

Abstract
Organellar acidification by the electrogenic vacuolar proton-ATPase is coupled to anion uptake and cation efflux to preserve electroneutrality. The defective organellar pH regulation, caused by impaired counterion conductance of the mutant cystic fibrosis transmembrane conductance regulator (CFTR), remains highly controversial in epithelia and macrophages. Restricting the pH-sensitive probe to CFTR-containing vesicles, the counterion and proton permeability, and the luminal pH of endosomes were measured in various cells, including genetically matched CF and non-CF human respiratory epithelia, as well as cftr+/+ and cftr−/− mouse alveolar macrophages. Passive proton and relative counterion permeabilities, determinants of endosomal, lysosomal, and phagosomal pH-regulation, were probed with FITC-conjugated transferrin, dextran, and Pseudomonas aeruginosa, respectively. Although CFTR function could be documented in recycling endosomes and immature phagosomes, neither channel activation nor inhibition influenced the pH in any of these organelles. CFTR heterologous overexpression also failed to alter endocytic organellar pH. We propose that the relatively large CFTR-independent counterion and small passive proton permeability ensure efficient shunting of the proton-ATPase–generated membrane potential. These results have implications in the regulation of organelle acidification in general and demonstrate that perturbations of the endolysosomal organelles pH homeostasis cannot be linked to the etiology of the CF lung disease.
INTRODUCTION
Dynamic networks of secretory and endocytic organelles are responsible for the biosynthesis, sorting, and delivery of protein and lipid cargo, as well as the cellular uptake of nutrients and a variety of pathogens (Mukherjee et al., 1997; Wu et al., 2001). The physiological functions of the secretory and endocytic pathway require progressively decreasing luminal pH from the endoplasmic reticulum (ER) to secretory vesicles and from endocytic vesicles to lysosomes, respectively (Mellman et al., 1986; Mukherjee et al., 1997; Weisz, 2003a). Acidification of the Golgi complex, for example, is necessary to ensure the pH optimum for sialylation, sulfation, fucosylation of polypeptides and ligand dissociation from the KDEL and mannose-6 phosphate receptor family (Kim et al., 1998; Weisz, 2003a; Olson et al., 2008). Endolysosomal acidification is essential for ligand-receptor sorting, activation of hydrolytic enzymes, as well as homo- and heterotypic membrane fusion and fission to maintain endocytosis and recycling (Mellman et al., 1986; Mukherjee et al., 1997). Phagolysosomal acidification is a prerequisite for the efficient destruction of ingested bacteria and presentation of antigens as part of the innate and acquired immune response (Steinberg et al., 2007a).
Regulation of organellar acidification is dictated by the interplay of multiple ion transport systems, second messengers and cytoplasmic factors. One of the most important players is the V-type adenosine triphosphatase (v-ATPase) proton pump, a multisubunit enzyme complex assembling from the V0 and V1 subunit (Jefferies et al., 2008; Marshansky and Futai, 2008). The v-ATPase utilizes the energy of ATP hydrolysis to build up the organellar electrochemical proton gradient. Because the v-ATPase is an electrogenic pump, macroscopic proton translocation is accompanied by anion uptake or cation efflux to preserve electroneutrality (Marshansky and Futai, 2008). Severe reduction in the counterion permeability leads to the acidification defect by the development of large, positive inside membrane potential, imposing energetic barrier on proton uptake. Based on these considerations, counterion conductance was proposed to play a determinant role in organellar acidification (Mellman et al., 1986; Jentsch, 2007) and was supported by the acidification defect of cells deficient of members of the intracellular ClC chloride channel family (Hara-Chikuma et al., 2005a; Hara-Chikuma et al., 2005b; Scheel et al., 2005; Jentsch, 2007; Graves et al., 2008). Aside the counterion conductance, accumulating evidence indicate that both the passive proton leak (Chandy et al., 2001; Wu et al., 2001) and direct regulation of the v-ATPase (Marshansky and Futai, 2008) contribute to the development of organelle-specific pH.
Cystic fibrosis (CF), one of the most prevalent genetic diseases of the Caucasian population, is caused by the functional defect of the cystic fibrosis transmembrane conductance regulator (CFTR). CFTR, a member of the ABC transporter family, is a protein kinase A (PKA)-regulated chloride channel and regulator of numerous epithelial transporters (Riordan et al., 1989; Riordan, 2008). The multifaceted clinical manifestations of CF are, in part, attributed to the impaired transepithelial salt and water movement, culminating in recurrent bacterial infection, colonization, and hyperinflammation of the lung, representing the major cause of morbidity in CF (Boucher, 2007). The mechanism of the CF lung disease, though not completely elucidated, can be attributed to the combination of defective airway surface liquid hydration and mucocilliary clearance, excessive mucus secretion, and Na+ resorption (Boucher, 2007).
Based on the notion that counterion permeability is one of the determinants of organellar acidification (Forgac, 1998), the CFTR chloride channel was endowed with the capacity to regulate the acidification of the trans-Golgi, trans-Golgi network (TGN), endosome, phagosome, and lysosome in selected epithelia and alveolar macrophages (Barasch et al., 1991; Poschet et al., 2001, 2002; Di et al., 2006; Teichgraber et al., 2008). This paradigm offered a possible explanation for the metabolic and cellular pathology of CF respiratory epithelia as well as alveolar macrophages. Defective acidification of the TGN was proposed to result in undersialylation of glycoconjugates and to promote adhesion and colonization of Pseudomonas aeruginosa and Staphylococcus aureus to respiratory epithelia (Barasch et al., 1991; Poschet et al., 2001; Campodonico et al., 2008). Likewise, impaired acidification of the endolysosomes was invoked in membrane trafficking defect (Barasch et al., 1991), the hyperinflammatory state of respiratory epithelia (Teichgraber et al., 2008) and compromised destruction of bacteria in alveolar macrophages (Di et al., 2006). Intriguingly, hyperacidification of the TGN and endosomes was also reported in CF respiratory epithelia and explained by consequence of the hyperactive epithelial sodium channel (ENaC; Poschet et al., 2001, 2006). Finally, using CFTR overexpression systems and loss-of-function studies, other investigators reported that neither the secretory (Golgi and TGN) nor the endocytic organelles (endosomes, lysosomes, and phagosomes) display a CFTR-dependent acidification defect (Lukacs et al., 1992; Biwersi and Verkman, 1994; Dunn et al., 1994; Root et al., 1994; Seksek et al., 1996; Machen et al., 2001; Haggie and Verkman, 2007, 2009b; Lamothe and Valvano, 2008). Given these discrepancies, improved methodologies and better understanding of the biophysical basis of organellar acidification are required to rationalize the possible contribution of CFTR activity to the pH regulation of intracellular organelles. These considerations led us to determine the intrinsic, passive proton permeability and CFTR-independent and -dependent relative counterion permeability of endosomes, lysosomes, and phagosomes in several cellular models, including genetically matched CF and non-CF respiratory epithelia and alveolar macrophages.
Using single-cell fluorescence ratio image analysis (FRIA), we show that the CFTR-independent and overall counterion permeability was remarkably higher than the passive proton permeability of endosomes, and therefore, CFTR activation cannot interfere with the endosomal pH regulation in CF respiratory epithelia and alveolar macrophages, as well as other heterologous expression systems. A similar phenomenon was discovered in lysosomes and phagolysosomes of respiratory epithelia and primary or immortalized mouse macrophages, respectively, with the caveat that recycling compromised the channel accumulation in both lysosomes and phagosomes as well. Our results have implications for the pH regulation of endocytic organelles and document that other factor than the acidification defect of endocytic organelles accounts for the hyperinflammatory CF lung phenotype.
MATERIALS AND METHODS
DNA Constructs and Chemicals
The extracellular triple hemagglutinin (3HA) tag, consisting of amino acid residues of SLEYPYDVPDY-ASYPYDVPDYAYPYDVPD, was inserted into the fourth extracellular loop after residue 897 in the G551D CFTR, as described for the wild-type (wt) and ΔF508 CFTR (Sharma et al., 2004; Pedemonte et al., 2005). The G551D CFTR cDNA was kindly provided by Dr. J. Rommens (Hospital for Sick Children, Toronto). All chemicals of the highest available purity were obtained from Sigma-Aldrich (Oakville, ON, Canada) except when indicated. Bafilomycin A1 (Baf) was from LC Laboratories (Woburn, MA).
Cells
HeLa, Madin-Darby canine kidney (MDCK) and RAW264.7 cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) in a thermostated cell culture incubator in 5% CO2 at 37°C. Baby hamster kidney (BHK) cells were grown in DMEM/F12 containing 5% FBS. IB3 cells (kindly provided by Dr. P. Zeitlin, Johns Hopkins University) were grown in LHC-8 basal medium (Invitrogen, Carlsbad, CA) containing 5% FBS. The IB3 cells have ΔF508/W1282X genotype (Zeitlin et al., 1991). The human bronchial epithelial cell line derived from a CF patient with a ΔF508/ΔF508 genotype (CFBE41o-, designated as CFBE) have been characterized (Cozens et al., 1994) and were cultured in MEM with GlutaMAX (Invitrogen, Carlsbad, CA) supplemented with 10% FBS serum at 37°C in a 5% CO2-humidified incubator as described (Gruenert et al., 1995). To allow differentiation of the MDCK and CFBE, epithelial cells were cultured at confluence for 3–5 d. Primary macrophages were obtained by intraperitoneal and bronchoalveolar lavages as previously described (Guilbault et al., 2008). Macrophages were isolated by centrifugation and resuspended in RPMI 1640 with 10% FBS and seeded on polylysine (Sigma)-coated glass coverslips. Coverslips were coated according to manufacturer's instructions. Experiments were performed 24–48 h after plating the cells.
All cell types used were either transiently or stably transfected with wt or G551D CFTR-3HA harboring triple hemagglutinin tags in the fourth extracellular loop (Sharma et al., 2004). BHK cell expressing the wt and G551D CFTR-3HA was generated as previously described and clones were selected in the presence of 500 μM methotrexate (Sharma et al., 2001).
CFTR variants were stably expressed in the CFBE and HeLa cells using lentiviral vectors by Dr. J. Wakefield (Tranzyme, Birmingham, AL) and selected in the presence of 5 μg/mP puromycin as described (Bebok et al., 2005). IB3 bronchial epithelia, expressing endogenous ΔF508 and W1282X CFTR at undetectable levels, were stably transfected with the pCEP4 expression plasmid encoding the wt or G551D CFTR-3HA. A mixture of clones were selected in hygromycin B. Transfected cells were maintained in LHC-8 containing 5% FBS and 100 μg · ml−1 hygromycin B. RAW cells were transiently transfected with CFTR encoding pNut-CFTR using FuGENE6 as DNA complexing agent (Roche, Basel, Switzerland) according to the manufacturer's recommendation and analyzed after 48 h. MDCK cells were infected with retrovirus encoding the CFTR-3HA variants as described previously (Benharouga et al., 2003).
Animals
Inbred C57BL/6-Cftr+/− heterozygous mice were maintained and bred in the Animal Facility of the McGill University Health Center Research Institute. All pups were genotyped between 12 and 14 d of age. The animals were kept in cages with sterile corn bedding (Anderson, Bestmonro, LA) and maintained in ventilated racks (Lab Products, Seaford, DE). Age-matched C57BL/6-Cftr+/+ mice and C57BL/6-Cftr−/− mice were maintained in murine pathogen–, Helicobacter-, and parasite-free conditions. They were housed (1–4 animals/cage), bred, and maintained in a facility under specific pathogen-free conditions. Mice were fed with either the NIH-31–modified, irradiated mouse diet for wt mice (Harlan Teklad, Indianapolis, IN) or a liquid diet starting at 14 d of age for knockout mice (Peptamen liquid diet; Nestlé Canada, Brampton, ON, Canada). The liquid diet was freshly prepared each morning and provided in 50-ml centrifuge tubes (Fisher Scientific, Nepean, ON, Canada). Mice used for experiments were between 19 and 22 wk of age. Experimental procedures with mice were conducted in accordance with the Canadian Council on Animal Care guidelines and with the approval of the Facility Animal Care Committee of the Montreal General Hospital Research Institute (Montreal, PQ, Canada).
Labeling of Endocytic Organelles with pH-sensitive Dyes
The luminal pH of early endosomes, recycling endosomes, lysosomes, and phagosomes was routinely determined after the selective labeling of the respective compartment with fluorescein isothiocyanate transferrin (FITC)-conjugated cargo (e.g., CFTR, transferrin, dextran, and P. aeruginosa PAO1) by FRIA as described for CFTR and other cargo molecules (Sharma et al., 2004; Barriere et al., 2007; Kumar et al., 2007; Barriere and Lukacs, 2008; Duarri et al., 2008; Varghese et al., 2008; Glozman et al., 2009).
To label internalized CFTR, cells were incubated with anti-HA (1:500 dilution equivalent to 10 μg/ml, MMS101R, Covance Laboratories, Madison, WI) primary Ab and FITC-conjugated goat anti-mouse secondary Fab (1:500 dilution, Jackson ImmunoResearch Laboratories, West Grove, PA) by incubating primary for 1 h at 37°C. Cells were then washed (140 mM NaCl, 5 mM KCl, 20 mM HEPES, 10 mM glucose, 0.1 mM CaCl2, and 1 mM MgCl2, pH 7.3) and chased for indicated time at 37°C. Fluid-phase Ab uptake was not detectable in mock-transfected cells (data not shown). When indicated, cell surface–resident CFTR was labeled on ice by successive incubation with the primary anti-HA Ab and secondary FITC-Fab.
