

Abstract
N-methylpurine-DNA glycosylase (MPG), a ubiquitous DNA repair enzyme, initiates excision repair of several N-alkylpurine adducts, induced by alkylating chemotherapeutics, and deaminated and lipid peroxidation-induced purine adducts. We have generated monoclonal antibodies (moAbs) against human MPG. Twelve independent hybridoma clones were characterized, which, except 520-16A, are identical based on epitope exclusion assay. Four moAbs, including 520-2A, 520-3A, 520-16A, and 520-26A, have high affinity (KD∼0.3-1.6 nM), and their subtypes were IgG2a, IgG1, IgG2a, and IgG2b, respectively. moAb 520-3A recognizes the sequence 52AQAPCPRERCLGPP66T, an epitope exclusively present in the N-terminal extension of human MPG. We found that moAb 520-3A significantly inhibited MPG's enzymatic activity towards different substrates, such as hypoxanthine, 1,N6ethenoadenine and methylated bases, which represent different classes of DNA damage, however, with different efficiencies. Real-time binding experiments using surface plasmon resonance (SPR) spectroscopy showed that the pronounced inhibition of activity was not in the substrate binding step. Single turnover kinetics (STO) revealed that the inhibition was at the catalytic step. Since we found that this antibody has an epitope in the N-terminal tail, the latter appears to have an important role in substrate discrimination, however, with a differential effect on different substrates.
1. Introduction
Cellular DNA is continuously exposed to endogenous or exogenous chemical or physical agents that induce DNA lesions. DNA base damage threatens genomic stability and cellular viability. Multiple DNA repair pathways exist in all organisms—from bacteria to humans—to preserve the integrity of the genome [1]. Damaged bases, if not repaired, can be mutagenic [2] and/or trigger cell death [3].
In all organisms, repair of DNA-containing small adducts, as well as altered and abnormal bases, occurs primarily via the base excision repair (BER) pathway, beginning with cleavage of the base by a DNA glycosylase [1,2]. Mechanistically, DNA glycosylases are classified as: mono- or bifunctional DNA glycosylases. Monofunctional DNA glycosylases, such as MPG and Uracil DNA-glycosylase, use an activated water molecule as a nucleophile to generate an apurinic or apyrimidinic (AP) site in DNA. Bifunctional DNA glycosylases/AP lyases, such as NTH1 and OGG1, use an activated amino group (Lys) or imino group (Pro) as the nucleophile to create a Schiff-base intermediate that coordinates base removal and subsequent strand incision (AP lyase) 3′ to the AP site [4,5]. Mammalian MPG is known to excise at least 17 structurally diverse modified bases from DNA, which are induced by alkylating chemotherapeutics, deamination and lipid peroxidation [6]. The lesion substrates primarily include the purine derivatives, such as 3-alkylpurines, 7-alkylguanine, 1,N6-ethenoadenine (εA), N2,3-ethenoguanine, and hypoxanthine (Hx) [7-12]. Moreover, the base alterations are located in both the major and minor grooves of duplex DNA. Mammalian MPG orthologs in Escherichia coli (AlkA) and yeast (MAG) have overlapping, but not identical, substrate ranges. Nonetheless, in spite of this functional similarity, mammalian MPG and E. coli (AlkA) do not share significant sequence similarity or structural homology [13,14], despite 3-methyladenine being a preferred substrate for both. MPG excises εA and Hx more efficiently than AlkA and MAG [11], but unlike AlkA, it cannot excise O2-alkylpyrimidines [15,16] and oxidized bases, such as 5-formyluracil and 5-hydroxymethyluracil [17] from DNA. MAG does not excise O2-methylthymine either [6,18]. Although MPG can excise different modified bases, the specificity of MPG towards all substrates is not the same. In our previous study, we showed that MPG is organized into three distinct domains with a protease hypersensitive region at the amino terminus [19]. The non-conserved, N-terminal extension plays a role in excision of some alkylation damage and 1,N2-ethenoguanine (1,N2-εG), although 1,N2-εG is yet to be detected in genomic DNA [9,20]. Studies with hybrid recombinant proteins containing N- and C-terminal halves of human and mouse glycosylases showed that the N-terminal extension of MPG could be critical for its recognition of 3-methylguanine and 7-methylguanine adducts in DNA [9]. Hx and εA are two other very important substrates of MPG. Hx was shown to be significantly mutagenic [21,22]. Besides all of this, how distinct those substrates are in respect to MPG is not yet known. We attempted to generate some highly specific and characterized antibodies against MPG to be used as probes for exploring substrate specificity, catalytic mechanisms and structure-function relationships.
Here we report that we raised several anti-MPG monoclonal antibodies and characterized them for binding specificity towards MPG. These antibodies significantly inhibited MPG's enzymatic activity. Surface plasmon resonance studies and single turnover studies showed that inhibition is not in the binding step but in the chemistry step. However, using three different DNA substrates which represent reaction products of three important different endogenous and environmental DNA damaging agents, we found that the degree of inhibition varied for different substrates. Furthermore, the antigenic epitope for the antibodies resides at the N-terminal extension of MPG and thus raises the possibility for the N-terminal playing a critical role in MPG's substrate specificity.
