

Abstract
Background
Thermostable enzymes from thermophilic microorganisms are playing more and more important roles in molecular biology R&D and industrial applications. However, over-production of recombinant soluble proteins from thermophilic microorganisms in mesophilic hosts (e.g. E. coli) remains challenging sometimes.
Results
An open reading frame TM0438 from a hyperthermophilic bacterium Thermotoga maritima putatively encoding 6-phosphogluconate dehydrogenase (6PGDH) was cloned and expressed in E. coli. The purified protein was confirmed to have 6PGDH activity with a molecular mass of 53 kDa. The kcat of this enzyme was 325 s-1 and the Km values for 6-phosphogluconate, NADP+, and NAD+ were 11, 10 and 380 μM, respectively, at 80°C. This enzyme had half-life times of 48 and 140 h at 90 and 80°C, respectively. Through numerous approaches including expression vectors, hosts, cultivation conditions, inducers, and codon-optimization of the 6pgdh gene, the soluble 6PGDH expression levels were enhanced to ~250 mg per liter of culture by more than 500-fold. The recombinant 6PGDH accounted for >30% of total E. coli cellular proteins when lactose was used as a low-cost inducer. In addition, this enzyme coupled with glucose-6-phosphate dehydrogenase for the first time was demonstrated to generate two moles of NADPH per mole of glucose-6-phosphate.
Conclusion
We have achieved a more than 500-fold improvement in the expression of soluble T. maritima 6PGDH in E. coli, characterized its basic biochemical properties, and demonstrated its applicability for NADPH regeneration by a new enzyme cocktail. The methodology for over-expression and simple purification of this thermostable protein would be useful for the production of other thermostable proteins in E. coli.
Background
Enzyme-based biocatalysis has become an attractive alternative to chemical catalysis because of its higher reaction selectivity and more modest reaction conditions [1,2]. But most enzymes are not suitable for industrial applications due to their relatively poor stability and biocatalyst re-use. The former can be addressed by protein engineering [3-5], enzyme immobilization [6,7], utilization of stable enzymes from extremophilic microorganisms [8,9], or their combinations [10-12]. The latter can be solved through enzyme immobilization [6,7]. For example, immobilized thermostable glucose isomerase has been used in the food industry to convert glucose to fructose at ~60°C for several months before its deactivation [10].
Discovery and utilization of thermoenzyme from (hyper)thermophilic microorganisms is of great interest for numerous applications. Thermotoga maritima is an anaerobic, rod-shaped eubacterium, originally isolated from geothermally heated marine sediment at Valcano, Italy. It has an optimum growth temperature of ~80°C [13]. T. maritima is regarded as an invaluable source of intrinsically thermostable enzymes [14,15]. The open reading frame (ORF) TM0438 was annotated to be a 6-phosphogluconate dehydrogenase (6PGDH, E.C.1.1.1.44) [16], but its biochemical function has not yet been confirmed.
E. coli is a common prokaryotic microorganism for genetic manipulation and for the production of recombinant proteins because of its fast cell growth in inexpensive media, rapid accumulation of cellular mass, amenability to high cell-density fermentation, simple scale-up, and relatively simple protein purification [17]. But E. coli often produces recombinant protein in the form of insoluble, inactive inclusion bodies. It is estimated that less than 20% of the ORFs in other genomes are likely to be expressed as soluble active proteins in E. coli [18]. A number of approaches have been explored to improve the expression of soluble recombinant proteins in E. coli. With regard to the expression vector, the heterologous protein could be fused with a protein-folding partner (e.g. thioredoxin, cellulose-binding module) [19-21] or with a secretory protein fragment (e.g. outer-membrane protein A) that aids protein folding in a less-reducing periplasmic environment [17]. Expression hosts can be chosen according to different approaches, such as (i) mitigating codon bias in a host containing a second plasmid expressing the E. coli rare tRNA genes [22], (ii) enhancing protein folding in a host co-expressing folding modulators, such as chaperons [17,23], (iii) decreasing formation of disulfide bond in some special host's cytoplasm [24], and (iv) repressing basal expression of a toxic protein in a host with a repressor [25]. In addition, cultivation conditions, such as expression temperature, medium composition, timing of induction, inducer concentration, and inducer type, can be optimized for over-expression of a soluble protein [26,27]. Recently, synthetic codon-optimized genes have been more adapted for heterologous protein expression [28-30]. But over-expression of soluble heterologous proteins, especially for hyperthermophilic ones, in E. coli still remains on a trial-and-test stage [22,31,32].
6-phosphogluconate dehydrogenase is responsible for converting 6-phosphogluconate to ribulose-5-phosphate and CO2, along with one NADPH generation from NADP+ [33-35]. Thermostable 6PGDH has some potential applications, such as generation of high-yield hydrogen from sugars [36,37] and biosynthesis of chiral alcohols. For the production of a third generation biofuel – hydrogen, utilization of thermostable enzymes would increase production rates and stabilize the enzyme at elevated temperature [12,36,37]. Biosynthesis of chiral alcohols mediated by enzymes requires low-cost regeneration of NAD(P)H [38-41]. Glucose-6-phosphate dehydrogenase (G6PDH) has been applied to generate one NADPH per glucose-6-phospahte [38,42,43]. The combination of G6PDH and 6PGDH may double NADPH yield from costly glucose-6-phosphate, but no such study has been reported.
In this study, we cloned the ORF TM0438 encoding a putative T. maritima 6PGDH and purified and characterized the enzyme. Using different approaches, we increased its expression levels in E. coli from hardly-detectable to more than 250 mg per liter of culture. Also, we demonstrated that two moles of NADPH per mole of glucose-6-phosphate were generated by using an enzyme cocktail containing G6PDH and 6PGDH.
Results
Expression of wild-type 6pgdh gene
The expression plasmid pET-trx-wt6pgdh was constructed based on a pET102-TOPO plasmid. This construct encodes a fusion protein with a N-terminal thioredoxin, which is well-known to enhance expression of soluble heterologous proteins in E. coli [21]. The E. coli BL21(DE3) harboring plasmid pET-trx-wt6pgdh did not produce a detectable protein band corresponding to the size of the fusion protein under various experimental conditions (expression temperatures from 15, 20, 30 to 37°C and IPTG concentrations from 20, 100 to 500 μM). Figure 1A shows a typical result of the cell lysate of E. coli BL21(DE3) induced by 500 μM IPTG at 20°C.
Figure 1.

