

Abstract
According to Khan et al. [Khan, A. U., Kovacic, D., Kolbanovskiy, A., Desai, M., Frenkel, K. & Geacintov, N. E. (2000) Proc. Natl. Acad. Sci. USA 97, 2984–2989], peroxynitrite (ONOO−) decomposes after protonation to singlet oxygen (1ΔgO2) and singlet oxonitrate (nitroxyl, 1NO−) in high yield. They claimed to have observed nitrosyl hemoglobin from the reaction of NO− with methemoglobin; however, contamination with hydrogen peroxide gave rise to ferryl hemoglobin, the spectrum of which was mistakenly assigned to nitrosyl hemoglobin. We have carried out UV–visible and EPR experiments with methemoglobin and hydrogen peroxide-free peroxynitrite and find that no NO− is formed. With this peroxynitrite preparation, no light emission from singlet oxygen at 1270 nm is observed, nor is singlet oxygen chemically trapped; however, singlet oxygen was trapped when hydrogen peroxide was also present, as previously described [Di Mascio, P., Bechara, E. J. H., Medeiros, M. H. G., Briviba, K. & Sies, H. (1994) FEBS Lett. 355, 287–289]. Quantum mechanical and thermodynamic calculations show that formation of the postulated intermediate, a cyclic form of peroxynitrous acid (trioxazetidine), and the products 1NO− and 1ΔgO2 requires Gibbs energies of ca. +415 kJ⋅mol−1 and ca. +180 kJ⋅mol−1, respectively. Our results show that the results of Khan et al. are best explained by interference from contaminating hydrogen peroxide left from the synthesis of peroxynitrite.
Peroxynitrite [oxoperoxonitrate(1−)] is an inorganic oxidant of biological importance that is produced from superoxide and nitrogen monoxide (1). The mechanisms by which peroxynitrite or peroxynitrous acid [pKa = 6.7 at 37°C (2)] oxidizes biomolecules are being investigated by various groups. There are two types or reaction: reactions between biomolecules and peroxynitrite (or peroxynitrous acid) that are first-order in peroxynitrite and in the biomolecule, and reactions that are first-order in peroxynitrite and zero-order in the biomolecule. In the latter case, either peroxynitrous acid undergoes homolysis to nitrogen dioxide and the hydroxyl radical (3–7), or it forms an activated intermediate (2, 8). In the absence of oxidizable molecules the radicals recombine to nitrate, or the activated intermediate undergoes an internal isomerization. The rate constant for this process, which is first-order in peroxynitrite, is 3.9 s−1 at 37°C (2). Whether radicals are intermediates is not relevant for the present discussion. All involved agree that at acid pH and at low temperatures peroxynitrous acid isomerizes to nitrate quantitatively, whereas at higher pH nitrite and dioxygen in a 2 to 1 ratio also are found.
Recently, a mechanism was proposed, in which peroxynitrite forms, after protonation, a cyclic intermediate, which dissociates to form oxonitrate(1−)—the commonly used name “nitroxyl” is outdated—and singlet oxygen (9):
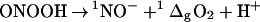 |
1 |
These products have been postulated before by Khan (10). As described in the more recent article (9), Khan et al. used methemoglobin (MetHb) to trap oxonitrate(1−), and 9,10-diphenylanthracene and 2,3-dimethyl-2-butene to trap singlet oxygen. Their mechanism does not explain the quantitative formation of nitrate, and we therefore investigated how these results (9) could have been obtained. We show here that singlet oxygen is not formed when peroxynitrite free of hydrogen peroxide is used, that the evidence for oxonitrate(1−) is based on a misinterpretation of the hemoglobin UV–visible spectra, and that the formation of these products and the postulated cyclic precursor, 4-hydrido-4-azy-1,2,3-trioxy[04]cycle, or trioxazetidine, is thermodynamically unlikely. The experimental evidence presented by Khan et al. (9) is best explained by the presence of hydrogen peroxide formed during the synthesis of peroxynitrite.
Materials and Methods
Chemicals.
