

Abstract
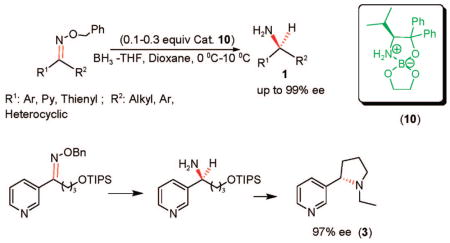
An asymmetric synthesis for the preparation of nonracemic amines bearing heterocyclic and heteroaromatic rings is described. A variety of important enantiopure thionyl and arylalkyl primary amines were afforded by the borane-mediated enantioselective reduction of O-benzyl ketoximes using 10% of catalyst 10 derived from (S)-diphenylvalinol and ethylene glycol with excellent enantioselectivity, in up to 99% ee. The optimal condition for the first asymmetric reduction of 3- and 4-pyridyl-derived O-benzyl ketoxime ethers was achieved using 30% of catalytic loading in dioxane at 10 °C. (S)-N-ethylnornicotine (3) was also successfully synthesized from the TIPS-protected (S)-2-amino-2-pyridylethanol in 97% ee.
Introduction
Nicotinic acetylcholine receptors (nAChRs) are a group of ligand-gated ion channel receptors that play a vital role in different biological processes, in particular, those related to the electrical transmission at the neuromuscular junction and central nervous system (CNS) functions.1–6 Several studies have demonstrated that (S)-nicotine (2 in Figure 1) and analogues display potent biological activity in mammals by modulation of nAChRs2 which, in addition, regulate the release of other important neurotransmitters, such as catecholamines (dopamine, norepinephrine), glutamate, γ-aminobutyric acid (GABA), and serotonin (5-hydroxytriptamine). In the past decade, intense research efforts in the area of neurobiology and pharmacology of nicotine and related nAChR agonists and antagonists have led to exciting developments in drug discovery for the treatment of Parkinson’s and Alzheimer’s (AD) diseases, schizophrenia, attention deficit/hyperactivity, and Tourette’s syndrome.3,4 Agonist SIB-1508Y (5) and ABT-418 (6), as well as other potentially active drugs, are presently in clinical trial for AD and Parkinson’s diseases.5 Several nicotine-derived drug candidates are also being developed for the treatment of nicotine addition, as analgesic and anesthetic agents.1c,6 Novel competitive antagonist N-n-nicotinium analogues (7) have been, recently, found to possess selective binding to nAChR subtypes depending on the lipophilicity of the alkyl groups, opening a new area of selective antagonists.7 Moreover, (S)-nicotine has been, recently, demonstrated to inhibit amyloid formation by β-peptides.5a,8 Consequently, new nonaddictive nicotine analogues that can prevent or retard the development of Alzheimer’s disease will be of interest in the future. (S)-Nicotine is more bioactive than the (R) enantiomer,9 as it is also observed for related alkaloid compounds. Surprisingly, appropriate methods for the enantioselective synthesis of nicotinic derivatives are scarce,3e,f and usually, the desired enantiomer is obtained by costly resolution methods.2a Hence, convenient and versatile asymmetric synthetic methods that permit the introduction of a chiral pyrrolidine-based ring skeleton, offering diverse structural features in the design of new enantiopure nicotinic compound, are needed.
Figure 1.

Nicotine and analogues.
In general, enantioenriched compounds with a stereogenic carbon center α to the amino group are being extensively used as key intermediaries in the synthesis of a large variety of pharmaceuticals,10 chiral auxiliaries,11 catalysts,12 and resolving agents.13 Accordingly, the development of facile and efficient synthetic strategies that provide highly enantiopure amines at low cost has received, recently, an increasing amount of attention.14 Organoborane reagents, especially oxazaborolidine–borane complexes, have achieved wide recognition for the asymmetric reduction of ketones due to their outstanding enantioselectivity, predictable absolute stereochemistry, and low environmental impact.12 Noteworthy, the use of these chiral boron-based reagents for the C=N reduction under catalytic conditions remains limited due to their relatively modest enantioselectivity and lack of reproducibility in some cases.12–17 Although the asymmetric reduction of oxime ethers with borane-based catalysts offers a facile and direct approach to obtain enantioenriched primary amines, more than an stoichiometric amount of in situ prepared oxazaborolidine has been employed to obtain a high degree of enantioselectivity.12,14–17 Itsuno et al.16 described the first catalytic reduction of acetophenone O-benzyl oxime using the B-H oxazaborolidine–borane complex prepared in situ by the reaction of 10 mol % of (S)-diphenylvalinol with 1 equiv of borane, achieving only 52% ee of (S)-1-phenylethanamine. Fontaine et al.17c used 2.5 equiv of diphenylvalinol–B-H oxazaborolidine for the reduction of arylalkyl ketoxime O-benzyl ethers, achieving excellent enantioselectivity and good yield. When the amount of diphenylvalinol was lowered to 1 equiv in the reduction of p-fluoro-2-cyclopropylacetophenone O-benzyl oxime, the yield of the corresponding amine decreased to 52%. In addition, previous reports in the literature have demonstrated that the in situ prepared B-H oxazaborolidines present unusual side products that can affect the enantioselectivity of the catalyst.18 Recently, stable spiroborate esters, derived from (R)- or (S)-1,1′-bi-2-naphthol and (S)-proline, were employed as chirality transfer agents in the borane-mediated reduction of arylalkyl ketoxime ethers.17e Nevertheless, 1 equiv of the expensive chiral reagents was necessary to achieve a high degree of stereoselectivity.
We recently disclosed a series of air- and moisture-stable crystalline spiroborate esters, derived from nonracemic 1,2-amino alcohols and ethylene glycol, for the enantioselective reduction of ketones that were fully characterized by optical and spectroscopic methods.19 Catalysts 8–12 (Figure 2) provide a high degree of enantioselectivity in the borane–DMS reduction of aromatic and aliphatic prochiral ketones, at room temperature, in less than 1 h.19b In a related study, enantiopure alcohols containing pyridyl and other heterocyclic fragments were obtained with an excellent yield in up to 99% ee using, in some cases, only 1 mol % of catalyst 12.19c
Figure 2.

Spiroborate esters derived from nonracemic amino alcohols and ethylene glycol.
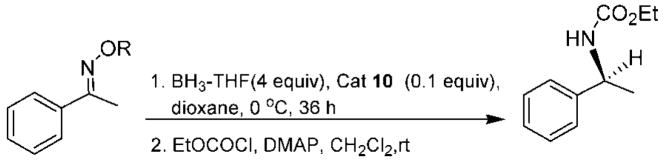
A preliminary study of catalysts 8–12 (Figure 2) in the borane-mediated reduction of aryl ketoximes as catalytic systems for the synthesis of enantiopure primary amines was recently reported.20 After several optimization studies using different equivalents of borane and borane sources, in addition to a variety of solvents and different temperatures, the asymmetric reduction of (E)-acetophenone O-benzyl oxime ether was accomplished, with only 10 mol % of catalyst 10 and 4 equiv of borane–THF in dioxane at 0 °C, affording α-methylbenzylamine with an excellent yield and 97% ee. After screening the catalysts presented in Figure 2, it was observed that the reactivity of catalysts 8, 9, and 11 was rather low at 0 °C. At room temperature, complete reduction was achieved with catalysts 8 and 9 but provided only 59 and 65% ee, respectively, while catalyst 11 derived from 1-amino-2-indanol afforded the (R)-benzylamine enantiomer in 88% ee. Interestingly, 10% of spiroborate 12 derived from diphenylprolinol offers outstanding enantioselectivity for acetophenone reduction, affording the 1-phenylethanol in 99% ee, while it provides modest enantioselectivity (77% ee) for the reduction of its corresponding benzyl oxime ether to the primary amine. Substituents at the ketoxime oxygen play a key role in the reduction stereoselectivity, as illustrated in Table 1. The (E)-acetophenone O-4-(trifluoromethy)benzyl oxime imparts outstanding enantioselectivity (99% ee), although with a modest yield (entry 4). Excellent enantioselectivity and good chemical yield (entry 5) were achieved for the 2-nitrobenzyl oxime. Unfortunately, N-2-nitrobenzyl diphenylvalinol was also isolated from the reaction mixture, possibly formed by the reaction of the 2-nitrobenzylic borate intermediate with the amino group, destroying the expensive amino alcohol. Consequently, benzylic oximes were selected as more adequate substrates since they can also provide excellent enantioselectivity (entry 2) and their synthesis affords pure products using the less expensive benzyl bromide. Besides, the diphenylvalinol can be recovered almost quantitatively after the reduction process. Other representative aromatic and cyclic O-benzyl oximes were prepared and reduced, giving excellent enantioselectivities (up to 99% ee) and demonstrating the capability of our developed spiroborate–borane method for an effective asymmetric catalytic reduction of oxime ethers.
Table 1.
Catalyzed Asymmetric Reduction of Acetophenone O-Substituted Oxime Ethers
 | ||||
|---|---|---|---|---|
| entry | oxime | R | yield (%)a,b | ee (%)c |
| 1 | a | Me | 85 | 95 |
| 2 | b | Bn | 77 | 97 |
| 3 | c | 4-MeOBn | 70 | 97 |
| 4 | d | 4-CF3Bn | 60 | 99 |
| 5 | e | 2-NO2Bn | 95 | 97 |
Borane was stabilized with <0.005 M N-isopropyl N-methyl tert-butylamine.
Isolated yield after purification by column chromatography.
The ee was analyzed using a Crompack Chirasil-Dex-CB GC column.