SDS-PAGE analysis of the expression of the wt6pgdh gene. (A) pET-trx-wt6pgdh in E. coli BL21(DE3), (B) pET-trx-wt6pgdh in E. coli BL21(DE3)-RIL, (C) pET-ci-wt6pgdh in E. coli BL21(DE3) and (D) pET-ci-wt6pgdh in E. coli BL21(DE3)-RIL. BI, before induction; T, total cellular proteins; S, soluble proteins; P, purified protein. Cells were grown at 37°C until A600 of 0.6. The induction condition was 20 μM IPTG at 20°C for 6 h.
Since the codon usage of E. coli BL21(DE3) is drastically different from that of T. maritima, E. coli BL21(DE3)-RIL that carries a ColE1-compatible vector encoding extra copies of tRNA genes of argU, ileY, and leuW was used to over-express soluble 6PGDH. E. coli BL21(DE3)-RIL/pET-trx-wt6pgdh produced a weak protein band corresponding to the size of the fusion protein under the conditions (500 μM IPTG and 20°C), but a majority of the fusion protein was insoluble (Figure 1B). Decreasing IPTG concentration to 20 μM did not improve the expression of soluble 6PGDH (data not shown), although lowering IPTG concentrations often improved the expression of soluble protein [27].
Because cellulose-binding module (CBM) tags have been reported to enhance heterologous protein expression and folding in E. coli [20,44], we attempted to express 6PGDH by replacing the thioredoxin tag with a Clostridium thermocellum family 3 CBM tag linked with an intein. Regardless of IPTG concentrations (20 or 500 μM IPTG), the E. coli BL21(DE3)/pET-trx-wt6pgdh did not produce any obvious soluble or insoluble protein bands corresponding to the right size (Figure 1C, 20 μM IPTG). But BL21(DE3)-RIL bearing the expression plasmid pET-trx-wt6pgdh produced some soluble 6PGDH (Figure 1D, 20 μM IPTG, 20°C). These results suggested that E. coli BL21(DE3)-RIL enhanced expression of the wt6pgdh and the CBM tag helped expression of soluble 6PGDH more efficiently than did thioredoxin.
Through affinity adsorption of CBM-tagged 6PGDH on regenerated amorphous cellulose followed by intein self-cleavage [44], approximately eight mg of 6PGDH was purified per liter of the culture. But the purified 6PGDH was composed of several small-size proteins (Lane P, Figure 1D), suggesting possible proteolysis or incomplete translation. The first cause was eliminated because addition of a protease inhibitor phenylmethanesulfonyl fluoride during cell disruption and protein purification did not change the composition of the small-size proteins (data not shown).
Codon analysis and optimization
Figure 2 shows the wt6pgdh DNA sequence, the deduced amino acids, and the codon-optimized DNA sequence (co6pgdh). The wt6pgdh gene contains 47 rare E. coli codons, accounting for ~10% of the entire sequence. They are 20 AGA, 5 AGG, 19 AUA, and 3 CUA. The AGA(Arg) and AUA(Ile) codon frequencies in the wt6pgdh gene are 4.3% and 4.0%, but are only 0.24% and 0.5% in E. coli http://www.kazusa.or.jp/codon, respectively. Moreover, two rare codons formed clusters AUA(Ile97)-AUA(Ile98) and AGA(Arg306)-AGA(Arg307). Following site-directed mutagenesis to remove these two rare-codon clusters, there were no noticeable changes in the SDS-PAGE patterns of the purified 6PGDHs before and after site-directed mutagenesis (data not shown). Therefore, the entire wt6pgdh DNA sequence was optimized to remove all 47 rare codons by using frequently-used E. coli codons, based on several rules: (i) keeping the GC ratio around 50%; (ii) avoiding cis-acting DNA sequences (internal TATA-boxes, chi-sites, and ribosomal entry sites; AT-rich or GC-rich sequence stretches; repeat sequences; and RNA secondary structures); (iii) precluding cutting sites of frequently-used restriction enzymes, and (iv) adding two stop TAATAA to ensure efficient termination of translation, and (v) using a strong terminator in the expression vector for enhancing mRNA stability. The overall GC content for the codon-optimized 6PGDH was 49% (Figure 2).
Figure 2.

The wild-type 6pgdh sequence, the deduced amino acid sequence, and the codon-optimized 6pgdh sequence. WT, wt6pgdh; CO, co6pgdh. The highlighted codons among the wt6pgdh gene are the rare codons in E. coli. The codon clusters in blue are the rare codons sites for site-directed mutagenesis. The underlined nucleotides in co6pgdh are the changed ones corresponding to wt6pgdh.
Expression of the codon-optimized 6pgdh gene
Plasmid pET-ci-co6pgdh encoding the fusion protein CBM-intein-6PGDH was expressed in E. coli BL21(DE3) and BL21(DE3)-RIL, separately. Figure 3 shows that both hosts produced soluble target proteins at similar levels, suggesting that E. coli BL21(DE3)-RIL was not necessary for expression of the codon-optimized gene. After purification, no small-size protein fragments accompanied with the purified 6PGDH were observed (Figure 3), suggesting that the rare codons mainly caused the incomplete translation (Figure 1D). Approximately 15–17 mg of 6PGDH was purified per liter of culture for both hosts.
Figure 3.

SDS-PAGE analysis of the expression and purification of T. maritima 6PGDH from plasmid pET-ci-co6pgdh in the E. coli BL21(DE3) (A) and E. coli BL21(DE3)-RIL (B). M, protein marker; BI, before induction; T, total cellular proteins; S, soluble proteins; P, purified protein. Cells were grown at 37°C until A600 of 0.6. The induction condition was 20 μM IPTG at 20°C for 6 h.
Basic biochemical characterization of T. maritima 6PGDH
The cleaved T. maritima 6PGDH through RAC adsorption and intein cleavage was purified and characterized. The pH effects on 6PGDH activity were studied in 50 mM citric acid/sodium citrate buffers (pH 5.0 and 6.0), a Bis-Tris (pH 6.5), Tris-HCl buffers (pH 7.0, 7.5 8.0, 8.5, and 9.0), and a Hepes buffer (pH 7.5). The optimum pH was found to be around pH 7.0 (Figure 4A). About 70% of 6PGDH activities remained at pH 6.0 and 9.0. The enzyme had the similar activities in the Hepes and Tris buffers (pH 7.5).
Figure 4.