Peroxynitrite was synthesized from sodium nitrite (0.6 M) and hydrogen peroxide (0.65 M) in a quenched-flow reactor (11); excess hydrogen peroxide was used to minimize nitrite contamination (12). To eliminate excess hydrogen peroxide, ONOO− was treated with manganese dioxide. Synthesized ONOO− contained low levels of contaminating hydrogen peroxide (<5 μM) and nitrite (10–30%) that were determined by the titanyl method and by absorbance measurements at 354 nm (ɛ = 24.6 M−1⋅cm−1), respectively (13). Peroxynitrite used for the UV–visible experiments was prepared according to ref. 14 and also contained very low levels of the contaminants hydrogen peroxide and nitrite. The concentration of the ONOO− stock solutions was determined spectrophotometrically at 302 nm with an extinction coefficient of 1,670 M−1⋅cm−1 (15). Sodium trioxodinitrate (Na2N2O3) was purchased from Cayman Chemicals; a stock solution of this salt was prepared in deoxygenated sodium hydroxide (0.01 M) and kept on ice. MetHb was prepared by oxidizing bovine hemoglobin (Sigma) with 20% excess potassium hexacyanoferrate(III) in 0.2 M sodium phosphate buffer, pH 7.4. Hexacyanoferrate(II) and excess hexacyanoferrate(III) were removed by desalting on a G-25 Sephadex column. Heme concentrations were determined spectrophotometrically at 406 nm (ɛ = 154 mM−1⋅cm−1) (16). The water-soluble disodium salt of anthracene-9,10-diyldiethyl disulfate (EAS) and the endoperoxide of N,N′-di(2,3-dihydroxypropyl)-1,4-naphthalenedipropanamide (DHPNO2) were synthesized and used according to refs. 17 and 18 and ref. 19, respectively. Sodium hypochlorite was analyzed iodometrically. All solutions were prepared with distilled water purified with a Millipore Milli-Q system.
UV–Visible Spectra of MetHb and Peroxynitrite.
Absorption spectra were collected with a UVIKON 820 spectrophotometer. An 80 μM MetHb solution in 0.1 M sodium phosphate buffer, pH 7, was placed in a sealable cuvette (path length 1 cm, total volume 3 ml) and deoxygenated by gently bubbling N2 through it for at least 30 min. Peroxynitrite was added from a deoxygenated stock solution (30 mM in 0.1 M NaOH) by using a gas-tight Hamilton syringe. Hydrogen peroxide was added analogously from a deoxygenated 880 mM stock solution in water.
Electron Paramagnetic Resonance (EPR) Measurements.
EPR spectra were recorded with a Bruker EMX instrument at 77 K; this temperature was attained with a fingertip liquid-nitrogen Dewar (20). Peroxynitrite was added to a nitrogen-bubbled solution of MetHb (final volume 0.7 ml), and after 5-min incubation the mixture was transferred into a 1-ml plastic syringe (4 mm inside diameter) and frozen in dry ice/ethanol. Just before EPR analysis, the syringe tip was removed and the hand-warmed sample was pushed to a fingertip Dewar containing liquid nitrogen. The same protocol was used for the reactions with Na2N2O3 except that the mixtures were incubated for 20 min at 37°C before freezing. The yield of nitrosyl hemoglobin (HbFeIINO) was determined by double integration of the measured EPR signal and subsequent comparison with the doubly integrated signals of samples with known concentrations of HbFeIINO prepared by adding a 5-fold excess of Na2N2O3 to MetHb. Under these conditions 99% conversion of MetHb to HbFeIINO was confirmed by the disappearance of the EPR signal of MetHb at g = 6.
Monomol Emission of Singlet Oxygen.
The infrared photoemission of singlet oxygen at 1,270 nm was measured with a liquid nitrogen-cooled germanium photodetector sensitive in the spectral region from 800 to 1,800 nm with a detector area of 0.25 cm2 and a sapphire window as described (17). A band-pass filter at 1,270 nm with a 10-nm half-bandwidth was used.
Chemical Trapping of 1ΔgO2 and HPLC/Mass Spectrometry Analysis.
EAS endoperoxide (EASO2) was separated from EAS by HPLC with a system consisting of LC 10ADVP pumps connected to an automatic SIL-10ADVP injector (Shimadzu, Tokyo). For analytical purposes, the system was equipped with a 150 mm × 4.6 mm inside diameter (particle size 5 μm) Supelco C18 reverse-phase column and a 20 × 2.1 mm (particle size 5 μm) Supelco C18 guard precolumn (Supelco, Bellefonte, PA), connected to a Shimadzu SPD-M10AVVP diode array. The samples were diluted 1:10 and 30 μl was injected. Solvent A was 25 mM ammonium formate, and solvent B was acetonitrile. The compounds were eluted by an increasing linear gradient of B from 15% to 17% during the first 8 min and by keeping it constant thereafter. The flow rate was 1.0 ml/min. Data acquisition was performed by means of Shimadzu class-vp software, version 5.3. Electrospray ionization (ESI) mass spectrometry analyses in the negative mode were performed with a Platform II mass spectrometer (Micromass, Altricham, U.K.). A Shimadzu LC-10AD pump was used to infuse the eluent, a 1:1 mixture of water and acetonitrile, at a final flow rate of 10 μl/min. The samples (20 μl) were injected through a 20-μl Rheodyne loop (Rheodyne; Cotati, CA). The source temperature was maintained at 80°C, and flow rates of the drying and nebulizing gas (nitrogen) were optimized at 300 and 29 liters/h, respectively. The cone voltage was maintained at 10 V, and the capillary and high-voltage electrode potentials were at 3.50 and 0.69 kV, respectively. Full-scan data were acquired over a mass range of 100–400 Da. The data were processed and transformed into values of molecular masses on mass scale by means of the Mass Lynx NT data system 3.20 version (Micromass).