2. Materials and methods
2.1 Construction of expression plasmids for human MPG lacking variable first exon
MPG has two splice variants: Exon I is variable with 7 (Ia) or 13 (Ib) amino acid residues, while the rest of the exons are identical. We intended to express and purify Exon I less hMPG in order to use it for raising antibody that may recognize hMPG in human cells independent of splice variance. The plasmid pRR15, harboring full-length cDNA human MPG, was used as a template for PCR to generate a 184 bp human MPG cDNA fragment with the deletion of the first 7 amino acid residues constituting Exon Ia at the N-terminus and inclusion of BamHI and NdeI sites at the 5′ end. The 3′ of the PCR product ended with MPG's internal Eco47III site and had no change in sequences. The PCR product was digested with BamHI and Eco47III and ligated in the plasmid, pPG23 [23], linearized with BamHI and Eco47III. The resulting construct was digested again with NdeI and EcoRI, and the MPG cDNA fragment, encoding all the exons except I, was religated into pRSETB linearized with NdeI and EcoRI to generate the final expression construct, pRR17, whose identity was confirmed by sequencing.
2.2 Purification of human MPG lacking the variable first exon
E. coli BL21(DE3) carrying pRR17 was grown in magnificient broth (MacConnell Research, CA) at 37 °C until the absorbance at 600 nm reached 0.6. The culture was cooled to 25°C and, after the addition of IPTG to 0.5 mM, was grown at 25 °C for 16 h, prior to chilling to 0°C. All subsequent procedures were carried out at 4 °C. After the bacteria were harvested by centrifugation, they were resuspended in buffer A (50 mM Tris-HCl, pH 8.0, 50 mM NaCl, and 0.1% Triton X-100, 5% glycerol) and then sonicated (10 × 45 s) on ice at full power using a Braun-Sonic U. After centrifugation of the cell lysate (2×30 min at 15,000 ×g), the supernatant was applied to ion-exchange columns, Q Sepharose (5 ml) and SP Sepharose (1 ml) (Amersham Pharmacia Biotech, Piscataway, NJ) connected in tandem. The columns were pre-equilibrated with buffer A. After washing with buffer A, the Q Sepharose column was removed, and the protein bound with SP Sepharose was eluted with a linear gradient of NaCl (50 to 450 mM) in buffer A. The electrophoretically pure peak fractions were pooled and kept at -80°C in aliquots. On average, 8-10 mg of human MPG was recovered from 1 L of E. coli cells.
2.3 Monoclonal antibody production and subtyping
The purified hMPG lacking variable first exon was used for immunization. The protein was dialyzed against phosphate buffered saline and emulsified in either complete Freunds (first immunization) or incomplete Freunds (subsequent immunizations) adjuvant before subcutaneous injection of Balb/c mice. A mouse with high titer was selected for monoclonal antibody production as described previously [24, 25]. Cultures were screened using an ELISA assay with the immunizing protein as the target [26]. Positive cultures were recloned by limit dilution, and the clones were tested for stable production of antibody. Antibodies were purified from ascites fluid by ammonium sulfate precipitation followed with ion exchange chromatography on DEAE cellulose (DE52) [26]. Antibodies were analyzed for IgG subclasses using a commercial capture-detection ELISA kit.
2.4 Epitope competition assay and binding affinity (KD) analysis
Purified monoclonal antibodies were radioiodinated and tested for direct binding to the immunizing protein attached to 96 well format Immulon 4 snap apart wells. Experiments were performed with 1 μg target protein per well, and multiple concentrations of radioiodinated antibody, diluted in PBS containing 5 mg/mL BSA, were tested for binding (1 hr at 25°C in duplicate). Wells were washed and counted in a gamma scintillation counter. The KD values were calculated from double reciprocal plots [27]. Competition binding experiments to assess epitope competition were performed as described previously [28] using the same binding format. Briefly, 50 ng of radioiodinated monoclonal antibody was mixed with different unlabelled antibodies in 50-fold molar excess. Binding data were collected, and the amount of competition for binding determined.
2.5 SDS-PAGE and Western Blot Analysis
Purified MPG (50 ng) or nuclear extracts from human or mouse cells (50 μg soluble protein) were separated by SDS-PAGE (15% polyacrylamide) and stained with Coomassie brilliant blue. For Western Blot analysis, proteins were transferred to a nitrocellulose membrane, and the MPG bands were visualized with the anti-human MPG monoclonal antibodies, 1:500-1000 dilution for cell extracts or 500 ng for purified protein, using enhanced chemi-luminescence protocol (Amersham Life Sciences, Piscataway, NJ).
2.6 Identification of Epitope Sequence Recognized by 520-3A moAb
To determine the hMPG epitope recognized by the monoclonal antibody 520-3A, we generated a library of bacterial clones expressing short peptides derived from hMPG. The library was constructed using DNase I in the presence of Mn2+ to cause double-strand breaks in the hMPG cDNA generating fragments, averaging 50 to 100 bp according to a published method [29]. Following standard colony hybridization techniques [30], 34 positive colonies were identified using 520-3A monoclonal antibody (1:1000 dilution) and enhanced chemi-luminescence protocol. Six out of 34 colonies yielded DNA inserts, and the cell-free extracts from all 6 colonies were tested by Western analysis using 520-3A antibody. Four of them showed expression of MPG peptides. All 4 of those DNA inserts were then sequenced for epitope sequence determination.