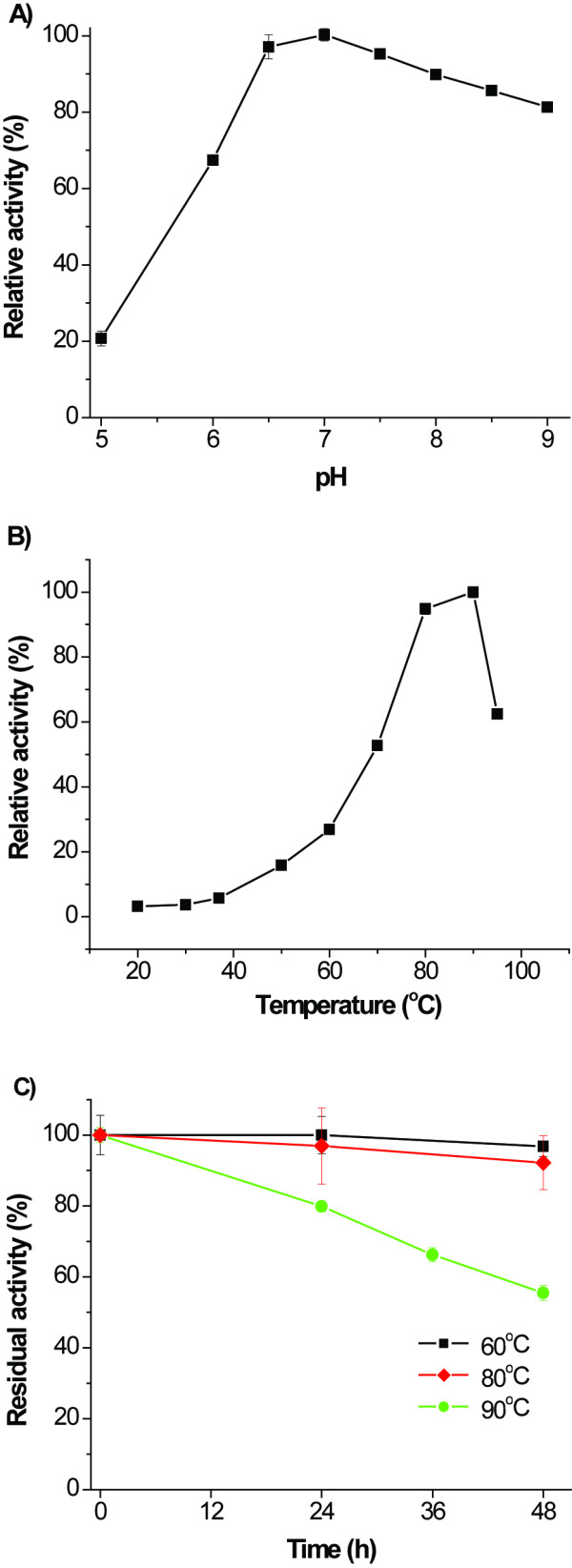
The reaction conditions of pH (A) and temperature (B) as well as thermostability (C) at an enzyme concentration of 0.24 mg/mL.
The effects of temperature on T. maritima 6PGDH activities were measured from 20 to 95°C (Figure 4B). The optimum temperature was around 90°C. The 6PGDH had an approximately 90% of its maximum activity at 80°C, but retained only ~2 and ~20% of its maximum activity at 30 and 60°C, respectively. The activation energy was 51.4 kJ/mol at a temperature range of 20–80°C, based on the Arrhenius plot. The 6PGDH was highly thermostable (Figure 4C) in a 50 mM Hepes buffer (pH 6.8) containing 500 mM NaCl, 1 mM EDTA, and 5 mM β-mercaptoethanol. It retained more than 90% activity for 48 h at 60 and 80°C and retained ~50% enzymatic activity after 48 h at 90°C. This enzyme had half-life times of 48 and 140 h at 90 and 80°C, respectively.
The kinetics properties of T. maritima 6PGDH followed the Michaelis-Menten equation. The apparent Km values were 11, 10, and 380 μM on 6-phosphogluconate, NADP+, and NAD+, respectively and kcat value was 325 s-1. Clearly, this enzyme preferred NADP+ to NAD+ as an electron acceptor.
Overexpression of co6pgdh and simple purification
Codon analysis indicated that the C. thermocellum CBM and the Synechocystis intein contained 21 rare codons. In addition, self-cleavage of intein during protein expression and cell disruption may result in some loss of the desired protein even at a decreased cultivation temperature [44]. Expression vector pET-co6pgdh-his was constructed to express a co6pgdh gene with a C-terminal His-tag. Figure 5A shows the SDS-PAGE analysis of 6PGDH expression. More than 200 mg of 6PGDH-His was purified per liter of culture. But the specific enzymatic activity of the 6PGDH-His was approximately 80% of that of 6PGDH without the His tag.
Figure 5.

SDS-PAGE analysis of the expression and purification of T. maritima 6PGDH from the E. coli BL21(DE3)/pET-co6pgdh-his (A) and E. coli BL21(DE3)/pET-co6pgdh (B). Cells were grown at 37°C until A600 of 0.6. The induction condition was 500 μM IPTG at 37°C for 4 h.
Since T. maritima 6PGDH was extremely thermostable (Figure 4C), heat precipitation was chosen for simplifying protein purification. Plasmid pET-co6gpdh was constructed for expressing the co6gpdh gene without a His-tag. The cell lysate was treated at 90 or 100°C with time lengths ranging from 15 minute to 6 hours. The highest 6PGDH yield was obtained under heat treatment conditions (90°C for 30 min), where a 6PGDH purity was around 85%, as judged by SDS-PAGE analysis (Figure 5B). The overall recovery yield was 90% according to 6PGDH activity. Approximately 190 mg of 6PGDH was obtained from the cells harvested after 4-hour induction at 37°C.
Figure 6 shows the fermentation profiles of E. coli BL21(DE3)/pET21-co6pgdh in 200 mL of LB medium in a 1-L Erlenmeyer flask at a constant cultivation temperature of 37°C. When the A600 reached 0.6, 500 μM of IPTG was added. The highest A600 was 3.9, and the total cell protein was 790 mg per liter at hour 12 (9.5 hours after IPTG induction). The 6PGDH content rose from ~10% before induction to 38% at hour 4.5 and then decreased to levels of ~30% for the remaining cultivation period. Up to 230 mg of 6PGDH was produced from one liter of the LB-grown culture.
Figure 6.

Cultivation profiles of cell growth, total cellular protein, and T. maritima 6PGDH content by E. coli BL21(DE3)/pET-co6pgdh in the LB medium at 37°C.