Ab Initio Calculations.
Calculations were performed with the gaussian 94 suite of programs (21) that apply density functional theory methods and a standard 6–311+G(d,p) basis set. We performed gradient-corrected calculations with the functional developed by Becke (22) for the exchange and by Lee et al. (23) for the correlation part (blyp). We also used the hybrid method Becke3-lyp, which combines Becke's three-parameter mixing of exchange terms (24) with the nonlocal part of the lyp correlation functional (b3lyp). To test basis set convergence dependence, additional calculations were performed with the program cpmd (25), which also uses the blyp functional. Valence orbitals were expanded in a basis of plane waves up to a kinetic energy cutoff of 70 rydberg (150 aJ). Only valence electrons were treated explicitly. To account for the effect of the inner electrons, we applied norm-conserving pseudopotentials of the Martins–Trouiller type (26), with cut-off radii of 0.5 atomic unit (au; 1 au = 0.529⋅10−10 m) for hydrogen, 1.12 au for nitrogen, and 1.11 au for oxygen.
Results and Discussion
MetHb Experiments.
The principal evidence for the formation of oxonitrate(1−) by Khan et al. (9) was the putative formation of nitrosyl hemoglobin by the addition of peroxynitrite to MetHb. However, we (27) and others (28, 29) have previously observed that ONOO− does not react with the iron(III) center of MetHb or metmyoglobin under similar conditions, which we repeated here (Fig. 1, trace 2). Khan et al. (9) prepared peroxynitrite by adding solid potassium superoxide to a nitrogen monoxide-saturated solution. In our experience, such solutions are invariably contaminated with nitrite and with hydrogen peroxide—formed from the spontaneous dismutation of superoxide—at concentrations that exceed the concentration of peroxynitrite. They did not report any efforts to remove hydrogen peroxide, nor did they measure hydrogen peroxide. We found that the simple addition of hydrogen peroxide, which forms ferryl hemoglobin, resulted the spectral features interpreted by Khan et al. as resulting from nitrosyl hemoglobin. Indeed, the experiment depicted in figure 1 of the cited paper does not support the generation of NO−. First, the absorbance spectrum measured after the reaction of MetHb with 2 equivalents of the NO− source Na2N2O3 (spectrum e of figure 1 of ref. 9) does not represent the spectrum of pure HbFeIINO, but rather that of a mixture of MetHb and HbFeIINO. This conclusion is based on the presence of the characteristic MetHb absorbance band around 500 nm and was not unexpected. As shown by Hollocher and coworkers (30), 5 equivalents of Na2N2O3 are needed to quantitatively generate HbFeIINO from MetHb, because of the fast parallel dimerization and dehydration reaction of HNO to yield N2O. Thus, if this spectrum is considered to be identical to that obtained from the reaction of MetHb with 1 equivalent of peroxynitrite (spectrum d of figure 1 in ref. 9), the yield of NO− from peroxynitrite cannot be considered quantitative. Second, the two spectra mentioned above obtained by reacting either Na2N2O3 or peroxynitrite with MetHb (spectra d and e of figure 1 of ref. 9), are not identical: the maximum around 500 nm is present only in the former. The absorbance changes observed by Khan et al. (9) upon addition of peroxynitrite to MetHb may derive from the partial formation of oxoiron(IV)Hb (HbFeIV⩵O), generated from the reaction of H2O2 with MetHb. As nitrite is known to reduce HbFeIV⩵O to MetHb (27) and to bind to MetHb, it is not possible to estimate accurately the amounts of nitrite and hydrogen peroxide present in the peroxynitrite solutions used by Khan et al.
Figure 1.

Absorbance spectra of a solution of 80 μM bovine MetHb in 0.1 M sodium phosphate buffer (pH 7.0) at room temperature under N2. Trace 1, MetHb solution; trace 2, MetHb + 160 μM ONOO−; trace 3, MetHb + 160 μM ONOO− + 400 μM H2O2; trace 4, MetHb + 160 μM ONOO− + 960 μM H2O2.
