

Abstract
Slipped strand mispairing during DNA synthesis is one proposed mechanism for microsatellite or short tandem repeat (STR) mutation. However, the DNA polymerase(s) responsible for STR mutagenesis have not been determined. In this study, we investigated the effect of the Escherichia coli dinB gene product (Pol IV) on mononucleotide and dinucleotide repeat stability, using an HSV-tk gene episomal reporter system for microsatellite mutations. For the control vector (HSV-tk gene only) we observed a statistically significant 3.5-fold lower median mutation frequency in dinB- than dinB+ cells (p<0.001, Wilcoxon Mann Whitney Test). For vectors containing an in-frame mononucleotide allele ([G/C]10) or either of two dinucleotide alleles ([GT/CA]10 and [TC/AG]11) we observed no statistically significant difference in the overall HSV-tk mutation frequency observed between dinB+ and dinB- strains. To determine if a mutational bias exists for mutations made by Pol IV, mutational spectra were generated for each STR vector and strain. No statistically significant differences between strains were observed for either the proportion of mutational events at the STR or STR specificity among the three vectors. However, the specificity of mutational events at the STR alleles in each strain varied in a statistically significant manner as a consequence of microsatellite sequence. Our results indicate that while Pol IV contributes to spontaneous mutations within the HSV-tk coding sequence, Pol IV does not play a significant role in spontaneous mutagenesis at [G/C]10, [GT/CA]10, or [TC/AG]11 microsatellite alleles. Our data demonstrate that in a wild type genetic background, the major factor influencing microsatellite mutagenesis is the allelic sequence composition.
Keywords: Microsatellite Mutation, dinB, Dinucleotide, Mononucleotide
1. Introduction
Microsatellite sequences of 1-5 nucleotides per unit repeat are found throughout the human genome [1-8]. Microsatellite sequences are often hotspots for mutation, and an increase in mutagenesis is observed as the length of the repeated sequence increases [9,10]. The predominant mutation in mono- and dinucleotide microsatellite sequences are single-unit insertions or deletions [9,11-13]. One proposed mechanism to explain instability in these microsatellites is the polymerase slippage model [14,15], also referred to as slipped strand mispairing [16]. In this model, DNA misalignments in the primer-template are used as substrates for DNA synthesis. Whether the misalignment creates a bulge on the template or primer strand dictates whether a deletion or insertion event will result. Two major mechanisms exist to correct misaligned DNA intermediates resulting from strand slippage: 3’ to 5’ exonuclease activity of the polymerase and post replicative mismatch repair (MMR) [12]. For short microsatellites, both mechanisms have been reported. However, in longer microsatellite tracts, MMR becomes the favored mechanism [12,17] because proofreading ability decreases as the length of the repeat increases [12,17-19]. Although polymerase utilization of slipped DNA intermediates is a requisite step in this model, the cellular polymerase(s) responsible for microsatellite mutagenesis are not known. We have used Escherichia coli (E. coli) as a genetic model system to investigate polymerase(s) responsible for microsatellite mutagenesis.
The E. coli dinB gene encodes DNA Polymerase IV (Pol IV) [20]. Pol IV does not require recA, umuDC, uvrA, polA or polB for activity [21,22] and errors made by Pol IV are correctable by MMR, suggesting Pol IV contributes to replication errors on undamaged templates [21]. Expression of the dinB gene has been shown to cause an increase in -1 frameshifts [21,23] as well as single nucleotide base substitutions with a preference toward G:C bases. Conversely, deletion of this gene has been shown to cause a significant decrease in the number of -1 frameshifts and single nucleotide base substitutions in both wild type cells and cells that have a mutant replicative polymerase [23]. Under non-SOS conditions, there are ~250 molecules of Pol IV present and upon up-regulation of the SOS system there are ~2500 molecules [24]. Based on these results, it has been speculated that Pol IV may bind more frequently at a stalled replication fork than the other SOS polymerases, making it a “default” bypass polymerase in E. coli [24]. Pol IV has also been suggested to be an auxiliary polymerase involved in spontaneous mutation [23].
In the present study, we examined spontaneous mutagenesis at three different microsatellite alleles, [G/C]10, [GT/CA]10, and [TC/AG]11 in isogenic dinB+ and dinB- E. coli cells. The strategy involves comparison of microsatellite mutations relative to an internal coding region control in the Herpes Simplex Virus thymidine kinase (HSV-tk) gene. Although the presence of Pol IV affected mutagenesis of the HSV-tk gene, no significant result was observed for any of the microsatellite alleles in the presence of Pol IV. Mutational specificity comparisons of the three microsatellite alleles showed a significant difference as a function of the sequence, with the [G/C]10 mononucleotide repeat conferring a majority of insertion events and the [GT/CA]10 and [TC/AG]11 dinucleotide repeats favoring deletion events, independent of the dinB status of the cells.
2. Materials and Methods
2.1. Escherichia coli Strains
Strain FT334 is derived from HB101 [25] with the genotype: tdk, upp, thi1, hsd20, supE44, lacY1, proA2, ara14, galK2, xyl5, mtl1, leuB6, rpsL20, recA13. Strain PP102 is mismatch repair deficient and is isogenic to strain FT334 with exception the of the following alleles: recA306, srl∷Tn10, mutL∷Tn5[13].
E. coli strains SMR 4562 and SMR 5830 were provided by Dr. Susan Rosenberg, Baylor College of Medicine. SMR 4562 is an independent isolate of FC40 [26]. SMR 5830 is isogenic to SMR4562, and is dinB10 [F’dinB10] [27]. Spontaneous 5-flourouracil-resistant, 5-fluoro-2’deoxythymidine (FUdR)-resistant isolates of both strains were isolated by selective plating as previously described [28]. The usual mechanism for FUdR-resistance has been shown to be mutations at the tdk locus [29]. The dinB+, tdk- isolate obtained from SMR 4562 was designated FCT21; the dinB10 [F’dinB10], tdk- isolate obtained from SMR 5830 was designated FTK44.
