Dega-Szafran Z, Dulewicz E. J Chem Soc Perkin Trans. 1983;II:345–352. [Google Scholar]
- 89.Jencks WP, Regenstein J. In: Handbooks of Biochemistry and Molecular Biology. Fasman GD, editor. CRC Press; Cleveland: 1976. pp. 305–351. [Google Scholar]
- 90.Johnson CD, Katritzky AR, Shakir N. J Chem Soc B. 1967:1235–1237. [Google Scholar]
- 91.Harris TK, Mildvan AS. Proteins. 1999;35:275–282. doi: 10.1002/(sici)1097-0134(19990515)35:3<275::aid-prot1>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 92.Kumar GA, Pan Y, Smallwood CJ, McAllister MA. J Comp Chem. 1998;19:1345–1352. [Google Scholar]
- 93.Kolthoff IM, Chantoon Mk. J Am Chem Soc. 1969;91:4621. [Google Scholar]
- 94.Torrance JW, Holliday GL, Mitchell JBO, Thornton JM. J Mol Biol. 2007;369:1140–1152. doi: 10.1016/j.jmb.2007.03.055. [DOI] [PubMed] [Google Scholar]
- 95.Wells TNC, Fersht AR. Biochemistry. 1986;25:1881–1886. doi: 10.1021/bi00356a007. [DOI] [PubMed] [Google Scholar]
- 96.Leatherbarrow RJ, Fersht AR, Winter G. Proc Natl Acad Sci. 1985;82:7840–7844. doi: 10.1073/pnas.82.23.7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Miller BG, Wolfenden R. Annu Rev Biochem. 2002;71:847–885. doi: 10.1146/annurev.biochem.71.110601.135446. [DOI] [PubMed] [Google Scholar]
- 98.Robertus JD, Alden RA, Birktoft JJ, Kraut J, Powers JC, Wilcox PE. Biochemistry. 1972;11:2439–2449. doi: 10.1021/bi00763a009. [DOI] [PubMed] [Google Scholar]
- 99.Joseph-McCarthy D, Rost LE, Komives EA, Petsko GA. Biochemistry. 1994;33:2824–2829. doi: 10.1021/bi00176a011. [DOI] [PubMed] [Google Scholar]
- 100.Debler EW, Ito S, Seebeck FP, Heine A, Hilvert D, Wilson IA. Proc Natl Acad Sci. 2005;102:4984–4989. doi: 10.1073/pnas.0409207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Raines RT, Sutton EL, Straus DR, Gilbert W, Knowles JR. Biochemistry. 1986;25:7142–7154. doi: 10.1021/bi00370a057. [DOI] [PubMed] [Google Scholar]
- 102.Alter GM, Casazza JP, Wang Z, Nemeth P, Srere PA, Evans CT. Biochemistry. 1990;29:7557–7563. doi: 10.1021/bi00485a003. [DOI] [PubMed] [Google Scholar]
- 103.Lawson SL, Wakarchuk WW, Withers SG. Biochemistry. 1996;35:10110–10118. doi: 10.1021/bi960586v. [DOI] [PubMed] [Google Scholar]
- 104.Seebeck FP, Hilvert D. J Am Chem Soc. 2005;127:1307–1312. doi: 10.1021/ja044647l. [DOI] [PubMed] [Google Scholar]
- 105.Polgar L, Bender ML. Biochemistry. 1967;6:610–620. doi: 10.1021/bi00854a032. [DOI] [PubMed] [Google Scholar]
- 106.Neet KE, Nanci A, Koshland DE., Jr J Biol Chem. 1968;243:6392–6401. [PubMed] [Google Scholar]
- 107.Zhang YL, Hollfelder F, Gordon SJ, Chen L, Keng YF, Wu L, Herschlag D, Zhang ZY. Biochemistry. 1999;38:12111–12123. doi: 10.1021/bi990836i. [DOI] [PubMed] [Google Scholar]
- 108.Brautigam CA, Steitz TA. J Mol Biol. 1998;277:363–377. doi: 10.1006/jmbi.1997.1586. [DOI] [PubMed] [Google Scholar]