To further assess whether any trace of nitrosyl hemoglobin could be detected we used low-temperature EPR. No HbFeIINO can be detected by low-temperature EPR of mixtures of 1 mM MetHb and 1 mM ONOO− under low oxygen tensions (Fig. 2, spectra A and B). As expected, mixtures of 1 mM MetHb and Na2N2O3 produced an EPR signal composite of those of hexa- and pentacoordinated HbFeIINO complexes (Fig. 2, spectra C and D). As previously reported, the yields of the detected complexes increased with increasing HN2O3− concentration (30). Under our experimental conditions, addition of either 0.1 or 1 mM HN2O3− to 1 mM MetHb generated 60 and 430 μM HbFeIINO, respectively. Consequently, even if only 10% of 1 mM ONOO− decayed to produce 100 μM NO−, the EPR signal of the subsequently formed HbFeIINO would be clearly detectable as it was in mixtures of 0.1 mM HN2O3− and 1 mM MetHb (Fig. 2C).
Figure 2.

Representative low-temperature EPR spectra obtained from mixtures of 1 mM MetHb and ONOO− or HN2O3− under reduced oxygen tensions. A, MetHb; B, MetHb + 1 mM ONOO−; C, MetHb + 0.1 mM HN2O3−; and D, MetHb + 1 mM HN2O3−. The incubation mixtures were prepared and EPR analyses were performed as described in Materials and Methods. Instrumental conditions: power, 20 mW; modulation amplitude, 5 G; scan rate, 7.4 G/s; gain, 1.42 × 104, except for D, where the gain was 2.83 × 103.
Both UV–visible and EPR experiments demonstrate that virtually no oxonitrate(1−) is produced during peroxynitrite decay.
Singlet Oxygen Detection.
We could also find no evidence for the formation of singlet oxygen produced by peroxynitrite decay unless there was hydrogen peroxide present. First, we monitored directly the monomol emission of singlet oxygen at 1,270 nm. Addition of a solution of acetic acid to a 3-ml solution of peroxynitrite (25 mM; pH 14) gave no signal (data not shown), whereas there was a strong signal at 1,270 nm when tert-butyl hydroperoxide (25 mM) (see figure 3 of ref. 31) or hydrogen peroxide (32, 33) was also present.
Detection of 1ΔgO2 from the decay of high concentrations of peroxynitrite in a two-phase system by Khan et al. (9) could be due to the reaction between peroxynitrite and contaminant hydrogen peroxide (33). Indeed, experiments with the water-soluble 1ΔgO2 chemical probe EAS (Reaction 2) (17, 18) provided no evidence for the formation of singlet oxygen during decay of peroxynitrite when hydrogen peroxide was absent (Fig. 3).
 |
2 |
Figure 3.

Chemical detection of 1ΔgO2 by formation of EASO2. (A–E) Gradient reverse-phase HPLC. (A) 0.09 mM EAS and 0.033 mM EASO2. (B) Trace a, 10 mM DHPNO2 and 8 mM EASO2; trace b, same as trace a after incubation at 40°C for 2 h in water. (C) Six aliquots of 10 μl of both 10 mM OCl− and H2O2 were added to 8 mM EAS in 150 mM sodium phosphate buffer, pH 7.4, such that the final concentrations of hypochlorite and hydrogen peroxide (if unreacted) were 1 mM each. (D) Trace a, 1 mM ONOO− and 8 mM EAS; trace b, 15 mM ONOO− and 8 mM EAS; and trace c, 15 mM ONOO−, 8 mM EAS, and 0.012 mM EASO2, all in 150 mM phosphate buffer, pH 7.4. (E) 100 mM H2O2, 15 mM ONOO−, and 8 mM EAS. (F) Mass spectra of EAS (a) and EASO2 (b). For the experiments described in D, trace b, and E, a 138 mM solution of ONOO− was infused at a rate of 8 μl/min into a mixture containing 8 mM EAS in 150 mM phosphate buffer, pH 7.4, with (E) or without (D, trace b) 100 mM H2O2. When ONOO− and EAS were mixed (final concentrations 1 and 8 mM, respectively) in 150 mM sodium acetate buffer, pH 5.5, no EASO2 was detected (data not shown).