To confirm that the primary and secondary Ab remains bound to CFTR during FRIA experiments, the pH resistance of Ab binding was measured by immunoperoxidase assay. After anti-HA– and HRP-conjugated secondary Ab binding to CFTR-expressing HeLa cells, the extracellular medium pH was adjusted to pH 7.2, 5.0, and 2.5 for 5 min (van Kerkhof et al., 2001; 0.15 M NaCl, 50 mM glycine, 0.1% BSA, 20 mM MES, for pH 2.5 and 5.0, 10 mM HEPES for pH 7.2). The amount of bounded HRP-conjugated Ab was measured by Amplex-Red as substrate, using a POLARstar OPTIMA fluorescence plate-reader (BMG Labtech, Offenburg, Germany; Barriere et al., 2006). The Ab binding was virtually unaltered at pH 5.0, but reduced by 50% at pH 2.5 (Supplemental Figure S1A).
Recycling endosomes were labeled with FITC-transferrin (Tf; 15 μg/ml, 45-min loading after 45 min serum-depletion at 37°C) and chased for 0–3 min. Lysosomal pH was measured with similar results on cells labeled by overnight fluid-phase uptake of FITC-dextran alone or in combination with Oregon Green 488-dextran (50 μg/ml, MW 10 kDa, Molecular Probes, Eugene, OR) and chased for >3 h.
Phagosomal pH was monitored following the uptake of FITC- or FITC/TRITC-conjugated P. aeruginosa (PAO1 strain, kindly provided by Dr. M. Parsek, University of Washington, Seattle). From an overnight culture ∼5 × 108 bacteria were opsonized in 20% FBS-PBS for 30 min at 37°C with agitation. Cells were washed with PBS and labeled in the presence of 1 mg/ml FITC, TRITC, or a combination of both in PBS at pH 8.0 for 30 min at room temperature under rotation. Excess of fluorescent dyes was removed by repeated centrifugation in ice-cold PBS. Bacteria were snap-frozen and stored at −80°C.
Organellar pH Measurement
FRIA of endocytic organelles was performed on an Axiovert 100 inverted fluorescence microscope (Carl Zeiss MicroImaging, Toronto, ON, Canada) at room temperature equipped with a Hamamatsu ORCA-ER 1394 (Hamamatsu, Japan) cooled CCD camera and a Planachromat (63× NA 1.4) objective essentially as described previously (Sharma et al., 2004; Barriere et al., 2007; Barriere and Lukacs, 2008; Glozman et al., 2009). Image acquisition and FRIA were performed with MetaFluor software (Molecular Devices, Downingtown, PA). Images were acquired at 490 ± 5 and 440 ± 10-nm excitation wavelengths, using a 535 ± 25-nm emission filter. To determine the initial rates of acidification of endosomes (see Figure 4E), CFTR was labeled with anti-HA and FITC-conjugated goat anti-mouse Fab sequentially for 1 h on ice and chased at 37°C for the indicated times. The cell surface remaining Abs were acid-stripped before image acquisition (van Kerkhof et al., 2001).
Figure 4.

CFTR complementation of CF respiratory epithelia and CFTR overexpression in HeLa and MDCK cells has no effect on the endosomal pH. (A and B) Measurement of endosomal pH dissipation rates in wt and G551D CFTR-3HA complemented CFBE and IB3 CF respiratory epithelia and overexpressing HeLa and MDCK cells. CFTR labeling was performed as described in Figure 2B. The endosomal pH dissipation rates were determined as described in Figure 2, E and F. (C) Vesicular pH of CFTR-containing vesicle was measured after 3 min of PKA stimulation as in Figure 2E in the indicated cell lines. Data were analyzed by two-tailed unpaired t tests and indicated as follows: * p = 0.0347 for CFTR G551D control versus forskolin in HeLa. (D) Vesicular pH of FITC-Tf containing vesicle before and after 3 min of PKA stimulation with the agonist cocktail. Means ± SEM; n = 3–5. FITC-Tf loading was described in Materials and Methods. (E) Initial acidification rates of endosomes were measured in wt and G551D CFTR-3HA–expressing HeLa and IB3 cells. CFTR labeling was performed on ice as described in Figure 2A and monitored by FRIA after increasing the temperature to 37°C for the indicated time in the presence or absence of PKA activators. Means ± SEM from three independent experiments. The pH of ∼10,000 and 8000 vesicles was measured in studies depicted on the top and bottom panels, respectively. (F) Initial acidification rates were measured after 4 min CFTR internalization as in E in the indicated cell lines. Means ± SEM; n = 3.
In situ calibration curves, describing the relationship between the fluorescence ratio values and endosome, lysosome, or phagosome pH, served to calculate the luminal pH of individual vesicles after fluorescence background subtraction at both excitation wavelengths (e.g., Supplemental Figure S1, B–D). In situ calibration was performed by clamping the vesicular pH between 4.5 and 7.4 in K+-rich medium (135 mM KCl, 10 mM NaCl, 20 mM HEPES, or 20 mM MES, 1 mM MgCl2, and 0.1 mM CaCl2) with 10 μM nigericin, 10 μM monensin, 0.4 μM Baf, and 20 μM carbonyl cyanide 3-chlorophenylhydrazone (CCCP; Sigma-Aldrich) and recording the fluorescence ratios. Calibration curves were obtained for each cargo molecules and repeated at regular intervals. As an internal control, one-point calibration was performed on each coverslip by clamping the organellar pH to 6.5 with 10 μM monensin and nigericin, 0.4 μM Baf, and 20 μM CCCP. In each experiment the pH of 200–800 endosomes/lysosomes and 100–400 phagosomes was determined. Mono- or multipeak Gaussian distributions of vesicular pH values were obtained with Origin 7.0 software (OriginLab, Northampton, MA), and the results of individual experiments were illustrated. The mean pH of each vesicle population was calculated as the arithmetic mean of the data in each individual experiment using 200–800 vesicles from 15 to 60 cells. At least three independent experiments were performed for each condition.
Determination of the Relative Counterion Permeability and Buffer Capacity of Organelles
Rapid dissipation of the organellar pH gradient by the proton pump inhibitor (Baf) and the protonophore (CCCP) was used to determine the relative counterion permeability of the organelles. Cells were labeled as described above and the organellar pH was continuously monitored by FRIA for the indicated time. pH dissipation was measured in the presence of 0.4 μM Baf and 20 μM CCCP. To measure the passive proton leak, only Baf was added. Both Baf and CCCP were used at saturating concentrations (Lukacs et al., 1990, 1991; Hackam et al., 1997; Steinberg et al., 2007b). When indicated, CFTR was activated with the PKA agonist cocktail (20 μM forskolin, 0.5 mM 8-(4-chlorophenyl-thio)adenosine 3′,5′-cyclic monophosphate sodium salt [CPT-cAMP], and 0.2 mM isobutylmethylxanthine [IBMX]) for 3 min before Baf+CCCP addition. FRIA was performed as described above. The pH dissipation rate was calculated from the initial slope of the fluorescence ratio change. The buffer capacity of endocytic organelles was calculated from the extent of rapid alkalinization after the addition of 0.5–2 mM NH4Cl in the presence or absence of Baf. The calculation was based on the formula from Roos and Boron (Roos and Boron, 1981; Sonawane and Verkman, 2003).
The Passive Proton Permeability Determination
The passive proton permeability of recycling endosomes, lysosomes, and phagosomes was calculated according to the following equation:
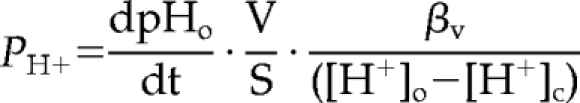 |
where PH+ is the organellar passive proton permeability in cm · s−1, dpHo/dt is the organellar proton flux in pH · s−1, V is the organellar volume in cm3, S is the organellar surface in cm2, βv is the organellar buffer capacity in M · pH−1, and ([H+]o − [H+]c), is the transmembrane proton gradient between the organelle lumen and the cytosol (Chandy et al., 2001; Grabe and Oster, 2001). The passive proton flux was measured by monitoring the organellar alkalinization rate immediately after 400 nM Baf addition as shown in Figure 2D. The buffer capacity was measured by monitoring the rapid alkalinization of organelles after NH4Cl addition in the presence of Baf as detailed in Materials and Methods and in Chandy et al. (2001). The surface and volume of endosomes and lysosomes were obtained from published data (Griffiths et al., 1989). The phagosome volume and surface calculation was based on the assumption that average phagosome diameter is 3 μm and the distance between the bacterial wall and the inner leaflet of the phagosomal membrane is 100 nm, based on EM observations (Hart et al., 1987; Chastellier, 2008). In light of the high counterion conductance of the endocytic organelles and the modest membrane potential of phagolysosomes (<25 mV including the Donnan potential effect; Steinberg et al., 2007b), we assumed that the membrane potential has modest contribution to the electrochemical proton driving force in endocytic organelles. Therefore, the passive proton permeability was calculated by taking into consideration only the chemical proton driving force, and thus it likely represents an overestimate. The cytoplasmic pH was assumed to be constant (pH 7.3) during the proton efflux measurements (Mukherjee et al., 1997).
Figure 2.

Monitoring wt and G551D CFTR-3HA endocytic sorting and pH regulation by FRIA in BHK cells. (A) Determining the sorting pathway of internalized CFTR by vesicular pH (pHv) measurement. Anti-HA antibody and FITC-conjugated secondary Fab was bound to CFTR-expressing BHK cells for 1 h at 0°C. Then the temperature was raised to 37°C for 0–1 h, and the pHv was measured by FRIA. Data are expressed as frequency of pHv and means (± SEM) pHv of the major endosomal population. The number of vesicles analyzed in a single experiment is indicated. (B) Wt CFTR was labeled with anti-HA primary Ab and FITC-conjugated secondary Fab for 1 h at 37°C and was chased for 30 min before FRIA. (C) G551D CFTR was labeled as described in B. (D) Determination of the passive proton and relative counterion permeability of wt CFTR-containing endosomes. Right panel, to unravel the CFTR dependent counterion permeability, protonophere (CCCP) was used to rapidly dissipate the pH gradient in the presence of Baf. H+ egress generated a negative inside membrane potential that was dissipated by Cl− efflux via CFTR and uptake of cations. Left panel, the endosomal pH was monitored as a function of time as described in B. For each experiment, 30–50 vesicles were tracked simultaneously. The vacuolar H+-ATPase was inhibited with 0.4 μM Baf to unmask the passive proton permeability. CCCP (20 μM), a protonophore, induced a rapid dissipation of the endosomal pH gradient, indicating the presence of constitutively active counterion conductance in endosomes. Addition of NH4Cl (20 mM) dissipated the pH gradient. Wt CFTR was labeled as in B. (E) CFTR activation enhances the counterion permeability of wt but not G551D CFTR-expressing endosomes in BHK cells. Endosomes were labeled as described in B and the pH dissipation rate was measured after addition of 0.4 μM Baf and 20 μM CCCP by FRIA. CFTR was activated with the PKA agonist cocktail (20 μM forskolin, 0.2 mM IBMX, and 0.5 mM CPT-cAMP cocktail [FK]) for 2–3 min before the pH dissipation was induced. Traces obtained in the presence of activated PKA are labeled by +FK. (F) Quantification of pH dissipation rate (left panel) and vesicular pH (right panel) after PKA activation. The pH dissipation rate was measured in experiments described in E. (G) No significant variation in late and early endosomal pH of wt CFTR-containing vesicles was detectable. The PKA sensitivity of a selected subpopulation of CFTR-containing vesicles with distinct initial pH was determined as in B. Means ± SEM; n = 3–5.
Immunofluorescence Microscopy
CFTR subcellular distribution was determined after internalization of anti-HA Ab for 1 h in DMEM and chased for 0.5 h. CFTR was visualized by FITC-conjugated goat anti-mouse Fab or TRITC-conjugated goat anti-mouse after cell fixation. During the last 45 min, cells were labeled with FITC-Tf or TRITC-Tf (15 μg/ml) after 45-min serum depletion to visualize recycling endosomes. Early endosome marker 1 (EEA1) and Rab5 were detected by indirect immunostaining using rabbit polyclonal anti-EEA-1 and anti-Rab5 antibodies from Abcam (Cambridge, United Kingdom) and Santa Cruz Biotechnology, (Santa Cruz, CA), respectively. Lysosomes were labeled with FITC-dextran (50 μg/ml, MW 10 kDa) as described for the pH determination. In some experiments, the lysosome was stained with mouse monoclonal anti-Lamp-2 Ab (H4B4 Ab was developed by Dr. J. Thomas August and Dr. James E. K. Hildreth and was obtained from the Developmental Studies Hybridoma Bank maintained by The University of Iowa, Department of Biological Sciences, Iowa City, IA). Single optical sections were collected by Zeiss LSM510 laser confocal fluorescence microscope, equipped with a Plan-Apochromat 63X/1.4 (Carl Zeiss Microimaging, Thornwood, NY) as described (Lechardeur et al., 2004). Images were processed with Adobe Photoshop (Adobe Systems, San Jose, CA) software. CFTR phagosomal colocalization was measured by internalizing anti-HA Ab complexed with TRITC-conjugated goat anti-mouse IgG for 1 h in DMEM and chased for 0.5 h, and then FITC-PAO1 bacteria were phagocytosed for the indicated time. Colocalization was performed using the “Colocalization” feature in the Volocity 4.1.0 (Improvision software; Molecular Devices, Sunnyvale, CA) software as described in the Supplemental Materials.