As mentioned previously, asymmetric synthesis of nonracemic amines with heterocyclic and heteroaromatic fragments is a key step in the preparation of many biological active compounds. Optically active pyridine-derived amines have attracted a strong interest,1–9 primarily, due to their existence in naturally occurring compounds, such as tobacco alkaloids,3a or as potential drug candidates.4 To our knowledge, there is a lack of direct methods for the enantioselective synthesis of these compounds. Herein, we describe the first catalytic asymmetric reduction of pyridyl alkyl and heterocyclic O-benzyl oxime ethers to obtain amino derivatives with a high degree of enantiopurity and good yield using a simple and convenient process and a further application of the method to the stereoselective synthesis of N-ethylnornicotine.
Results and Discussion
It is well-known that the enantioselectivity in the reduction of C=N bonds depends not only on the chirality’s transfer agent but also on the E/Z isomeric purity.14,15 In a comparative study, Sakito and co-worker15i studied the borane-mediated reduction of E and Z aromatic and aliphatic ketoxime ethers in the presence of (−)-norephedrine, demonstrating that each geometric isomer affords the chiral amine with the opposite absolute configuration (Scheme 1). Since (E)-ketoximes are easier to purify by crystallization or column chromatography than their benzylated ethers, our first aim was to prepare pure (E)-oximes. Most ketoximes were initially prepared using Na2CO3 in EtOH/water at 60–70 °C (method A), affording, mainly, the E isomer (>99%). However, in the case of certain pyridyl ketoximes, a significant amount of the Z isomer was observed, about 25% for oxime of compound 19 (Scheme 2), possibly formed by E/Z isomerization under the heating conditions. To obtain enriched (E)-pyridyl ketoxime isomers, pyridine was used as base at room temperature, attaining high yields of the product (90% for oxime of 19) in a relative short time (3 h) (method B).21 These oximes were carefully purified by recrystallization or column chromatography to obtain only the E isomer.
Scheme 1.

Stereoselectivity in the Borane Reduction of Oxime Ethers with (S)-Norephedrine
Scheme 2.

Synthesis of (E)-O-Benzyl Ketoxime Ethers
Excellent yields of the desired (E)-oxime benzyl ethers (Scheme 2) were obtained after oxime treatment with NaH and benzyl bromide at low temperature (0 °C) and purified by column chromatography on silica. We have observed that some benzylic oximes isomerize or decompose under vacuum distillation at temperatures over 150 °C.22,23 Oxime and benzyl oxime isomeric purity was carefully assessed by TLC, NMR, and GC/MS analysis.
(E)-Heteroaryl and Heterocyclic O-Benzyl Oxime Reduction
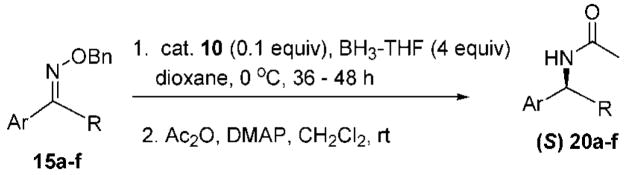
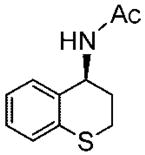
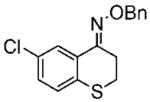
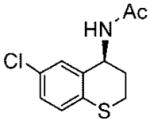
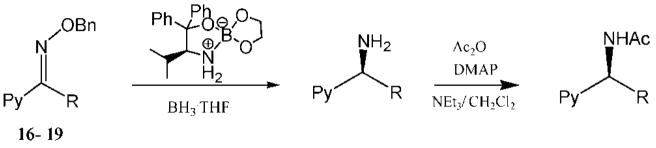
The heteroaryl and heterocyclic benzyl oximes (15a–f, Table 2) were reduced at 0 °C with 0.1 equiv of catalyst 10 and 4 equiv of borane in dioxane until the conversion was completed. After an acidic work up, the corresponding (S) primary amines were acetylated with acetic anhydride in the presence of triethylamine and DMAP in dichloromethane. All of the products (20a–f) were purified by column chromatography, and the enantiomeric excess was determined by GC with a Crompack Chirasil-Dex-CB column, using the optimal condition established with the racemic amide derivative. The enantioselectivity of the reactions was excellent, up to 99% ee for thiochroman-4-amine (entry 5), and with good to high yields of the isolated pure amides.
Table 2.
Asymmetric Reduction of (E)-Heteroaryl and Heterocyclic O-Benzyl Oximes
 | ||||
|---|---|---|---|---|
| entry | 15 | 20a | yield (%)b | ee (%)c |
| 1 |
 15a |
 20a |
85 | 98 |
| 2 |
 15b |
 20b |
92 | 96 |
| 3 |
 15c |
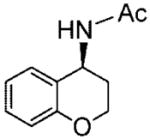 20c |
76 | 94 |
| 4 |
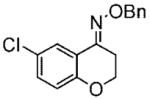 15d |
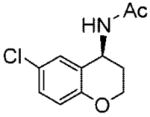 20d |
73 | 95 |
| 5 |
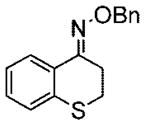 15e |
 20e |
71 | 99 |
| 6 |
 15f |
 20f |
70 | 94 |
The reactions were carried out using 4 equiv of borane stabilized with NaBH4.
Isolated yield of amides purified by column chromatography.
Determined by GC of acetyl derivatives on chiral column (CP-Chirasil-Dex-CB).
Pyridyl Ethanone O-Benzyl Oximes Reduction
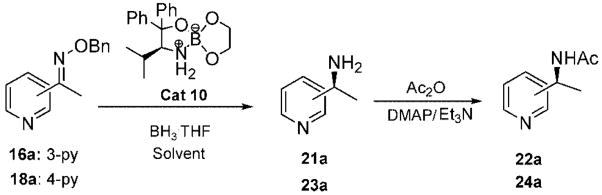
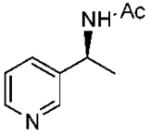
Initially, the reduction of (E)-1-(3-pyridyl)ethanone O-benzyl oxime (16a) in THF with 0.1 mol % of catalyst 10 and 5 equiv of BH3 · THF for 72 h at 0 °C was carried out according to our previously developed protocol.20 One equivalent of borane was required for the boron coordination to the pyridyl nitrogen. The extraction of (S)-1-(3-pyridyl)ethylamine (21a) in ether was unsuccessful, due to the high solubility of the amine in water, and hence the workup procedure was modified. The reaction was first quenched with methanol at 0 °C and then refluxed overnight to hydrolyze the boron amino complex. The desired product 21a was obtained by a simple solvent removal under vacuum and purification by column chromatography. However, the amine’s chemical yield and optical purity were assessed from the acetylated derivative due to the amides’ stability and facile resolution by GC. Initially, the corresponding amide (22a) was obtained in 92% ee, but the isolated yield was low (entry 1, Table 3). This result encouraged us to further optimize the reaction conditions to improve both the enantioselectivity and chemical yield. Dioxane was the best solvent, although THF and t-butyl methyl ether afforded also good selectivity (entries 1–4). By increasing the amount of catalyst to 0.3 equiv and the temperature at 10 °C in dioxane, the reaction time decreased and, fortunately, the amine 22a was obtained with 75% yield and 98% ee (entry 6). Under similar conditions, the reduction of the 1-(4-pyridyl)ethanone O-benzyl oxime (18a) gave excellent enantioselectivity (99% ee), with 84% yield of amine 24a (entry 10). A larger catalytic load did not improve the enantioselectivity or the chemical yield (entry 11).
Table 3.
Optimization Studies for the Asymmetric Reduction of Pyridyl Ethanone O-Benzyl Oximes
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | benzyl oxime | cat 10 (equiv) | T (°C) | solvent | time (h) | yield (%)a | ee (%)b |
| 1 | 16a | 0.1 | 0 | THF | 72 | 46 | 92 |
| 2 | 16a | 0.1 | 25 | THF | 48 | 53 | 89 |
| 3 | 16a | 0.1 | 0 | t-BuOMe | 72 | 42 | 93 |
| 4 | 16a | 0.1 | 0 | dioxane | 72 | 38 | 98 |
| 5 | 16a | 0.3 | 0 | dioxane | 72 | 50 | 99 |
| 6 | 16a | 0.3 | 10 | dioxane | 48 | 75 | 98 |
| 7 | 18a | 0.1 | 0 | THF | 72 | 38 | 99 |
| 8 | 18a | 0.2 | 10 | dioxane | 72 | 64 | 94 |
| 9 | 18a | 0.1 | 25 | THF | 48 | 63 | 90 |
| 10 | 18a | 0.3 | 10 | dioxane | 48 | 84 | 99 |
| 11 | 18a | 0.5 | 10 | dioxane | 48 | 79 | 98 |
The reactions were carried out using 5 equiv of borane stabilized with NaBH4. Isolated yield of amides by column chromatography.
Determined by GC of acetyl derivatives on a chiral column (CP-Chirasil-Dex-CB).
Our attention was directed to the synthesis of 2-pyridylalkylamine. Initially, 1-(2-pyridyl)ethanone O-benzyl oxime was reduced in the presence of 10 mol % of catalyst and THF as solvent, with 5 equiv of BH3 · THF, but the reaction provided a low enantioselectivity (entry 1, Table 4), due to the uncatalyzed reaction that takes place by hydrogen transfer from the borane coordinated to the pyridine nitrogen (Scheme 3). Increasing the amount of borane and catalyst produced modest results. In the presence of 1.0 equiv of catalyst and 4.0 equiv of borane–THF in dioxane at 10 °C, compound 27 was achieved in 73% ee and 79% yield. Attempts to protect the pyridine nitrogen with BEt3 provided unsatisfactory results (entry 6), and protection with BF3 afforded only 54% ee, although the yield increased to 73% (entry 7).
Table 4.