EAS and EASO2 were analyzed by gradient reverse-phase HPLC (Fig. 3A) and electrospray tandem mass spectrometry (Fig. 3F). The mass spectrum of EAS recorded in the negative mode exhibits a major [M − 2H]2− ion at m/z = 212 (Fig. 3Fa), corresponding to the doubly charged molecule. The spectrum of EASO2 exhibits, as expected, an intense [M − 2H]2− ion at m/z = 228 (Fig. 3Fb). For comparison, we showed that EASO2 is formed from the reaction of EAS with singlet oxygen generated either by thermal decomposition of the endoperoxide DHPNO2 (10 mM) (19) (Fig. 3B) or by 1 mM OCl−/H2O2 system (Fig. 3C). In contrast, neither 1 mM nor 15 mM ONOO− led to the formation of detectable amounts of EASO2 (Fig. 3D, trace a, and trace b, respectively) as proved by co-injection of EASO2 in the 15 mM ONOO− assay (Fig. 3D trace c). Peroxynitrite in the presence of high concentrations of H2O2 produced EASO2 (Fig. 3E), as already described by Di Mascio et al. (32, 33).
Theoretical Considerations.
Khan et al. (9) propose a strained four-membered structure, 4-hydrido-4-azy-1,2,3-trioxy[04]cycle, or trioxazetidine, for the peroxynitrous acid that then undergoes a simultaneous double homolysis to form 1HNO and 1O2. We estimated the energy of this four-membered structure with three ab initio calculations. All identify the—nonplanar—ring structure as a local minimum of the potential energy surface, albeit at a very high energy compared with the ground state of ONOOH: The g94/blyp, g94/b3lyp, and cpmd/blyp differences between the ring structure and cis-peroxynitrous acid are +433, +429, and +406 kJ⋅mol−1, respectively. Although the results apply to the gas phase, these differences should be good approximations for the aqueous phase. These values are nearly 4 times higher than the experimentally measured activation enthalpy of +86.5 kJ⋅mol−1 (34).
A thermodynamic analysis shows that formation of oxonitrate(1−) and singlet oxygen from peroxynitrous acid, Reaction 1, is most unlikely. The Gibbs energies of formation of ONOO−, 1NO−, and 1ΔgO2 have been calculated as +59 to +71 kJ⋅mol−1 (2, 35), +136 kJ⋅mol−1 (36), and +112 kJ⋅mol−1 (37), respectively. The latter value includes a Gibbs solvation energy, which was taken to be the same as that of triplet oxygen. This results in a Gibbs energy of reaction between +176 and +188 kJ⋅mol−1. These results are shown in Fig. 4.
Figure 4.

Gibbs energy differences between the postulated intermediates (9), nitrate and cis-peroxynitrous acid. The experimentally observed energetics of the isomerization pathway to nitrate are also indicated (2, 34).
From 1NO− and 1O2 to NO3−.
The proposed mechanism, which involves the oxidation of oxonitrate(1−) to nitrite, does not explain the quantitative formation of nitrate, observed when peroxynitrous acid isomerizes at low pH and low temperature. As Khan et al. (9) themselves point out, oxidizing agents would be required, whereas experimentally there is no need for such agents. Furthermore, oxonitrate(1−) would react with itself to yield dinitrogen monoxide, which has not been observed.
Singlet Dioxygen Formation.
The simple explanation for the results of Khan et al. (9) is that hydrogen peroxide was present in the peroxynitrite preparation. The formation of singlet oxygen from peroxynitrite and hydrogen peroxide was described in 1994 (32). Radi and coworkers (38) have found that the reaction of hydrogen peroxide and peroxynitrous acid or peroxynitrite yields dioxygen. To increase the yield of the latter a large excess of hydrogen peroxide had to be used. The mechanism of dioxygen formation (39, 40) involves the one-electron oxidation of hydrogen peroxide by peroxynitrous acid (Reaction 3), the formation of dioxoperoxonitrate(1−) (O2NOO−) from nitrogen dioxide and superoxide (Reaction 4), followed by heterolysis to produce nitrite and dioxygen (Reaction 5):
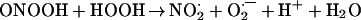 |
3 |
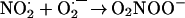 |
4 |
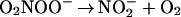 |
5 |
Given the Gibbs energy of formation of dioxoperoxonitrate(1−), +39 kJ⋅mol−1 (41), nitrite, −32.2 kJ⋅mol−1 (42), and of singlet oxygen, +94.6 kJ⋅mol−1 (37), formation of singlet oxygen in Reaction 5 is not favorable, ΔrxnG° = +23.0 kJ⋅mol−1. However, a direct oxidation of superoxide (ΔfG° = +31.8 kJ⋅mol−1; ref. 37) by nitrogen dioxide (ΔfG° = +63.0 kJ⋅mol−1; ref. 36), Reaction 6,
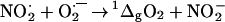 |
6 |
without dioxoperoxonitrate(1−) as an intermediate would allow thermodynamically the formation of singlet oxygen, with a Gibbs energy change of −32.4 kJ⋅mol−1.