Western Blotting
CFTR expression level of cell lines was determined by immunoblotting using anti-HA (MMS101R, Covance) for epithelial cells or anti-CFTR antibodies (M3A7 and L12B4, Chemokine, East Orange, NJ) for macrophages. Cell lysates were prepared using RIPA buffer containing 10 μg/ml leupeptin, pepstatin, 100 μM phenylmethylsulfonyl fluoride, 10 μM MG132, and 10 mM N-ethylmaleimide, and immunoblotting of CFTR was performed as described previously using enhanced chemiluminescence (Sharma et al., 2004).
Iodide Efflux Assay
The plasma membrane cAMP-dependent halide conductance of transfected and parental cells was determined with the iodide efflux as described (Sharma et al., 2001). The parental cells have no or negligible amount of cAMP-activated halide conductance. The CFTR inhibitor MalH2 (kindly provided by Dr. A. Verkman, University of California, San Francisco) was added simultaneously with the PKA agonist cocktail containing 20 μM forskolin, 0.5 mM CPT-cAMP, and 0.2 mM IBMX. The pH sensitivity of the iodide efflux was determined after incubating the cells at pH 5 during the last 10 min of the iodide loading and the efflux measurement in 20 mM MES-supplemented loading and efflux buffer, respectively. The iodide-selective electrode was calibrated in MES containing buffer at pH 5.
Statistical Analysis
Experiments were repeated at least three times or as indicated. Data are means ± SEM. Significance was assessed by calculating two-tailed p values at 95% confidence level with unpaired t test, using Prism software (GraphPad Software, San Diego, CA).
RESULTS
Considering the low plasma membrane density and slow metabolic turnover rate of CFTR in various expression systems (Heda et al., 2001; Sharma et al., 2004), it is conceivable that only a small population of secretory and newly formed endocytic vesicles contain the channel. Previous studies, including our own, used pH probes (e.g., cellubrevin-phluorin, synaptobrevin-GFP-phluorin, FITC-transferrin, and FITC-dextran) that labeled organelles in CFTR-independent manner (Barasch et al., 1991; Lukacs et al., 1992; Biwersi and Verkman, 1994; Dunn et al., 1994; Seksek et al., 1996; Botelho et al., 2000; Chandy et al., 2001; Poschet et al., 2002; Machen et al., 2003; Haggie and Verkman, 2007, 2009). Here we introduced two novel approaches to monitor the CFTR-dependent endosomal pH regulation with high specificity. First, we implemented a method that restricted the pH-sensitive probe to CFTR-containing vesicles (Figure 1A; Sharma et al., 2004; Glozman et al., 2009). Second, to control CFTR-independent endosomal pH changes provoked by the activation of cAMP-dependent PKA, a direct regulator of the v-ATPase and Na+/H+ exchanger activity (Marshansky and Futai, 2008), we utilized the G551D CFTR-3HA, a class IV CF mutation with severely impaired channel activity (Becq et al., 1994, 2007). In the following experiments, first we validated the use of G551D CFTR-3HA and the pH measurement methodology as well as the counterion and passive proton permeability determination in nonpolarized BHK cells. Subsequent studies were performed on genetically matched CF and non-CF human respiratory epithelia (IB3 and CFBE) as well as cftr−/− and cftr+/+ primary mouse macrophages and other heterologous CFTR overexpression systems. These studies not only allowed us to assess the effect of CFTR deficiency in genetically matched respiratory epithelia and alveolar macrophages, but also to determine whether the endogenous counterion permeability can limit the acidification in CFTR-independent cellular models.
Figure 1.

Selective labeling of CFTR-3HA–containing endosomes with pH-sensitive probes. (A) Schematic comparison of the subcellular distribution of CFTR and various endosomal pH probes. It is predicted that CFTR has partially overlapping localization with the fluid-phase marker dextran-FITC (left panel) and cellubrevin or synaptobrevin coupled with GFP-Phluorin (middle panel). Completely overlapping distribution of the pH-probe and endosomal CFTR is anticipated by labeling the exofacial 3HA-tag of CFTR with anti-HA Ab and FITC-Fab. (B) Immunoblot analysis of wt and G551D CFTR-3HA expression in stably transfected BHK cells. Equal amounts of cell lysates were immunoblotted with anti-HA Ab. Core- and complex-glycosylated CFTR are labeled, indicated by empty and filled arrowhead, respectively. (C) Plasma membrane halide conductance was measured by the iodide efflux assay in BHK monolayers expressing wt or G551D CFTR. PKA was stimulated with 20 μM forskolin, 0.2 mM IBMX, and 0.5 mM CPT-cAMP cocktail (+FK) at the indicated time at 22°C. Data are means of triplicate determinations from a single representative experiment. (D) Subcellular localization of internalized CFTR in BHK cells. Internalized wt and G551D CFTR-3HA was labeled with anti-HA Ab and visualized by TRITC-conjugated secondary Fab. Lysosomes and recycling endosomes were labeled with 50 μg/ml FITC-dextran (loaded overnight and chased for 3 h) and 15 μg/ml FITC-Tf (loaded for 45 min in serum-free medium), respectively. Single optical sections were obtained by laser confocal fluorescence microscopy. Bar, 10 μm.
Comparison of the Biosynthetic and Endocytic Membrane Trafficking of wt and G551D CFTR-3HA
To utilize the G551D CFTR-3HA for monitoring the vesicular pH without conferring PKA-dependent chloride permeability to endocytic organelles, first we assessed the membrane trafficking pathway of the G551D CFTR in relation to its wt counterpart. BHK cells were stably transfected with G551D and wt CFTR-3HA. The steady-state expression level of the core- and complex-glycosylated G551D and wt CFTR was comparable, measured by immunoblotting (Figure 1B). Likewise, the cell surface expression of the wt and G551D CFTR-3HA was similar, determined by anti-HA Ab-binding assay (Supplemental Figure S2A). PKA activation by forskolin, CPT-cAMP, and IBMX failed to activate G551D CFTR contrary to the wt channel, monitored by the iodide efflux assay (Figure 1C). The biosynthetic processing efficiency, metabolic stability, and internalization rates of the complex-glycosylated wt and G551D CFTR, measured by pulse-chase experiments, and anti-HA Ab uptake assay, respectively, were similar (Supplemental Figure S2, B and C, and data not shown).
To determine the postendocytic fate of the G551D CFTR, internalized channels were colocalized with the Tf receptor (Tf-R), a marker of recycling endosomes (Mukherjee et al., 1997). CFTR was labeled by in vivo anti-HA Ab capture at 37°C for 1 h. Tf-Rs were visualized by FITC-Tf. Quantitative colocalization of CFTR, on micrographs obtained by fluorescence laser confocal microscopy (FLCM), revealed that 85 ± 2 and 80 ± 4% (mean ± SEM, n = 4 experiments) of internalized Tf was colocalized with G551D and wt CFTR, respectively. Conversely, 52 ± 4% of wt and 62 ± 3% of G551D CFTR were confined to Tf-positive endosomes (Supplemental Figure S3A). Confinement of internalized G551D and wt CFTR to early endosomes was confirmed with their colocalization with rab5 and EEA1 (Supplemental Figure S3B and data not shown) and exclusion from FITC-dextran–loaded lysosomes (Figure 1D), an observation confirmed on other cells (see below). These results, jointly, indicate that the wt and G551D CFTR have overlapping postendocytic membrane trafficking that was further validated by vesicular pH measurements (see below).
Monitoring the Postendocytic Fate of G551D CFTR by Vesicular pH Determination
Based on the characteristic pH of the endolysosomal compartment (Mukherjee et al., 1997), the postendocytic sorting of G551D and wt CFTR could be inferred from the luminal pH of internalized CFTR-containing vesicles, as has been shown for a variety of cargo molecules (Barriere et al., 2007; Kumar et al., 2007; Duarri et al., 2008; Varghese et al., 2008). Cell-surface CFTR-3HA was labeled with primary anti-HA and FITC-conjugated secondary Fab on ice. After the removal of the excess extracellular Ab, internalization was initiated by raising the temperature to 37°C (Figure 2A). The luminal pH of individual endocytic vesicles was measured by FRIA, using 450- and 490-nm excitation wavelengths with an in situ calibration technique, and was plotted as their frequency distribution (Figure 2, A–C, Barriere et al., 2007; Barriere and Lukacs, 2008; Glozman et al., 2009). Although the cell-surface–bound probe (0-h chase) was exposed to the extracellular medium pH (∼7.4), more than 90% of vesicles acidified to pH ∼6.3–6.5 after a 0.5–1-h chase at 37°C (Figure 2A). Comparable results were obtained by continuous labeling of CFTR at 37°C for 1 h to enhance the signal-to-noise ratio (Figure 2B). Importantly, G551D, similar to the wt CFTR, was targeted to mildly acidic recycling endosomes after 1-h chase (pH ∼ 6.52, Figure 2C). Considering that the recycling endosomes mean pH is 6.4–6.5, measured in FITC-Tf–loaded BHK cells (Sharma et al., 2004), these results indicate that G551D like its wt counterpart recycles back to the cell surface and largely avoids lysosomal delivery. This conclusion is also in line with the limited (<8%) colocalization of the wt and G551D CFTR with Lamp2- and dextran-loaded lysosomes, determined by the Volocity program (see Figures 1D and 3C and Supplemental Figure S3D). CFTR was also confined to recycling endosomes after 4-h chase (data not shown). No significant dissociation of Ab from CFTR was observed at pH 5 (see Materials and Methods). Furthermore, the metabolic stability of the Ab-bound CFTR complex remained unaltered (Sharma et al., 2004), consistent with the notion that Ab binding did not provoke premature lysosomal degradation of the channel. Thus the G551D CFTR could serve as a negative control for evaluating the contribution of wt CFTR activation to the endosomal pH regulation.
Figure 3.

Wt and G551D CFTR expression, function, and postendocytic localization in CF respiratory epithelia and HeLa cells. (A) CFTR expression was probed by immunoblot analysis in mock, wt, and G551D CFTR-3HA expressing CFBE and IB3 respiratory epithelia, as well as in HeLa cells. Equal amounts of cell lysates were immunoblotted with anti-HA Ab. (B) Wt and G551D CFTR activity was measured by the iodide efflux assay as described in Figure 1C. Data are means of triplicate determinations from a representative experiment. (C) Localization of internalized wt and G551D CFTR in CFBE and IB3 cells. Internalized CFTR was labeled as described in Figure 1D. Endosomes were visualized by 15 μg/ml FITC-Tf loading or by indirect immunostaining of EEA1 and rab5. Lysosomes were identified by 50 μg/ml FITC-dextran loading as described in Materials and Methods or by indirect Lamp2 immunostaining. Single optical sections were obtained by laser confocal fluorescence microscopy. Bar, 10 μm.
The Effect of CFTR Activation on the Counterion Conductance and the Steady-State pH of Endosomes In Situ
To assess the relative magnitude of CFTR-dependent counterion permeability and the passive proton leak of individual endosomes, determinants of the endosomal pH, we modified a technique developed to measure these parameters in cell suspension of mouse peritoneal macrophages and Chinese hamster ovary (CHO) cells (Lukacs et al., 1990, 1992). The assay is based on the assumption that the dissipation rate of vesicular pH gradient by high concentration of protonophore (20 μM FCCP or CCCP) is rate-limited by the endosomal counterion conductance in the presence of the H+-ATPase inhibitor, Baf (Lukacs et al., 1990, 1992). Under these conditions, activation of CFTR should enhance the initial rate of Baf+CCCP–induced H+-efflux by the provision of additional Cl− conductance and dissipation of the inside negative endosomal membrane potential generated by the H+ efflux (Figure 2D, right panel). This was a reasonable assumption, considering that the luminal Cl− concentration of newly formed endosomes was rapidly reduced from 150 to 10–50 mM (Hara-Chikuma et al., 2005b).
The pH of individual, CFTR-3HA–containing endosomes labeled by primary anti-HA– and FITC-conjugated secondary Fab, was determined by FRIA as a function of time. After the inhibition of the vacuolar H+-ATPase activity by Baf, slow endosomal alkalinization, caused by the passive H+ efflux along the proton electrochemical gradient, was detected (Figure 2D). The observation that saturating concentration of protonophore CCCP (20 μM) accelerated the Baf-induced pH dissipation rate by nearly 10-fold (Figure 2D) suggests that early endosomes have a relatively small passive proton and large counterion conductance.
Incomplete inhibition of the H+-ATPase cannot account for the slow pH dissipation rate, because the Baf dose–response curve indicated that the v-ATPase was fully inhibited at 0.5 μM Baf concentration in vivo (data not shown; Lukacs et al., 1990). These observations also imply that a relatively slow H+-ATPase activity can maintain the endosomal pH gradient at steady state. Similar results were observed in parental BHK cells, ruling out the possibility that CFTR expression is responsible for the high constitutive counterion conductance and limited proton leak of endosomes (see Supplemental Figure S3E).
Activation of wt CFTR by PKA stimulation increased the CCCP+Baf–induced endosomal pH dissipation rate by threefold (Figure 2, E and F). In sharp contrast, the pH dissipation remained unaltered in G551D CFTR-expressing cells (Figures 2, E and F, and 4B). These results confirmed that wt, but not the G551D CFTR, is susceptible to PKA activation in endosomes (Becq et al., 1994).