2.7 Competetion of the hMPG Antibody with a Synthetic Epitope Peptide Corresponding to Residues 52-82 of hMPG
Individual membrane strips containing 50ng of purified hMPG protein were processed for Western Blot analysis by incubating with 500ng of 520-3A moAb, which was preincubated with varying amounts (0-9 μg) of peptide corresponding to residues 52-82 or 9 μg of control peptide containing unrelated sequence at 25°C for 3 hrs.
2.8 Preparation of Substrates
Hx or εA containing 50-mer oligonucleotides with the sequence 5′-TCGAGGATCCTGAGCTCGAGTCGACGXTCGCGAATTCTGCGGATCCAAGC-3′ (where X represents Hx, or εA) were purchased from Operon Technologies (Alameda, CA) and Gene Link (Hawthorne, NY). The complementary oligonucleotide containing T opposite Hx was synthesized by the Recombinant DNA Laboratory Core Facility at the University of Texas Medical Branch (Galveston, TX). The oligonucleotides were purified on a polyacrylamide sequencing gel. The Hx oligonucleotide was labeled at the 5′ end using T4 polynucleotide kinase and γ32P-ATP and annealed to complementary oligonucleotide to prepare 32P-end-labeled duplex oligonucleotide as described previously [31]. [3H]-labeled methylated calf thymus DNA substrate (∼370 c.p.m./μg, 109 fmol 7-methylguanine and 20 fmol 3-methyladenine in a total of 960 pmol adenine and 640 pmol guanine per μg DNA) was prepared as described previously [9].
2.9 MPG-Mediated Excision Activity Assay
The hMPG protein (5-10 nM) was incubated in the presence of MPG (0-29 nM) and APE (control, 10 nM) antibodies. The standard reaction mixtures contained 25 mM HEPES-KOH, pH 7.9, 0.5 mM DTT, 150 mM NaCl, 10 μg/ml nuclease-free BSA, and 10% glycerol in a total volume of (20-200 μl). Incubation was at 37°C for 10 min, if not otherwise stated. For methylated substrates, the reaction was terminated by the addition of 200 μg of carrier calf thymus DNA and the large DNA fragments' precipitation with cold ethanol in the presence of 0.3M sodium acetate. The radio-labeled base adducts in the ethanol-soluble fraction were quantified directly by liquid scintillation counting [9]. For Hx and εA, the reaction was stopped by inactivating the enzyme at 75°C for 5, min and the products were analyzed as described previously [32].
2.10 Binding Studies Using Surface Plasmon Resonance
We examined the modulation of MPG binding to Hx or εA by hMPG moAb using a Biacore-1000 (Biacore, Uppsala, Sweden). A 50-mer duplex oligonucleotide, containing an Hx, εA or adenine (control) at the 26th position from the 5′ end of one strand, was used for measuring enzyme-substrate DNA interactions. Oligonucleotides were biotinylated and immobilized on streptavidin-coated Biacore chips. Previously, we showed Mg2+ can inhibit the binding of Hx towards MPG by surface plasmon resonance [32]. Under similar conditions of MPG-Hx interactions, we tested the effect of 520-3A antibody on MPG's (40-125 nM) binding towards Hx, εA or unmodified oligonucleotide in the presence of various concentrations of antibody (0-1000 nM).
2.11 Single Turnover (STO) Kinetic Study
The hMPG (40-62.5 nM) was incubated with various concentrations (0-60 nM) of 520-3A antibody and a fixed concentration (2 nM) of 5′ 32P-labeled Hx or εA-containing duplex oligonucleotide substrates at 37°C in an assay buffer containing 25 mM HEPES-KOH, pH 7.9, 10 μg/ml nuclease-free BSA, 0.5 mM DTT, 150 mM NaCl and 10% glycerol in a total volume of 100 μl. Aliquots of 5 μl were removed at different times (0-20 min) and heat inactivated at 80°C in a preheated microcentrifuge tube. The products containing the AP-sites were quantitatively cleaved into smaller fragments, followed by resolving on denaturing gels, and radioactivity in the incised oligonucleotide was also quantified as described previously [32].
3. Results
3.1 Purification of human MPG lacking variable first exon
The ΔExon I hMPG protein was purified to homogeneity as shown by SDS-PAGE (Fig. 1). The human enzyme showed cleavage of Hx with specific activity similar to that of mouse MPG as described earlier [9]. The human protein was dialyzed in phosphate buffered saline and used for immunization and antibody production.
Figure 1. Purification of ΔExon I hMPG.

The ΔExon I hMPG protein was purified to near homogeneity. The details of the purification are described in “Materials and Methods.”