Asymmetric Reduction of 25 under Different Reaction Conditions
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | cat 10 (equiv) | BH3–THF (equiv)a | T (°C) | solvent | time (h) | isolated yield (%)b | ee (%)c |
| 1 | 0.1 | 5.0 | 0 | THF | 72 | 50 | 8 |
| 2 | 1.0 | 5.0 | 0 | THF | 96 | 46 | 13 |
| 3 | 1.0 | 2.0 | 10 | dioxane | 72 | 57 | 60 |
| 4 | 1.0 | 3.0 | 10 | dioxane | 48 | 71 | 57 |
| 5 | 1.0 | 4.0 | 10 | dioxane | 48 | 79 | 73 |
| 6 | 0.3 | 5.0 | 10 | dioxane | 72 | 34 | 4d |
| 7 | 1.0 | 2.0 | 10 | dioxane | 48 | 73 | 54e |
Stabilized with NaBH4.
Isolated yield of amide.
Determined by GC of acetyl derivatives on a chiral column (CP-Chirasil-Dex-CB).
Triethyl borane (1.0 equiv) was added.
BF3 (1.0 equiv) was added.
Scheme 3.

Intramolecular Uncatalyzed Reduction of 25
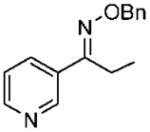
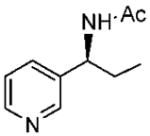
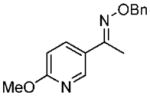
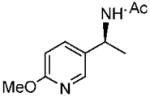
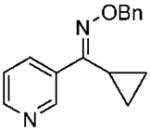
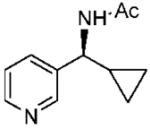
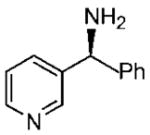
Under optimized conditions, a variety of other pyridylalkyl and pyridylphenyl O-benzyl oxime ethers were tested to investigate the generality of the reaction. The asymmetric reduction of 16–19 was conducted with 30 mol % of catalyst and 5 equiv of borane in dioxane at 10 °C for 2 days. As illustrated in Table 5, excellent enantioselectivities (95–99% ee) were obtained with good to excellent yields of isolated pure products by column chromatography. The enantiomeric excess values of the amine or their acetylated derivatives were determined by GC or HPLC analysis, using suitable chiral stationary phases. The (S)-cyclopropyl 3- and 4-pyridyl methanamines (entries 4 and 8) were afforded in 96 and 98% ee, respectively. Noteworthy, the reduction of the 3-pyridyl phenyl oxime ether provided the (S)-amine 31 (entry 5) in excellent enantiopurity (95% ee), with the pyridine ring being the larger group and the phenyl the smaller group in the face selectivity. In addition, both E and Z isomers of cyclopropyl-4-pyridyl O-benzylketoxime (18c) were separated by chromatography on a silica column and reduced by our established protocol. The E isomer produced the (S)-amine in 98% ee (entry 8); however, the minor Z isomer afforded the (R)-amine in only 71% ee.
Table 5.
Asymmetric Reduction of Representative Pyridyl O-Benzyl Oxime Ethers
 | ||||
|---|---|---|---|---|
| entry | benzyloxime | producta | yield (%)b | ee (%)c |
| 1 |
 16a 16a
|
 22a 22a
|
75 | 98 |
| 2 |
 16b 16b
|
 28 28
|
89 | 99 |
| 3 |
 16c 16c
|
 29 29
|
88 | 98 |
| 4 |
 16d 16d
|
 30 30
|
83 | 96d |
| 5 |
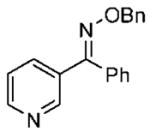 17 17
|
 31 31
|
82 | 95 |
| 6 |
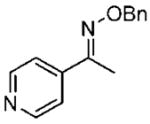 18a 18a
|
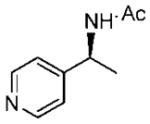 24a 24a
|
84 | 99 |
| 7 |
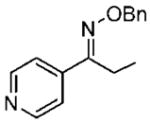 18b 18b
|
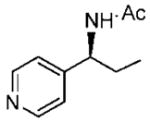 32 32
|
85 | 96 |
| 8 |
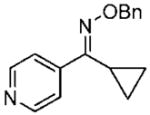 18c 18c
|
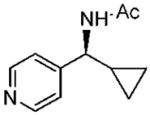 33 33
|
91 | 98 |
| 9 |
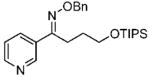 19 19
|
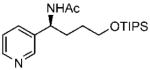 34 34
|
95 | 95e |
The reactions were carried out using 1 equiv of oxime ether, 0.3 equiv of catalyst 10 and 5 equiv of borane stabilized with NaBH4 in dioxane at 10 °C until 100% amine conversion.
Isolated yield of products after column chromatography.
Determined by GC of acetyl derivatives on a chiral column (CP-Chirasil-Dex-CB).
Determined by chiral HPLC (Chiralcel OD-H column).
Determined by chiral HPLC (Chiralcel IB column).
To demonstrate the capability of our methodology for the synthesis of (S)-nicotine analogues, we decided to prepare (S)-N-ethylnornicotine (3) for its biological activity as a potential selective nAChR agonist. Initially, an attempt was made to synthesize racemic nornicotine (4) from amine 35 by first removing the protecting TIPS group using TBAF in THF at 0 °C to obtain the amino alcohol 36, followed by an intramolecular Mitsunobu24 reaction, as indicated in Scheme 4. However, the cyclization step gave a complex mixture of products. As an alternative, (S)-N-ethylnornicotine (3) was successfully prepared from compound (S)-34, previously synthesized in 95% ee (HPLC analysis). The amino alcohol (S)-37 was afforded in 89% yield, after deprotection with TBAF in THF at 0 °C and subsequent reduction with borane providing N-ethylamine 38 in 86% yield. The desired pyrrolidine derivative 3 was furnished in 84% yield after a successful intramolecular cyclization of (S)-38 with Ph3P/DIAD.25 In summary, the synthesis of (S)-N-ethylnornicotine was achieved with excellent enantiopurity (97% ee determined by GC) and 64% overall yield. This approach opens the door to the highly enantioselective synthesis of other important biological targets in a facile and efficient way.
Scheme 4.

Enantioselective Synthesis of the (S)-Nicotine Analogue
Conclusion
We have investigated the effect of heteroaryl and heterocyclic groups in the asymmetric borane reduction of O-benzyl oxime ethers with the novel spiroborate 10 derived from diphenylvalinol and ethylene glycol as catalyst. A true catalytic and practical synthesis of a variety of chiral primary amines containing heterocyclic and heteroaryl rings with a high degree of enantioselectivity with only 10% of spiroborate 10 was developed. After several optimization studies, the first borane reduction of a variety of pyridyl benzyl oximes afforded enantiopure amines in up to 99% ee, using 30% of 10 at 10 °C. The (S)-nicotine analogue 3 was prepared in 97% ee from enantiopure 34 with 64% overall yield. Considering the convenience and generality of the method and the abundance of natural products that contain the heterocyclic amine functionality, this method should find wide interest in both academic research and industry.
Experimental Section
Catalyst, 19 (R)-2-amino-1,1,2-triphenylethanol,26 (S)-2-amino-1,1,2-triphenylethanol, 26 (S)-2-amino-3-methyl-1,1-diphenylbutan-1-ol,27 cyclopropylpyridin-3yl methanone,28 cyclopropylpyridin-4yl methanone,28 and 4-hydroxy-1-pyridin-3-yl butan-1-one29 were synthesized according to literature procedures. BH3 · THF (1 M solution in THF, stabilized with <0.005 M NaBH4) and other starting materials and chemical reagents were purchased and used without purification unless otherwise noted.
General Procedure for the Preparation of O-Benzyl Oximes
To a suspension of NaH (1.1 equiv) in DMF was added dropwise a solution of hydroxyl oxime (1.0 equiv) and maintaining the temperature at 0 °C. After the addition, the reaction mixture was stirred for 1 h. Then, BnBr (1.05 equiv) in DMF was added dropwise at 0 °C. The resulting mixture was stirred overnight at rt and then quenched with saturated aqueous NH4Cl solution and extracted with ether. The organic phases were combined and dried over anhydrous Na2SO4. The solvents were evaporated under vacuum, and the residue was purified by flash silica gel column chromatography.
(E)-1-(Thiophen-3-yl)ethanone O-Benzyl Oxime (15a)
Purified by column chromatography on silica gel/hexane: AcOEt (30:1) as a colorless oil; 94% (6.1 g); 1H NMR (400 MHz, CDCl3) δ 2.30 (s, 3H), 5.27 (s, 2H), 7.3–7.5 (m, 8H) ppm; 13C NMR (100 MHz, CDCl3) δ 13.2, 76.7, 123.4, 125.4, 126.1, 127.8, 128.2, 128.4, 138.2, 139.0, 151.3 ppm; IR ν (cm−1) 3031, 2922, 1601, 1454, 1363, 1015, 734, 755; GC-MS m/z 231.0 (M+). Anal. Calcd for C13H13NOS: C, 67.50; H, 5.66; N, 6.06. Found: C, 67.67; H, 5.71; N, 6.17.
(E)-1-(2,5-Dimethylthiophen-3-yl)ethanone O-Benzyl Oxime (15b)
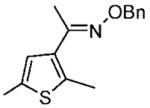
Purified by column chromatography on silica gel/hexane: AcOEt (30:1) as a colorless oil; yield 92% (2.10 g); 1H NMR (400 MHz, CDCl3) δ 2.24 (s, 3H), 2.44 (s, 3H), 2.46 (s, 3H), 5.24 (s, 2H), 6.72 (s, 1H), 7.3–7.5 (m, 5H) ppm; 13C NMR (100 MHz, CDCl3) δ 15.0, 15.1, 15.2, 75.9, 125.5, 127.7, 128.1, 128.3, 133.3, 135.4, 135.8, 138.5, 153.0 ppm; IR ν (cm−1) 3031, 2919, 2857, 1604, 1496, 1454, 1364, 1266, 1208, 1181, 1143, 1081, 1048, 1012, 985, 924, 882, 833, 733, 696; GC-MS m/z 259.0 (M+). Anal. Calcd for C15H17NOS: C, 69.46; H, 6.61; N, 5.40. Found: C, 69.71; H, 6.66; N, 5.41.