To determine the consequence of CFTR activation on the steady-state endosomal pH, the luminal pH values were plotted before and after 3-min stimulation by PKA agonists. The endosomal pH of PKA-stimulated cells remained unaltered, regardless whether wt or G551D CFTR was expressed (Figure 2, F, right panel, and G).
A small fraction of internalized CFTR was confined to vesicles with pH < 6 and pH > 6.6 after 1-h chase, likely representing channels in late endosomes en route to lysosomes and in endocytic carrier vesicles, respectively (Figure 2, A and B; Sharma et al., 2004). Analysis of the luminal pH of these vesicles revealed that PKA-dependent CFTR activation was unable to influence the steady-state pH of late endosomes/lysosomes and endocytic carrier vesicles (Figure 2G).
Endosomal pH Regulation Is Not Influenced by CFTR Ablation or Overexpression in CF Epithelia and Heterologous Cell Models, Respectively
The inability of CFTR functional overexpression to hyperacidify BHK endosomes suggested that the relatively high endogenous counterion permeability in the presence of a small passive proton leak cannot limit the proton accumulation by the v-ATPase in cells that have no endogenous CFTR. This inference was tested by overexpressing wt CFTR in HeLa cells and polarized MDCK epithelia.
The loss of endogenous CFTR on the endosomal pH homeostasis was examined using genetically matched CF and non-CF respiratory epithelia. To this end, wt and G551D CFTR-3HA were stably expressed in IB3 and CFBE cells, widely used models of CF respiratory epithelia lacking functional CFTR (Gruenert et al., 1995; Bruscia et al., 2002). IB3 and CFBE cells were derived from the bronchial epithelia of CF patients with ΔF508/W1282X and ΔF508/ΔF508 CFTR genotypes, respectively (Zeitlin et al., 1991; Cozens et al., 1994) and have no detectable CFTR expression by immunoblotting (Figure 3A). Although IB3 cells are nonpolarized, both CFBE and MDCK cells were differentiated into polarized monolayers to ensure selective labeling of apical endosomes (see Materials and Methods).
Heterologous expression of wt and G551D CFTR-3HA was verified by immunoblotting (Figure 3A) and cell surface anti-HA Ab-binding assay (Supplemental Figure 2A). Iodide efflux assay revealed that wt but not G551D CFTR expression conferred PKA-stimulated plasma membrane halide conductance in HeLa, IB3, and CFBE cells (Figure 3B). None of the parental cells had detectable endogenous CFTR- and PKA-activated halide conductance (Figure 3, A and B, and data not shown).
Internalized wt and G551D CFTR were primarily targeted to early endosomes and excluded from lysosomes in IB3, CFBE, and HeLa cells, visualized by colocalization with FITC-Tf, EEA1, or rab5 and exclusion from dextran- or Lamp2-containing lysosome (Figure 3C, Supplemental Figure S3, C and D, and data not shown). These results indicate that the recycling propensity of endocytosed G551D CFTR is independent of the cellular expression system, as observed previously for wt CFTR (Sharma et al., 2004; Gentzsch et al., 2007; Varga et al., 2008).
The PKA-induced acceleration of the pH dissipation rate in the presence of CCCP+Baf showed that wt CFTR can function as a chloride conductive efflux pathway to facilitate H+ egress from early endosomes of CFBE, IB3, HeLa, and MDCK cells (Figure 4, A and B). This conclusion was confirmed by the inability of PKA to activate H+ efflux from endosomes of G551D CFTR-expressing cells (Figure 4B). Importantly, despite full activation of wt CFTR by the agonist cocktail in 1.5–2 min at room temperature (see Supplemental Figure S4B), no significant change in the steady-state pH of wt CFTR-expressing endosomes, including the CFBE and IB3 respiratory epithelia, was observed relative to that in G551D CFTR-expressing cells (Figure 4C). Likewise, we were unable to detect significant changes in the initial acidification rate of early endosomes, after the synchronized internalization of Ab-labeled CFTR from the cell surface (Figure 4, E and F). These data strongly suggest that the CFTR-independent counterion permeability is sufficient to ensure unrestricted proton accumulation during the acidification and at steady state by the v-ATPase. Finally, the HeLa and MDCK data suggest that the endosomal acidification is not limited by their endogenous counterion permeability, because provision of exogenous CFTR chloride conductance at an inwardly directed electrochemical chloride gradient (Sonawane and Verkman, 2003) could not hyperacidify endosomes.
CFTR Effect on the pH Homeostasis of Recycling Endosomes
Although the anti-HA and FITC-Fab labeling of CFTR enabled us to restrict the pH measurements to CFTR-containing vesicles, we wanted to establish whether the Ab binding interferes with the channel function. To assess the potential effect of Ab labeling on CFTR transport activity, CFTR-3HA–expressing BHK cells were incubated with saturating concentration of anti-HA Ab (37°C for 30 min). The mean PKA-dependent whole cell current density was reduced by 42% (from 35.1 ± 8.0 to 20.5 ± 6.7 pA/pF) in the presence of anti-HA Ab measured by the patch-clamp technique (Supplemental Figure S4A). The activation kinetics of CFTR by PKA agonists remained unaltered (Supplemental Figure S4B). Although these observations suggested that the CFTR-Ab complex retains significant activity, we sought an alternative assay to evaluate the CFTR-dependent endosomal pH regulation in the presence of fully functional channels.
On the basis of the overlapping subcellular distribution of FITC-Tf with wt and G551D CFTR (Figure 1D and Supplemental Figure S3A), we followed the endosomal pH after labeling the recycling endosomes with FITC-Tf. The Baf+CCCP–induced pH dissipation rates of recycling endosomes were determined in IB3 respiratory epithelia, as well as in BHK and HeLa cells. The endosomal counterion conductance was stimulated by about twofold with PKA agonists in wt, but not in G551D CFTR or parental cells (Supplemental Figure S3E). This suggests that the anti-HA Ab binding did not significantly limit the CFTR-dependent counterion flux in early or recycling endosomes. The steady-state endosomal pH was not, or only marginally affected by the wt CFTR activation relative to that of the G551D CFTR (Figure 4D), supporting our conclusion that neither ablation nor overexpression of CFTR influences the endosomal pH regulation, presumably due to the relatively high CFTR-independent counterion conductance.
Internalized CFTR Traverses the Immature Phagosome
Although there is no direct evidence available for the functional expression of CFTR in phagosomes, recent observations suggested that the phagolysosomal acidification of CFTR-deficient alveolar macrophages was severely compromised (Di et al., 2006). These results could not be confirmed by Haggie and coworkers (Haggie and Verkman, 2007). Considering that the phagosomal membrane undergoes substantial compositional change during maturation (Steinberg and Grinstein, 2007), it was plausible to assume that CFTR may traverse the limiting membrane of phagosomes and facilitate acidification by provision of chloride as a counterion. To assess CFTR localization and impact on the phagosomal proton and counterion permeability, first we used transiently transfected RAW264.7 mouse peritoneal macrophages. We were unable to detect endogenous CFTR by immunoblotting and iodide efflux assay in RAW cells (Figure 5, A and B, and data not shown). Transient expression of CFTR was verified by immunoblotting and functional assay (Figure 5, A and B). Immunostaining showed that the internalized channel was targeted to Tf-labeled recycling endosomes (∼90% colocalization) and largely excluded from dextran-labeled lysosomes (∼10% colocalization) after the labeling of endocytosed CFTR by anti-HA Ab capture in vivo (Figure 5C).
Figure 5.

Expression, activity, and membrane trafficking of CFTR-3HA in RAW macrophages. (A) CFTR plasma membrane channel activity was measured by the iodide efflux assay in transiently transfected RAW cells as described in Figure 1C. Data are means of triplicate determinations. (B) CFTR expression in transiently transfected RAW macrophages was visualized by immunoblotting using M3A7 and L12B4 anti-CFTR Ab. Equal amounts of cell lysates were immunoblotted. (C) Subcellular localization of internalized wt CFTR-3HA in transiently transfected RAW cells and the maturation of phagosomes. Top panels, internalized CFTR was colocalized with recycling endosomes and excluded from lysosomes. FITC-Tf and FITC-dextran labeling of recycling endosomes and lysosomes, respectively, was performed as described in Figure 1D. Single optical sections were obtained by fluorescence laser confocal microscopy (FLCM). Lower panels, phagosomal maturation was monitored by the colocalization of synchronously ingested, FITC-conjugated P. aeruginosa (PAO1) with TRITC-Tf– and TRITC-dextran–labeled lysosomes as a function of incubation time at 37°C. The labeling of recycling endosomes and lysosomes are described in Materials and Methods. Single optical sections were obtained by FLCM. Bar, 10 μm. (D) Colocalization of FITC-conjugated P. aeruginosa with Ab-labeled CFTR in RAW macrophages. Transiently expressed CFTR-3HA was labeled in vivo by Ab capture for 90 min without chase with anti-HA Ab and TRITC-conjugated secondary Fab at 37°C. Synchronously initiated phagocytosis of FITC-conjugated P. aeruginosa was performed for the indicated time before fixing the cells with paraformaldehyde. Single optical sections were obtained by FLCM. Bar, 10 μm.
To assess whether the activation of the phagocytic signaling cascade influences the subcellular targeting of CFTR, the postendocytic fate of the channel was determined in cells ingesting fluorophore-labeled P. aeruginosa (PAO1). Control experiments showed that after the engulfment of the FITC-labeled P. aeruginosa, newly formed phagosomes matured into phagolysosomes in ∼20 min, as shown by their fusion with TRITC-dextran loaded lysosomes (Figure 5C, bottom panels). Colocalization of bacteria with labeled Tf-R confirmed that the assay can reproduce the transient recruitment of Tf-receptors into immature phagosomes (Figure 5C, bottom panels) as reported earlier (Botelho et al., 2000).
CFTR recruitment to phagosomes was followed after labeling the endocytic CFTR-3HA pool with anti-HA Ab for 1–2 h at 37°C. P. aeruginosa was then bound to the plasma membrane at 4°C, and phagocytosis was initiated by raising the temperature to 37°C. Limited colocalization of CFTR with newly formed phagosomes was observed after 5 min of incubation by FLCM (Figure 5D). In sharp contrast, CFTR was largely undetectable in mature phagosomes 15–30 min after P. aeruginosa ingestion (Figure 5D). Similar results were obtained upon labeling CFTR on ice for 1 h before phagocytosis and initiating both internalization and phagocytosis simultaneously (data not shown). These results suggest that CFTR and ingested bacteria have only transiently overlapping postendocytic trafficking pathways and CFTR is virtually eliminated from mature phagosomes in RAW macrophages. Another possibility is that the anti-HA Ab complex is susceptible to rapid degradation in the proteolytically active phagosomes. This is unlikely to be the case because the labeled CFTR-Ab complex remained detectable after 90-min chase with comparable staining intensity, and inhibition of phagolysosomal proteases did not prevent the removal of Ab-labeled CFTR from phagosomes (see Figure 6F and data not shown). To further support the observation that CFTR is only transiently confined to immature phagosomes, we compared the luminal pH of CFTR-containing vesicles in phagocytosing RAW cells.
Figure 6.

The phagosomal acidification is CFTR-independent in RAW macrophages. (A) The pH distribution profile of wt CFTR-containing endosomes in transiently transfect RAW macrophages. FRIA of internalized CFTR-3HA was performed as described in Figure 2B. (B) The pH dissipation rate of FITC-Tf– or FITC-CFTR–containing endosomes was measured in mock or wt CFTR-3HA–transfected RAW cells as indicated. The pH dissipation was initiated by Baf+CCCP addition as described in Figure 2E and also shown in C. MalH2, 20 μM, was added to inhibit CFTR during labeling and the measurement. (C) The CFTR-dependent PKA-stimulated endosomal pH dissipation was inhibited by MalH2. The pH dissipation of FITC-Tf–loaded endosomes was measured in the absence or presence of PKA stimulation (+FK) as in B. MalH2 (20 μM) was used during the labeling and the measurement to inhibit CFTR activity. (D) Vesicular pH of FITC-Tf– or FITC-CFTR–containing vesicle in the absence or presence of PKA activation (+FK) for 3 min. Vesicular pH measurements were performed as in C. When indicated, 20 μM MalH2 was included. (E) Phagosomal acidification kinetics of RAW macrophages. Phagosomal pH was measured after synchronous ingestion of FITC-conjugated P. aeruginosa (PAO1) by FRIA in control and transiently transfected RAW cells with CFTR-3HA (+CFTR). CFTR-expressing cells were identified by labeling the channel with anti-HA Ab and TRITC-conjugated Fab capture before phagocytosis. MalH2 was added to the medium as in B. Means ± SEM; n = 4. (F) Effect of phagocytosis on the pH of CFTR-containing vesicles in RAW cells. The postendocytic trafficking of synchronously internalized CFTR was measured by FRIA as in Figure 2A. CFTR was labeled with anti-HA Ab and FITC-conjugated Fab on ice. TRITC-conjugated P. aeruginosa was adsorbed to the cell surface at 4°C. pH measurements were performed on double-stained cells after raising the temperature to 37°C for the indicated time. Means ± SEM; n = 4. (G) Vesicular pH and pH dissipation rates of CFTR-containing vesicles after 5- or 30-min chase in RAW cells transiently expressing wt CFTR-3HA. CFTR was labeled as described in F. (H) The steady-state pH of CFTR-containing endosomes and PAO1 ingested phagosomes are insensitive to PKA stimulation. The vesicular pH was determined before and after 3 min of stimulation with the PKA agonist cocktail (+FK) by FRIA. Means ± SEM; n = 4.