To decrease the inducer cost, different concentrations of IPTG (20, 100 or 500 μM) as well as lactose (100 μM) were further investigated. Nearly all cells had similar growth patterns except that 500 μM IPTG slightly inhibited cell growth during the first six-hour induction. After cell lysis and heat precipitation, approximately 194, 224, 208, and 250 mg of the 6PGDH protein were obtained under the conditions of 20, 100, and 500 μM IPTG, and 100 μM lactose, respectively. The largest amount of 6PGDH was obtained when lactose was used as the inducer, suggesting that low-cost lactose was an effective inducer and worked as a supplementary carbon source for protein synthesis.
NADPH regeneration
Coupling of 6PGDH and G6PDH was believed to generate one more mole of NADPH per mole of glucose-6-phosphate relative to G6PDH alone. Xylitol can be produced from xylose and NADPH mediated by xylose reductase [45,46]. Figure 7 shows kinetics of xylitol synthesis with glucose-6-phosphate as the NADPH regeneration substrate and G6PDH in the presence and absence of 6PGDH. In the case of three enzyme cocktails (G6PDH, 6PGDH, and xylose reductase), 35.5 mM xylitol was produced at hour six, nearly twice that of the two-enzyme system (19.5 mM, G6PDH and xylose reductase). The yields of xylitol synthesized were 175% and 97%, respectively, relative to glucose-6-phosphate consumed for the reactions mediated by the three enzymes and by the two enzymes.
Figure 7.
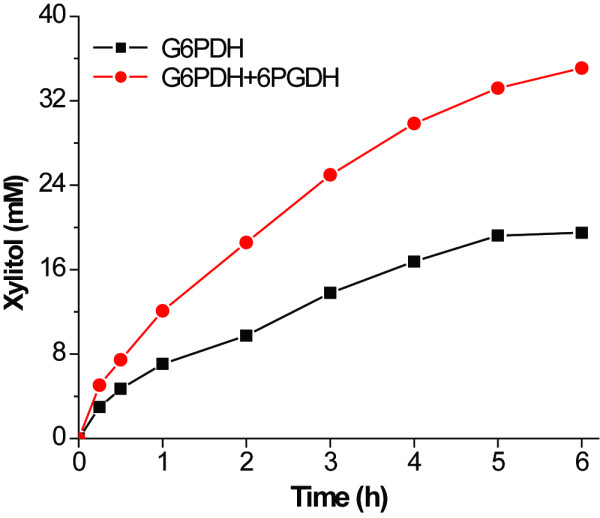
Profile of xylitol synthesis coupled with NADPH regeneration reaction from glucose-6-phosphate mediated by G6PDH alone or coupled with 6PGDH.
Discussion
The ORF TM0438 was confirmed to encode a hyperthermophilic 6PGDH. Through different approaches (expression vectors, hosts, cultivation conditions, inducer type, and gene sequence), we increased expression levels of the soluble T. maritima 6PGDH from nearly-undetectable to more than 250 mg per liter of the LB-grown culture, as summarized in Figure 8. Several lessons were learned from this study. (A) The codon bias between the rare codon-rich wt6pgdh gene and the expression host E. coli was the largest cause for its low expression. It can be addressed (i) mainly by a codon-optimized synthetic gene (Figure 5) or (ii) partially by using E. coli BL21(DE3)-RIL (Figure 1). (B) The C. thermocellum CBM tag enhanced T. maritima 6PGDH folding more efficiently than did thioredoxin (Figure 1Dvs Figure 1B). (C) Decreasing cultivation temperature and/or inducer concentration possibly decreased formation of inclusion body. But it was not necessary for the expression of the codon-optimized 6pgdh gene (Figure 5). (D) The 6PGDH was induced by a low level of IPTG or lactose. Low-cost lactose was highly recommended because it was used as the carbon source for supporting cell growth and protein synthesis (low cost plus high protein yield). (E) More than 30% of the E. coli cellular protein was soluble T. maritima 6PGDH after numerous approaches. (F) Heat precipitation was effective to obtain relatively pure thermostable enzymes (Figure 5B), suggesting that feasibility of ultra-low-cost mass production of this thermostable enzyme.
Figure 8.

Production and purification of T. maritima 6PGDH. The 6PGDH amounts were calculated based on their activity relative to the purified 6PGDH specific activity. The amount of total T. maritima 6PGDH proteins produced by E. coli were estimated from the band intensities on the SDS-PAGE gels relative to those contained in the soluble fraction. Expression host was E. coli BL21(DE3) except where ** denoted E. coli BL21(DE3)-RIL. The data from left to right were obtained from Figure 1A, Figure 1C, Figure 1D, Figure 3A, Figure 3B, Figure 5A, and the expression by the E. coli BL21(DE3)/pET-co6pgdh (induced by 100 μM lactose at 37°C for 10 h).
Few studies have been conducted pertaining to cloning and characterization of 6-phosphogluconate dehydrogenase [47,48]. The T. maritima 6PGDH is the most thermostable 6PGDH characterized so far, with half-life times of 48 h and ~140 h at 90°C and 80°C, respectively. This enzyme was far more thermostable than the Bacillus stearothermophilus 6PGDH with a half-life time of about 15 min at 70°C [49]. The hyper-thermostability of T. maritima 6PGDH makes it possible to simplify its purification by heat precipitation, different from most protein purification technologies, such as chromatography or adsorption/desorption [19,44]. Heat precipitation is becoming a popular protein purification protocol for hyper-thermostable proteins [50,51].
NAD(P)H enzymatic regeneration can be conducted by combining a number of enzyme/substrates, for example, glucose dehydrogenase [52], formate dehydrogenase/formate [53], phosphate dehydrogenase/phosphite [46], G6PDH/glucose-6-phosphate [42,43,54], and so on. Starting from G6PDH and glucose-6-phosphate, the addition of 6PGDH doubled NADPH yield to two NADPH per glucose-6-phosphate, resulting in 1.8-fold xylitol production as compared to G6PDH alone (Figure 7). This new enzyme cocktail would make the NADPH regeneration from glucose-6-phosphate more economically feasible.
High expression levels of T. maritima 6PGDH in E. coli (> 30% of total cellular protein), simple purification by heat precipitation and its hyper-thermostability suggest great potential for decreasing protein costs associated with its production, separation, and use. Although costly LB medium was used for production of 6PGDH in flasks on a laboratory scale, the production cost of 6PGDH is anticipated to decrease greatly by using low-cost lean medium plus high-cell-density fermentation in bioreactors. This highly-thermostable 6PGDH would be invaluable for high-yield generation of hydrogen from polysaccharides and water mediated by cell-free synthetic pathway biotransformation (SyPaB) [12,36,37].