3.2 Production and characterization of anti-MPG monoclonal antibodies
Twelve independent hybridoma clones were isolated and extensively characterized. Among them, 4 moAbs (520-2A, 520-3A, 520-16A, and 520-26A) were of high affinity (KD=∼0.3-1.6 nM; Table 1), and the rest were of lower affinity. The high affinity moAbs were subtyped. The 520-2A, 520-3A, 520-16A, and 520-26A were IgG2a, IgG1, IgG2a, and IgG2b respectively (Table 1). Epitope competition assay showed that, unlike 520-16A, the other 3 moAbs competed for binding with each other (Table 1), and homologous competition always resulted in > 80% reduction in binding. Then we selected 520-3A, a high affinity antibody, for epitope identification. Upon sequencing, all 4 of those DNA inserts which showed expression of MPG peptides were found to be identical and matching the nucleotide sequences 156 to 246 from human MPG. moAb 520-3A recognizes the sequence 52QAPCPRERCLGPPT67TPGPYRSIYFSSPKGH82, which corresponds to the residues 156 to 246 in the hMPG coding sequence. In Western Blot analysis, the antibody (520-3A) cross-reacted with a polypeptide of 34 kDa in the cell-free extract of a variety of human cell lines (Fig. 2), and the protein sizes were similar to those reported by others using polyclonal antibodies [10]. However, this moAb does not cross-react with MPG in cell extracts from mouse cells (Fig. 2). Therefore, the segment 67T through 82H may be excluded from the above epitope sequence, and the epitope could be further narrowed down to 52AQAPCPRERCLGPP66T, which is a unique human sequence. Thus, it appears that the moAb 520-3A and possibly others require at least 15 residues to bind to MPG in human cells. However, an epitope peptide containing residues 52-82 successfully inhibited ∼70% of the antibody's MPG binding (Fig. 3), whereas the shorter peptide (52-66) failed to inhibit activity (data not shown). Probably the extra 16 residues at the carboxy terminus are required to retain the minimum conformation for the antibody to recognize the epitope.
Table 1.
Characterization of anti-human MPG monoclonal antibodies
| Hybridoma clones | Antigen Binding affinity (KD, nM) | Antibody subclass | Epitope recognition | Epitope sequence in hMPG (see text for details) |
|---|---|---|---|---|
| 520-2A | 1.0 | IgG2a | Same | |
| 520-3A | 0.32 | IgG1 | Same | 52AQAPCPRERCLGPP66T |
| 520-16A | 0.76 | IgG2a | Different | |
| 520-26A | 1.6 | IgG2b | Same |
Figure 2. Western Blot analysis of MPG by monoclonal antibody in different human and mouse cell lines.

(A) Cell-free extracts from human or mouse cells (50 μg of soluble protein) were subjected to Western Blot analysis. The details of the reaction conditions are described in “Materials and Methods.” The band intensities for MPG and β-actin were quantified by densitometric image analysis, and the data obtained for MPG were normalized with those of β-actin.
Figure 3. Competetion of the hMPG moAb, 520-3A with a synthetic epitope peptide corresponding to residues 52-82 of hMPG.

50 ng of purified protein was loaded onto the SDS-PAGE and the synthetic peptide inhibits ∼70% of the binding. The details of the experiments are described in “Materials and Methods.”
3.3 Inhibition of MPG activity by the antibody
The activity of the purified hMPG was measured in the presence of 0-29 nM antibody using different MPG substrates. The IC50 is strikingly different for εA compared to Hx or methylated substrates (Fig. 4 A-D). An unrelated control mouse moAb against human APE did not have any effect on MPG's activity (Fig. 4C). However, the antibody inhibits MPG's activity with ∼ 5 fold more efficiency towards Hx and methylated substrates than ∊A, despite the fact that Hx is neutral, whereas methylated bases are positively charged substrates. Moreover, unlike Hx or methylated adducts inhibition of εA removal follows a bi-phasic curve. Therefore, this antibody is particularly useful in understanding the discriminating properties of MPG's substrate specificity.
Figure 4. Modulation of MPG activity by 520-3A moAb.
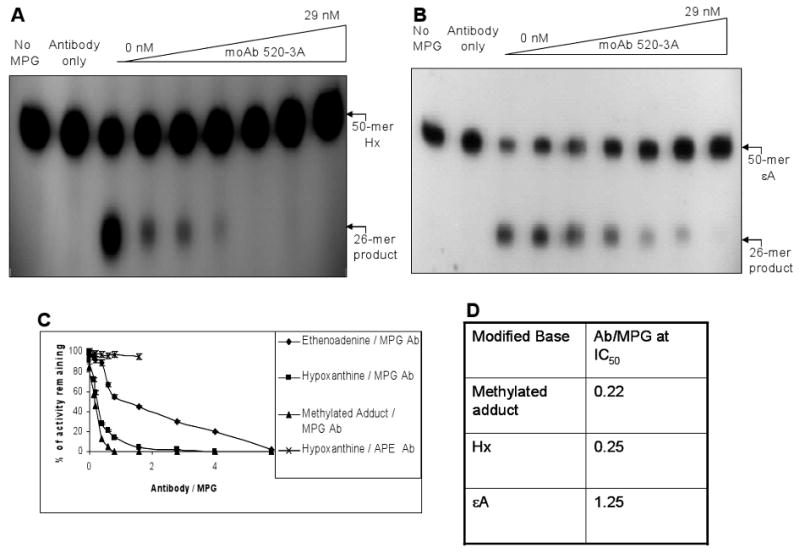
The activity of the purified hMPG (5 nM) was measured in the presence of 0-29 nM antibody concentration for Hx (A) and εA (B). A comparison of Hx and εA with methylated substrate measured by using [3H]-labeled methylated calf thymus DNA is shown in (C). Data taken from panel C are used for IC50 determination and presented in (D). IC50 denotes 50% of the remaining MPG activity. The details of the reaction conditions are described in “Materials and Methods.” Data represent mean values with standard error derived from at least three independent experiments.