2.2. Construction of Microsatellite Containing Vectors
All of the microsatellite sequences constructed were inserted in frame into the 5’ coding region of the HSV-tk gene as previously described [30]. The [GT/CA]9 and [TC/AG]9 dinucleotide repeats were inserted between bases 111 and 112 in the sequence context [GT (insert) TCTC]. Therefore, the total allele lengths of the dinucleotide microsatellites are [GT/CA]10 and [TC/AG]11. The [G/C]9 mononucleotide repeat was inserted between bases 110 and 111 in the sequence context [CG (insert) TC] to give a total allele length of [G/C]10. The [C/G]10 repeat allele was inserted between bases 109 and 110 in the sequence context [GC (insert) GT] for a total allele length of [C/G]10. The [G/C]10 and [C/G]10 microsatellite inserts were synthesized by an in vitro DNA polymerase reaction as previously described [13]. The oligonucleotides used in construction of the vectors were synthesized by Biosynthesis, Inc (Lewisville, TX). The restriction endonucleases used in construction were purchased from Invitrogen Life Technologies (Carlsbad, CA). The HSV-tk gene cassettes were subcloned into the pJY1 shuttle vector, which is a derivative of the low-copy plasmid pBR322 [30].
2.3. Analysis of Mutation Frequency
Each Escherichia coli strain was transformed with plasmids containing microsatellite repeats by electroporation. Transformation efficiency (CmR colonies/ng DNA) of each preperation of competent cells and plasmid DNA was determined in a preliminary study. Chloramphenicol (Cm) and 5-Fluoro-2’deoxyuridine (FUdR) were purchased from Sigma Chemical Co. (St. Louis, MO). Using the same cells and DNA, the electroporation was repeated, and after a 60 minute expression period, an aliquot of approximately 20 CmR transformed cells was used to inoculate multiple 3mL LB cultures containing 50μg/ml chloramphenicol. Overnight growth of the cultures at 37° C for 18-24 hours selected plasmid bearing populations of approximately 2-5×109 CmR viable cells/ml. The mutation frequency of the HSV-tk gene was determined by selective plating on VBA plates + 50 μg/ml chloramphenicol in the presence or absence of 40 μM FUdR as previously described [31]. The presence of FUdR selects for cells that have a HSV-tk deficient phenotype. This phenotype can be achieved through expansions or deletions at the microsatellite which change the reading frame, as well as frameshifts, base substitutions, and rearrangements throughout the length of the HSV-tk gene and its promoter region [28,30,31]. For each microsatellite containing plasmid in each E. coli strain, at least 10 independent overnight cultures were analyzed. The mutation frequency (MF) was calculated as the number of FUdRR, CmR viable cells/ml divided by the number of CmR viable cells/ml. The HSV-tk mutation frequencies were analyzed statistically using the nonparametric Wilcoxon-Mann-Whitney Test.
2.4. HSV-tk Mutational Specificity Analysis
The HSV-tk mutational target is carried on a multicopy ColE1 plasmid and the gene is under no selective pressure during growth of the bacterial culture. Therefore, each cell potentially contains more than one plasmid containing an inactivating HSV-tk mutation or a mixture of wild type and mutant plasmids. In order to accurately assess the distribution of mutations between the microsatellite allele and the HSV-tk coding sequence within a given plasmid-bearing population, we isolated total plasmid DNA from a given culture. This population of DNA molecules was then sampled for individual HSV-tk plasmids to derive a mutational spectrum. In this manner, we are able to recover inactivating mutations that might otherwise be masked in a cell with a mixed plasmid population.
Plasmid DNA was isolated from 5 independent overnight cultures of each strain and vector combination. This DNA population was introduced into E. coli strain FT334 by electroporation under conditions of 1 DNA molecule per cell, (data not shown) and immediately placed on ice. Each electroporation was then divided into 10-20 aliquots and each aliquot added to 1mL 1X VBA broth. After expression for 2 hours at 37°C the cultures were plated on selective medium and 1 FUdR-resistant mutant was picked from each plate [31]. This was done to ensure independence of the cultures and prevent the possibility of picking siblings for sequencing. The FUdR-resistant colonies were grown overnight in LB broth containing 50 μg/ml Cm and DNA was isolated from each the following day. Large insertions/deletions were detected by AvaI/BglII restriction enzyme digest [30]. DNA sequencing reactions were carried out according to Beckman Coulter (Fullerton, CA) protocol and were sequenced using a Beckman Coulter CEQ 8000 according to company approved procedure. Mutants from different overnight cultures arose independently, by definition. Among mutants isolated from a single culture, those that are of a different mutational event can also be considered independent. Because the types of independent events were similar across the populations, mutants from the five populations were combined to generate mutational spectra for each strain and plasmid. As an example, we isolated total plasmid DNA from 5 randomly picked cultures of dinB+ bacteria carrying the [G/C]10 vector. From each independent culture we isolated 20 FUdRR mutant plasmids. The sequences of these 100 mutants were combined to generate the mutational spectrum for this genotype/vector combination. The variation in the types of mutational events observed within one microsatellite vector or cell type, or between different cell types were statistically analyzed using Fisher’s Exact test (two tailed) with an α-value of 0.05.
3. Results
In this study we have inserted various mononucleotide and dinucleotide repeat sequences in-frame into the 5’ end of the HSV-tk gene. Using a forward assay we are able to detect mutations that occur in the gene, both in the artificial repeat we have inserted as well as in the entire length of the coding region of the HSV-tk gene itself.
3.1. Effect of Mismatch Repair on Microsatellite Mutagenesis
We previously reported that the quantitative effect of MMR deficiency on HSV-tk gene mutagenesis was relatively modest when the entire transformed population was sampled as a single unit [13]. In contrast, mutagenesis at a chromosomal locus (RifR) was elevated several orders of magnitude in the same MMR-deficient cultures. One potential reason for this differential effect is that a high frequency of pre-existing mutant plasmid DNAs are introduced into both MMR+ and MMR- populations during transformation. Alternatively, the number of cell generations between transformation and mutational sampling may have been insufficient to reveal the MMR effect in plasmid DNA. In the current study, we report the results of a modified mutagenesis protocol that was designed to increase the sensitivity of the plasmid-based assay for the detection of host cell genotype effects. The major change in our method is that we use only a small number of transformed cells to inoculate cultures, thereby minimizing the probability of pre-existing mutations while maximizing the number of cell generations.