Conclusion
In conclusion, we over-expressed more than 250 mg of T. maritima 6PGDH per liter of culture through numerous approaches, characterized its basic biochemical properties, and demonstrated its applicability for high-yield NADPH regeneration. This hyper-thermostable 6PGDH was easily purified by heat precipitation. The methodology for over-expression and simple purification of this thermostable protein would be useful for the production of other thermostable proteins in E. coli.
Methods
Chemicals, plasmids, and strains
All chemicals were of reagent grade, purchased from Sigma (St. Louis, MO) and Fisher Scientific (Pittsburgh, PA), unless otherwise noted. Regenerated amorphous cellulose (RAC) was prepared through cellulose dissolution by ice-cooled concentrated phosphoric acid followed by regeneration in water [55]. Pfx50 DNA polymerase, Champion™ pET102 Directional TOPO® Expression Kit with E. coli BL21 Star™ (DE3), and Ni-NTA agarose were purchased from Invitrogen (Carlsbad, CA). The T. maritima genomic DNA was purchased from the American Type Culture Collection (Manassas, VA). The strains, plasmids, and oligonucleotides used in this study are listed in Table 1.
Table 1.
The strains, plasmids, and oligonucleotides used in this study
| Description | Contents | Reference/sources |
| Strain | ||
| E. coli Bl21star(DE3) | B F- ompT hsdSB(rB-mB-) gal dcm rne131 (DE3) | Invitrogen |
| E. coli Bl21(DE3)-RIL | B F- ompT hsdS(rB-mB-) dcm Tetr gal λ(DE3) endA Hte [argU ileY leuW Camr] |
Stratagene |
| Plasmid | ||
| pCIP | Ampr, T7 promoter, lacO, ColE1 ori, parental DNA, replacing pgm gene with wt6pgdh, co6pgdh or g6pdh |
[44] |
| pET21a | Epoch Biolabs | |
| pET-trx-wt6pgdh | Ampr, T7 promoter, lacO, ColE1 ori, Trx-wt6pgdh | This study |
| pET-ci-wt6pgdh | wt6pgdh gene subcloned into pCIP | This study |
| pET-ci-co6pgdh | co6pgdh gene subcloned into pCIP | This study |
| pET-co6pgdh-his | co6pgdh gene with C-terminal (His)6 cloned into pET21a | |
| pET-co6pgdh | Removed C-terminal (His)6 from pET-co6pgdh-his | This study |
| pET-ci-g6pdhp | Expression of T. maritima g6pdh | This study |
| pET26b-xr | Expression of N. crassa xylose reductase | [57] |
| Primers* | Final plasmid | |
| Trx-F | 5'-caccatggtgaaatctcatattggtctcatcggtc-3' | pET-trx-wt6pgdh |
| Trx-R | 5'-tcatcctatctctccttcctcccagttg-3' | |
| CI-wt-F | 5'-ccagtctactcgaggtgaaatctcatattggtctcatcggtc-3' | pET-ci-wt6pgdh |
| CI-wt-R | 5'-ccagtctagtcgaccctatctctccttcctcccag-3' | |
| CI-co-F | 5'-ccagtcta ctcgagggctcttccatgaaatcccacattggcctgatc-3' | pET-ci-co6pgdh |
| CI-co-R | 5'-ccagtctaggatcctcaagtcgagccaatctccccctcctccc-3' | |
| NH-F | 5'-gaggagggggagattggctaacatcaccaccaccattaag-3' | pET-co6pgdh |
| NH-R | 5'-cttaatggtggtggtgatgttagccaatctccccctcctc-3' | |
| G6P-F | 5'-ccagtctactcgagggctcttcc atgaagtgcagtctgggattg-3' | pET-ci-g6pdh |
| G6P-R | 5'-ccagtctagtcgacagttttctccattttctacc-3' |
* Underlined nucleotide sequences indicate restriction endonuclease sites.
Construction of expression plasmids
Five expression plasmids were constructed for expressing wild-type 6pgdh (wt6pgdh) and codon-optimized 6pgdh (co6pgdh) genes under the control of T7 promoter (Table 2). Plasmid pET-trx-wt6pgdh encoding a fusion protein of thioredoxin (Trx) and 6PGDH was constructed by insertion of amplified wt6pgdh gene into pET102 Directional TOPO® . The wt6pgdh DNA fragment was PCR amplified using primers Trx-F and Trx-R from the T. maritima genomic DNA. Plasmid pET-ci-wt6pgdh was constructed by replacing the pgm gene in the plasmid pCIP [44] by the wt6pgdh gene. The whole 6pgdh DNA sequence was optimized based on the codon usage for E. coli B http://www.kazusa.or.jp, yielding the co6pgdh DNA sequence. The co6pgdh DNA sequence with a C-terminal His-tag was synthesized by Epoch Biolabs (Sugar Land, TX) and cloned into pET21a via the restriction endonuclease sites Nde1 and BamH1 to obtain pET-co6pgdh-his. Plasmid pET-ci-co6pgdh was constructed similarly to pET-ci-wt6pgdh using the co6pgdh DNA sequence. Plasmid pET-co6pgdh encoding 6PGDH protein without a His-tag was constructed based on pET-co6pgdh-his by replacing the first codon (CAT) of the His-tag with a stop codon (TAA). The site-directed mutagenesis was conducted by using primers NH-F and NH-R following QuikChange™ Site-Directed Mutagenesis (Stratagene, La Jolla, CA).
Table 2.
Expression plasmids for T. maritima 6PGDH with or without the tags
| Plasmid | Modular organization | Molecular mass |
| pET-trx-wt6pgdh | Trx-WT6PGDH | 66, 634 Da |
| pET-ci-wt6pgdh | CBM-intein-WT6PGDH | 91, 187 Da |
| pET-ci-co6pgdh | CBM-intein-CO6PGDH | 91, 187 Da |
| pET-co6pgdh-his | CO6PGDH-(His)6 | 53, 975 Da |
| pET-co6pgdh | CO6PGDH | 53, 152 Da |
Protein expression and purification
The E. coli strain harboring the expression plasmid was grown in a 200 mL LB medium in a 1-L flask with appropriate antibiotics at 37°C until the A600 reached ~0.6. After addition of the inducer (isopropyl β-D-1-thiogalactopyranoside – IPTG or lactose), the culture were grown at 37°C or a lower temperature (e.g. 20°C). For the CBM-tag proteins, they were purified through affinity binding on RAC followed by intein self-cleavage [44]. For the His-tag proteins, the cells were disrupted by sonicator in a 50 mM Tris-HCl (pH 7.5) containing 500 mM NaCl, 5 mM imidazol, and 20% (w/v) glycerol. After centrifugation at 8,000 g for 5 min, the supernatant was collected and purified by using affinity binding on Ni-NTA resin. For purification through heat precipitation, the soluble fraction of E. coli cell lysate was incubated at 90°C for 30 min in a 50 mM Tris-HCl (pH 7.5) buffer containing 100 mM NaCl and 20% (w/v) glycerol. After centrifugation, the supernatant contained relatively pure 6PGDH.