3.4 Mechanism of antibody inhibition of MPG reaction
To elucidate the mechanism of inhibition, we tested various reaction steps of MPG. Unlike alkylated calf thymus DNA, Hx and εA are more defined substrates, and, therefore, we used this oligo substrate to analyze the detailed mechanisms of antibody-mediated inhibition of MPG activity using pre-steady-state kinetics. We have discussed previously [32] how pre-steady-state kinetic analysis provides the opportunity to identify the intermediate step(s) that might be affected by the antibody. Briefly, the effect of moAb was assessed on substrate binding [measured by surface plasmon resonance (SPR)] and chemistry [catalysis; measured by single turn over (STO) kinetics] steps.
3.5 Surface plasmon resonance (SPR) to analyze substrate binding
The 50-mer oligonucleotide containing Hx or εA was biotinylated and immobilized on streptavidin-coated chips. An unmodified oligonucleotide with a similar sequence was used as a control. Using SPR, one can follow real-time DNA-protein interactions compared with other commonly used methods, such as gel shift assay. We established the binding conditions for oligonucleotides as before [32]. Then, we tested the effect of antibody on MPG's binding to the Hx- or εA- containing oligonucleotide and found an increase in MPG's affinity for Hx at lower concentrations of antibody and a small decrease at higher moAb concentrations. For εA we found a steady increase at lower concentrations, and it reached near saturation at higher concentrations (Fig. 5B and C). The antibody, at a 1:2 molar ratio with MPG, enhanced binding of the enzyme onto Hx and εA, while inhibiting its activity significantly. Notably, the antibody also increased the nonspecific binding of MPG towards the control oligo (Fig 5A). These results suggest that in order to affect MPG's activity the antibody does not abrogate MPG's substrate binding step.
Figure 5. Effect of 520-3A moAb on MPG binding to control (A), Hx (B) and εA (C) -containing oligonucleotides.

The DNA–protein interactions were carried out using (40-125) nM MPG in the presence of different moAb concentrations (0-1000 nM). The details of the experiments are described in “Materials and Methods.”
3.6 STO Kinetics
Prompted by the observation that antibody can inhibit product formation, we tested whether antibody affected its catalytic reaction other than the binding step. We conducted STO kinetics with MPG proteins to measure the kchem. The reaction was performed at substrate and enzyme concentrations of 2 and 40-62.5 nM, respectively, with or without antibody (0-60 nM). Data were analyzed using the first-order rate equation:
| (1) |
where A0 represents the amplitude of the exponential phase, and kobs is the observed rate constant associated with the reaction process. Under the STO conditions ([E]≫ [S]), all the substrate molecules should remain bound by enzymes. The binding step should not affect the rate of product formation; hence, under these conditions kobs can be considered as kchem. In fact, at [S] : [E] of 1:20 or 1:30, the kchem did not alter, ensuring the enzyme reactions are following the STO conditions properly (Fig 6 A and B). Under similar conditions, incubation of antibody with hMPG showed pronounced inhibition (Fig. 6 C, D and E), indicating that the antibody was inhibiting the catalytic reaction at the chemistry step of MPG reaction. The kchem for MPG-mediated Hx or εA catalysis decreased with increasing antibody concentration, albeit, Hx removal was more affected. Thus, the new MPG moAb, which is extensively characterized for its immuonologic properties, neutralizes MPG activity by impeding its chemistry (catalysis) step without abrogating the binding step.
Figure 6. Effect of 520-3A moAb on MPG reaction with Hx (A) and εA (B) under single turnover conditions.

The reaction was performed using substrate and enzyme concentrations of 2 and 40 - 62.5 nM, respectively, as described in “Materials and Methods.” Effect of moAb on pre–steady-state kinetic parameters under single turnover conditions for Hx (C) or εA (D). (E) Data derived from Panel (C) or (D) were analyzed using the first-order rate equation: [P]t = A0{ 1- exp (-kobs t)} as described in “Results.” kchem, catalytic constant at the chemistry step.