In order to test the validity of this new method, we quantitated the cumulative effect of MMR loss. Plasmid vectors containing artificial microsatellite sequences were introduced into isogenic mutL+ (FT334) and mutL- (PP102) recA- E. coli cells by electroporation. Approximately 20 transformed cells were then used to inoculate several independent cultures, the cultures were grown overnight, and plated under FUdR selection to determine the HSV-tk mutation frequency. For the control vector (containing no microsatellite) the HSV-tk mutation frequency was increased ~700-fold for the mutL deficient strain compared to the mutL proficient strain (Figure 1). This value is very similar to the magnitude of the effect observed for the loss of MMR at the chromosomal RifR locus (900-fold) in the same strains [13]. This result demonstrates that the effect of MMR on mutagenesis at the plasmid-encoded HSV-tk gene target is more effectively quantitated by this experimental approach. In-frame insertion of the [G/C]10 repeat allele into the 5’ region of the HSV-tk gene resulted in an ~18,000-fold increase in the mutation frequency in the MMR deficient cells, compared to MMR proficient cells. The difference in the median mutation frequencies observed for the two strains was statistically significant (Wilcoxon Mann Whitney Test, p=0.002). Insertion of the [GT/CA]10 and [TC/AG]11 alleles resulted in an ~500-fold and ~3,800-fold increases, respectively, in the mutation frequency of the mutL- cells compared to mutL+ (p<0.001, p<0.001). In MMR deficient cells, the addition of microsatellite repeats had an effect of the same or greater magnitude than the HSV-tk only control vector, confirming that these STR sequences truly are acted upon by the MMR proteins. A hierarchy was detected for the relative mutagenesis between the mutL- and mutL+ strain, with the mononucleotide repeat [G/C]10 having the greatest quantitative difference (Figure 1), followed by the two dinucleotide repeats, in the order [TC/AG]11 then [GT/CA]10. In addition, pairwise comparisons of the mutation frequencies for the three microsatellite containing vectors in the mutL- strain were all statistically significant (p≤0.04). Overall, these results indicate that the effect of MMR at the STR alleles is sequence specific.
Figure 1. Effect of MMR on mutation frequencies of microsatellite containing vectors.

Vectors were introduced into mutL+ (FT334) and mutL- (PP102) cells and the mutation frequencies were determined for the HSV-tk only as well as microsatellite containing vectors as described in Materials and Methods. The ratios of mutL-/mutL+ mutation frequencies as determined for 10-20 cultures of each strain and vector combination are depicted in the graph. The median mutation frequencies for mutL+ strain are: 3.6×10-8, control; 4.6×10-8, [G/C]10; 22×10-8, [GT/CA]11; 5.0×10-8, [TC/AG]11. The median mutation frequencies for the mutL- strain are: 2.5×10-5, control; 85×10-5, [G/C]10; 12×10-5, [GT/CA]11; 19×10-5, [TC/AG]11
3.2. Effect of Pol IV on Spontaneous Mutation Frequency
To determine the effect of Pol IV on spontaneous mutagenesis, plasmid vectors were introduced into isogenic E. coli strains FCT21 (dinB+) and FTK44 (dinB-) and the HSV-tk mutation frequencies determined. Growth curve analyses performed under conditions of the mutagenesis assay indicated that the cells were in stationary phase at the time of sampling for mutation frequency analyses (data not shown). For the control vector, the median HSV-tk mutation frequency was 6.8×10-8 in the presence of dinB and 2.0 ×10-8 in the absence of dinB (Table 1). This 3.5-fold higher mutation frequency observed in the presence of dinB is statistically significant, indicating that Pol IV plays a role in spontaneous mutagenesis at the episomal HSV-tk gene (Wilcoxon Mann Whitney Test, p<0.001). To determine the effect of dinB on microsatellite mutagenesis, vectors containing different mononucleotide and dinucleotide repeats were introduced into dinB+ and dinB- E. coli cells and the mutation frequency for each culture was determined (Figure 2; summarized in Table 1). In our vector nomenclature, the leading/lagging template strands are indicated as first the leading strand, then the lagging strand. For example, [G/C]10 indicates that the 10 G’s are the leading strand template and the 10 C’s are the lagging strand template. For the mononucleotide [G/C]10 vector, the median mutation frequency was 6.9 ×10-7 in the presence of dinB and 4.6×10-7 in its absence. This difference in the median mutation frequency of the [G/C]10 allele in the presence of Pol IV was not statistically significant (p=0.17). To examine whether there is a replication strand bias for STR mutagenesis, we also measured mutation frequencies for a [C/G]10 allele in each strain. The median mutation frequency was 7.1×10-7 in the dinB+ strain and 7.6×10-7 in the dinB- strain. The result of statistical analysis indicated that there was no significant difference in the mutation frequency between the two cell types (p=0.84). The [TC/AG]11 dinucleotide repeat produced similar results to the mononucleotide allele. The median mutation frequency was 4.7×10-7 in dinB+ cells and 3.3×10-7 in dinB- cells, a result that was again not significant (p=0.2). The second dinucleotide repeat, [GT/CA]10 produced a median mutation frequency of 2.3×10-7 in cells containing dinB and 3.4×10-7 in cells that were deficient of dinB. These median mutation frequencies produced an opposite effect of those observed with the control vector or other microsatellite alleles, in that the presence of Pol IV decreased the mutation frequency 1.5-fold at the HSV-tk target compared to cells that lack the polymerase. This opposing effect was not found to be statistically significant (p=0.46). Overall, these results indicate that the absence of Pol IV does not affect the mutation frequency observed for microsatellite containing vectors.
Table 1.
Summary of the Effect of dinB on Spontaneous Mutagenesis
| Median HSV-tk Mutation Frequency ×10-8 | ||||
|---|---|---|---|---|
| STR Allele | dinB+ (FCT21)a | dinB- (FTK44)a | Ratiob | p-valuec |
| None | 6.8 | 2.0 | 3.5 | 0.0012 |
| [G/C]10 | 69 | 46 | 1.5 | 0.17 |
| [C/G]10 | 71 | 76 | 0.93 | 0.84 |
| [GT/CA]10 | 23 | 34 | 0.68 | 0.46 |
| [TC/AG]11 | 47 | 33 | 1.4 | 0.20 |
FCT21 is a derivative of SMR4562
FTK44 is a derivative of SMR5830
dinB+/dinB-
Wilcoxon Mann Whitney Test for 10-62 independent cultures of each strain and vector combination
Figure 2. Effect of Pol IV on mutation frequencies of microsatellite-containing vectors.