CFTR-independent Endosomal and Phagosomal Acidification in RAW Macrophages
The postendocytic trafficking of transiently expressed CFTR-3HA was determined by FRIA in RAW macrophages. The pH distribution profile of internalized wt CFTR-3HA indicated that the channels were primarily targeted to recycling endosomes in accord with immunolocalization data (Figure 6A). The PKA-dependent phosphorylation augmented the Baf+ CCCP–induced endosomal pH dissipation by threefold, confirming that the channel is functional in endosomes (Figure 6, B and C). Remarkably, preincubating the cells with the water-soluble MalH2, a specific inhibitor of CFTR (Muanprasat et al., 2004; Sonawane et al., 2008), fully suppressed the PKA-dependent pH dissipation of recycling endosomes (Figure 6, B and C).
Next, the PKA sensitivity of the endogenous counterion permeability of Tf-labeled recycling endosomes was measured. The Baf+CCCP–induced pH dissipation assay revealed that recycling endosomes have PKA-stimulated and MalH2-sensitive endogenous counterion conductance (Figure 5B). This observation suggests that RAW macrophages express a low level of endogenous CFTR that is below the detection limit of immunoblotting (Figure 6B). Neither PKA-activation nor MalH2 inhibition caused any significant change in the early endosomal pH of parental and CFTR-expressing RAW cells (Figure 6D).
It has been accepted that the rapid acidification of newly formed phagosomes is mediated by the vacuolar proton ATPase in macrophages (Lukacs et al., 1990, 1991; Hackam et al., 1997; Yates et al., 2005) and precedes the phagolysosomal fusion (Geisow et al., 1981). CFTR-dependent chloride uptake may promote proton accumulation by shunting the membrane potential of immature phagosomes. To address this scenario, macrophages were allowed to engulf FITC-labeled P. aeruginosa synchronously, in parental and CFTR overexpressing RAW cells, and the phagosomal pH was monitored by FRIA. CFTR expressors were identified by anti-HA Ab and TRITC-Fab staining. Neither overexpression nor inhibition of CFTR by MalH2 influenced the rapid, early phase of the Baf-sensitive acidification of newly formed phagosomes (Figure 6E). After 30 min of bacterial engulfment, the phagosomal H+ concentration reached pH ∼5 regardless of CFTR activity (Figure 6E).
The possible influence of phagocytosis on the postendocytic CFTR sorting next was assessed using FRIA. Both the cell surface binding of P. aeruginosa and CFTR labeling by anti-HA and FITC-Fab was performed on ice. Internalization and phagocytosis was initiated by shifting the temperature to 37°C. After transient confinement of wt CFTR to vesicles with luminal pH ∼6.0, likely representing sorting endosomes and immature phagosomes, the channel was transferred into recycling endosomes, characterized by more alkaline luminal pH (∼6.3–6.5; Figure 6F). The CFTR transfer to recycling endosomes coincided with the progressive acidification of phagosomes and the formation of the mature phagolysosomes with luminal pH ∼5.0, determined in separate samples (Figure 6E). These observations underscore the distinct trafficking route of CFTR and bacteria in RAW cells. The phagosomal acidification was blocked by Baf and reversed by NH4Cl (Figure 6, E and G). CFTR activation had no measurable effect on the steady-state pH of CFTR-containing endosomes and early or mature phagosomes (Figure 6, G and H). On the basis of the immunochemical localization and pH measurements, however, we could not rule out that a small fraction of CFTR reached the lysosomes. Next, we assessed CFTR function in lysosomal pH regulation.
Acidification of Endolysosomes and Phagosomes Is CFTR-independent in Primary Alveolar and Peritoneal Macrophages
CFTR was proposed to be indispensable for the normal acidification and bactericidal effect of phagolysosomal compartment in alveolar macrophages (Di et al., 2006). Functional expression of CFTR was measured by the pH dissipation assay in primary alveolar and peritoneal macrophages harvested from cftr+/+ and cftr−/− mice. The pH dissipation rate of FITC-Tf–labeled recycling endosomes revealed that only cftr+/+, but not cftr−/− macrophages possessed PKA-activated and MalH2-sensitive counterion conductance (Figure 7, A and B), providing strong evidence for CFTR expression in endosomes in line with the electrophysiological detection of the channel at the plasma membrane (Di et al., 2006).
Figure 7.

Acidification of phagosomes in CFTR-deficient primary alveolar (AM) and peritoneal (PM) macrophages is preserved. (A and B) CFTR is active in early endosomes, but not in lysosomes, of primary alveolar and peritoneal mouse macrophages from cftr+/+ mice. pH dissipation rates were measured in FITC-Tf– or FITC/Oregon488-dextran–loaded endosomes and lysosomes, respectively in macrophages isolated from cftr+/+ or cftr−/− mice. Tf and dextran labeling was described in Materials and Methods. MalH2, 20 μM, was added to inhibit CFTR during labeling, and the measurement. pH dissipation rates were measured after Baf and CCCP addition as described in Figure 2E. (C and D) CFTR can be activated in immature phagosomes of cftr+/+ macrophages. pH dissipation rates of phagosomes ingesting FITC-labeled PAO1 was measured after 5 and 30 min of infection. MalH2, 20 μM, was added during phagocytosis. (E) Phagosomal acidification kinetics of cftr+/+ and cftr−/− alveolar macrophages are similar. Acidification of phagosomes, synchronously ingesting FITC-PAO1, was monitored by FRIA. Inset, the variation of immature and mature phagosomal pH after 3 min PKA stimulation is reported as the function of the initial pH. (F) Comparison of phagosomal pH of cftr+/+ and cftr−/− peritoneal macrophages by FRIA of FITC or FITC- and TRITC-labeled PAO1. Phagocytosis was initiated synchronously and the duration (5–45 min) is indicated. Means ± SEM; n = 4 mice. (G) Protonophore-induced increase of the pH dissipation rate of endosomes and lysosomes relative to the passive proton efflux rate as an indicator of counterion permeability. The pH dissipation rates of FITC-Tf– and FITC-dextran–loaded recycling endosomes and lysosomes, respectively, were measured by FRIA in the presence of Baf alone and Baf+ CCCP as in Figure 2, D and E. CFTR-expressing HeLa, RAW and respiratory epithelia (CFBE and IB3) were used. Baf+CCCP simultaneous addition yielded results similar to those obtained by consecutive addition with 1-min delay. Results expressed as fold increase of the CCCP+Baf–induced proton efflux rate over that of the passive proton release. (H) Protonophore-induced increase of the pH dissipation rate relative to the passive proton efflux rate of endocytic organelles in primary macrophages. Baf- and Baf+CCCP–induced pH dissipation rates of recycling endosomes, lysosomes as well as immature and mature phagosomes were measured by the FRIA using the indicated pH-sensitive probe as described in Materials and Methods and in G. Means ± SEM; n = 4 mice.
Remarkably, immature phagosomes also displayed PKA-activated counterion permeability after 5-min ingestion of FITC-conjugated P. aeruginosa in cftr+/+ alveolar and peritoneal macrophages (Figure 7, C and D). Because this counterion permeability was inhibited by the MalH2 and was undetectable in cftr−/− cells, we concluded that functional CFTR is associated with immature phagosomes in primary macrophages (Figure 7, C and D). Maturation of phagosomes, however, led to the loss of PKA-stimulated counterion permeability, measured after 30 min of phagosome formation (Figure 7, C and D). The assay pH sensitivity was preserved, as demonstrated by the rapid alkalinization of phagosomes by NH4Cl (data not shown).
Neither the initial rate of acidification nor the steady-state pH of mature phagosomes was influenced by CFTR expression in alveolar and peritoneal macrophages (Figure 7, E and F). Furthermore, no significant change in the steady-state pH of immature and mature phagosomes was detected upon PKA activation in cftr+/+ and cftr−/− macrophages (Figure 7E, insert).
Because lysosomal fusion was proposed to play a pivotal role in phagosomal maturation (Di et al., 2006), CFTR contribution to the lysosomal pH homeostasis was also assessed using FITC-dextran– and Oregon488-dextran–labeled lysosomes. No lysosomal CFTR activity was measurable by the Baf+CCCP–induced pH dissipation assay (Figure 7, A and B). This could be attributed to the channel efficient recycling, inactivation, and degradation in lysosomes or a combination of these processes. Finally, no difference in the lysosomal pH of control or PKA-stimulated primary macrophages isolated from cftr−/− and cftr+/+ mice could be detected (Supplemental Figure S6A). These observations support the notion that the phagosomal and lysosomal acidifications are CFTR-independent processes in primary macrophages.
The Endogenous Counterion Permeability of Endophagocytic Compartments Does Not Restrict the Proton Pump Activity, the Role of Passive Proton Permeability
The counterion flux required for the dissipation of the vacuolar H+-ATPase–induced membrane potential is determined by the proton accumulation rate. To achieve the set point of organellar pH, the proton accumulation rate should be adjusted according to the buffer capacity and the proton leak (Wu et al., 2001). Considering that CFTR overexpression, activation, and ablation failed to change the organellar acidification in endolysosomes and phagosomes, we hypothesized that the endogenous, CFTR-independent counterion permeability is sufficiently high to dissipate the membrane potential (Steinberg et al., 2007b) and thereby cannot impede the proton translocation rate. This assumption implies that the passive proton permeability of endocytic organelles is small in comparison with their counterion permeability. To support this prediction, we assessed the counterion and passive proton permeabilities of endocytic organelles in respiratory epithelia, macrophages, and other heterologous expression systems, assuming that the passive pH dissipation rate is proportional with the proton permeabilities.
The Baf-induced proton efflux reflects the passive proton leak that is compensated by the v-ATPase activity at the steady-state pH. Comparison of the Baf- and Baf+CCCP–induced pH dissipation rates revealed that the CFTR-independent counterion permeability was ∼5–10 times higher than the passive proton permeability both in recycling endosomes and lysosomes of IB3 and CFBE epithelia (Figure 7G). Similar data were obtained in primary macrophages (Figure 7H). These results strongly suggest that the counterion permeability is sufficiently high to support the acidification of organelles in CFTR-deficient respiratory epithelia and primary macrophages.
Decreasing passive proton permeability along the secretory pathway was proposed as a critical determinant of the progressively increasing luminal acidification (Wu et al., 2001). An analogous role of the passive proton permeability may prevail along the endocytic pathway. To determine the passive proton permeability of endocytic organelles, first the buffer capacity of endosomes, lysosomes, and phagosomes was measured, using the ammonium chloride pulse technique (Roos and Boron, 1981; Figure 8, A and B). The passive proton permeability was calculated based on the Baf-induced proton efflux rate and the assumption that the predominant driving force is the transmembrane pH gradient at a constant cytoplasmic pH 7.3 as described in Materials and Methods. The passive proton permeability of lysosomes was at least twofold lower than in recycling endosomes in BHK, HeLa, IB3, and CFBE cells (Figure 8C), supporting the notion that down-regulation of the passive proton leak may contribute to the progressive acidification along the endocytic pathway. For comparison, the passive proton permeability of the ER and Golgi compartment was also plotted, as determined in previous publications (Llopis et al., 1998; Chandy et al., 2001; Wu et al., 2001; Weisz, 2003b).
Figure 8.

Determination of the passive proton permeability of endocytic organelles. (A) Measurement of the buffer capacity of endocytic organelles. The lysosomal compartment of BHK cells were loaded with dextran as described in Materials and Methods, and the organellar pH was monitored by FRIA. The extent of lysosome alkalinization was measured after the addition of small amounts of NH4Cl (e.g., 0.1–2 mM) in the presence of Baf to prevent the compensatory activation of the v-ATPase. The number indicates the final concentration of NH4Cl in mM. The buffer capacity was calculated from the alkalinization, induced by the 0.5–1 mM NH4Cl as described in Roos and Boron (1981) and Chandy et al. (2001). (B) The buffer capacity of recycling endosomes, lysosomes, and phagosomes was measured on FITC-Tf– and FITC-dextran–loaded organelles as described in A. The early and mature phagosomal buffer capacities were measured after 5 and 30 min phagocytosis of FITC-conjugated P. aeruginosa. (C) The passive proton permeability of recycling endosomes, lysosomes, and phagosomes. The passive proton efflux rate from the indicated organelles was measured in the presence of 400 nM Baf by FRIA after their selective labeling as described in Materials and Methods. The passive proton permeability was calculated as described in Materials and Methods. For comparison, the passive proton permeability of the Golgi compartment and the ER was indicated obtained from previous publications (Llopis et al., 1998; Chandy et al., 2001; Wu et al., 2001; Weisz, 2003b).