Relevance of Isomerization Intermediates to Biology.
The main sinks for peroxynitrite in a biological milieu are the bimolecular reactions with carbon dioxide (43, 44) sulfur- or selenium-containing compounds (45–48), and metalloproteins (27, 49–51). In these reactions peroxynitrite is estimated to disappear at rates in excess of 70–80 s−1, approximately 40 times faster than the isomerization, which involves either an activated intermediate, radicals, or, according to Khan et al. (9), 1NO− and singlet oxygen. Thus, these species do not contribute significantly to the biological reactivity of peroxynitrite, except for environments of low pH, such as those found in the phagosomes of phagocytic cells.
Conclusions
We presented above seven arguments against the conclusion of Khan et al. (9) that singlet oxonitrate(1−) and singlet oxygen are intermediates in the isomerization of peroxynitrous acid to nitrate and a proton. Furthermore, the formation of these, or any other intermediates, is of limited relevance to biology.
This publication on the formation of singlet oxygen from peroxynitrite is reminiscent of a previous erroneous claim by Khan and Kasha (52) in 1994 that singlet oxygen is formed from the reaction of superoxide with hydrogen peroxide. This reaction, too, has a positive Gibbs energy, but more importantly, in 1947 George (53), and later several other groups (54–59) showed that, compared with the rate of superoxide dismutation (60), the reaction, even with triplet oxygen as a product, is too slow to be relevant.
We found another criticism (61) of the work of Khan et al. (9) while this paper was under review. It mentions two of the seven arguments discussed above. Although these authors, too, come to the conclusion that hydrogen peroxide was present during the experiments of Khan et al. (9), they could not account for the formation of singlet oxygen, although its formation from peroxynitrite and hydrogen peroxide has been described in the literature (32, 33) and one of their own works (41) provided a valuable clue for a possible mechanism of this reaction.
Acknowledgments
We thank Profs. K. Shank and R. Radi for helpful discussions and Dr. P. L. Bounds for a critical reading of the manuscript. This work was supported by grants from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; Brazil), the Conselho Nacional para o Desenvolvimento Científico e Tecnológico (CNPq; Brazil), the Programa de Apoio aos Núcleos de Excelência (PRONEX/FINEP; Brazil), the Deutsche Forschungsgemeinschaft (SFB 503/B1; Germany), the National Foundation for Cancer Research (Bethesda, MD), and the Eidgenössische Technische Hochschule, Zürich.
Abbreviations
- EAS
disodium salt of anthracene-9,10-diyldiethyl disulfate
- EASO2
EAS endoperoxide
- DHPN
N,N′-di(2,3-dihydroxypropyl)-1,4-naphthalenedipropanamide
- MetHb
methemoglobin
- HbFeIINO
nitrosyl hemoglobin
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Present address: Institut für Ernährungsphysiologie, Bundesforschungsanstalt für Ernährung, Haid-und-Neu-Strasse 9, D-76131 Karlsruhe, Germany.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.190256897.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.190256897
References
- 1.Beckman J S, Koppenol W H. Am J Physiol Cell Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 2.Koppenol W H, Kissner R. Chem Res Toxicol. 1998;11:87–90. doi: 10.1021/tx970200x. [DOI] [PubMed] [Google Scholar]
- 3.Pfeiffer S, Gorren A C F, Schmidt K, Werner E R, Hansert B, Bohle D S, Mayer B. J Biol Chem. 1997;272:3465–3470. doi: 10.1074/jbc.272.6.3465. [DOI] [PubMed] [Google Scholar]