Mutation frequency data were analyzed by box plot [39] for each vector and cell type combination. The gray boxes represent the dinB+ (FCT21) values and the white speckled boxes represent dinB- (FTK44) values. Each box represents the 95% confidence interval for one allele and genetic background calculated from the number of independent cultures shown in parentheses. Horizontal bars represent median mutation frequencies. Verticle bars represent the range of values included in the statistic. Outliers exist for each individual vector and cell type combination, but were removed from the graph for simplicity purposes ([G/C]10 dinB+: 20, 26; [G/C]10 dinB-: 16; [C/G]10 dinB+: 19.4; [GT/CA]10 dinB+: 9.1, 9.4; [GT/CA]10 dinB-: 8.5; [TC/AG]11 dinB+: 13, 14, 16, 17, 22, 147; [TC/AG]11 dinB-: 18, 27, 46). Non-parametric statistical analysis showed no statistical difference between strains in median mutation frequencies for any of the repeat alleles tested.
3.3. Mutational Specificity of the [G/C]10 Microsatellite Allele
The results of the mutation frequency analysis indicated that the presence of dinB had no significant effect on mutagenesis of various vectors containing microsatellite alleles. This said, there is still a possibility that a mutational bias exists in the types of mutations that occur in cells that contain or lack the polymerase. To determine if a mutational bias does exist for mutations made by Pol IV, mutational spectra analyses were completed. As explained in the methods section, total plasmid DNA was isolated from five overnight cultures. Each population was sampled for mutations by re-introducing this DNA into tdk-,recA- E. coli strain FT334 under conditions of one molecule per cell. This HSV-tk mutational system detects mutation events that occur both at the microsatellite allele and also throughout the entire coding and promoter region of the HSV-tk gene [10]. The data in Table 2 summarizes the mutational events that occurred in plasmids recovered from both dinB+ and dinB- cells. In the dinB+ spectrum, 61% of the mutational events occurred at the [G/C]10 allele; similarly, 72% occurred at the [G/C]10 allele in dinB- cells, a difference that is not statistically significant (p=0.11, Fishers Exact Test). These populations correspond to a median mutation frequency at the [G/C]10 allele of 3.8×10-7 for the five dinB+ cultures, compared to 1.7 ×10-7 for the five dinB- cultures. The mutational events at the microsatellite allele were expansions and deletions of 1 unit, with a bias toward expansions in both strains (77%, dinB+; 65%, dinB-) (Figure 3). No statistical difference was observed between the number of expansions and deletions at the microsatellite in the presence or absence of dinB (p=0.16). Overall, our results indicate that Pol IV does not play a significant role in spontaneous mutagenesis at the [G/C]10 allele.
Table 2.
Mutational Events Within the [G/C]10 Vector in dinB+ and dinB- Strains
| Mutation Class | Number of Events (Proportion of Region) | |
|---|---|---|
| dinB+ (FCT21) | dinB- (FTK44) | |
| STR [G/C]10 | ||
| Median Mutation Frequencya | 3.8×10-7 | 1.7×10-7 |
| Number | 61 | 55 |
| Expansion | 47 (.77) | 36 (.65) |
| Deletion | 14 (.23) | 19 (.35) |
| HSV-tk Coding | ||
| Median Mutation Frequencya | 2.4×10-7 | 0.64×10-7 |
| Number | 39 | 21 |
| Base Substitutions | 11 (.28) | 1 (.05) |
| Frameshifts | 8 (.21) | 4 (.19) |
| Complex | 2 (.05) | 2 (.10) |
| Large Insertions or Deletionsb | 18 (.46) | 14 (.67) |
| Total Sequenced Events | 100 | 76 |
Median mutation frequency for 5 independent cultures of each strain
Determined by AvaI/BglII restriction enzyme digest
Figure 3. Range of unit changes at the [G/C]10, [GT/CA]10, and [TC/AG]11 alleles.
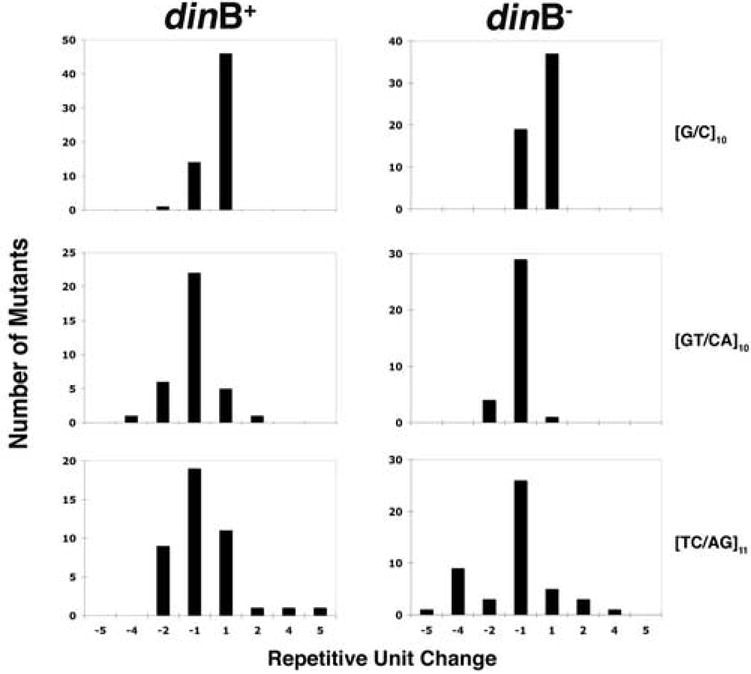
The x-axis represents the range of unit changes observed among the vectors. The y-axis represents the number of mutants containing that mutational event in each spectrum. dinB+ is strain FCT21. dinB- is strain FTK44
The remaining mutational events observed occurred at different locations through the coding region of the HSV-tk gene. As summarized in Table 2, the median HSV-tk coding region mutation frequency was 24×10-8 for the dinB+ populations and 6.4×10-8 for the dinB- populations. This 3.75-fold difference is similar to what was observed for the control plasmid (Table 1). The coding region point mutations for each strain containing the [G/C]10 vector are given in Tables 3 and 4. Overall, there was no large mutational hotspot observed in either cell type. The frameshift events observed were a fairly equal mix of insertions and deletions. In cells containing Pol IV, a similar percentage of frameshifts (21%) and base substitutions (28%) were observed. In the absence of Pol IV, a higher proportion of frameshifts (19%) over base substitutions (5%) was observed. These results correlate with the results from the HSV-tk vector only control, showing that the HSV-tk gene functions as an internal control for the microsatellite-containing vector.