(E)-6-Chlorochroman-4-one O-Benzyl Oxime (15d)
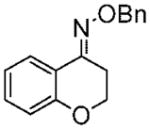
Purified by column chromatography on silica gel/hexane: AcOEt (9:1) as an oil with a slight yellow color; yield: 84% (1.62 g);1H NMR (400 MHz, CDCl3) δ 2.98 (t, 2H, J = 6.0 Hz), 4.24 (t, 2H, J = 6.0 Hz), 5.29 (s, 2H), 6.9 (m, 1H), 7.39 (m, 1H), 7.43 (m, 5H), 7.92 (s, 1H) ppm; 13C NMR (CDCl3) δ 24.0, 65.1, 76.7, 119.1, 119.8, 123.8, 126.6, 128.0, 128.2, 128.4, 130.7, 137.6, 147.7, 155.1 ppm; IR ν (cm−1) 3031, 2928, 1618, 1475, 1452, 1425, 1364, 1284, 1250, 1216, 1094, 1068, 1039, 1014, 962, 814, 732; GC/MS m/z 287.0 (M+), 91.1 (PhCH2+). Anal. Calcd for C16H14ClNO2: C, 66.79; H, 4.90; N, 4.87. Found: C, 67.03; H, 4.89; N, 4.85.
(E)-Thiochroman-4-one O-Benzyl Oxime (15e)
Purified by column chromatography on silica gel/hexane: AcOEt (9:1) as an oil with a slight yellow color; yield 84% (1.89 g); 1H NMR (400 MHz, CDCl3) δ 2.99 (t, 2H, J = 6.0 Hz), 3.21 (t, 2H, J = 6.0 Hz), 5.32 (s, 2H), 7.2 (m, 1H), 7.3 (m, 2H), 7.44 (m, 5H), 8.0 (m, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 26.0, 26.9, 76.6, 25.4, 126.2, 127.9, 128.2, 128.3 128.4, 129.1, 129.8, 135.9, 137.9, 152.3 ppm; IR ν (cm−1) 3059, 2921, 1598, 1495, 1468, 1433, 1364, 1330, 1286, 1245, 1208, 1007, 931, 907, 751, 695; GC/MS m/z 269.0 (M+), 91.1 (PhCH2+). Anal. Calcd for C16H15NOS: C, 71.34; H, 5.61; N, 5.21. Found: C, 71.45; H, 5.66; N, 5.30.
(E)-6-Chlorothiochroman-4-one O-Benzyl Oxime (15f)
Purified by column chromatography on silica gel/hexane: AcOEt (9:1) as an oil with a slight yellow color; yield 82% (2.82 g);1H NMR (400 MHz, CDCl3) δ 2.98 (t, 2H, J = 6.0 Hz), 3.18 (t, 2H, J = 6.0 Hz), 5.32 (s, 2H), 7.32 (m, 2H), 7.41 (m, 5H), 8.06 (s, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 25.9, 26.4, 77.0, 125.8, 128.1, 128.2, 128.5, 129.0, 129.4, 131.1, 131.2, 134.2, 137.6, 151.2 ppm; IR ν (cm−1) 2922, 1573, 1496, 1455, 1394, 1364, 1305, 1240, 1099, 1048, 1009, 940, 889, 796, 725, 695; GC/MS m/z 303.0 (M+), 91.1 (PhCH2+). Anal. Calcd for C16H14ClNOS: C, 63.25; H, 4.64; N, 4.61. Found: C, 63.50; H, 4.66; N, 4.67.
(E)-1-(Pyridine-3-yl)propan-1-one O-Benzyl Oxime (16b)
Purified by column chromatography on silica gel/hexane: AcOEt (2: 1) as a colorless oil; yield 80% (0.96 g); 1H NMR (400 MHz, CDCl3) δ 1.19 (t, 3H, J = 7.6 Hz), 2.86 (q, 2H, J = 7.6 Hz), 5.29 (s, 2H), 7.3–7.5 (m, 6H), 7.98 (m, 1H), 8.6 (m, 1H), 8.9 (m, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.9, 19.9, 76.5, 123.3, 127.9, 128.2, 128.4, 131.3, 133.5, 137.9, 147.7, 150.0, 157.6 ppm; IR ν (cm−1) 3032, 2972, 2937, 2877, 1604, 1454, 1412, 1365, 1018, 982, 951, 905, 876, 808, 747; GC-MS m/z 240.2 (M+). Anal. Calcd for C15H16N2O: C, 74.97; H, 6.71; N, 11.66. Found: C, 75.06; H, 6.76; N, 11.63.
(E)-1-(6-Methoxypyridin-3-yl)ethanone O-Benzyl Oxime (16c)
Purified by column chromatography on silica gel/hexane: AcOEt (2:1) as a colorless oil; yield 90% (1.41 g); 1H NMR (400 MHz, CDCl3) δ 2.29 (s, 3H), 4.00 (s, 3H), 5.27 (s, 2H), 6.8 (m, 1H), 7.4–7.5 (m, 5H), 7.97 (m, 1H), 8.4 (m, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 12.4, 53.6, 76.0, 110.7, 125.9, 127.8, 128.2, 128.4, 136.2, 138.0, 144.8, 152.4, 164.6 ppm; IR ν (cm−1) 3026, 2947, 2897, 1726, 1601, 1499, 1452, 1376, 1285, 1020, 928, 900, 833, 738, 698; GC-MS m/z 256.1 (M+). Anal. Calcd for C15H16N2O2: C, 70.29; H, 6.29; N, 10.93. Found: C, 70.01; H, 6.27; N, 10.56.
(E)-Cyclopropyl(pyridine-3-yl)methanone O-Benzyl Oxime (16d)
Purified by column chromatography on silica gel/hexane: AcOEt (2:1) as a colorless oil; yield 88% (0.89 g); 1H NMR (400 MHz, CDCl3) δ 0.69 (m, 2H), 1.0 (m, 2H), 2.3 (m, 1H), 5.28 (s, 2H), 7.3–7.5 (m, 6H), 7.8 (m, 1H), 8.6 (m, 1H), 8.7 (m, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 5.8, 9.77, 76.3, 122.9, 127.9, 128.1, 128.4, 130.5, 135.6, 137.8, 149.3, 149.7, 158.2 ppm; IR ν (cm−1) 3087, 3031, 2924, 2869, 1592, 1564, 1497, 1475, 1455, 1412, 1365, 1327, 1083, 1022, 981, 938, 808,734; GC-MS m/z 252.1 (M+). Anal. Calcd for C16H16N2O: C, 76.16; H, 6.39; N, 11.10. Found: C, 76.02; H, 6.39; N, 11.00.
(E)-1-(Pyridine-4-yl)propan-1-one O-Benzyl Oxime (18b)
Purified by column chromatography on silica gel/hexane: AcOEt (2:1) as a colorless oil; yield 90% (1.08 g); 1H NMR (400 MHz, CDCl3) δ 1.19 (t, 3H, J = 7.6 Hz), 2.8 (q, 2H, J = 7.6 Hz), 5.31 (s, 2H), 7.4–7.5 (m, 5H), 7.6 (m, 2H), 8.7 (m, 2H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.9, 19.5, 76.7, 120.4, 128.0, 128.2, 128.4, 137.7, 142.9, 150.2, 157.7 ppm; IR ν (cm−1) 3031, 2976, 2936, 2877, 1591, 1454, 1409, 1366, 1015, 992, 979, 922, 824, 740; GC-MS m/z 240.2 (M+). Anal. Calcd for C15H16N2O: C, 74.97; H, 6.71; N, 11.66. Found: C, 75.21; H, 6.78; N, 11.65.
(E)-Cyclopropyl(pyridine-4-yl)methanone O-Benzyl Oxime (18cE)
Purified by column chromatography on silica gel/hexane: AcOEt (2:1) as a colorless oil; yield 94% (1.32 g); 1H NMR (400 MHz, CDCl3) δ 0.73 (m, 2H), 1.0 (m, 2H), 2.2 (m, 1H), 5.29 (s, 2H), 7.3–7.5 (m, 7H), 8.6 (m, 2H) ppm; 13C NMR (100 MHz, CDCl3) δ 6.1, 9.4, 77.0, 122.5, 127.9, 128.1, 128.4, 137.7, 142.6, 149.8, 158.1 ppm; IR ν (cm−1) 3066, 3031, 2930, 2876, 1588, 1541, 1496, 1454, 1408, 1365, 1210, 1083, 984, 940, 909, 820, 697; GC-MS m/z 252.2 (M+). Anal. Calcd for C16H16N2O: C, 76.16; H, 6.39; N, 11.10. Found: C, 76.08; H, 6.40; N, 11.04.
(E)-1-(Pyridine-3-yl)-4-(triisopropylsilyloxy)butan-1-one O-Benzyl Oxime (19)
Purified by column chromatography on silica gel/hexane: AcOEt (2:1) as a colorless oil; yield 93% (1.98 g); 1H NMR (400 MHz, CDCl3) δ 1.04–1.14 (m, 21H), 1.85 (m, 2H), 2.93 (m, 2H), 3.77 (t, 2H, J = 6.4 Hz), 5.29 (s, 2H), 7.3–7.5 (m, 6), 8.02 (m, 1H), 8.63 (m, 1H), 8.96 (d, 1H, J = 2.0 Hz) ppm; 13C NMR (100 MHz, CDCl3) δ 12.0, 18.0, 23.2, 29.7, 62.8, 76.3, 123.2, 127.8, 128.2, 128.4, 131.6, 133.6, 137.9, 147.8, 149.9, 156.5 ppm; IR ν (cm−1) 3065, 3031, 2928, 2877, 1619, 1475, 1455, 1425, 1283, 1250, 1216, 1095, 1040, 1014, 962, 815, 696. Anal. Calcd for C25H38N2O2Si: C, 70.38; H, 8.98; N, 6.57. Found: C, 70.11; H, 9.02; N, 6.38.