The T. maritima glucose-6-phosphate dehydrogenase (G6PDH) was produced by E. coli BL21(DE3)-RIL/pET-ci-g6pdh. The G6PDH was purified through affinity binding on RAC followed by intein self-cleavage [44] and characterized as described [56]. The Neurospora crassa xylose reductase was expressed and purified as described elsewhere [57].
6PGDH activity assays
T. maritima 6PGDH activity was measured in a 50 mM Hepes buffer (pH 7.5) containing 2 mM 6-phosphogluconate, 1 mM NADP+, 5 mM Mg2+, 0.5 mM Mn2+, and 0.5 mg bovine serum albumin per mL at 80°C for 5 min. The reaction product NADPH was measured at 340 nm by DU® 800 UV/visible spectrophotometer (Beckman Coulter, Fullerton, CA). The enzyme unit was defined as one μmole of NADPH produced per min. For determining enzyme kinetic parameters, the Km of 6-phosphogluconate was measured in a 50 mM Hepes (pH 7.5) buffer containing 1 mM NADP+, 5 mM Mg2+, 0.5 mM Mn2+, along with 2.5 to 50 μM 6-phosphogluconate; the Km of NADP+ was measured in the same buffer 2 mM 6-phosphogluconate with various concentrations of NADP+ from 2.5 to 50 μM. The Km of NAD+ was measured using 50 to 1000 μM NAD+.
Protein assays
Concentration of soluble protein was measured by the Bio-Rad Bradford protein kit with bovine serum albumin (BSA) as a standard protein. Total cellular protein was measured as described previously [58].
Xylitol production with NADPH regeneration
Synthesis of xylitol from xylose mediated by xylose reductase was conducted in a 200 μL reaction volume in the presence of G6PDH or G6PDH/6PGDH at 25°C. The reaction mixture contained a 50 mM Hepes buffer (pH 7.5) with 50 mM xylose, 1 mg xylose reductase/mL, 2 mM NADP+, 20 mM glucose-6-phospahte, 0.16 mg T. maritima 6PGDH/mL, 0.3 mg T. maritima 6PGDH/mL, 1 mg BSA/mL, 0.5 mM MnCl2 and 5 mM MgCl2. A 10 μL of the sample was withdrawn and diluted 20-fold in 5 mM H2SO4. Xylitol was measured by a HPLC equipped with the Bio-Rad Aminex HPX-87H column [59].
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
YW and YHPZ designed the experiment. YW performed the experiments. YW and YHPZ wrote the manuscript. Both authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
This work was supported to YHPZ mainly by the Air Force Office of Scientific Research (FA9550-08-1-0145) and partially by ICTAS. We thank Dr. Huiming Zhao for providing plasmid pET26b-xr containing a xylose reductase gene.
Contributor Information
Yiran Wang, Email: yiran@vt.edu.
Y-H Percival Zhang, Email: ypzhang@vt.edu.
References
- Schmid A, Dordick JS, Hauer B, Kiener A, Wubbolts M, Witholt B. Industrial biocatalysis today and tomorrow. Nature. 2001;409:258–268. doi: 10.1038/35051736. [DOI] [PubMed] [Google Scholar]
- Blumer-Schuette SE, Kataeva I, Westpheling J, Adams MW, Kelly RM. Extremely thermophilic microorganisms for biomass conversion: status and prospects. Curr Opin Biotechnol. 2008;19:210–217. doi: 10.1016/j.copbio.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Liu W, Hong J, Bevan DR, Zhang YH. Fast identification of thermostable beta-glucosidase mutants on cellobiose by a novel combinatorial selection/screening approach. Biotechnol Bioeng. 2009 doi: 10.1002/bit.22340. [DOI] [PubMed] [Google Scholar]
- Lehmann M, Wyss M. Engineering proteins for thermostability: the use of sequence alignments versus rational design and directed evolution. Curr Opin Biotechnol. 2001;12:371–375. doi: 10.1016/s0958-1669(00)00229-9. [DOI] [PubMed] [Google Scholar]
- Giver L, Gershenson A, Freskgard PO, Arnold FH. Directed evolution of a thermostable esterase. Proc Natl Acad Sci USA. 1998;95:12809–12813. doi: 10.1073/pnas.95.22.12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancor L, Luckarift HR. Bioinspired enzyme encapsulation for biocatalysis. Trends Biotechnol. 2008;26:566–572. doi: 10.1016/j.tibtech.2008.06.009. [DOI] [PubMed] [Google Scholar]
- De Cordt S, Vanhoof K, Hu J, Maesmans G, Hendrickx M, Tobback P. Thermostability of soluble and immobilized alpha-amylase from Bacillus licheniformis. Biotechnol Bioeng. 1992;40:396–402. doi: 10.1002/bit.260400309. [DOI] [PubMed] [Google Scholar]
- Blumer-Schuette SE, Kataeva I, Westpheling J, Adams MW, Kelly RM. Extremely thermophilic microorganisms for biomass conversion: status and prospects. Curr Opin Biotechnol. 2008;19:210–217. doi: 10.1016/j.copbio.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Adams MW, Perler FB, Kelly RM. Extremozymes: expanding the limits of biocatalysis. Biotechnology . 1995;13:662–668. doi: 10.1038/nbt0795-662. [DOI] [PubMed] [Google Scholar]
- Bhosale SH, Rao MB, Deshpande VV. Molecular and industrial aspects of glucose isomerase. Microbiol Rev. 1996;60:280–300. doi: 10.1128/mr.60.2.280-300.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson HR, Hooker JM, Francis MB, Clark DS. Solubilization and stabilization of bacteriophage MS2 in organic solvents. Biotechnol Bioeng. 2007;97:224–234. doi: 10.1002/bit.21245. [DOI] [PubMed] [Google Scholar]
- Zhang Y-HP. A sweet out-of-the-box solution to the hydrogen economy: is the sugar-powered car science fiction? Energy Environ Sci. 2009;2:272–282. [Google Scholar]
- Huber R, Langworthy TA, Konig H, Thomm M, Woese CR, Sleytr UB, Stetter KO. Thermotoga maritima sp. nov. represents a new genus of unique extremely thermophilic eubacteria growing up to 90°C. Arch Microbiol. 1986;144:324–333. [Google Scholar]
- Adams MW, Kelly RM. Finding and using hyperthermophilic enzymes. Trends Biotechnol. 1998;16:329–332. doi: 10.1016/s0167-7799(98)01193-7. [DOI] [PubMed] [Google Scholar]