Table 3.
Mutational Spectra of the HSV-tk coding region in dinB+ cells (FCT21)
| Mutation | Locationa | Sequenceb | Mutation | Location | Sequence |
|---|---|---|---|---|---|
| +G/C | -56 | AGTGGGACC | AT→GC | 241 | GGGAAAA |
| 291 | TATCGTC | 671 | TGCTTGG | ||
| 571 | TGCTACC | ||||
| AT→CG | 1042 | GATTACG | |||
| -G/C | 512 - 515 | ATGCCCCGCC | |||
| 605 - 610 | TGACCCCCCAGG | GC→CG | 427 | AAGCGCC | |
| 577c | CCGGCCG | ||||
| +A/T | 255 - 256 | CGCAACTG | 622 | CTGGCGT | |
| 268 - 269 | TGGCCCT | 1072 | GACGCCC | ||
| 785 | TGCCAAT | ||||
| GC→AT | 256 | ACGCAAC | |||
| 656 | CCGGCAC |
Nucleotide position of the mutated base in the HSV-tk gene. Numbering of the HSV-tk sequence begins at the G residue within the BglII recognition site [31].
Wild-type sequence of the sense strand (5’ to 3’ direction). The underlined base(s) indicated the sequence context of the mutation. Insertions occur within or preceding the underlined base(s) and deletions or base substitutions occur at or within the underlined base(s).
3 mutations observed
Table 4.
Mutational Spectra of the coding region of the HSV-tk gene in dinB- cells (FTK44)
| Mutation | Locationa | Sequenceb | Mutation | Location | Sequence |
|---|---|---|---|---|---|
| -G/C | 486 – 493c | ATCGGGGGGGAGG | GC→CG | -16 | TGCCGGG |
| +G/C | -66 | CGGCACC | |||
| -A/T | 241 - 244 | GGGAAAACCA |
Nucleotide position of the mutated base in the HSV-tk gene. Numbering of the HSV-tk sequence begins at the G residue within the BglII recognition site [31].
Wild-type sequence of the sense strand (5’ to 3’ direction). The underlined base(s) indicated the sequence context of the mutation. Insertions occur within or preceding the underlined base(s) and deletions or base substitutions occur at or within the underlined base(s).
2 mutation observed
3.4. Mutational Specificity at [GT/CA]10 and [TC/AG]11 Microsatellite Alleles
Following the mutation frequency analysis of the two dinucleotide vectors, ten mutants from each of five populations per cell type were screened to determine the location of the mutational event, either at the microsatellite allele or in the HSV-tk coding region (Table 5). For the [GT/CA]10 allele, 80% of the mutation events were at the microsatellite in dinB+ cells, compared to 71% in the dinB- cells. Statistical analysis showed no significant difference in the proportion of mutations at the [GT/CA]10 allele versus the HSV-tk coding region (p=0.45, Fisher’s Exact Test). The majority of mutational events for this vector were 1-unit deletions (meaning loss of a [GT/CA] unit) in both cell types (Figure 3), and the types of mutational events at the repeat allele were not statistically significant between strains (p=0.11). For the [TC/AG]11 allele, a similar trend to that of the [GT/CA]10 allele was observed. We detected 88% of the mutational events to be at the microsatellite in dinB+ cells and 98% in dinB- cells (Table 5). Statistical analysis of the proportion of mutational events at the microsatellite allele versus the HSV-tk coding region was not statistically significant (p=0.06). For this microsatellite allele the majority of mutational events in both strains were alterations of 1 unit (Figure 3), similar to what was observed with the [GT/CA]10 dinucleotide vector.
Table 5.
Mutational Analyses of [GT/CA]10 and [TC/AG]11 Vectors
| Mutation Class | Number of Events (Proportion of Region) | |
|---|---|---|
| dinB+ (FCT21) | dinB- (FTK44) | |
| [GT/CA]10 | ||
| Median Mutation Frequency | 2.3 ×10-7 | 3.4 ×10-7 |
| Events in Coding | 9 | 14 |
| Events at STR | 35 | 34 |
| Expansion at STR | 6 (0.17) | 1 (0.03) |
| Deletion at STR | 29 (0.83) | 33 (0.97) |
| [TC/AG]11 | ||
| Median Mutation Frequency | 4.7 ×10-7 | 3.3 ×10-7 |
| Events in Coding | 6 | 1 |
| Events at STR | 42 | 48 |
| Expansion at STR | 14 (0.33) | 9 (0.20) |
| Deletion at STR | 28 (0.67) | 39 (0.80) |
3.5. Microsatellite Length and Sequence Specificity
Given the variability in the location of mutational events between the different microsatellite vectors, further analyses of the data were performed comparing all three microsatellite repeat alleles. Chi Square statistical analyses were used to compare the three STR-containing vectors within a given strain to determine if the observed mutational spectra were affected by either allele unit length or allele sequence composition. The first analysis compared the distribution of mutations within the microsatellite allele versus the HSV-tk coding region for the three STR containing vectors, in either the dinB+ or dinB- strain. The results were statistically significant irrespective of cell type (p=0.0016, dinB+; p=0.0007, dinB-, Chi Square). The next analysis examined the type of mutational events that occurred within each microsatellite region. For each strain, the mutational specificity among the alleles differed in a statistically significant manner (p<0.001 both backgrounds, Chi Square). As mentioned previously, the [G/C]10 mononucleotide allele showed a large proportion of expansion events occurring at the repeat region (Table 2 and Figure 3). This result is opposite to what was observed in the [GT/CA]10 and [TC/AG]11 dinucleotide alleles (Table 5).
The final test was to determine of there was a difference in the unit change of the mutational event at the repeat allele (gain or loss of 1-5 repeat units). The data in this case showed a hierarchy for the number of different unit changes observed, in the order of [TC/AG]11 with the largest range, then [GT/AC]10, and finally [G/C]10 (Figure 3). For the [G/C]10 allele, we detected almost 100% of the events to be insertions or deletions of only 1 unit. This however was not the case for the two dinucleotide alleles. Both the [GT/CA]10 and [TC/AG]11 vectors had events other than 1-unit changes, with the limits of the range extending as far as gain or loss of 5 units. The results of statistical analysis were again significant (p<0.001, Chi Square) regardless of the background of the cells. Overall, these results indicate that both the composition of the repeat allele and the unit length of the repeat play a role in microsatellite mutagenesis.