General Procedure for Asymmetric Reduction of Thiofuranyl and Heterocyclic O-Benzyl Oximes with Spiroborate 10
To a 25 mL two-necked flask under N2 was added catalyst 10 (33 mg, 0.1 mmol). Then, anhydrous dioxane (10 mL) was introduced, and BH3 · THF (4 mL, 1 M in THF stabilized with <0.005 M NaBH4) was added in one portion. The resulting mixture was stirred at rt for 30 min until a transparent solution was observed. The solution was cooled at 0 °C, and the benzyl oxime (1 mmol) in dioxane (5 mL) was added dropwise during 1.5 h by a syringe pump. The resulting mixture was stirred at 0 °C until the conversion was completed in about 48 h. Then, the reaction was quenched with 6 N HCl and then 6 N NaOH until the solution was basic. It was extracted with ether, and the combined organic phase was washed with a saturated NaCl solution and dried over anhydrous Na2SO4. The solvent was removed under vacuum, and the residue was analyzed by GC for amine conversion and then used directly to form the acetylated derivatives for GC analysis on a chiral column. The acetamide derivatives of racemic amines were first prepared by reduction of the benzyl oximes with borane and used as standard samples for chiral GC analysis. Generally, the ee was determined using a Crompack Chirasil-Dex-CB GC column (30 m × 0.25 mm × 0.25μm), with a He flow at a 1.0 mL/min rate and an initial temperature of 70 °C for 10 min, followed by a 5 °C/min ramp until 190 °C, and maintaining this temperature for about 25 min, depending on the particular compound.
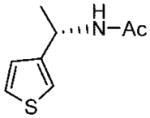
(S)-N-(1-(Thiophen-3-yl)ethyl)acetamide (20a)
Purified by column chromatography on silica gel/hexane: AcOEt (1:1) as a white solid; yield 85% (144 mg); mp 69–71 °C; 98% ee; [α]20D = −120° (c 1.05, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.56 (d, 3H J = 6.8 Hz), 2.03 (s, 3H), 5.3 (m, 1H), 5.73 (s, 1H), 7.19 (m, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 21.3, 23.5, 44.1, 120.7, 126.3, 126.5, 144.4, 169.0 ppm; IR ν (cm−1) 3285, 3095, 3079, 2968, 1629, 1545, 1446, 1417.18, 1367, 1274, 1214, 1167, 1121, 1050 971, 862, 784, 731, 693; GC/MS m/z 169.0 (M+); HRMS m/z 170.0631 (M + H)+.
(S)-N-(1-(2,5-Dimethylthiophen-3-yl)ethyl)acetamide (20b)
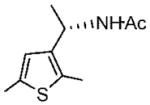
Purified by column chromatography on silica gel/hexane: AcOEt (1: 1) as a white solid; yield 92% (169 mg); mp 102–104 °C; 96% ee; [α]20D = −117.5° (c 1.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.46 (d, 3H, J = 6.8 Hz), 1.99 (s, 3H), 2.41 (s, 3H), 2.45 (s, 3H), 5.1 (m, 1H), 5.58 (s, 1H), 6.62 (s, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 12.8, 15.2, 21.7, 23.4, 43.0, 123.1, 132.7, 136.4, 138.3, 168.7; IR ν (cm−1) 3278, 3064, 2972, 2919, 2869, 1635, 1544, 1442, 1368, 1334, 1311, 1274, 1222, 1132, 1108, 1035, 968, 824, 733; GC-MS m/z 197.0 (M+). HRMS m/z 198.0944 (M + H)+.
(S)-N-(6-Chlorochroman-4-yl)acetamide (20d)
Purified by column chromatography on silica gel/hexane: AcOEt (1:1) as a white solid; yield 73% (0.161 g); mp 188–190 °C; 95% ee; [α]20D=−56.4° (c 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ 2.0–2.1 (m, 1H), 2.09 (s, 3H), 2.1 (m, 2H), 4.2–4.3 (m, 2H), 5.2 (m, 1H), 5.80 (s, 1H), 6.8 (m, 1H), 7.2 (m, 1H), 7.19 (s, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 23.4, 28.8, 43.6, 63.6, 118.7, 123.6, 125.6, 128.6, 129.3, 153.8, 169.4 ppm; IR ν (cm−1) 3265, 3083, 2984, 1634, 1537, 1483, 1411, 1370, 1261, 1222, 1193, 1107, 1022, 993, 878, 814, 751, 677; GC/MS m/z 225.1 (M+), 167.0 (M+ – NHAc); HRMS m/z 226.0627 (M + H)+.
(S)-N-(Thiochroman-4-yl)acetamide (20e)
Purified by column chromatography on silica gel/hexane: AcOEt (1:1) as a white solid; yield 71% (0.150 g); mp 186–188 °C; 99% ee; [α]20D = −134.6° (c 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ 2.04 (s, 3H), 2.15 (m, 1H), 2.4 (m, 1H), 3.0–3.14 (m, 2H), 5.22 (s, 1H), 5.91, (s, 1H), 7.1 (m, 1H), 7.26 (m, 2H), 7.2 (m, 1H) ppm; 13C NMR (CDCl3) δ 22.7, 23.4, 28.1, 46.8, 124.5, 125.5, 126.8, 130.5, 132.6, 133.5, 169.1 ppm; IR ν (cm−1) 3191, 3034, 2944, 2837, 1632, 1532, 1476, 1431, 1369, 1322, 1277, 1199, 1095, 944, 796, 753; GC/MS 207.0 (M+), 149.0 (M+ – NHAc); HRMS m/z 208.0788 (M + H)+.
(S)-N-(6-Chlorothiochroman-4-yl)acetamide (20f)
Purified by column chromatography on silica gel/hexane: AcOEt (1:1) as a white solid; yield 70% (0.167 g); mp 190–192 °C; 94% ee; [α]20D =−107.6° (c 1.00, CHCl3);1H NMR (400 MHz, CDCl3) δ 2.07 (s, 3H), 2.1–2.3 (m, 1H), 3.0 (m, 2H), 5.2 (m, 1H), 5.91 (s, 1H), 7.1(m, 1H), 7.14 (m, 1H), 7.3 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 22.9, 23.4, 28.1, 46.6, 76.7, 128.1, 128.3, 129.9, 132.1, 134.2, 169.2; IR ν (cm−1) 3269, 3048, 2940, 1635, 1532, 1463, 1427, 1367, 1186, 1098, 1054, 944, 887, 811, 730; GC/MS m/z 241.1 (M+), 183.0 (M+ – NHAc); HRMS m/z 242.0399 (M + H)+.
General Procedure for Asymmetric Reduction of Pyridyl O-Benzyl Oximes with Catalyst 10
To a dried 50 mL reaction tube under N2 was added catalyst 10 (50 mg, 0.15 mmol, 0.3 equiv) at room temperature. Then, anhydrous dioxane (4 mL) was introduced, and BH3 · THF (2.5 mL, 1.0 M in THF stabilized with <0.005 M NaBH4) was added. The resulting mixture was stirred at room temperature for 1 h until a clear solution formed. The oxime benzyl ether (0.5 mmol, 1.0 equiv) in 4 mL of dioxane was added dropwise by syringe pump for 1 h. The resulting mixture was stirred for 2 days at 10 °C under nitrogen. The reaction mixture was quenched with methanol (5 mL) at 0 °C and then refluxed overnight. The solvents were evaporated under vacuum, and the residue was directly acetylated, in most cases, to form the amide derivatives. To a solution of crude amine in anhydrous CH2Cl2 (10 mL) were added DMAP (13 mg, 10%), Et3N (0.2 mL, 1 mmol, 2.0 equiv) and acetic anhydride (0.11 mL, 1.0 mmol, 2.0 equiv). The resulting mixture was stirred for 3 h. The solvent was removed under water pump and then under high vacuum. The residue was purified directly by flash chromatography on a silica gel column, eluted first by ether and then by CH2Cl2/CH3OH (10/1), giving the corresponding products. As indicated previously, the enantiomeric excess was determined by GC with a chiral column, using a Crompack Chirasil-Dex-CB GC column (30 m × 0.25 mm × 0.25 μm), with He flow at a 1.0 mL/min rate isothermal at 140–160 °C or with a gradient at an initial temperature of 80–120 °C for 5–15 min, followed by a 2–5 °C/min ramp until 130–170 °C, and maintaining this temperature for about 35–100 min, depending on the particular compound. Generally, the HPLC enantiomeric analysis was carried out with a Chiralcel-IB or OD-H columns (0.46 cm × 25 cm) with a 0.5 mL/min flow of 90% hexane/10% 2-propanol for 100 min or at 0.4 mL/min flow with 95% hexane/5% 2-isopropanol for 60 min, using a UV detector at 254 nm.
(S)-N-(1-(Pyridine-3-yl)propyl)acetamide (28)
Purified by column chromatography on silica gel/CH2Cl2: CH3OH (10:1) as a colorless oil; yield 89% (80 mg); 99% ee; [α]20D = −109.7° (c 1.30, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.96 (t, 3H, J = 7.6 Hz), 1.88 (m, 2H, J = 7.6 Hz), 2.05 (s, 3H), 4.95 (m, 1H, J = 7.6 Hz), 6.10 (s, 1H), 7.29 (m, 1H), 7.6 (m, 1H), 8.5 (m, 1H), 8.60 (m, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.7, 23.3, 28.8, 53.0, 123.5, 134.5, 137.8, 148.4, 148.7, 169.5; IR ν (cm−1) 3259, 3060, 2968, 2933, 2877, 1647, 1544, 1373, 1301, 1137, 1027, 795, 693 ppm; GC-MS m/z 178.1 (M+); HRMS m/z 179.1175 (M + H)+.