- Bronnenmeier K, Kern A, Liebl W, Staudenbauer WL. Purification of Thermotoga maritima enzymes for the degradation of cellulosic materials. Appl Environ Microbiol. 1995;61:1399–1407. doi: 10.1128/aem.61.4.1399-1407.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Nelson WC, Ketchum KA, et al. Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature. 1999;399:323–329. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- Baneyx F, Mujacic M. Recombinant protein folding and misfolding in Escherichia coli. Nat Biotechnol. 2004;22:1399–1408. doi: 10.1038/nbt1029. [DOI] [PubMed] [Google Scholar]
- Jenney FE, Jr, Adams MW. The impact of extremophiles on structural genomics (and vice versa) Extremophiles. 2008;12:39–50. doi: 10.1007/s00792-007-0087-9. [DOI] [PubMed] [Google Scholar]
- Hong J, Ye X, Wang Y, Zhang YH. Bioseparation of recombinant cellulose-binding module-proteins by affinity adsorption on an ultra-high-capacity cellulosic adsorbent. Anal Chim Acta. 2008;621:193–199. doi: 10.1016/j.aca.2008.05.041. [DOI] [PubMed] [Google Scholar]
- Murashima K, Kosugi A, Doi RH. Solubilization of cellulosomal cellulases by fusion with cellulose-binding domain of noncellulosomal cellulase engd from Clostridium cellulovorans. Proteins. 2003;50:620–628. doi: 10.1002/prot.10298. [DOI] [PubMed] [Google Scholar]
- LaVallie ER, DiBlasio EA, Kovacic S, Grant KL, Schendel PF, McCoy JM. A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cytoplasm. Biotechnology (N Y) . 1993;11:187–193. doi: 10.1038/nbt0293-187. [DOI] [PubMed] [Google Scholar]
- Gustafsson C, Govindarajan S, Minshull J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004;22:346–353. doi: 10.1016/j.tibtech.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Martinez-Alonso M, Vera A, Villaverde A. Role of the chaperone DnaK in protein solubility and conformational quality in inclusion body-forming Escherichia coli cells. FEMS Microbiol Lett. 2007;273:187–195. doi: 10.1111/j.1574-6968.2007.00788.x. [DOI] [PubMed] [Google Scholar]
- Sorensen HP, Mortensen KK. Soluble expression of recombinant proteins in the cytoplasm of Escherichia coli. Microb Cell Fact. 2005;4:1. doi: 10.1186/1475-2859-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan KA, Xu Z, Watkins RE, Brennan RG, Skurray RA, Brown MH. Optimized production and analysis of the staphylococcal multidrug efflux protein QacA. Protein Expr Purif. 2009;64:118–124. doi: 10.1016/j.pep.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Hettwer S, Sterner R. A novel tryptophan synthase beta-subunit from the hyperthermophile Thermotoga maritima. Quaternary structure, steady-state kinetics, and putative physiological role. J Biol Chem . 2002;277:8194–8201. doi: 10.1074/jbc.M111541200. [DOI] [PubMed] [Google Scholar]
- Kopetzki E, Schumacher G, Buckel P. Control of formation of active soluble or inactive insoluble baker's yeast alpha-glucosidase PI in Escherichia coli by induction and growth conditions. Mol Gen Genet. 1989;216:149–155. doi: 10.1007/BF00332244. [DOI] [PubMed] [Google Scholar]
- Gustafsson C, Govindarajan S, Minshull J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004;22:346–353. doi: 10.1016/j.tibtech.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Angov E, Hillier CJ, Kincaid RL, Lyon JA. Heterologous protein expression is enhanced by harmonizing the codon usage frequencies of the target gene with those of the expression host. PLoS ONE. 2008;3:e2189. doi: 10.1371/journal.pone.0002189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen HP, Mortensen KK. Advanced genetic strategies for recombinant protein expression in Escherichia coli. J Biotechnol. 2005;115:113–128. doi: 10.1016/j.jbiotec.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Esposito D, Chatterjee DK. Enhancement of soluble protein expression through the use of fusion tags. Curr Opin Biotechnol. 2006;17:353–358. doi: 10.1016/j.copbio.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Adams MW, Kelly RM. Finding and using hyperthermophilic enzymes. Trends Biotechnol. 1998;16:329–332. doi: 10.1016/s0167-7799(98)01193-7. [DOI] [PubMed] [Google Scholar]
- Selig M, Xavier KB, Santos H, Schonheit P. Comparative analysis of Embden-Meyerhof and Entner-Doudoroff glycolytic pathways in hyperthermophilic archaea and the bacterium Thermotoga. Arch Microbiol. 1997;167:217–232. doi: 10.1007/BF03356097. [DOI] [PubMed] [Google Scholar]
- Sundaramoorthy R, Iulek J, Barrett MP, Bidet O, Ruda GF, Gilbert IH, Hunter WN. Crystal structures of a bacterial 6-phosphogluconate dehydrogenase reveal aspects of specificity, mechanism and mode of inhibition by analogues of high-energy reaction intermediates. FEBS J. 2007;274:275–286. doi: 10.1111/j.1742-4658.2006.05585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rippa M, Giovannini PP, Barrett MP, Dallocchio F, Hanau S. 6-Phosphogluconate dehydrogenase: the mechanism of action investigated by a comparison of the enzyme from different species. Biochim Biophys Acta . 1998;1429:83–92. doi: 10.1016/s0167-4838(98)00222-2. [DOI] [PubMed] [Google Scholar]
- Zhang Y-HP, Evans BR, Mielenz JR, Hopkins RC, Adams MW. High-yield hydrogen production from starch and water by a synthetic enzymatic pathway. PLoS ONE. 2007;2:e456. doi: 10.1371/journal.pone.0000456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Wang Y, Hopkins RC, Adams MW, Evans BR, Mielenz JR, Zhang Y-HP. Spontaneous high-yield production of hydrogen from cellulosic materials and water catalyzed by enzyme cocktails. ChemSusChem. 2009;2:149–152. doi: 10.1002/cssc.200900017. [DOI] [PubMed] [Google Scholar]