4. Discussion
E. coli polymerase IV is a distributive polymerase that lacks a 3’to 5’ exonuclease (proofreading) activity and is prone to elongation of misaligned primer/template structures [20]. The overespression of the polymerase causes -1 frameshifts in polypurine runs of G in undamaged DNA [22]. When looking at spontaneous mutagenesis, previous studies have shown a statistically significant decrease in the number of frameshift and base substitution events in the absence of dinB [23,24,32] while overexpression of the dinB gene results in a significant increase in the mutation frequency [21,22]. Pol IV was chosen as a candidate to analyze for strand slippage within microsatellite alleles because a role for the polymerase in spontaneous mutagenesis was acknowledged, as was its role in mutagenesis at homoploymeric G sequences. In this study, we examined the effect of dinB on spontaneous mutagenesis at the HSV-tk gene and three different microsatellite alleles. The mutation frequency for the HSV-tk only control vector was 3.5-fold lower in the absence of dinB. Although we observed a significant decrease in mutation frequency with the loss of dinB at the HSV-tk coding sequence, we did not observe a significant effect with the loss of dinB on the mutation frequency of any of the three artificial microsatellite alleles examined.
4.1. Pol IV and HSV-tk Mutagenesis
The effect of Pol IV on spontaneous mutagenesis is complex, and dependent on at least two factors: the protein level of Pol IV and the location of the mutational target. A higher expression of Pol IV is detected from the F factor compared to expression from the chromosomal copy of the dinB gene [24]. Deletion of dinB from the chromosomal location has a negligible effect on mutagenesis [24]; however deletion from the episome produced a significant effect on mutagenesis [24,32]. Pol IV protein levels also are dependent on the growth phase of the culture. Levels of Pol IV protein are transcriptionally induced in late stationary phase [33], and Pol IV does not contribute to spontaneous mutations during exponential growth when expressed at endogenous levels [34].
A second factor that influences the involvement of Pol IV in spontaneous mutagenesis is the location of the mutational target. There are many reports in the literature in which both chromosomally and episomally located targets were used to look at a role for Pol IV in mutagenesis. Many of these studies were performed using a chromosomal target and resulted in no difference in the mutation frequency in the presence of dinB [32,34,35], while a few did observe a decrease in mutagenesis in the absence of dinB [21,23]. One of these studies also examined an episomal mutational target and found that it was more susceptible to mutagenesis than any of the chromosomal targets tested [32]. A study completed previous to this had established that an episomal target was very highly prone to mutagenesis in the presence of dinB [21].
In this study we have used a dinB+ E. coli strain carrying the F factor containing dinB, as well as the chromosomal copy. The dinB- strain carries a nonfunctional dinB10 allele in both a chromosomal and episomal location. Both cultures were sampled during early stationary phase of growth. We have used the HSV-tk gene target, which is episomally located, to show a 3.5-fold decrease in spontaneous mutation frequency in the absence of Pol IV (Table 1). Mutational analyses at the coding region of the HSV-tk gene for the [G/C]10 vector produced results that were very similar to the HSV-tk gene only vector. An approximate 4-fold difference in the spontaneous mutation frequency at the HSV-tk coding region was detected for dinB+ vs. dinB- cells (Table 2). Overall, our results are consistent with a role for pol IV in mutagenesis of an episomal target.
4.2. Pol IV and Microsatellite Mutagenesis
The mutational analyses of the [G/C]10 mononucleotide region showed no statistically significant differences in mutation frequency of the dinB+ and dinB- strains (Table 2 and Figure 2). This indicates that Pol IV does not play a role in the spontaneous mutagenesis at the [G/C]10 repeat allele. Further analyses showed no strain difference in the types of mutational events at the repeat allele. This result was surprising to us, as Pol IV was shown to create -1 frameshifts in homopolymeric G6 runs [22]. The difference between our study and the previous study [22] could be due to sequence context effects (lac gene vs. HSV-tk gene) or the length of the repetitive target (6 vs. 10 nucleotides).
Recently, Kuban et al. reported that dinB was found to make more errors on the lagging strand during synthesis than on the leading strand [36]. In our [G/C]10 vector, the mononucleotide G repeat is located on the leading strand of synthesis. To test whether replication strand bias may have been one of the reasons that we did not observe a significant difference in the mutation frequency at the mononucleotide allele, we constructed the opposite allele [C/G]10, in which the G repeat is located on the lagging strand. The results of mutation frequency analyses of this vector showed no statistically significant difference with dinB status, just as was observed in the [G/C]10 vector (Table 1 and Figure 2).
The analyses of the spontaneous mutation frequencies of the two dinucleotide repeat vectors also did not emulate that of the HSV-tk only control. In the [TC/AG]11 vector, we observed a similar trend to the control; however the observed decrease in mutation frequency was not significant. In the second dinucleotide vector, [GT/CA]10, we observed a non-significant increase in the mutation frequency in the absence of dinB. The reason for this opposite effect on mutation frequency with the [GT/CA]11 vector may be sequence composition. Overall, these results indicate that Pol IV does not play a role in the spontaneous mutagenesis of the dinucleotide regions, as was observed for the mononucleotide repeat allele.
One possible mechanism to explain a role of Pol IV in coding region but not microsatellite region mutagenesis is MMR efficiency. We have shown in this study that each of the microsatellite alleles is highly subjected to MMR. Errors made by Pol IV at the cII gene of lambda phage were shown to be correctable by MMR [21]. Therefore, it is possible that mutational intermediates generated by Pol IV at the microsatellite versus the coding region are differentially repaired during DNA replication. In this model, MMR more effectively repairs the polymerase errors made in the microsatellite alleles than those that are made in non-repetitive tracts (in our assay, the errors made in the HSV-tk coding region). This model is consistent with the trend we observed for lower microsatellite mutation frequencies in the absence of dinB that did not reach statistical significance (Tables 2 and 5). Our data also showed a larger number of insertion events in the mononucleotide [G/C]10 allele (Table 2). This result is consistent with both this proposed model and previous literature that showed MMR removed -1 frameshift intermediates more efficiently than +1 frameshift intermediates in a 10bp mononucleotide G run [37].