DISCUSSION
The paucity of our understanding of CFTR function in organellar pH homeostasis is illustrated by the fact that CFTR is invoked in facilitating (Barasch et al., 1991; Di et al., 2006; Teichgraber et al., 2008), inhibiting (Poschet et al., 2002), or having no effect on the acidification of exocytic and endocytic organelles (Lukacs et al., 1992; Dunn et al., 1994; Root et al., 1994; Seksek et al., 1996; Gibson et al., 2000; Machen et al., 2001; Haggie and Verkman, 2007, 2009b; Lamothe and Valvano, 2008). Comparison of these studies is hampered by different cellular models and pH detection methodologies used as recently reviewed by Haggie and Verkman (2009a). Because organellar acidification has fundamental influence on various cellular functions with proposed relevance to the hyperinflammatory CF lung infection (Konstan et al., 1994; Pier et al., 1996; Rowe et al., 2005; Sagel et al., 2007), our primary goal was to assess the role of counterion conductance in general, and CFTR in particular, in the pH regulation of endocytic compartments in well-defined cellular models.
The Endogenous Counterion Conductance Is Not Rate-limiting for Endosomal Acidification of CF Respiratory Epithelia
During the past decade, three major determinants of organellar acidification have been established: the counterion conductance, the intrinsic proton leak, and the v-ATPase activity (Weisz, 2003b). The major counterion conductance of endolysosomes consists of members of two branches of the ClC gene family; ClC-3, -4, and -5, and ClC-6 and -7 (Marshansky et al., 2002; Jentsch et al., 2005; Picollo and Pusch, 2005; Jentsch, 2007). Subcellular localization of the ClC-3, -4, and -5 in concert with functional data suggest that these ClC transporters are permissive for the acidification of early endosomes and synaptic vesicles by providing a significant electrical shunt pathway (Stobrawa et al., 2001; Suzuki et al., 2006; Maritzen et al., 2008). Ablation of ClC-4 and -5 impairs the acidification of endosomes (Gunther et al., 1998; Hara-Chikuma et al., 2005a), whereas ClC-3 depletion causes defective acidification of synaptic vesicles (Stobrawa et al., 2001; Maritzen et al., 2008) as well as early endosomes (Li et al., 2002; Hara-Chikuma et al., 2005b). Although additional Cl− transport pathway may be involved (Schenck et al., 2009). ClC-4, -5, and -7 are now known to operate as voltage-dependent Cl−/H+ antiporters (Picollo and Pusch, 2005; Scheel et al., 2005; Graves et al., 2008). If two Cl− ions are exchanged for one H+, as determined for the ClC-7, a single transport cycle of the ClC-3, -4, and -5 ensures charge neutralization of two protons uptake at the cost of one proton efflux. Impaired functional expression of ClC-3, -4, or -5, therefore, impedes the v-ATPase–mediated proton accumulation by increasing the inside positive membrane potential (Marshansky et al., 2002). Although these data provide supportive evidence for the permissive role of ClC-3, -4, and -5 in endosomal acidification, they are short on demonstrating that the overall counterion permeability limits the proton pump activity in vivo, a prerequisite for CFTR to directly influence the endosomal pH regulation.
To compare the CFTR-independent and CFTR-dependent counterion and proton permeabilities, endosomes containing functional (wt) or nonfunctional (G551D) CFTR were labeled with the pH-sensitive fluorophore, restricting the probe to CFTR-expressing vesicles. Using saturating concentration of Baf in the absence or in combination with a protonophore, we determined the relative magnitude of passive proton leak and the maximum rate of pH dissipation, an indicator of the lower limit of endosomal counterion permeability. The CFTR-independent counterion permeability was several-fold larger than the passive proton permeability in apical endosomes of polarized bronchial epithelia (CFBE) derived from CF patient. Importantly, although CFTR complementation of CFBE and IB3 conferred a threefold increase in counterion permeability (Figure 4B and Supplemental Figure S3E), it failed to alter the passive proton permeability and the initial acidification rate, important determinants of organellar pH (Figure 4, E and F, and data not shown; Chandy et al., 2001; Machen et al., 2003). These results are complemented with studies on cells (HeLa, BHK, and MDCK) where organellar acidification is independent of CFTR. The channel overexpression failed to hyperacidify the endolysosomal compartments (Figure 4, C and D, and Supplemental Figure S6C), implying that the v-ATPase activity is not limited by endogenous counterion permeability. Similar results were obtained in IB3 respiratory epithelia and HeLa cells using FITC-Tf labeling of recycling endosomes and jointly support the notion that the endosomal pH cannot be regulated by modest alteration of the relatively large counterion conductance under physiological conditions.
Identification of two ubiquitously expressing endolysosomal cation efflux pathways supports the inference that the CFTR-independent counterion permeability is significant and cannot restrict the relatively slow endosomal proton accumulation. The TRPV2, a member of the transient receptor potential channel family, was confined to early endosomes (Saito et al., 2007). Its limited selectivity toward Ca2+ over K+, activation by luminal acidification and low chloride concentration makes it a plausible candidate to contribute to cation efflux accompanying proton accumulation similar to the TRPML1 (mucolipin1) channel, localized to late endosomes and lysosomes and invoked in iron release (Dong et al., 2008).
CFTR-independent Lysosomal Acidification
The lysosomal acidification defect (pH ∼5.9) was implicated in ceramide accumulation, which in turn caused excessive secretion of the proinflammatory cytokine interleukin-1 (IL-1) and the keratinocyte-derived chemokine (the mouse homologue of IL-8), cell death, and susceptibility to P. aeruginosa infection of respiratory epithelia (Bruscia et al., 2008; Teichgraber et al., 2008). Importantly, alkalinization of lysosomes to pH ∼5.9 was accounted for the phagosomal acidification defect and intracellular proliferation of P. aeruginosa in cftr−/− alveolar macrophages (Di et al., 2006). We were unable to reproduce any alkalinization of lysosomes in CF respiratory epithelia (CFBE or IB3) compared with their CFTR-complemented counterparts (Figure 4C, and Supplemental Figure S6C). Likewise, we could not resolve significant change in the lysosomal pH of alveolar or peritoneal macrophages derived from cftr−/− and cftr+/+ mouse or after the channel activation by PKA agonist. These results confirm and expand recent observations indicating that the phagosomal/lysosomal acidification of ΔF508-CFTR alveolar and CFTR-deficient bone marrow mouse macrophages, as well as primary respiratory epithelia are preserved (Haggie and Verkman, 2007, 2009b; Lamothe and Valvano, 2008).
Albeit CFTR could be activated in endosomes, no channel function was detected in lysosomes of CFTR complemented CFBE and IB3 epithelia by the pH dissipation assay (Supplemental Figure S6B) and no difference in the lysosomal pH could be established (Supplemental Figure S6C). Similar data were obtained in RAW, BHK, and HeLa cells (Figure 4, C and D, and data not shown). These results cannot be attributed to a significant increase of the electrochemical chloride gradient across the lysosomal membrane, because the luminal chloride concentration of late endosomes/lysosomes was found to be 40–50 mM (Hara-Chikuma et al., 2005b).
Cell type differences and pH measurement methodology may explain the discordant data on lysosomal pH. Teichgraber et al. (2008) monitored the lysosomal pH of freshly isolated respiratory epithelia with uncharacterized cellular composition using lysosensor-green D189 that labeled acidic organelles nonselectively. The calibration was performed on permeabilized cells, based on the correlation between the luminal pH and the fluorescence intensity of the dye. Cell permeabilization in extracellular medium (containing 132 mM NaCl and 1 mM CaCl2) likely induced significant changes in organelle composition and adsorption of lysosensor-green to cytosolic constituents. Because the fluorescence intensity of lysosensor-green was measured in areas of 1.5 × 1.5 mm and not in individual vesicles (Teichgraber et al., 2008), nonspecific changes during the calibration could not be ruled out. Our measurements were performed on well-characterized bronchial epithelia (CFBE and IB3) that have been used extensively (Zeitlin et al., 1991; Cozens et al., 1994; Gruenert et al., 1995; Moyer et al., 1999; Bebok et al., 2005; Swiatecka-Urban et al., 2005; Varga et al., 2008). We relied on ratiometric pH determination of FITC/Oregon488-dextran–labeled individual lysosomes that allowed concentration-independent, in situ calibration, a standard assay to monitor endolysosomal pH that could also resolve small pH changes in subpopulations of organelles (Haggie and Verkman, 2009b; see Supplemental Figure S1, B–D). Experiments were also carried out on FITC/TMR-dextran labeled lysosomes, to validate the assay pH sensitivity, with identical results (data not shown).
Several considerations support our conclusion that lysosomal acidification is independent of CFTR in respiratory epithelia and macrophages. 1) Internalized CFTR constitutively recycles back to the cell surface and only <10% of complex-glycosylated channels are targeted to lysosomes in 1–2 h. Highly efficient recycling preserves the long metabolic half-life of CFTR in post-Golgi compartments and was observed in a wide variety of heterologous expression systems, including respiratory epithelia (Varga et al., 2008). 2) CF mutations provoked ubiquitination and rapid down-regulation of CFTR from the cell surface by lysosomal proteolysis (Sharma et al., 2004). We assume that a similar mechanism is responsible for the nonnative wt CFTR turnover (Sharma et al., 2004). Indeed, CFTR immunoreactive degradation intermediates were detectable in purified lysosomes upon inhibition of lysosomal protease (Benharouga et al., 2001). Therefore, CFTR channel density in the lysosomal membrane is limited and immunolocalization studies could visualize partially degraded, nonfunctional channels (Di et al., 2006). 3) Even if low copy number of CFTR is inserted into the lysosomal membrane, the acidic pH inhibits by >50% the channel activity. This inference is based on the inhibitory effect of pH 5 on the PKA-activated plasma membrane halide conductance in BHK cells (Supplemental Figure S5). 4) Finally, and perhaps most importantly, we showed that the CFTR-independent endogenous counterion permeability of lysosomes was 5–10-fold higher than the passive proton permeability in CFBE and IB3 respiratory epithelia and alveolar macrophages (Figure 7, G and H). Remarkably, the lysosomal passive proton permeability appears to be <1 μm/s, the lowest among acidic organelles of the endocytic and secretory pathway (Figure 8C), maximizing the acidification efficiency, whereas minimizing the counterion flux requirement. These observations imply that the transmembrane potential generated by lysosomal proton accumulation is largely dissipated by CFTR-independent counterion movements, mediated by the ubiquitously expressed ClC6 and ClC7 as Cl/H exchangers and a subset of cation-proton exchangers and cation channels (Marshansky et al., 2002; Jentsch, 2007; Orlowski and Grinstein, 2007; Dong et al., 2008).
CFTR Expression and Activation Does Not Influence Phagosomal Acidification
It was proposed that CFTR is essential for the bactericidal activity of alveolar macrophages by ensuring phagolysosomes acidification (Di et al., 2006). Ingested bacteria in CFTR-deficient alveolar macrophages remained in mildly acidic phagosomes because of the lysosomal acidification defect (Di et al., 2006). Importantly, no phagosomal acidification defect was reported in alveolar macrophages from ΔF508 cftr homozygous mice or by inhibiting the wt channel with the channel blocker, CFTRinh-172 (Haggie and Verkman, 2007). Likewise, the phagosomal acidification of bone marrow macrophages obtained from cftr−/− mouse was unaffected (Lamothe and Valvano, 2008). To address whether expression of ΔF508-CFTR and cell-specific differences could explain the contrasting results, our studies were performed on CFTR-deficient alveolar and peritoneal macrophages. No difference in the phagosomal acidification rate and extent was documented in CFTR-deficient and control macrophages (Figure 7, E and F). Importantly, similar results were obtained upon inhibition of the endogenous CFTR activity by MalH2 in RAW macrophages. Analysis of CFTR membrane trafficking, as well as the relative proton and counterion permeabilities, provided compelling rationale for the inability of CFTR to regulate phagosomal acidification in these models.
CFTR was only transiently confined to newly formed phagosomes and eliminated from mature phagosomes, demonstrated by CFTR immunolocalization (Figure 5D), as well as by vesicular pH determination of FITC-labeled CFTR in RAW macrophages (Figure 6, D and F). Exogenous overexpression of CFTR-3HA cannot account for this phenomenon, because endogenous CFTR was identified in the immature phagosome of RAW, alveolar, and peritoneal macrophages by the PKA- and MalH2-sensitive pH dissipation (Figures 6B and 7, A and B). The rapid loss of CFTR during phagosomal maturation parallels the Tf-R retrieval for recycling (Figure 6, C and D), demonstrated to be accomplished by COPI-coated vesicle budding (Botelho et al., 2000).
Finally, we showed that the CFTR-independent counterion permeability of both the immature and mature phagosome is several-fold higher than its endogenous proton permeability. The relatively high counterion permeability of CFTR-deficient and wt primary macrophages (as well as RAW cells) is comparable to that observed in thioglycolate elicited peritoneal and RAW macrophages (Lukacs et al., 1990). The modest phagosomal membrane potential (12–27 mV) of RAW macrophages is in accord with efficient electrical shunting (Steinberg et al., 2007a). We envision that similarly low phagosomal membrane potential develops in primary CF alveolar macrophages in the presence of the endogenous counterion permeability. Therefore, neither CFTR complementation nor overexpression influences the phagosomal pH homeostasis during the fast acidification phase or at steady state (Haggie and Verkman, 2007; Figure 6, D and F).