4. Discussion
We have identified a specific antibody inhibitor of MPG, a DNA glycosylase, with recognition and cleavage specificity for a wide variety of structurally diverse alkylated, deaminated, and etheno DNA adducts. Despite the importance of DNA repair glycosylases in repairing mutagenic and toxic DNA base adducts caused by endogenous and exogenous sources, such as oxidative and nitrosative stress, replication errors, and cigarette smoke, there are not many well-characterized inhibitors for this important class of BER enzymes. UGI, a viral protein, has been shown to inhibit UDG, which excises uracils in DNA generated from replication misincorporations [33]. Speina et al. [34] screened a battery of compounds, including various base analogs and tryptophan pyrolysate (Trp-P-1), a mutagenic DNA intercalator heterocyclic amine found in cooked food, targeting a number of E. coli and human DNA glycosylases. The 2-thioxanthine was effective for E. coli Fpg protein and was shown to inhibit its excision activity, whereas Trp-P-1 inhibited multiple DNA glycosylases, such as AlkA, TagA, MPG, NTH, and Fpg by altering the DNA secondary structures.
In a previous paper, we showed that Mg2+ could inhibit MPG's activity by abrogating its substrate binding and decreasing the active enzyme concentration [32]. However, Mg2+-mediated inhibition might be better considered as a regulator for balanced BER than an inhibitor as all the downstream BER enzymes, other than MPG, require Mg2+ for their optimum repair activity.
We have shown before, by systematic deletion analysis of MPG from N- and C-termini, that a minimally sized polypeptide (NΔ100CΔ18) lacking 100 and 18 amino acid residues from the amino and carboxyl termini, respectively, and wild-type enzyme had similar kinetic and binding properties for εA [7]. Since then, there were several reports on the crystallographic structures of the similarly truncated protein in complex with εA or control DNA [13, 14, 35]. Using recombinant chimeric proteins, containing N- and C-terminal halves of human and mouse MPG, we found that the N-terminal half is critical for the recognition of 3-methylguanine and 7-methylguanine [9]. Dr. Jacques Laval's group also showed that N-terminal tail is important for the catalysis of 1,N2-εG-DNA (20). From this study, however, it is apparent that MPG's modes of action towards different substrates are strikingly different. As εA appears less inhibited at lower concentrations of antibody compared to higher concentrations, it clearly shows clearly a bi-phasic reaction, which indicates the possibility of two different mechanisms for the inhibitions. We also do not rule out the possibility of inhibition of catalysis by unspecific binding of the antibody at higher concentrations. Nonetheless, the structural information of εA bound to truncated MPG available from literature [13, 14, 35] does not adequately reveal the full scenario for other DNA adducts, including Hx and methylated bases.
MPG, being a broad substrate enzyme, must be flexible for DNA binding in order to recognize DNA lesions of varied structures. In fact, for similar reasons T. Ellenberger and his colleagues proposed that a “nonspecific catalytic mechanism” must be met for an enzyme to succeed as a generalist as one of the major criteria, which comes “at the expense of catalytic power” [23]. For this study, we have compared three different substrates which represent the products of three major classes of DNA damaging agents of both endogenous and environmental origins: alkylating chemotherapeutics (methylated substrates), deamination (Hx) and lipid peroxidation (εA) -induced purine adducts. So, from the major differences in the IC50s for inhibition of at least three different substrates by an antibody with an epitope in the N-terminal tail it is clearly indicated that the mechanism of reaction varies for structurally different substrates. This inhibition can be attributed to two different scenarios: (1) the N-terminal tail is important and involved in MPG reaction, but its importance may depend on the substrate type, as the excision of methylated substrates and Hx are 5 times more inhibited than εA; (2) The antibody when binding to the N-terminal tail may affect the structural integrity and perhaps the catalytic core of MPG by posing spatial steric hindrance with different degrees for different substrate types. Thus, this affects their excision accordingly and generates substrate specificity. In fact, the first 70 amino acid residues comprising the N-terminal extension of hMPG are indispensable for 1,N2-εG excision reaction [20]. The differences in IC50s for N-terminal-specific antibody-mediated inhibition of three different substrates for human MPG again suggest that the N-terminal tail can be a critical regulatory domain for the activity of human MPG towards different substrates which remains to be tested. Since the N-terminal extension is present in all the MPG's from higher eukaryotes, it suggests an evolutionarily conserved regulatory function of the N-terminal tail for MPG's activity.
Acknowledgments
We thank Dr. Aykut Üren from Biacore Molecular interaction Shared Resources facility of the Lombardi Cancer Center for SPR studies. We thank Ms. Karen Howenstein for expert editorial and secretarial help. The work was supported by NIH grants RO1 CA 92306 (RR) and RO1 CA 53791 (SM).