- 4.Gatti R M, Alvarez B, Vasquez-Vivar J, Radi R, Augusto O. Arch Biochem Biophys. 1998;349:36–46. doi: 10.1006/abbi.1997.0451. [DOI] [PubMed] [Google Scholar]
- 5.Merényi G, Lind J, Goldstein S, Czapski G. Chem Res Toxicol. 1998;11:712–713. doi: 10.1021/tx980043h. [DOI] [PubMed] [Google Scholar]
- 6.Richeson C E, Mulder P, Bowry V W, Ingold K U. J Am Chem Soc. 1998;120:7211–7219. [Google Scholar]
- 7.Coddington J W, Hurst J K, Lymar S V. J Am Chem Soc. 1999;121:2438–2443. [Google Scholar]
- 8.Koppenol W H. In: Metal Ions in Biological Systems. Sigel A, Sigel H, editors. New York: Dekker; 1999. pp. 597–619. [PubMed] [Google Scholar]
- 9.Khan A U, Kovacic D, Kolbanovskiy A, Desai M, Frenkel K, Geacintov N E. Proc Natl Acad Sci USA. 2000;97:2984–2989. doi: 10.1073/pnas.050587297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khan A U. J Biolumin Chemilumin. 1995;10:329–333. doi: 10.1002/bio.1170100604. [DOI] [PubMed] [Google Scholar]
- 11.Beckman J S, Beckman T W, Chen J, Marshall P A, Freeman B A. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saha A, Goldstein S, Cabelli D, Czapski G. Free Radical Biol Med. 1998;24:653–659. doi: 10.1016/s0891-5849(97)00365-1. [DOI] [PubMed] [Google Scholar]
- 13.Kissner R, Nauser T, Bugnon P, Lye P G, Koppenol W H. Chem Res Toxicol. 1997;10:1285–1292. doi: 10.1021/tx970160x. [DOI] [PubMed] [Google Scholar]
- 14.Koppenol W H, Kissner R, Beckman J S. Methods Enzymol. 1996;269:296–302. doi: 10.1016/s0076-6879(96)69030-2. [DOI] [PubMed] [Google Scholar]
- 15.Hughes M N, Nicklin H G. J. Chem. Soc. A. 1968. 450–452. [Google Scholar]
- 16.Augusto O, Cilento G. Arch Biochem Biophys. 1975;168:549–556. doi: 10.1016/0003-9861(75)90286-6. [DOI] [PubMed] [Google Scholar]
- 17.Di Mascio P, Sies H. J Am Chem Soc. 1989;111:2909–2914. [Google Scholar]
- 18.McCall D B. Ph.D. thesis. Detroit: Wayne State Univ.; 1984. [Google Scholar]
- 19.Pierlot C, Aubry J M, Briviba K, Sies H, Di Mascio P. Methods Enzymol. 2000;319:3–20. doi: 10.1016/s0076-6879(00)19003-2. [DOI] [PubMed] [Google Scholar]
- 20.Giorgio S, Linares E, Ischiropoulos H, Von Zuben F J, Yamada A, Augusto O. Infect Immun. 1998;66:807–814. doi: 10.1128/iai.66.2.807-814.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frisch M J, Trucks G W, Schlegel H B, Gill P M W, Johnson B G, Robb M A, Cheeseman J R, Keith T A, Petersson G A, Montgomery J A, et al. gaussian 94. Pittsburgh: Gaussian; 1995. , Revision C.3. [Google Scholar]
- 22.Becke A D. Phys Rev A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 23.Lee C, Yang W, Parr R G. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 24.Becke A D. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 25.Hutter J, Ballone P, Bernasconi M, Focher P, Fois E, Goedecker S, Parrinello M, Tuckerman M. cpmd. Stuttgart, and IBM Research Laboratory Zürich: Max-Planck-Institut für Festkörperforschung; 1998. [Google Scholar]
- 26.Trouiller N, Martins J L. Phys Rev B. 1991;43:1993–2006. doi: 10.1103/physrevb.43.1993. [DOI] [PubMed] [Google Scholar]
- 27.Exner M, Herold S. Chem Res Toxicol. 2000;13:287–293. doi: 10.1021/tx990201k. [DOI] [PubMed] [Google Scholar]
- 28.Alayash A I, Ryan B A, Cashon R E. Arch Biochem Biophys. 1998;349:65–73. doi: 10.1006/abbi.1997.0449. [DOI] [PubMed] [Google Scholar]
- 29.Minetti M, Scorza G, Pietraforte D. Biochemistry. 1999;38:2078–2087. doi: 10.1021/bi982311g. [DOI] [PubMed] [Google Scholar]
- 30.Bazylinski D A, Goretski J, Hollocher T C. J Am Chem Soc. 1985;107:7986–7989. [Google Scholar]
- 31.Di Mascio P, Briviba K, Sasaki S T, Catalani L H, Medeiros M H G, Bechara E J H, Sies H. Biol Chem. 1997;378:1071–1074. [PubMed] [Google Scholar]