(S)-N-(1-(6-Methoxypyridine-3-yl)ethyl)acetamide (29)
Purified by column chromatography on silica gel/CH2Cl2: CH3OH (10: 1) as a white solid; yield 88% (85 mg); mp 66–67 °C; 98% ee; [α]20D = −63.5° (c 1.85, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.50 (d, 3H, J = 6.8 Hz), 2.01 (s, 3H), 3.95 (s, 3H), 5.1 (m, 1H), 5.92 (s, 1H), 6.7 (m, 1H), 7.6 (m, 1H), 8.16 (d, 1H, J = 2.0 Hz) ppm; 13CNMR (100 MHz, CDCl3) δ 21.3, 23.4, 46.3, 53.5, 110.9, 131.4, 137.2, 144.6, 163.6, 169.2 ppm; IR ν (cm−1) 3277, 2988, 1629, 1539, 1500, 1430, 1384, 1293, 1258, 1120, 1019, 972, 826, 744; GC-MS m/z 194.1 (M+); HRMS m/z 195.1123 (M + H)+.
(S)-N-(Cyclopropyl(pyridine-3-yl)methyl)acetamide (30)
Purified by column chromatography on silica gel/CH2Cl2: CH3OH (10: 1) as a white solid; yield 83% (157 mg); mp 122–123 °C; 96% ee; [α]20D = −32.5° (c 1.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.41 (m, 1H), 0.52 (m, 1H), 0.69 (m, 2H), 1.19 (m, 1H), 2.07 (s, 3H), 4.4 (m, 1H, CH), 6.2 (s, 1H), 7.3 (m, 1H), 7.7 (m, 1H), 8.5 (d, 1H, J = 3.6 Hz), 8.68 (s, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 3.9, 4.2, 16.4, 23.3, 55.5, 123.4, 134.4, 137.7, 148.3, 148.6, 169.4 ppm; IR ν (cm−1) 3244, 3062, 3004, 2928, 2849, 1630, 1546, 1426, 1372, 1298, 1194, 1162, 1104, 1091, 1020, 948, 845, 807, 715; GC-MS m/z 190.1 (M+); HRMS m/z 191.1174 (M + H)+.
(S)-N-(1-(Pyridine-4-yl)propyl)acetamide (32)
Purified by column chromatography on silica gel/CH2Cl2: CH3OH (10:1) as a colorless oil; yield 85% (76 mg); 96% ee; [α]20D = −114° (c 1.15, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.97 (t, 3H, J = 7.6 Hz), 1.84 (m, 2H, J = 7.6 Hz), 2.08 (s, 3H), 4.93 (m, 1H, J = 7.6 Hz), 5.87 (s, 1H), 7.23 (dd, 2H, J = 1.6, 6.0 Hz), 8.6 (dd, 2H, J = 1.6, 6.0 Hz) ppm; 13C NMR (100 MHz, CDCl3) δ 10.5, 23.3, 28.6, 54.0, 121.7, 150.1, 169.8 ppm; IR ν (cm−1) 3259, 3056, 2968, 2936, 2877, 1648, 1601, 1543, 1415, 1372, 1299, 1178, 999, 825, 789; GC-MS m/z 178.1 (M+); HRMS m/z 179.1175 (M + H)+.
(S)-N-(Cyclopropyl(pyridine-4-yl)methyl)acetamide (33)
Purified by column chromatography on silica gel/CH2Cl2: CH3OH (10: 1) as a white solid; yield 91% (176 mg); mp 78–80 °C; 98% ee; [α]20D = −15.6° (c 1.25, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.49 (m, 2H), 0.72 (m, 2H), 1.14 (m, 1H), 2.10 (s, 3H), 4.37 (m, 1H), 6.20 (s, 1H), 7.32 (d, 2H, J = 6.0 Hz), 8.60 (dd, 2H, J = 1.6, 6.0 Hz) ppm; 13C NMR (100 MHz, CDCl3) δ 3.77, 4.42, 16.3, 23.3, 56.7, 121.7, 150.0, 151.0, 169.5 ppm; IR ν (cm−1) 3276, 3080, 3006, 1649, 1598, 1542, 1432, 1410, 1371, 1312, 1289, 1221, 1200, 1105, 1052, 1023, 957, 850, 820, 808, 737; GC-MS m/z 190.2 (M+); HRMS m/z 191.1174 (M + H)+.
(S)-N-(1-(Pyridine-3-yl)-4-(triisopropylsilyloxy)butyl)acetamide (34)
Purified by column chromatography on silica gel/CH2Cl2: CH3OH (10:1) as a colorless oil; yield 91% (331 mg); 95% ee by HPLC; [α]20D = −109.7° (c 1.9, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.07–1.18 (m, 21H), 1.56 (m, 2H), 1.94 (m, 2H), 2.04 (s, 3H), 3.75 (m, 2H), 5.04 (m, 1H), 6.15 (s, 1H), 7.3 (m, 1H), 7.6 (m, 1H), 8.53 (t, 1H), 8.6 (d, 1H, J = 2.0 Hz) ppm; 13C NMR (100 MHz, CDCl3) δ 12.0, 18.0, 23.3, 29.3, 32.1, 51.4, 62.5, 123.5, 134.3, 138.0, 148.3, 148.6, 169.4 ppm; IR ν (cm−1) 3278, 3054, 2942, 2865, 1648, 1546, 1463, 1428, 1372, 1292, 1099, 1068, 1012, 995, 881, 796, 714, 680; GC-MS m/z 364.2 (M+); HRMS m/z 365.2614 (M + H)+, calcd for C20H37O2N228Si m/z 365.2619.
(S)-N-(4-Hydroxy-1-(pyridine-3-yl)butyl)acetamide (37)
To a round-bottom flask was added a solution of 34 (364 mg, 1.0 mmol) in THF (10 mL). The solution was cooled with an ice-bath, and Bu4NF (1.5 mL, 1.0 M in THF) was added dropwise. The mixture was stirred for 2 h until 37 was consumed. Solvents were removed under vacuum, and the residue was purified by chromatography on a silica gel column, eluted with CH2Cl2: CH3OH (5: 1). The product was obtained as a colorless oil; yield 89% (186 mg); [α]20D = −92° (c 1.2, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.56 (m, 2H), 1.92 (m, 2H), 1.99 (s, 3H), 3.41 (br, 1H), 3.67 (m, 2H), 5.01 (m, 1H), 7.28 (m, 2H), 7.68 (d, 1H, J = 7.6 Hz), 8.47 (d, 1H, J = 4.0 Hz), 8.57 (s, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 23.1, 29.0, 32.5, 51.3, 61.7, 123.7, 134.8, 138.5, 148.0, 148.2, 170.2 ppm; IR ν (cm−1) 3265, 3056, 2931, 2861, 1647, 1543, 1479, 1428, 1372, 1299, 1102, 1058, 1041, 806, 747, 713; GC-MS m/z 208.1 (M+); HRMS m/z 209.1281 (M + H)+, calcd for C11H17O2N2 m/z 209.1284.
(S)-4-(N-Ethylamino)-4-(pyridin-3-yl)butan-1-ol (38)
To a dried two-neck flask at room temperature under nitrogen was added 37 (180 mg, 0.87 mmol) in 10 mL of THF and BH3 · THF (2.6 mL, 1.0 M in THF, stabilized with <0.005 M NaBH4). The solution was refluxed for 3.5 h. Then, the mixture was cooled with an ice-bath, and 5 mL of MeOH was added dropwise to quench the borane. The resulting mixture was refluxed overnight. The solvents were evaporated under reduced pressure, and the residue was directly purified by chromatography on a silica gel column eluted with CH2Cl2:CH3OH (4:1). The product was obtained as a colorless oil; yield 86% (145 mg); [α]20D = −37° (c 2.2, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.09 (t, 3H, J = 7.2 Hz), 1.67 (m, 2H), 1.85 (m, 2H), 2.53 (q, 2H, J = 7.2 Hz), 3.0 (br, 1H), 3.7 (m, 3H), 7.32 (m, 1H), 7.6 (m, 1H), 8.5 (m, 2H) ppm; 13C NMR (100 MHz, CDCl3) δ 15.0, 30.5, 36.2, 41.6, 60.9, 62.7, 123.6, 134.3, 139.3, 148.7, 148.9 ppm; IR ν (cm−1) 3268, 2932, 2861, 1655, 1578, 1426, 1379, 1320, 1268, 1104, 1060, 1027, 928, 808, 715; GC-MS m/z 194.0 (M+); HRMS m/z 195.1490 (M + H)+, calcd for C11H19O1N2 195.1492.
(S)-3-(1-Ethylpyrrolidin-2-yl)pyridine30 (3)
To a dried three-neck round-bottom flask containing PPh3 (393 mg, 1.5 mmol), Et3N · HCl (104 mg, 0.75 mmol), and compound 38 (145 mg, 0.75 mmol) in anhydrous CH2Cl2 (30 mL) under nitrogen was added a solution of diisopropyl azodicarboxilato (DIAD) (303 mg, 1.5 mmol) in anhydrous CH2Cl2 (10 mL) dropwise at −20 °C. The reaction was monitored by TLC until the starting material was totally consumed (around 3 h). When the reaction was completed, water (15 mL) was added and the aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic phases were dried over anhydrous Na2SO4. The solvents were removed under vacuum, and the residue was purified by chromatography on silica gel eluted with CH2Cl2:CH3OH (95:5). The product was obtained as a colorless oil; yield 84% (110 mg); 97% ee by GC; [α]20D = −139° (c 2.2, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.04 (t, 3H, J = 7.2 Hz), 1.73 (m, 1H), 1.85 (m, 1H), 2.0 (m, 1H), 2.12 (m, 1H), 2.25 (m, 2H), 2.64 (m, 1H), 3.27 (m, 1H), 3.4 (m, 1H), 7.28 (m, 1H), 7.7 (m, 1H), 8.5 (m, 1H), 8.6 (m, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 13.8, 22.5, 35.1, 48.2, 53.2, 67.5, 123.5, 134.9, 139.6, 148.5, 149.6 ppm; GC-MS m/z 176.1 (M+).