In summary, our results, obtained on genetically matched respiratory epithelial cell lines and primary alveolar macrophages, indicate that CFTR has no role in the acidification of endosomes, lysosomes, and phagosomes because of the relatively large counterion and limited proton permeability of these organelles. Therefore, other factors than the acidification defect must be invoked in defining the connection between CF mutations and the severe lung pathology. At a more general level, our results showed that the counterion permeability of endocytic organelles exceeds several fold the requirement for shunting the v-ATPase generated membrane potential in all cell types examined. Thus, limited attenuation or elevation of the counterion permeability cannot play a decisive role in the organellar acidification, consistent with the emerging paradigm suggesting that subunit composition of the v-ATPase and the passive proton leak are the most critical factors in adjusting the luminal pH of acidic organelles (Hurtado-Lorenzo et al., 2006). The smallest passive proton permeability of lysosomes among acidic organelles is consistent with this hypothesis (Figure 8C).
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Johanna Rommens for the G511D CFTR cDNA, Dr. Matt Parsek (University of Seattle) for the P. aeruginosa PAO1 strain, Dr. Alan Verkman (University of California San Francisco) for the MalH2 inhibitor and Dr. Pamela Zeitlin (Johns Hopkins University) for the IB3 cells. We thank Hadi Salah for generating the CFTR-transfected IB3 cells. This work was funded by the Canadian Institute of Health Research (CIHR), by the National Institutes of Health National Institute of Diabetes, and Digestive, and Kidney Diseases, by the Canada Foundation for Innovation to G.L., and by the CIHR and the Canadian Cystic Fibrosis Foundation to D.R. G.L. also holds a Canada Research Chair in the Molecular Cell Biology of Cystic Fibrosis.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E09-01-0061) on May 6, 2009.
REFERENCES
- Barasch J., Kiss B., Prince A., Saiman L., Gruenert D., al-Awqati Q. Defective acidification of intracellular organelles in cystic fibrosis. Nature. 1991;352:70–73. doi: 10.1038/352070a0. [DOI] [PubMed] [Google Scholar]
- Barriere H., Lukacs G. L. Analysis of endocytic trafficking by single-cell fluorescence ratio imaging. Curr. Protoc. Cell Biol. 2008 doi: 10.1002/0471143030.cb1513s40. Chapter 15, Unit 15 13. [DOI] [PubMed] [Google Scholar]
- Barriere H., Nemes C., Du K., Lukacs G. L. Plasticity of polyubiquitin recognition as lysosomal targeting signals by the endosomal sorting machinery. Mol. Biol. Cell. 2007;18:3952–3965. doi: 10.1091/mbc.E07-07-0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barriere H., Nemes C., Lechardeur D., Khan-Mohammad M., Fruh K., Lukacs G. L. Molecular basis of oligoubiquitin-dependent internalization of membrane proteins in mammalian cells. Traffic. 2006;7:282–297. doi: 10.1111/j.1600-0854.2006.00384.x. [DOI] [PubMed] [Google Scholar]
- Bebok Z., Collawn J. F., Wakefield J., Parker W., Li Y., Varga K., Sorscher E. J., Clancy J. P. Failure of cAMP agonists to activate rescued deltaF508 CFTR in CFBE41o- airway epithelial monolayers. J. Physiol. 2005;569:601–615. doi: 10.1113/jphysiol.2005.096669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becq F., Jensen T. J., Chang X. B., Savoia A., Rommens J. M., Tsui L. C., Buchwald M., Riordan J. R., Hanrahan J. W. Phosphatase inhibitors activate normal and defective CFTR chloride channels. Proc. Natl. Acad. Sci. USA. 1994;91:9160–9164. doi: 10.1073/pnas.91.19.9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benharouga M., Haardt M., Kartner N., Lukacs G. L. COOH-terminal truncations promote proteasome-dependent degradation of mature cystic fibrosis transmembrane conductance regulator from post-Golgi compartments. J. Cell Biol. 2001;153:957–970. doi: 10.1083/jcb.153.5.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benharouga M., Sharma M., So J., Haardt M., Drzymala L., Popov M., Schwapach B., Grinstein S., Du K., Lukacs G. L. The role of the C terminus and Na+/H+ exchanger regulatory factor in the functional expression of cystic fibrosis transmembrane conductance regulator in nonpolarized cells and epithelia. J. Biol. Chem. 2003;278:22079–22089. doi: 10.1074/jbc.M301030200. [DOI] [PubMed] [Google Scholar]
- Biwersi J., Verkman A. S. Functional CFTR in endosomal compartment of CFTR-expressing fibroblasts and T84 cells. Am J. Physiol. 1994;266:C149–C156. doi: 10.1152/ajpcell.1994.266.1.C149. [DOI] [PubMed] [Google Scholar]
- Bompadre S. G., Sohma Y., Li M., Hwang T. C. G551D and G1349D, two CF-associated mutations in the signature sequences of CFTR, exhibit distinct gating defects. J. Gen. Physiol. 2007;129:285–298. doi: 10.1085/jgp.200609667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho R. J., Hackam D. J., Schreiber A. D., Grinstein S. Role of COPI in phagosome maturation. J. Biol. Chem. 2000;275:15717–15727. doi: 10.1074/jbc.M910068199. [DOI] [PubMed] [Google Scholar]
- Boucher R. C. Cystic fibrosis: a disease of vulnerability to airway surface dehydration. Trends Mol. Med. 2007;13:231–240. doi: 10.1016/j.molmed.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Bruscia E., Sangiuolo F., Sinibaldi P., Goncz K. K., Novelli G., Gruenert D. C. Isolation of CF cell lines corrected at DeltaF508-CFTR locus by SFHR-mediated targeting. Gene Ther. 2002;9:683–685. doi: 10.1038/sj.gt.3301741. [DOI] [PubMed] [Google Scholar]
- Bruscia E. M., Zhang P. X., Ferreira E., Caputo C., Emerson J. W., Tuck D., Krause D. S., Egan M. E. Macrophages directly contribute to the exaggerated inflammatory response in CFTR-/- mice. Am J. Respir. Cell. Mol. Biol. 2009;40:295–304. doi: 10.1165/rcmb.2008-0170OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campodonico V. L., Gadjeva M., Paradis-Bleau C., Uluer A., Pier G. B. Airway epithelial control of Pseudomonas aeruginosa infection in cystic fibrosis. Trends Mol. Med. 2008;14:120–133. doi: 10.1016/j.molmed.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandy G., Grabe M., Moore H. P., Machen T. E. Proton leak and CFTR in regulation of Golgi pH in respiratory epithelial cells. Am J. Physiol. Cell Physiol. 2001;281:C908–C921. doi: 10.1152/ajpcell.2001.281.3.C908. [DOI] [PubMed] [Google Scholar]
- Chastellier C. D. EM analysis of phagosomes. Methods Mol. Biol. 2008;445:261–285. doi: 10.1007/978-1-59745-157-4_17. [DOI] [PubMed] [Google Scholar]
- Cozens A. L., Yezzi M. J., Kunzelmann K., Ohrui T., Chin L., Eng K., Finkbeiner W. E., Widdicombe J. H., Gruenert D. C. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am J. Respir. Cell. Mol. Biol. 1994;10:38–47. doi: 10.1165/ajrcmb.10.1.7507342. [DOI] [PubMed] [Google Scholar]
- Di A., et al. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat. Cell Biol. 2006;8:933–944. doi: 10.1038/ncb1456. [DOI] [PubMed] [Google Scholar]
- Dong X. P., Cheng X., Mills E., Delling M., Wang F., Kurz T., Xu H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature. 2008;455:992–996. doi: 10.1038/nature07311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarri A., et al. Molecular pathogenesis of megalencephalic leukoencephalopathy with subcortical cysts: mutations in MLC1 cause folding defects. Hum Mol. Genet. 2008;17:3728–3739. doi: 10.1093/hmg/ddn269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn K. W., Park J., Semrad C. E., Gelman D. L., Shevell T., McGraw T. E. Regulation of endocytic trafficking and acidification are independent of the cystic fibrosis transmembrane regulator. J. Biol. Chem. 1994;269:5336–5345. [PubMed] [Google Scholar]
- Forgac M. Structure, function and regulation of the vacuolar (H+)-ATPases. FEBS Lett. 1998;440:258–263. doi: 10.1016/s0014-5793(98)01425-2. [DOI] [PubMed] [Google Scholar]
- Geisow M. J., D'Arcy Hart P., Young M. R. Temporal changes of lysosome and phagosome pH during phagolysosome formation in macrophages: studies by fluorescence spectroscopy. J. Cell Biol. 1981;89:645–652. doi: 10.1083/jcb.89.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentzsch M., Choudhury A., Chang X. B., Pagano R. E., Riordan J. R. Misassembled mutant DeltaF508 CFTR in the distal secretory pathway alters cellular lipid trafficking. J. Cell Sci. 2007;120:447–455. doi: 10.1242/jcs.03350. [DOI] [PubMed] [Google Scholar]
- Gibson G. A., Hill W. G., Weisz O. A. Evidence against the acidification hypothesis in cystic fibrosis. Am J. Physiol. Cell Physiol. 2000;279:C1088–C1099. doi: 10.1152/ajpcell.2000.279.4.C1088. [DOI] [PubMed] [Google Scholar]
- Glozman R., Okiyoneda T., Mulvihill C. M., Rini J. M., Barriere H., Lukacs G. L. N-glycans are direct determinants of CFTR folding and stability in secretory and endocytic membrane traffic. J. Cell Biol. 2009;184:847–862. doi: 10.1083/jcb.200808124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabe M., Oster G. Regulation of organelle acidity. J. Gen Physiol. 2001;117:329–344. doi: 10.1085/jgp.117.4.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves A. R., Curran P. K., Smith C. L., Mindell J. A. The Cl−/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature. 2008;453:788–792. doi: 10.1038/nature06907. [DOI] [PubMed] [Google Scholar]
- Griffiths G., Back R., Marsh M. A quantitative analysis of the endocytic pathway in baby hamster kidney cells. J. Cell Biol. 1989;109:2703–2720. doi: 10.1083/jcb.109.6.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenert D. C., Finkbeiner W. E., Widdicombe J. H. Culture and transformation of human airway epithelial cells. Am J. Physiol. 1995;268:L347–L360. doi: 10.1152/ajplung.1995.268.3.L347. [DOI] [PubMed] [Google Scholar]
- Guilbault C., et al. Fenretinide corrects newly found ceramide deficiency in cystic fibrosis. Am J. Respir Cell. Mol. Biol. 2008;38:47–56. doi: 10.1165/rcmb.2007-0036OC. [DOI] [PubMed] [Google Scholar]
- Gunther W., Luchow A., Cluzeaud F., Vandewalle A., Jentsch T. J. ClC-5, the chloride channel mutated in Dent's disease, colocalizes with the proton pump in endocytotically active kidney cells. Proc. Natl. Acad. Sci. USA. 1998;95:8075–8080. doi: 10.1073/pnas.95.14.8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackam D. J., Rotstein O. D., Zhang W. J., Demaurex N., Woodside M., Tsai O., Grinstein S. Regulation of phagosomal acidification. Differential targeting of Na+/H+ exchangers, Na+/K+-ATPases, and vacuolar-type H+-ATPases. J. Biol. Chem. 1997;272:29810–29820. doi: 10.1074/jbc.272.47.29810. [DOI] [PubMed] [Google Scholar]
- Haggie P. M., Verkman A. S. Cystic fibrosis transmembrane conductance regulator-independent phagosomal acidification in macrophages. J. Biol. Chem. 2007;282:31422–31428. doi: 10.1074/jbc.M705296200. [DOI] [PubMed] [Google Scholar]
- Haggie P. M., Verkman A. S. Defective Organellar Acidification as a Cause of Cystic Fibrosis Lung Disease—Re-Examination of a Recurring Hypothesis. Am J. Physiol. Lung Cell Mol. Physiol. 2009a;296:L859–L867. doi: 10.1152/ajplung.00018.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggie P. M., Verkman A. S. Unimpaired lysosomal acidification in respiratory epithelial cells in cystic fibrosis. J. Biol. Chem. 2009b;284:7681–7686. doi: 10.1074/jbc.M809161200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara-Chikuma M., Wang Y., Guggino S. E., Guggino W. B., Verkman A. S. Impaired acidification in early endosomes of ClC-5 deficient proximal tubule. Biochem. Biophys. Res. Commun. 2005a;329:941–946. doi: 10.1016/j.bbrc.2005.02.060. [DOI] [PubMed] [Google Scholar]
- Hara-Chikuma M., Yang B., Sonawane N. D., Sasaki S., Uchida S., Verkman A. S. ClC-3 chloride channels facilitate endosomal acidification and chloride accumulation. J. Biol. Chem. 2005b;280:1241–1247. doi: 10.1074/jbc.M407030200. [DOI] [PubMed] [Google Scholar]
- Hart P. D., Young M. R., Gordon A. H., Sullivan K. H. Inhibition of phagosome-lysosome fusion in macrophages by certain mycobacteria can be explained by inhibition of lysosomal movements observed after phagocytosis. J. Exp. Med. 1987;166:933–946. doi: 10.1084/jem.166.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heda G., Tanwani M., Marino C. The ΔF508 mutation shortens the biochemical half-life of plasma membrane CFTR in polarized epithelial cells. Am J. Physiol. Cell Physiol. 2001;49:C166–C174. doi: 10.1152/ajpcell.2001.280.1.C166. [DOI] [PubMed] [Google Scholar]