- Wong CH, Whitesides GM. Enzyme-catalyzed organic-synthesis – NAD(P)H cofactor regeneration by using glucose-6-phosphate and the glucose-6-phosphate-dehydrogenase from Leuconostoc-Mesenteroides. J Am Chem Soc. 1981;103:4890–4899. [Google Scholar]
- Groger H, Chamouleau F, Orologas N, Rollmann C, Drauz K, Hummel W, Weckbecker A, May O. Enantioselective reduction of ketones with "designer cells" at high substrate concentrations: highly efficient access to functionalized optically active alcohols. Angew Chem Int Ed Engl. 2006;45:5677–5681. doi: 10.1002/anie.200503394. [DOI] [PubMed] [Google Scholar]
- Shaw NM, Robins KT, Kiener A. Lonza: 20 years of biotransformations. Adv Synth Catal. 2003;345:425–435. [Google Scholar]
- Hummel W. Large-scale applications of NAD(P)-dependent oxidoreductases: recent developments. Trends Biotechnol. 1999;17:487–492. doi: 10.1016/s0167-7799(98)01207-4. [DOI] [PubMed] [Google Scholar]
- Zhang H, Witholt B, Li Z. Efficient NADPH recycling in enantioselective bioreduction of a ketone with permeabilized cells of a microorganism containing a ketoreductase and a glucose 6-phosphate dehydrogenase. Adv Synth Catal. 2006;348:429–433. [Google Scholar]
- Hanson RL, Goldberg S, Goswami A, Tully TP, Patel RN. Purification and cloning of a ketoreductase used for the preparation of chiral alcohols. Adv Synth Catal. 2005;347:1073–1080. [Google Scholar]
- Hong J, Wang Y, Ye X, Zhang Y-HP. Simple protein purification through affinity adsorption on regenerated amorphous cellulose followed by intein self-cleavage. J Chromatogr A. 2008;1194:150–154. doi: 10.1016/j.chroma.2008.04.048. [DOI] [PubMed] [Google Scholar]
- Nidetzky B, Neuhauser W, Haltrich D, Kulbe KD. Continuous enzymatic production of xylitol with simultaneous coenzyme regeneration in a charged membrane reactor. Biotechnol Bioeng. 1996;52:387–396. doi: 10.1002/(SICI)1097-0290(19961105)52:3<387::AID-BIT4>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Johannes TW, Woodyer RD, Zhao H. Efficient regeneration of NADPH using an engineered phosphite dehydrogenase. Biotechnol Bioeng. 2007;96:18–26. doi: 10.1002/bit.21168. [DOI] [PubMed] [Google Scholar]
- Barrett MP, Phillips C, Adams MJ, Le Page RW. Overexpression in Escherichia coli and purification of the 6-phosphogluconate dehydrogenase of Trypanosoma brucei. Protein Expr Purif. 1994;5:44–49. doi: 10.1006/prep.1994.1006. [DOI] [PubMed] [Google Scholar]
- Chooback L, Price NE, Karsten WE, Nelson J, Sundstrom P, Cook PF. Cloning, expression, purification, and characterization of the 6-phosphogluconate dehydrogenase from sheep liver. Protein Expr Purif. 1998;13:251–258. doi: 10.1006/prep.1998.0896. [DOI] [PubMed] [Google Scholar]
- Pearse BM, Harris JJ. 6-Phosphogluconate dehydrogenase from Bacillus stearothermophilus. FEBS letters. 1973;38:49–52. doi: 10.1016/0014-5793(73)80510-1. [DOI] [PubMed] [Google Scholar]
- Pluthero FG. Rapid purification of high-activity Taq DNA-polymerase. Nucleic Acids Res. 1993;21:4850–4851. doi: 10.1093/nar/21.20.4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Marco A, Casatta E, Savaresi S, Geerlof A. Recombinant proteins fused to thermostable partners can be purified by heat incubation. J Biotechnol. 2004;107:125–133. doi: 10.1016/j.jbiotec.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Xu Z, Jing K, Liu Y, Cen P. High-level expression of recombinant glucose dehydrogenase and its application in NADPH regeneration. J Ind Microbiol Biotechnol. 2007;34:83–90. doi: 10.1007/s10295-006-0168-2. [DOI] [PubMed] [Google Scholar]
- Kula MR, Wandrey C. Continuous enzymatic transformation in an enzyme-membrane reactor with simultaneous NADH regeneration. Meth Enzymol. 1987;136:9–21. doi: 10.1016/s0076-6879(87)36004-5. [DOI] [PubMed] [Google Scholar]
- Wong CH, Whitesides GM. Enzyme-catalyzed organic-synthesis – NAD(P)H cofactor regeneration by using glucose-6-phosphate and the glucose-6-phosphate-dehydrogenase from Leuconostoc-Mesenteroides. J Am Chem Soc. 1981;103:4890–4899. [Google Scholar]
- Zhang Y-HP, Cui J, Lynd LR, Kuang LR. A transition from cellulose swelling to cellulose dissolution by o-phosphoric acid: evidence from enzymatic hydrolysis and supramolecular structure. Biomacromolecules. 2006;7:644–648. doi: 10.1021/bm050799c. [DOI] [PubMed] [Google Scholar]
- Hansen T, Schlichting B, Schonheit P. Glucose-6-phosphate dehydrogenase from the hyperthermophilic bacterium Thermotoga maritima: expression of the g6pd gene and characterization of an extremely thermophilic enzyme. FEMS Microbiol Lett. 2002;216:249–253. doi: 10.1111/j.1574-6968.2002.tb11443.x. [DOI] [PubMed] [Google Scholar]
- Woodyer R, Simurdiak M, Donk WA van der, Zhao H. Heterologous expression, purification, and characterization of a highly active xylose reductase from Neurospora crassa. Appl Environ Microbiol. 2005;71:1642–1647. doi: 10.1128/AEM.71.3.1642-1647.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y-H, Lynd LR. Quantification of cell and cellulase mass concentrations during anaerobic cellulose fermentation: development of an enzyme-linked immunosorbent assay-based method with application to Clostridium thermocellum batch cultures. Anal Chem. 2003;75:219–227. doi: 10.1021/ac020271n. [DOI] [PubMed] [Google Scholar]
- Zhang YHP, Lynd LR. Regulation of cellulase synthesis in batch and continuous cultures of Clostridium thermocellum. J Bacteriol. 2005;187:99–106. doi: 10.1128/JB.187.1.99-106.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]