Alternatively, a second model would be that the mutations in the microsatellite alleles arise primarily from mitotic recombination, implying that polymerase slippage is not playing a role in mutagenesis at the repeat tracts. In this model, the errors produced within the HSV-tk coding region would arise due to polymerase events, but the errors at the microsatellite alleles are produced by recombination events. This model would be consistent with the specificity results we obtained for the range of changes at the microsatellite allele for the dinucleotide vectors (Figure 3). The primary mutational event at a repetitive allele is gain or loss of 1 unit repeat. For the dinucleotide vectors we observed a large range, up to gain or loss of 5 units. These larger unit changes could be the result of a recombination event.
A final model to explain our observations is that Pol IV is not utilized during DNA synthesis within the microsatellite region, but is engaged in DNA synthesis within the coding region. Further experimentation will be required to differentiate among the models.
There are many reports in the literature showing that the sequence composition and length of the repeat allele plays a large role in the mutagenesis of microsatellites [9,11,37,38]. We compared various aspects of the mutational differences detected among the three microsatellite alleles. Chi Square analyses supported a significant effect of the allele sequence on the distribution, type, and range of mutational events observed. However, the background of the cells, proficient or deficient in dinB, does not effect mutagenesis. Each repeat is unique in composition and may be handled differently by the gauntlet of polymerases present in the cell. In the case of this study, we conclude that dinB does not affect the mutation frequency or specificity at the microsatellites examined. We also confirmed the differential mutagenesis of microsatellites that is dependent upon the composition of the repeat allele.
Acknowledgments
This research was supported by Public Health Service Grant R01CA10006 and generous donations to the Jake Gittlen Cancer Research Foundation.
We thank Guang Yan for her technical assistance, Suzanne Hile for critical evaluation of the data and manuscript, and Dr. Susan Rosenberg for the FC40 E. coli strains.
Abbreviations
- Cm
Chloramphenicol
- E. coli
Escherichia Coli
- FudR
5-Fluoro-2’deoxyuridine
- HSV-tk
Herpes Simplex Virus thymidine kinase
- MMR
Mismatch repair
- Pol
Polymerase
- Rif
Rifampicin
- STR
Short Tandem Repeat (microsatellite)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beckman JS, Weber JL. Survey of human and rat microsatellites. Genomics. 1992;12:627–631. doi: 10.1016/0888-7543(92)90285-z. [DOI] [PubMed] [Google Scholar]
- 2.Debrauwere H, Gendrel CG, Lechat S, Dutreix M. Differences and similarities between various tandem repeat sequences: minisatellites and microsatellites. Biochimie. 1997;79:577–586. doi: 10.1016/s0300-9084(97)82006-8. [DOI] [PubMed] [Google Scholar]
- 3.Tautz D, Renz M. Simple sequences are ubiquitous repetitive components of eukaryotic genomes. Nucleic Acids Res. 1984;12:4127–4138. doi: 10.1093/nar/12.10.4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vogt P. Potential genetic functions of tandem repeated DNA sequence blocks in the human genome are based on a highly conserved “chromatin folding code”. Hum Genet. 1990;84:301–336. doi: 10.1007/BF00196228. [DOI] [PubMed] [Google Scholar]
- 5.Stallings RL. Conservation and evolution of (CT)n/(GA)n microsatellite sequences at orthologous positions in diverse mammalian genomes. Genomics. 1995;25:107–113. doi: 10.1016/0888-7543(95)80115-3. [DOI] [PubMed] [Google Scholar]
- 6.Stallings RL, Ford AF, Nelson D, Torney DC, Hildebrand CE, Moyzis RK. Evolution and distribution of (GT)n repetitive sequences in mammalian genomes. Genomics. 1991;10:807–815. doi: 10.1016/0888-7543(91)90467-s. [DOI] [PubMed] [Google Scholar]
- 7.Katti MV, Ranjekar PK, Gupta VS. Differential distribution of simple sequence repeats in eukaryotic genome sequences. Mol Biol Evol. 2001;18:1161–1167. doi: 10.1093/oxfordjournals.molbev.a003903. [DOI] [PubMed] [Google Scholar]
- 8.Toth G, Gaspari Z, Jurka J. Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res. 2000;10:967–981. doi: 10.1101/gr.10.7.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wierdl M, Dominska M, Petes TD. Microsatellite instability in yeast: dependence on the length of the microsatellite. Genetics. 1997;146:769–779. doi: 10.1093/genetics/146.3.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eckert KA, Yan G, Hile SE. Mutation rate and specificity analysis of tetranucleotide microsatellite DNA alleles in somatic human cells. Mol Carcinog. 2002;34:140–150. doi: 10.1002/mc.10058. [DOI] [PubMed] [Google Scholar]
- 11.Sia EA, Kokoska RJ, Dominska M, Greenwell P, Petes TD. Microsatellite instability in yeast: dependence on repeat unit size and DNA mismatch repair genes. Mol Cell Biol. 1997;17:2851–2858. doi: 10.1128/mcb.17.5.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tran HT, Keen JD, Kricker M, Resnick MA, Gordenin DA. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol Cell Biol. 1997;17:2859–2865. doi: 10.1128/mcb.17.5.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eckert K, Yan G. Mutational analysis of dinucleotide and tetranucleotide microsatellite in Escherichia coli: influence of sequence on expansion mutagenesis. Nucleic Acids Research. 2000;28:2831–2838. doi: 10.1093/nar/28.14.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Streisinger G, Okada Y, Emrich J, Newton J, Tsugita A, Terzaghi E, Inouye M. Frameshift mutations and the genetic code. This paper is dedicated to Professor Theodosius Dobzhansky on the occasion of his 66th birthday. Cold Spring Harb Symp Quant Biol. 1966;31:77–84. doi: 10.1101/sqb.1966.031.01.014. [DOI] [PubMed] [Google Scholar]