Abbreviations
- BER
base excision repair
- AP
Apurinic / apyrimidinic
- MPG
N-Methylpurine DNA-glycosylase
- APE
Apurinic / apyrimidinic endonuclease
- Hx
Hypoxanthine
- εA
1,N6ethenoadenine
- moAb
monoclonal antibody
- m3A
3-methyl adenine
- m7G
7-methyl guanine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 2.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger TE. DNA Repair and Mutagenesis. American Society for Microbiology. 2006:169–226. [Google Scholar]
- 3.Larson K, Sahm J, Shenkar R, Strauss B. Methylation-induced blocks to in vitro DNA replication. Mutat Res. 1985;150:77–84. doi: 10.1016/0027-5107(85)90103-4. [DOI] [PubMed] [Google Scholar]
- 4.Breimer L, Lindahl T. DNA glycosylase activities for thymine residues damaged by ring saturation, fragmentation, or ring contraction are functions of endonuclease III in Escherichia coli. J Biol Chem. 1984;259:5543–5548. [PubMed] [Google Scholar]
- 5.Hatahet Z, Kow YW, Purmal AA, Cunningham RP, Wallace SS. New substrates for old enzymes. 5-Hydroxy-2′-deoxycytidine and 5-hydroxy-2′-deoxyuridine are substrates for Escherichia coli endonuclease III and formamidopyrimidine DNA N-glycosylase, while 5-hydroxy-2′-deoxyuridine is a substrate for uracil DNA N-glycosylase. J Biol Chem. 1994;269:18814–18820. [PubMed] [Google Scholar]
- 6.Singer B, Hang B. What structural features determine repair enzyme specificity and mechanism in chemically modified DNA? Chem Res Tox. 1997;10:713–732. doi: 10.1021/tx970011e. [DOI] [PubMed] [Google Scholar]
- 7.Roy R, Biswas T, Hazra TK, Roy G, Grabowski DT, Izumi T, Srinivasan G, Mitra S. Specific interaction of wild-type and truncated mouse N-methylpurine-DNA glycosylase with ethenoadenine-containing DNA. Biochemistry. 1998;37:580–589. doi: 10.1021/bi972313l. [DOI] [PubMed] [Google Scholar]
- 8.Roy R, Brooks C, Mitra S. Purification and biochemical characterization of recombinant N-methylpurine-DNA glycosylase of the mouse. Biochemistry. 1994;33:15131–15140. doi: 10.1021/bi00254a024. [DOI] [PubMed] [Google Scholar]
- 9.Roy R, Kennel SJ, Mitra S. Distinct substrate preference of human and mouse N-methylpurine-DNA glycosylases. Carcinogenesis. 1996;17:2177–2182. doi: 10.1093/carcin/17.10.2177. [DOI] [PubMed] [Google Scholar]
- 10.O' Connor TR. Purification and characterization of human 3-methyladenine-DNA glycosylase. Nucl Acids Res. 1993;21:5561–5569. doi: 10.1093/nar/21.24.5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saparbaev M, Laval J. Excision of hypoxanthine from DNA containing dIMP residues by the Escherichia coli, yeast, rat, and human alkylpurine DNA glycosylases. Proc Natl Acad Sci USA. 1994;91:5873–5877. doi: 10.1073/pnas.91.13.5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dosanjh MK, Roy R, Mitra S, Singer B. 1,N6-ethenoadenine is preferred over 3-methyladenine as substrate by a cloned human N-methylpurine-DNA glycosylase (3-methyladenine-DNA glycosylase) Biochemistry. 1994;33:1624–1628. doi: 10.1021/bi00173a002. [DOI] [PubMed] [Google Scholar]
- 13.Labahn J, Scharer OD, Long A, Ezaz-Nikpay K, Verdine GL, Ellenberger TE. Structural basis for the excision repair of alkylation-damaged DNA. Cell. 1996;86:321–329. doi: 10.1016/s0092-8674(00)80103-8. [DOI] [PubMed] [Google Scholar]
- 14.Lau AY, Scharer OD, Samson L, Verdine GL, Ellenberger T. Crystal structure of a human alkylbase-DNA repair enzyme complexed to DNA: mechanisms for nucleotide flipping and base excision. Cell. 1998;95:249–258. doi: 10.1016/s0092-8674(00)81755-9. [DOI] [PubMed] [Google Scholar]
- 15.Lindahl T, Sedgwick B, Sekiguchi M, Nakabeppu Y. Regulation and expression of the adaptive response to alkylating agents. Annu Rev Biochem. 1988;57:133–157. doi: 10.1146/annurev.bi.57.070188.001025. [DOI] [PubMed] [Google Scholar]
- 16.McCarthy TV, Karran P, Lindahl T. Inducible repair of O-alkylated DNA pyrimidines in Escherichia coli. EMBO J. 1984;3:545–550. doi: 10.1002/j.1460-2075.1984.tb01844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bjelland S, Birkeland NK, Benneche T, Volden G, Seeberg E. DNA glycosylase activities for thymine residues oxidized in the methyl group are functions of the AlkA enzyme in Escherichia coli. J Biol Chem. 1994;269:30489–30495. [PubMed] [Google Scholar]