- 109.Luo J, Bruice TC. Proc Natl Acad Sci. 2004;101:13152–13156. doi: 10.1073/pnas.0405502101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mazumder D, Kahn K, Bruice TC. J Am Chem Soc. 2003;125:7553. doi: 10.1021/ja030138s. [DOI] [PubMed] [Google Scholar]
- 111.Valina AL, Mazumder-Shivakumar D, Bruice TC. Biochemistry. 2004;43:15657–15672. doi: 10.1021/bi049025r. [DOI] [PubMed] [Google Scholar]
- 112.Wilmouth RC, Westwood NJ, Anderson K, Brownlee W, Claridge TD, Clifton IJ, Pritchard GJ, Aplin RT, Schofield CJ. Biochemistry. 1998;37:17506–17513. doi: 10.1021/bi9816249. [DOI] [PubMed] [Google Scholar]
- 113.Rupert PB, Massey AP, Sigurdsson ST, Ferre-D’Amare AR. Science. 2002;298:1421–1424. doi: 10.1126/science.1076093. [DOI] [PubMed] [Google Scholar]
- 114.Cho HS, Ha NC, Choi G, Kim HJ, Lee D, Oh KS, Kim KS, Lee W, Choi KY, Oh BH. J Biol Chem. 1999;274:32863–32868. doi: 10.1074/jbc.274.46.32863. [DOI] [PubMed] [Google Scholar]
- 115.Cho HS, Choi G, Choi KY, Oh BH. Biochemistry. 1998;37:8325–8330. doi: 10.1021/bi9801614. [DOI] [PubMed] [Google Scholar]
- 116.Lommerse JPM, Price SL, Taylor R. J Comp Chem. 1997;18:757–774. [Google Scholar]
- 117.Kortemme T, Morozov AV, Baker D. J Mol Biol. 2003;326:1239–1259. doi: 10.1016/s0022-2836(03)00021-4. [DOI] [PubMed] [Google Scholar]
- 118.Radzicka A, Wolfenden R. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- 119.Miller BG, Hassell AM, Wolfenden R, Milburn MV, Short SA. Proc Natl Acad Sci. 2000;97:2011–2016. doi: 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hur S, Bruice TC. Proc Natl Acad Sci. 2002;99:9668–9673. doi: 10.1073/pnas.142307099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Warshel A, Strajbl M, Villa J, Florian J. Biochemistry. 2000;39:14728–14738. doi: 10.1021/bi000987h. [DOI] [PubMed] [Google Scholar]
- 122.Gao JL. Curr Opin Struct Biol. 2003;13:184–192. doi: 10.1016/s0959-440x(03)00041-1. [DOI] [PubMed] [Google Scholar]
- 123.Begley TP, Appleby TC, Ealick SE. Curr Opin Struct Biol. 2000;10:711–718. doi: 10.1016/s0959-440x(00)00148-2. [DOI] [PubMed] [Google Scholar]
- 124.Gill SC, von Hippel PH. Anal Biochem. 1989;182:319. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 125.Otwinowski Z, Minor W. Macromolec Crystallogr A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 126.Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr, Sect D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 128.Emsley P, Cowtan K. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 129.Turner DL. J Magn Reson. 1983;54:146–148. [Google Scholar]
- 130.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. J Biomol NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 131.Smallcombe SH. J Am Chem Soc. 1993;115:4776–4785. [Google Scholar]
- 132.Goddard TD, Kneller DG. SPARKY. University of California; San Francisco: 2004. [Google Scholar]
- 133.Frisch MJ, et al. Gaussian 03, Revision C02. Gaussian, Inc.; Wallingford CT: 2004. [Google Scholar]