- 15.Streisinger G, Owen J. Mechanisms of spontaneous and induced frameshift mutation in bacteriophage T4. Genetics. 1985;109:633–659. doi: 10.1093/genetics/109.4.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levinson G, Gutman GA. Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol Biol Evol. 1987;4:203–221. doi: 10.1093/oxfordjournals.molbev.a040442. [DOI] [PubMed] [Google Scholar]
- 17.Kroutil LC, Register K, Bebenek K, Kunkel TA. Exonucleolytic proofreading during replication of repetitive DNA. Biochemistry. 1996;35:1046–1053. doi: 10.1021/bi952178h. [DOI] [PubMed] [Google Scholar]
- 18.Sagher D, Hsu A, Strauss B. Stabilization of the intermediate in frameshift mutation. Mutat Res. 1999;423:73–77. doi: 10.1016/s0027-5107(98)00227-9. [DOI] [PubMed] [Google Scholar]
- 19.Strand M, Prolla TA, Liskay RM, Petes TD. Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature. 1993;365:274–276. doi: 10.1038/365274a0. [DOI] [PubMed] [Google Scholar]
- 20.Wagner J, Gruz P, Kim S, Yamada M, Matsui K, Fuchs R, Nohmi T. The dinB gene encodes a novel E. coli DNA polymerase, DNA Pol IV, involved in mutagenesis. Molecular Cell. 1999;4:281–286. doi: 10.1016/s1097-2765(00)80376-7. [DOI] [PubMed] [Google Scholar]
- 21.Wagner J, Nohmi T. Escherichia coli DNA polymerase IV mutator activity: genetic requirements and mutational specificity. Journal of Bacteriology. 2000;182:4587–4595. doi: 10.1128/jb.182.16.4587-4595.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim S, Maenhaut-Michel G, Yamada M, Yamamoto Y, Matsui K, Sofuni T, Nohmi T, Ohmori H. Multiple pathways for SOS-induced mutagenesis in Escherichia coli: an overexperssion of dinB/dinP results in strongly enhancing mutagenesis in the absence of any exogenous treatment to damage DNA. Proc Nat Acad Sci USA. 1997;94:13792–13797. doi: 10.1073/pnas.94.25.13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strauss B, Roberts R, Francis L, Pouryazdanparast P. Role of the dinB gene product in spontaneous mutation in Escherichia coli with an impared replicative polymerase. Journal of Bacteriology. 2000;182:6742–6750. doi: 10.1128/jb.182.23.6742-6750.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim SR, Matsui K, Yamada M, Gruz P, Nohmi T. Roles of chromosomal and episomal dinB genes encoding DNA pol IV targeted and untargeted mutagenesis in Escherichia coli. Mol Genet Genomics. 2001;266:207–215. doi: 10.1007/s004380100541. [DOI] [PubMed] [Google Scholar]
- 25.Eckert KA, Drinkwater NR. recA-dependent and recA-independent N-ethyl-N-nitrosourea mutagenesis at a plasmid-encoded herpes simplex virus thymidine kinase gene in Escherichia coli. Mutat Res. 1987;178:1–10. doi: 10.1016/0027-5107(87)90079-0. [DOI] [PubMed] [Google Scholar]
- 26.McKenzie GJ, Harris RS, Lee PL, Rosenberg SM. The SOS response regulates adaptive mutation. Proc Natl Acad Sci U S A. 2000;97:6646–6651. doi: 10.1073/pnas.120161797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKenzie GJ, Lee PL, Lombardo MJ, Hastings PJ, Rosenberg SM. SOS mutator DNA polymerase IV functions in adaptive mutation and not adaptive amplification. Mol Cell. 2001;7:571–579. doi: 10.1016/s1097-2765(01)00204-0. [DOI] [PubMed] [Google Scholar]
- 28.Eckert KA, Ingle CA, Klinedinst DK, Drinkwater NR. Molecular analysis of mutations induced in human cells by N-ethyl-N-nitrosourea. Mol Carcinog. 1988;1:50–56. doi: 10.1002/mc.2940010111. [DOI] [PubMed] [Google Scholar]
- 29.Summers WC, Raksin P. A method for selection of mutations at the tdk locus in Escherichia coli. J Bacteriol. 1993;175:6049–6051. doi: 10.1128/jb.175.18.6049-6051.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hile S, Yan G, Eckert K. Somatic mutation rates and specificities at TC/AG and GT/CA microsatellite sequences in nontumorigenic human lymphoblastoid cells. Cancer Research. 2000;60:1698–1703. [PubMed] [Google Scholar]
- 31.Eckert K, Hile S, Vargo P. Development and use of an in vitro HSV-tk forward mutation assay to study eukaryotic DNA polymerase processing of DNA alkyl lesions. Nucleic Acids Research. 1997;25:1450–1457. doi: 10.1093/nar/25.7.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuban W, Jonczyk P, Gawel D, Malanowska K, Schaaper RM, Fijalkowska IJ. Role of Escherichia coli DNA polymerase IV in in vivo replication fidelity. J Bacteriol. 2004;186:4802–4807. doi: 10.1128/JB.186.14.4802-4807.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Layton JC, Foster PL. Error-prone DNA polymerase IV is controlled by the stress-response sigma factor, RpoS, in Escherichia coli. Mol Microbiol. 2003;50:549–561. doi: 10.1046/j.1365-2958.2003.03704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wolff E, Kim M, Hu K, Yang H, Miller JH. Polymerases leave fingerprints: analysis of the mutational spectrum in Escherichia coli rpoB to assess the role of polymerase IV in spontaneous mutation. J Bacteriol. 2004;186:2900–2905. doi: 10.1128/JB.186.9.2900-2905.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKenzie GJ, Magner DB, Lee PL, Rosenberg SM. The dinB operon and spontaneous mutation in Escherichia coli. J Bacteriol. 2003;185:3972–3977. doi: 10.1128/JB.185.13.3972-3977.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuban W, Banach-Orlowska M, Bialoskorska M, Lipowska A, Schaaper RM, Jonczyk P, Fijalkowska IJ. Mutator phenotype resulting from DNA polymerase IV overproduction in Escherichia coli: preferential mutagenesis on the lagging strand. J Bacteriol. 2005;187:6862–6866. doi: 10.1128/JB.187.19.6862-6866.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gragg H, Harfe BD, Jinks-Robertson S. Base composition of mononucleotide runs affects DNA polymerase slippage and removal of frameshift intermediates by mismatch repair in Saccharomyces cerevisiae. Mol Cell Biol. 2002;22:8756–8762. doi: 10.1128/MCB.22.24.8756-8762.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morel P, Reverdy C, Michel B, Ehrlich SD, Cassuto E. The role of SOS and flap processing in microsatellite instability in Escherichia coli. Proc Natl Acad Sci U S A. 1998;95:10003–10008. doi: 10.1073/pnas.95.17.10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samuels ML, Witmer JA. Statistics for the Life Sciences. Prentice Hall; Upper Saddle River: 2003. [Google Scholar]