- 32.Di Mascio P, Bechara E J H, Medeiros M H G, Briviba K, Sies H. FEBS Lett. 1994;355:287–289. doi: 10.1016/0014-5793(94)01224-5. [DOI] [PubMed] [Google Scholar]
- 33.Di Mascio P, Briviba K, Bechara E J H, Medeiros M H G, Sies H. Methods Enzymol. 1996;269:395–400. doi: 10.1016/s0076-6879(96)69040-5. [DOI] [PubMed] [Google Scholar]
- 34.Padmaja S, Kissner R, Bounds P L, Koppenol W H. Helv Chim Acta. 1998;81:1201–1206. [Google Scholar]
- 35.Merényi G, Lind J. Chem Res Toxicol. 1998;11:243–246. doi: 10.1021/tx980026s. [DOI] [PubMed] [Google Scholar]
- 36.Stanbury D M. Adv Inorg Chem. 1989;33:69–138. [Google Scholar]
- 37.Koppenol W H. In: Focus on Membrane Lipid Oxidation. Vigo-Pelfrey C, editor. Vol. 1. Boca Raton, FL: CRC; 1989. pp. 1–13. [Google Scholar]
- 38.Alvarez B, Denicola A, Radi R. Chem Res Toxicol. 1995;8:859–864. doi: 10.1021/tx00048a006. [DOI] [PubMed] [Google Scholar]
- 39.Goldstein S, Czapski G. J Am Chem Soc. 1998;120:3458–3463. [Google Scholar]
- 40.Hodges G R, Ingold K U. J Am Chem Soc. 1999;121:10695–10701. [Google Scholar]
- 41.Goldstein S, Czapski G, Lind J, Merényi G. Inorg Chem. 1998;37:3943–3947. doi: 10.1021/ic980051l. [DOI] [PubMed] [Google Scholar]
- 42.Wagman D D, Evans W H, Parker V B, Schumm R H, Halow I, Bailey S M, Churney K L, Nuttal R L. J Phys Chem Ref Data. 1982;11, Suppl. 2:64–72. [Google Scholar]
- 43.Lymar S V, Hurst J K. J Am Chem Soc. 1995;117:8867–8868. [Google Scholar]
- 44.Denicola A, Freeman B A, Trujillo M, Radi R. Arch Biochem Biophys. 1996;333:49–58. doi: 10.1006/abbi.1996.0363. [DOI] [PubMed] [Google Scholar]
- 45.Koppenol W H, Moreno J J, Pryor W A, Ischiropoulos H, Beckman J S. Chem Res Toxicol. 1992;5:834–842. doi: 10.1021/tx00030a017. [DOI] [PubMed] [Google Scholar]
- 46.Radi R, Beckman J S, Bush K M, Freeman B A. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 47.Quijano C, Alvarez B, Gatti R M, Augusto O, Radi R. Biochem J. 1997;322:167–173. doi: 10.1042/bj3220167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Briviba K, Kissner R, Koppenol W H, Sies H. Chem Res Toxicol. 1998;11:1398–1401. doi: 10.1021/tx980086y. [DOI] [PubMed] [Google Scholar]
- 49.Floris R, Piersma S R, Yang G, Jones P, Wever R. Eur J Biochem. 1993;215:767–775. doi: 10.1111/j.1432-1033.1993.tb18091.x. [DOI] [PubMed] [Google Scholar]
- 50.Castro L A, Robalinho R L, Cayota A, Meneghini R, Radi R. Arch Biochem Biophys. 1998;359:215–224. doi: 10.1006/abbi.1998.0898. [DOI] [PubMed] [Google Scholar]
- 51.Hausladen A, Fridovich I. J Biol Chem. 1994;269:29405–29408. [PubMed] [Google Scholar]
- 52.Khan A U, Kasha M. Proc Natl Acad Sci USA. 1994;91:12365–12367. doi: 10.1073/pnas.91.26.12365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.George P. Disc. Faraday Soc. 1947. , 196–222. [Google Scholar]
- 54.Ferradini C, Seide C. Int J Radiat Phys Chem. 1969;1:219–224. [Google Scholar]
- 55.Rigo A, Stevanato R, Finazzi-Agro A, Rotilio G. FEBS Lett. 1977;80:130–132. doi: 10.1016/0014-5793(77)80422-5. [DOI] [PubMed] [Google Scholar]
- 56.Koppenol W H, Butler J, van Leeuwen J W. Photochem Photobiol. 1978;28:655–660. [Google Scholar]
- 57.Melhuish W H, Sutton H C. J. Chem. Soc. Chem. Commun. 1978. 970–971. [Google Scholar]
- 58.Bors W, Michel C, Saran M. FEBS Lett. 1979;107:403–406. doi: 10.1016/0014-5793(79)80417-2. [DOI] [PubMed] [Google Scholar]
- 59.Weinstein J, Bielski B H J. J Am Chem Soc. 1979;101:58–62. [Google Scholar]
- 60.Bielski B H J. Photochem Photobiol. 1978;28:645–649. [Google Scholar]
- 61.Merényi G, Lind J, Czapski G, Goldstein S. Proc Natl Acad Sci USA. 2000;97:8216–8218. doi: 10.1073/pnas.150238197. [DOI] [PMC free article] [PubMed] [Google Scholar]