Supplementary Material
Acknowledgments
Financial support by the National Institutes of Health through their MBRS-SCORE (GM 08216) and PR-AABRE-INBRE (NC P20 RR-016470) grant is greatly appreciated. We express our special gratitude to the NSF-MRI (01–07) and NIH-MBRS programs to have made possible the acquisition of a 400 MHz NMR spectrometer. The NSF-ADVANCE (SBE-0123645), NIH-INBRE and NIH-RISE, NIH-MARC, NSF-AMP undergraduate student’s support is also gratefully acknowledged.
Footnotes
Supporting Information Available: Experimental procedures, physical properties and spectral data for all oximes, data characterization for known benzyl oximes and acetamides, and enantiomeric determination by chromatography for all racemic and nonracemic acetamides. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Sine SM. Neurobiology. 2002;53:431–446. doi: 10.1002/neu.10139. [DOI] [PubMed] [Google Scholar]; (b) Tonder JE, Olesen PH. Curr Med Chem. 2001;8:651. doi: 10.2174/0929867013373165. [DOI] [PubMed] [Google Scholar]; (c) Jensen AA, Frolund B, Liljefors T, Krogsgaard-Larsen P. J Med Chem. 2005;48:4705. doi: 10.1021/jm040219e. [DOI] [PubMed] [Google Scholar]
- 2.(a) Lloyd GK, Williams M. J Pharm Exp Ther. 2000;292:461. [PubMed] [Google Scholar]; (b) Cosford NDP, Blecher L, Herbaut A, Mccallum JS, Venier JM, Dawson H, Whitten JP, Adams P, Chavez-Noriega L, Correa LD, Crona JH, Mahaffy LS, Menzaghi LS, Rao TS, Reid R, Sacaan SI, Santori E, Stauderman KA, Whelan K, McDonald IA. J Med Chem. 1996;39:3235. doi: 10.1021/jm960328w. [DOI] [PubMed] [Google Scholar]; (c) Galzi JL, Changeux JP. Neuropharmacology. 1995;34:563. doi: 10.1016/0028-3908(95)00034-4. [DOI] [PubMed] [Google Scholar]
- 3.(a) Breining SR. Curr Top Med Chem. 2004;4:609. doi: 10.2174/1568026043451131. [DOI] [PubMed] [Google Scholar]; (b) Talley TT, Yalda S, Ho KY, Tor Y, Soti FS, Kem WR, Taylor P. Biochemistry. 2006;45:8894. doi: 10.1021/bi060534y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Abreo MA, Lin NH, Garvey DS, Gunn DE, Hettinger AM, Wasicak JT, Pavlik PA, Martin YC, Donnelly-Roberts DL, Anderson DJ, Sullivan JP, Williams M, Arneric SP, Holladay MW. J Med Chem. 1996;39:817. doi: 10.1021/jm9506884. [DOI] [PubMed] [Google Scholar]; (d) Newhouse PA, Potter A, Lenox RH. Med Chem Res. 1993;2:628. [Google Scholar]; (e) Felpin FX, Girard S, Vo-Thanh G, Robins RJ, Villiéras J, Lebreton J. J Org Chem. 2001;66:6305. doi: 10.1021/jo010386b. [DOI] [PubMed] [Google Scholar]; (f) Ayers JT, Xu R, Dwoskin LP, Crooks PA. AAPS J. 2005;7:E752. doi: 10.1208/aapsj070375. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Brennan MB. Chem Eng News. 2000;78:23. [Google Scholar]
- 4.(a) Holladay MW, Dart MJ, Lynch JK. J Med Chem. 1997;40:4169. doi: 10.1021/jm970377o. [DOI] [PubMed] [Google Scholar]; (b) Latli B, D’Amour K, Casida JE. J Med Chem. 1999;42:2227. doi: 10.1021/jm980721x. [DOI] [PubMed] [Google Scholar]; (c) Nielsen SF, Nielsen EO, Olsen GM, Liljefors T, Peters D. J Med Chem. 2000;43:2217. doi: 10.1021/jm990973d. [DOI] [PubMed] [Google Scholar]; (d) Mullen G, Napier J, Balestra M, DeCory T, Hale G, Macor J, Mack R, Loch J, Wu E, Kover A, Verhoest P, Sampognaro A, Phillips E, Zhu Y, Murray R, Griffith R, Blosser J, Gurley D, Machulskis A, Zongrone J, Rosen A, Gordon J. J Med Chem. 2000;43:4045. doi: 10.1021/jm000249r. [DOI] [PubMed] [Google Scholar]; (e) Chelucci G. Tetrahedron: Asymmetry. 2005;16:2353. [Google Scholar]
- 5.(a) Bachurin SO. Med Res Rev. 2003;23:48. doi: 10.1002/med.10026. [DOI] [PubMed] [Google Scholar]; (b) Vernier JM, El-Abdellaoui H, Holsenback H, Cosford NDP, Bleicher L, Barker G, Bontempi B, Chavez-Noriega L, Menzaghi F, Rao TS, Reid R, Sacaan AI, Suto C, Washburn M, Lloyd GK, McDonald IA. J Med Chem. 1999;42:1684. doi: 10.1021/jm990035d. [DOI] [PubMed] [Google Scholar]
- 6.(a) Schmitt JD, Bencherif M. In: In Annual Reports in Medicinal Chemistry. Doherty AM, editor. Vol. 35. Academic Press; New York: 2000. pp. 41–51. [Google Scholar]; (b) Pereira EFR, Hilmas C, Santos MD, Alkondon M, Maelicke A, Albuquerque EX. J Neurobiol. 2002;53:479. doi: 10.1002/neu.10146. [DOI] [PubMed] [Google Scholar]
- 7.Wilkins HL, Jr, Grinevich VP, Ayers JT, Crooks PA, Dwoskin LP. J Pharm Exp Ther. 2003:304. doi: 10.1124/jpet.102.043349. [DOI] [PubMed] [Google Scholar]
- 8.(a) Palmer AM. Trends Pharmacol Sci. 2003;23:426. doi: 10.1016/s0165-6147(02)02056-4. [DOI] [PubMed] [Google Scholar]; (b) Salomon AR, Marcinowski KJ, Friedland RP, Zagorski MG. Biochemistry. 1996;35:13568. doi: 10.1021/bi9617264. [DOI] [PubMed] [Google Scholar]; (c) Liu Q, Zhao B. Br J Pharmacol. 2004;141:746. doi: 10.1038/sj.bjp.0705653. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Dickerson TJ, Janda KD. Proc Natl Acad Sci USA. 2003;100:8182. doi: 10.1073/pnas.1332847100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aceto MD, Martin BR, Uwaydah IM, May EL, Harris LS, Izazola-Conde C, Deway WL, Bradshaw TJ, Vincek WC. J Med Chem. 1979;22:174. doi: 10.1021/jm00188a009. [DOI] [PubMed] [Google Scholar]
- 10.(a) Lawrence SA. Amines: Synthesis, Properties and Applications. Cambridge University Press; Cambridge, U.K: 2004. [Google Scholar]; (b) Breuer M, Ditrich K, Habicher T, Hauer B, Keâeler M, Sturmer R, Zelinski T. Angew Chem, Int Ed. 2004;43:788. doi: 10.1002/anie.200300599. [DOI] [PubMed] [Google Scholar]; (c) Hieber G, Ditrich K. Chim Oggi. 2001;19:16. [Google Scholar]; (d) Franchini C, Carocci A, Catalano A, Cavalluzzi MM, Corbo F, Lentini G, Scilimati A, Tortorella P, Camerino DC, Luca A. J Med Chem. 2003;46:5238. doi: 10.1021/jm030865y. [DOI] [PubMed] [Google Scholar]; (e) Abreo MA, Lin NH, Garvey DS, Gunn DE, Hettinger AM, Wasicak JT, Pavlik PA, Martin YC, Donnelly-Roberts DL, Anderson DJ, Sullivan JP, Stephen MW, Arneric SP, Holladay MW. J Med Chem. 1996;39:817. doi: 10.1021/jm9506884. [DOI] [PubMed] [Google Scholar]
- 11.(a) Henderson KW. Chem Commun. 2000:479. [Google Scholar]; (b) Denolf B, Mangelinckx S, Tornroos KW, Kimpe ND. Org Lett. 2006;8:3129. doi: 10.1021/ol0611245. [DOI] [PubMed] [Google Scholar]; (c) Tanuwidjaja J, Peltier HM, Ellman JA. J Org Chem. 2007;72:626. doi: 10.1021/jo0616512. [DOI] [PubMed] [Google Scholar]; (d) Bloch R. Chem Rev. 1998;98:1407. doi: 10.1021/cr940474e. [DOI] [PubMed] [Google Scholar]
- 12.(a) Corey EJ, Helal CJ. Angew Chem, Int Ed. 1998;37:1986. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]; (b) Hayashi Y, Gotoh H, Hayashi T, Shoji M. Angew Chem, Int Ed. 2005;44:4212. doi: 10.1002/anie.200500599. [DOI] [PubMed] [Google Scholar]; (c) Peelen TJ, Chi Y, Gellman SH. J Am Chem Soc. 2005;127:11598. doi: 10.1021/ja0532584. [DOI] [PubMed] [Google Scholar]; (d) Liu TY, Cui HL, Jiang K, Du W, He ZQ, Chen YC. Org Lett. 2007;9:3671. doi: 10.1021/ol701648x. [DOI] [PubMed] [Google Scholar]; (e) Bartoli G, Bosco M, Carlone A, Pesciaioli F, Sambri L, Mechiorre P. Org Lett. 2007;9:1403. doi: 10.1021/ol070309o. [DOI] [PubMed] [Google Scholar]; (f) Fache FSE, Tommasino ML, Lemaire M. Chem Rev. 2000;100:2159. doi: 10.1021/cr9902897. [DOI] [PubMed] [Google Scholar]; (g) Dickerson TJ, Lovell T, Meijler MM, Noodleman L, Janda KD. J Org Chem. 2004;69:6603. doi: 10.1021/jo048894j. [DOI] [PubMed] [Google Scholar]; (h) France S, Guerin DJ, Miller SJ, Lectka T. Chem Rev. 2003;103:2985. doi: 10.1021/cr020061a. [DOI] [PubMed] [Google Scholar]; (i) Glushkov VA, Tolstikov AG. Russ Chem Rev (Engl Transl) 2004;73:581. and refs. cited therein. [Google Scholar]; (j) Cho BT. Tetrahedron. 2006;62:7621. and refs. cited therein. [Google Scholar]