- 18.Bjoras M, Klungland A, Johansen RF, Seeberg E. Purification and properties of the alkylation repair DNA glycosylase encoded the MAG gene from Saccharomyces cerevisiae. Biochemistry. 1995;34:4577–4582. doi: 10.1021/bi00014a010. [DOI] [PubMed] [Google Scholar]
- 19.Roy R, Kumar A, Lee JC, Mitra S. The domains of mammalian base excision repair enzyme N-methylpurine-DNA glycosylase. Interaction, conformational change, and role in DNA binding and damage recognition. J Biol Chem. 1996;271:23690–23697. doi: 10.1074/jbc.271.39.23690. [DOI] [PubMed] [Google Scholar]
- 20.Saparbaev M, Langouet S, Privezentzev CV, Guengerich FP, Cai H, Elder RH, Laval J. 1,N(2)-ethenoguanine, a mutagenic DNA adduct, is a primary substrate of Escherichia coli mismatch-specific uracil-DNA glycosylase and human alkylpurine-DNA-N-glycosylase. J Biol Chem. 2002;277:26987–26993. doi: 10.1074/jbc.M111100200. [DOI] [PubMed] [Google Scholar]
- 21.Hill-Perkins M, Jones MD, Karran P. Site-specific mutagenesis in vivo by single methylated or deaminated purine bases. Mutat Res. 1986;162:153–163. doi: 10.1016/0027-5107(86)90081-3. [DOI] [PubMed] [Google Scholar]
- 22.Lindahl T. DNA glycosylases, endonucleases for apurinic/apyrimidinic sites, and base excision-repair. Prog Nucleic acid Res Mol Biol. 1979;22:135–163. doi: 10.1016/s0079-6603(08)60800-4. [DOI] [PubMed] [Google Scholar]
- 23.Chakravarti D, Ibeanu GC, Tano K, Mitra S. Cloning and expression in Escherichia coli of a human cDNA encoding the DNA repair protein N-methylpurine-DNA glycosylase. J Biol Chem. 1991;266:15710–15715. [PubMed] [Google Scholar]
- 24.Kennel SJ, Foote LJ, Lankford PK. Analysis of surface proteins of mouse lung carcinomas using monoclonal antibodies. Cancer Res. 1981;41:3465–3470. [PubMed] [Google Scholar]
- 25.de St Groth SF, Scheidegger DJ. Production of monoclonal antibodies: strategy and tactics. J Immunol Methods. 1980;35:1–21. doi: 10.1016/0022-1759(80)90146-5. [DOI] [PubMed] [Google Scholar]
- 26.Kennel SJ. Binding of monoclonal antibody to protein antigen in fluid phase or bound to solid supports. J Immunol Methods. 1982;26:1–12. doi: 10.1016/0022-1759(82)90070-9. [DOI] [PubMed] [Google Scholar]
- 27.Kennel SJ, Chen JP, Lankford PK, Foote LJ. Monoclonal antibodies from rats immunized with fragment D of human fibrinogen. Thromb Res. 1981;22:309–320. doi: 10.1016/0049-3848(81)90124-9. [DOI] [PubMed] [Google Scholar]
- 28.Davern SM, Lankford PK, Foote LJ, S J. Monoclonal antibodies to CD44 epitopes on mouse endothelium. Hybrid Hybridomics. 2002;21:339–349. doi: 10.1089/153685902761022689. [DOI] [PubMed] [Google Scholar]
- 29.Anderson S. Shotgun DNA sequencing using cloned DNase I-generated fragments. Nucleic Acids Res. 1981;9:3015–3027. doi: 10.1093/nar/9.13.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manniatis T, Foitsch EF, Sambrook J. A laboratory manual. New York: Cold Spring Harbor Laboratory Press; 1982. Molecular Cloning. [Google Scholar]
- 31.Roy R, Biswas T, Lee JC, Mitra S. Mutation of a unique aspartate residue abolishes the catalytic activity but not substrate binding of the mouse N-methylpurine-DNA glycosylase (MPG) J Biol Chem. 2000;275:4278–4282. doi: 10.1074/jbc.275.6.4278. [DOI] [PubMed] [Google Scholar]
- 32.Adhikari S, Toretsky JA, Yuan L, Roy R. Magnesium, essential for base excision repair enzymes, inhibits substrate binding of N-methylpurine-DNA glycosylase. J Biol Chem. 2006;281:29525–29532. doi: 10.1074/jbc.M602673200. [DOI] [PubMed] [Google Scholar]
- 33.Bennett SE, Mosbaugh DW. Characterization of the Escherichia coli uracil-DNA glycosylase inhibitor protein complex. J Biol Chem. 1992;267:22512–22521. [PubMed] [Google Scholar]
- 34.Speina E, Ciesla JM, Graziewicz M, Laval J, Kazimierczuk Z, Tudek B. Inhibition of DNA repair glycosylases by base analogs and tryptophan pyrolysate, Trp-P-1. Acta Biochimica Polonica. 2005;52:167–178. [PubMed] [Google Scholar]
- 35.Lau AY, Wyatt MD, Glassner BJ, Samson LD, Ellenberger T. Molecular basis for discriminating between normal and damaged bases by the human alkyladenine glycosylase, AAG. Proc Natl Acad Sci U S A. 2000;5:13573–13578. doi: 10.1073/pnas.97.25.13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Brien PJ, Ellenberger T. Dissecting the broad substrate specificity of human 3-methyladenine-DNA glycosylase. J Biol Chem. 2004;279:9750–9757. doi: 10.1074/jbc.M312232200. [DOI] [PubMed] [Google Scholar]