- 13.Kozma D. CRC Handbook of Optical Resolutions via Diatereomeric Salt Formation. 1. CRC Press LLC; Boca Raton, FL: 2001. [Google Scholar]
- 14.(a) Johansson A. Contemp Org Synth. 1995;66:393. and refs. cited therein. [Google Scholar]; (b) Deloux L, Srebnik M. Chem Rev. 1993;93:763. refs. cited therein. [Google Scholar]; (c) Davis FA, Zhou P, Chen BC. Chem Soc Rev. 1998;27:13. [Google Scholar]; (d) Fernández H, Hooper MW, Knight FI, Brown JM. Chem Commun. 1997:173. [Google Scholar]; (e) Fernández H, Maeda K, Hooper MW, Brown JM. Chem—Eur J. 2000;6:1840. doi: 10.1002/(sici)1521-3765(20000515)6:10<1840::aid-chem1840>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]; (f) Kobayashi S, Ishitani H. Chem Rev. 1999;99:1069. doi: 10.1021/cr980414z. [DOI] [PubMed] [Google Scholar]
- 15.(a) Bolm C, Felder M. Synlett. 1994:655. [Google Scholar]; (b) Lantos I, Flisak J, Liu L, Matsunoka R, Mendelson W, Stevenson D, Tubman K, Tucker L, Zhang WY, Adams J, Sorenson M, Garigipati R, Erhardt K, Ross S. J Org Chem. 1997;62:5385. [Google Scholar]; (c) Tillyer RD, Boudreau C, Tschaen D, Dolling UH, Reider PJ. Tetrahedron Lett. 1995;36:4337. [Google Scholar]; (d) Shimizu M, Kamei M, Fujisawa T. Tetrahedron Lett. 1995;36:8607. [Google Scholar]; (e) Shimizu M, Tsukamoto K, Matsutani T, Fujisawa T. Tetrahedron. 1998;54:10265. [Google Scholar]; (f) Masui M, Shioiri T. Tetrahedron Lett. 1998;39:5195. [Google Scholar]; (g) Cho BT, Ryu MH. Bull Korean Chem Soc. 1994;15:191. [Google Scholar]; (h) Demir AS. Pure Appl Chem. 1997;69:105–108. [Google Scholar]; (i) Sakito Y, Yoneyoshi Y, Suzukamo G. Tetrahedron Lett. 1988;29:223. [Google Scholar]
- 16.(a) Itsuno S, Sakurai Y, Ito K, Hirao A, Nakahama S. Bull Chem Soc Jpn. 1987;60:395. [Google Scholar]; (b) Itsuno S, Nakano M, Miyazaki K, Masuda H, Ito K. J Chem Soc, Perkin Trans 1. 1985:2039. [Google Scholar]; (c) Itsuno S, Sakurai Y, Shimizu K, Ito K. J Chem Soc, Perkin Trans 1. 1990:1859. [Google Scholar]; (d) Inoue T, Sato D, Komura K, Itsuno S. Tetrahedron Lett. 1999;40:5379. [Google Scholar]
- 17.(a) Itsuno S, Matsumoto T, Sato D, Inoue T. J Org Chem. 2000;65:5879. doi: 10.1021/jo0007837. [DOI] [PubMed] [Google Scholar]; (b) Sailes HE, Watts JP, Whiting A. J Chem Soc, Perkin Trans 1. 2000:3362. [Google Scholar]; (c) Fontaine E, Namane C, Meneyrol J, Geslin M, Serva L, Roussey E, Tissandie S, Maftouh M, Roger P. Tetrahedron: Asymmetry. 2001;12:2185. [Google Scholar]; (d) Krzeminski MP, Zaidlewicz M. Tetrahedron: Asymmetry. 2003;14:1463. [Google Scholar]; (e) Chu YB, Shan ZX, Liu DJ, Sun NN. J Org Chem. 2006;71:3998. doi: 10.1021/jo060123n. [DOI] [PubMed] [Google Scholar]
- 18.(a) Stepanenko V, Ortiz-Marciales M, Barnes CE, Garcia C. Tetrahedron Lett. 2006;47:7603. doi: 10.1016/j.tetlet.2008.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ortiz-Marciales M, De Jesus M, Gonzales E, Raptis RG, Baran P. Acta Crystallogr. 2004;C60:173. doi: 10.1107/S0108270104000666. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lang A, Noth H, Schmidt M. Chem Ber. 1997;130:241. [Google Scholar]; (d) Mathre DJ, Thompson AS, Douglas AW, Carroll JD, Corley EG, Grabowski EJJ. J Org Chem. 1993;58:2880. [Google Scholar]; (e) Tlahuext H, Santiesteban F, García-Báez E, Contreras R. Tetrahedron: Asymmetry. 1994;5:1579. [Google Scholar]; (f) Santiesteban F, Mancilla T, Klaébe A, Contreras R. Tetrahedron Lett. 1983;24:759. [Google Scholar]; (g) Brown JM, Guy C, Lloyd-Jones GC, Layzell TP. Tetrahedron: Asymmetry. 1993;4:2151. [Google Scholar]
- 19.(a) Stepanenko V, Ortiz-Marciales M, Correa W, De-Jesús M, Espinosa S, Ortiz L. Tetrahedron: Asymmetry. 2006;17:112–115. [Google Scholar]; (b) Ortiz-Marciales M, Stepanenko V, Correa W, De Jesús M, Espinosa S. 11/512,599. US Patent Application. 2006 Aug 30;; (c) Stepanenko V, Ortiz-Marciales M, De Jesús M, Correa W, Vázquez C, Ortiz L, Guzmán I, De la Cruz W. Tetrahedron: Asymmetry. 2007;18:2738. doi: 10.1016/j.tetasy.2007.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Huang X, Ortiz-Marciales M, Huang K, Stepanenko V, Merced FG, Ayala AM, De Jesús M. Org Lett. 2007;9:1793. doi: 10.1021/ol0704791. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ortiz-Marciales M, Huang X, Huang K, Stepanenko V, De Jesús M, Merced F. PCT/US07/76195. 2007 Aug 17;
- 21.Benjahad A, Croisy M, Monneret C, Bisagni E, Mabire D, Coupa S, Poncelet A, Csoka I, Guillemont J, Meyer C, Andries K, Pauwels R, de Béthune MP, Himmel DM, Das K, Arnold E, Nguyen CH, David S, Grierson DS. J Med Chem. 2005;48:1948. doi: 10.1021/jm0408621. [DOI] [PubMed] [Google Scholar]
- 22.Purification of oxime benzyl ethers by vacuum distillation should be avoided since they isomerize and decompose under heating by a reverse radical disproportionation.23
- 23.Blake JA, Ingold KU, Lin S, Mulder P, Pratt DA, Sheeller B, Walton JC. Org Biomol Chem. 2004;2:415. doi: 10.1039/b313491a. [DOI] [PubMed] [Google Scholar]
- 24.Mitsunobu O. Synthesis. 1981:1. [Google Scholar]
- 25.Bernotas RC, Cube RV. Tetrahedron Lett. 1991;32:161. [Google Scholar]
- 26.(a) Lawrence SA. In Amines: Synthesis, Properties and Applications. Cambridge University Press; Cambridge, U.K: 2004. [Google Scholar]; (b) Breuer M, Ditrich K, Habicher T, Hauer B, Keâeler M, Sturmer R, Zelinski T. Angew Chem, Int Ed. 2004;43:788. doi: 10.1002/anie.200300599. [DOI] [PubMed] [Google Scholar]; (c) Hieber G, Ditrich K. Chim Oggi. 2001;19:16. [Google Scholar]; (d) Franchini C, Carocci A, Catalano A, Cavalluzzi MM, Corbo F, Lentini G, Scilimati A, Tortorella P, Camerino DC, Luca A. J Med Chem. 2003;46:5238. doi: 10.1021/jm030865y. [DOI] [PubMed] [Google Scholar]; (e) Abreo MA, Lin NH, Garvey DS, Gunn DE, Hettinger AM, Wasicak JT, Pavlik PA, Martin YC, Donnelly-Roberts DL, Anderson DJ, Sullivan JP, Stephen MW, Arneric SP, Holladay MW. J Med Chem. 1996;39:817. doi: 10.1021/jm9506884. [DOI] [PubMed] [Google Scholar]
- 27.(a) Henderson KW. Chem Commun. 2000:479. [Google Scholar]; (b) Denolf B, Mangelinckx S, Tornroos KW, Kimpe ND. Org Lett. 2006;8:3129. doi: 10.1021/ol0611245. [DOI] [PubMed] [Google Scholar]; (c) Tanuwidjaja J, Peltier HM, Ellman JA. J Org Chem. 2007;72:626. doi: 10.1021/jo0616512. [DOI] [PubMed] [Google Scholar]
- 28.Edwards William B. J Heterocycl Chem. 1975;12:413. [Google Scholar]
- 29.Ohkawa S, Terao JS, Terashita ZI, Shibouta Y, Nishikawat K. J Med Chem. 1991;34:267. doi: 10.1021/jm00105a042. [DOI] [PubMed] [Google Scholar]
- 30.Damaj MI, Glassco W, Dukat M, May EL, Richard A, Martin BR. Drug Dev Res. 1996;38:177. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.