- Hurtado-Lorenzo A., et al. V-ATPase interacts with ARNO and Arf6 in early endosomes and regulates the protein degradative pathway. Nat. Cell Biol. 2006;8:124–136. doi: 10.1038/ncb1348. [DOI] [PubMed] [Google Scholar]
- Jefferies K. C., Cipriano D. J., Forgac M. Function, structure and regulation of the vacuolar (H+)-ATPases. Arch. Biochem. Biophys. 2008;476:33–42. doi: 10.1016/j.abb.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch T. J. Chloride and the endosomal-lysosomal pathway: emerging roles of CLC chloride transporters. J. Physiol. 2007;578:633–640. doi: 10.1113/jphysiol.2006.124719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch T. J., Poet M., Fuhrmann J. C., Zdebik A. A. Physiological functions of CLC Cl− channels gleaned from human genetic disease and mouse models. Annu. Rev. Physiol. 2005;67:779–807. doi: 10.1146/annurev.physiol.67.032003.153245. [DOI] [PubMed] [Google Scholar]
- Kim J. H., Johannes L., Goud B., Antony C., Lingwood C. A., Daneman R., Grinstein S. Noninvasive measurement of the pH of the endoplasmic reticulum at rest and during calcium release. Proc. Natl. Acad. Sci. USA. 1998;95:2997–3002. doi: 10.1073/pnas.95.6.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstan M. W., Hilliard K. A., Norvell T. M., Berger M. Bronchoalveolar lavage findings in cystic fibrosis patients with stable, clinically mild lung disease suggest ongoing infection and inflammation. Am J. Respir. Crit. Care Med. 1994;150:448–454. doi: 10.1164/ajrccm.150.2.8049828. [DOI] [PubMed] [Google Scholar]
- Kumar K. G., et al. Site-specific ubiquitination exposes a linear motif to promote interferon-alpha receptor endocytosis. J. Cell Biol. 2007;179:935–950. doi: 10.1083/jcb.200706034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamothe J., Valvano M. A. Burkholderia cenocepacia-induced delay of acidification and phagolysosomal fusion in cystic fibrosis transmembrane conductance regulator (CFTR)-defective macrophages. Microbiology. 2008;154:3825–3834. doi: 10.1099/mic.0.2008/023200-0. [DOI] [PubMed] [Google Scholar]
- Lechardeur D., Xu M., Lukacs G. L. Contrasting nuclear dynamics of the caspase-activated DNase (CAD) in dividing and apoptotic cells. J. Cell Biol. 2004;167:851–862. doi: 10.1083/jcb.200404105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Wang T., Zhao Z., Weinman S. A. The ClC-3 chloride channel promotes acidification of lysosomes in CHO-K1 and Huh-7 cells. Am J. Physiol. Cell Physiol. 2002;282:C1483–C1491. doi: 10.1152/ajpcell.00504.2001. [DOI] [PubMed] [Google Scholar]
- Llopis J., McCaffery J. M., Miyawaki A., Farquhar M. G., Tsien R. Y. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc. Natl. Acad. Sci. USA. 1998;95:6803–6808. doi: 10.1073/pnas.95.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs G. L., Chang X. B., Kartner N., Rotstein O. D., Riordan J. R., Grinstein S. The cystic fibrosis transmembrane regulator is present and functional in endosomes. Role as a determinant of endosomal pH. J. Biol. Chem. 1992;267:14568–14572. [PubMed] [Google Scholar]
- Lukacs G. L., Rotstein O. D., Grinstein S. Phagosomal acidification is mediated by a vacuolar-type H(+)-ATPase in murine macrophages. J. Biol. Chem. 1990;265:21099–21107. [PubMed] [Google Scholar]
- Lukacs G. L., Rotstein O. D., Grinstein S. Determinants of the phagosomal pH in macrophages. In situ assessment of vacuolar H(+)-ATPase activity, counterion conductance, and H+ “leak.”. J. Biol. Chem. 1991;266:24540–24548. [PubMed] [Google Scholar]
- Machen T. E., Chandy G., Wu M., Grabe M., Moore H. P. Cystic fibrosis transmembrane conductance regulator and H+ permeability in regulation of Golgi pH. J. Pancreas. 2001;2:229–236. [PubMed] [Google Scholar]
- Machen T. E., Leigh M. J., Taylor C., Kimura T., Asano S., Moore H. P. pH of TGN and recycling endosomes of H+/K+-ATPase-transfected HEK-293 cells: implications for pH regulation in the secretory pathway. Am J. Physiol. Cell Physiol. 2003;285:C205–C214. doi: 10.1152/ajpcell.00008.2003. [DOI] [PubMed] [Google Scholar]
- Maritzen T., Keating D. J., Neagoe I., Zdebik A. A., Jentsch T. J. Role of the vesicular chloride transporter ClC-3 in neuroendocrine tissue. J. Neurosci. 2008;28:10587–10598. doi: 10.1523/JNEUROSCI.3750-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshansky V., Ausiello D. A., Brown D. Physiological importance of endosomal acidification: potential role in proximal tubulopathies. Curr. Opin. Nephrol. Hypertens. 2002;11:527–537. doi: 10.1097/00041552-200209000-00009. [DOI] [PubMed] [Google Scholar]
- Marshansky V., Futai M. The V-type H+-ATPase in vesicular trafficking: targeting, regulation and function. Curr. Opin. Cell Biol. 2008;20:415–426. doi: 10.1016/j.ceb.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman I., Fuchs R., Helenius A. Acidification of the endocytic and exocytic pathways. Annu. Rev. Biochem. 1986;55:663–700. doi: 10.1146/annurev.bi.55.070186.003311. [DOI] [PubMed] [Google Scholar]
- Moyer B. D., et al. A PDZ-interacting domain in CFTR is an apical membrane polarization signal. J. Clin. Invest. 1999;104:1353–1361. doi: 10.1172/JCI7453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muanprasat C., Sonawane N. D., Salinas D., Taddei A., Galietta L. J., Verkman A. S. Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J. Gen Physiol. 2004;124:125–137. doi: 10.1085/jgp.200409059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S., Ghosh R. N., Maxfield F. R. Endocytosis. Physiol. Rev. 1997;77:759–803. doi: 10.1152/physrev.1997.77.3.759. [DOI] [PubMed] [Google Scholar]
- Olson L. J., Hindsgaul O., Dahms N. M., Kim J. J. Structural insights into the mechanism of pH-dependent ligand binding and release by the cation-dependent mannose 6-phosphate receptor. J. Biol. Chem. 2008;283:10124–10134. doi: 10.1074/jbc.M708994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlowski J., Grinstein S. Emerging roles of alkali cation/proton exchangers in organellar homeostasis. Curr. Opin. Cell Biol. 2007;19:483–492. doi: 10.1016/j.ceb.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedemonte N., Lukacs G. L., Du K., Caci E., Zegarra-Moran O., Galietta L. J., Verkman A. S. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J. Clin. Invest. 2005;115:2564–2571. doi: 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picollo A., Pusch M. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature. 2005;436:420–423. doi: 10.1038/nature03720. [DOI] [PubMed] [Google Scholar]
- Pier G. B., Grout M., Zaidi T. S., Goldberg J. B. How mutant CFTR may contribute to Pseudomonas aeruginosa infection in cystic fibrosis. Am J. Respir. Crit. Care Med. 1996;154:S175–S182. doi: 10.1164/ajrccm/154.4_Pt_2.S175. [DOI] [PubMed] [Google Scholar]
- Poschet J. F., Boucher J. C., Tatterson L., Skidmore J., Van Dyke R. W., Deretic V. Molecular basis for defective glycosylation and Pseudomonas pathogenesis in cystic fibrosis lung. Proc. Natl. Acad. Sci. USA. 2001;98:13972–13977. doi: 10.1073/pnas.241182598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poschet J. F., Fazio J. A., Timmins G. S., Ornatowski W., Perkett E., Delgado M., Deretic V. Endosomal hyperacidification in cystic fibrosis is due to defective nitric oxide-cylic GMP signalling cascade. EMBO Rep. 2006;7:553–559. doi: 10.1038/sj.embor.7400674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poschet J. F., Skidmore J., Boucher J. C., Firoved A. M., Van Dyke R. W., Deretic V. Hyperacidification of cellubrevin endocytic compartments and defective endosomal recycling in cystic fibrosis respiratory epithelial cells. J. Biol. Chem. 2002;277:13959–13965. doi: 10.1074/jbc.M105441200. [DOI] [PubMed] [Google Scholar]
- Riordan J. R., et al. CFTR function and prospects for therapy. Annu. Rev. Biochem. 2008;77:701–726. doi: 10.1146/annurev.biochem.75.103004.142532. [DOI] [PubMed] [Google Scholar]
- Riordan J. R. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Roos A., Boron W. F. Intracellular pH. Physiol. Rev. 1981;61:296–434. doi: 10.1152/physrev.1981.61.2.296. [DOI] [PubMed] [Google Scholar]
- Root K. V., Engelhardt J. F., Post M., Wilson J. W., Van Dyke R. W. CFTR does not alter acidification of L cell endosomes. Biochem. Biophys. Res. Commun. 1994;205:396–401. doi: 10.1006/bbrc.1994.2678. [DOI] [PubMed] [Google Scholar]
- Rowe S. M., Miller S., Sorscher E. J. Cystic fibrosis. N. Engl. J. Med. 2005;352:1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- Sagel S. D., Chmiel J. F., Konstan M. W. Sputum biomarkers of inflammation in cystic fibrosis lung disease. Proc. Am Thorac. Soc. 2007;4:406–417. doi: 10.1513/pats.200703-044BR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M., Hanson P. I., Schlesinger P. Luminal chloride-dependent activation of endosome calcium channels: patch clamp study of enlarged endosomes. J. Biol. Chem. 2007;282:27327–27333. doi: 10.1074/jbc.M702557200. [DOI] [PubMed] [Google Scholar]
- Scheel O., Zdebik A. A., Lourdel S., Jentsch T. J. Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature. 2005;436:424–427. doi: 10.1038/nature03860. [DOI] [PubMed] [Google Scholar]
- Schenck S., Wojcik S. M., Brose N., Takamori S. A chloride conductance in VGLUT1 underlies maximal glutamate loading into synaptic vesicles. Nat. Neurosci. 2009;12:156–162. doi: 10.1038/nn.2248. [DOI] [PubMed] [Google Scholar]
- Seksek O., Biwersi J., Verkman A. S. Evidence against defective trans-Golgi acidification in cystic fibrosis. J. Biol. Chem. 1996;271:15542–15548. doi: 10.1074/jbc.271.26.15542. [DOI] [PubMed] [Google Scholar]
- Sharma M., Benharouga M., Hu W., Lukacs G. L. Conformational and temperature-sensitive stability defects of the delta F508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J. Biol. Chem. 2001;276:8942–8950. doi: 10.1074/jbc.M009172200. [DOI] [PubMed] [Google Scholar]
- Sharma M., et al. Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J. Cell Biol. 2004;164:923–933. doi: 10.1083/jcb.200312018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonawane N. D., Verkman A. S. Determinants of [Cl−] in recycling and late endosomes and Golgi complex measured using fluorescent ligands. J. Cell Biol. 2003;160:1129–1138. doi: 10.1083/jcb.200211098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonawane N. D., Zhao D., Zegarra-Moran O., Galietta L. J., Verkman A. S. Nanomolar CFTR inhibition by pore-occluding divalent polyethylene glycol-malonic acid hydrazides. Chem. Biol. 2008;15:718–728. doi: 10.1016/j.chembiol.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg B. E., Grinstein S. Assessment of phagosome formation and maturation by fluorescence microscopy. Methods Mol. Biol. 2007;412:289–300. doi: 10.1007/978-1-59745-467-4_19. [DOI] [PubMed] [Google Scholar]
- Steinberg B. E., Huynh K. K., Grinstein S. Phagosomal acidification: measurement, manipulation and functional consequences. Biochem. Soc. Trans. 2007a;35:1083–1087. doi: 10.1042/BST0351083. [DOI] [PubMed] [Google Scholar]
- Steinberg B. E., Touret N., Vargas-Caballero M., Grinstein S. In situ measurement of the electrical potential across the phagosomal membrane using FRET and its contribution to the proton-motive force. Proc. Natl. Acad. Sci. USA. 2007b;104:9523–9528. doi: 10.1073/pnas.0700783104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stobrawa S. M., et al. Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron. 2001;29:185–196. doi: 10.1016/s0896-6273(01)00189-1. [DOI] [PubMed] [Google Scholar]
- Suzuki T., Rai T., Hayama A., Sohara E., Suda S., Itoh T., Sasaki S., Uchida S. Intracellular localization of ClC chloride channels and their ability to form hetero-oligomers. J. Cell. Physiol. 2006;206:792–798. doi: 10.1002/jcp.20516. [DOI] [PubMed] [Google Scholar]
- Swiatecka-Urban A., et al. The short apical membrane half-life of rescued ΔF508-cystic fibrosis transmembrane conductance regulator (CFTR) results from accelerated endocytosis of ΔF508-CFTR in polarized human airway epithelial cells. J. Biol. Chem. 2005;280:36762–36772. doi: 10.1074/jbc.M508944200. [DOI] [PubMed] [Google Scholar]
- Teichgraber V., et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat. Med. 2008;14:382–391. doi: 10.1038/nm1748. [DOI] [PubMed] [Google Scholar]
- van Kerkhof P., Sachse M., Klumperman J., Strous G. J. Growth hormone receptor ubiquitination coincides with recruitment to clathrin-coated membrane domains. J. Biol. Chem. 2001;276:3778–3784. doi: 10.1074/jbc.M007326200. [DOI] [PubMed] [Google Scholar]
- Varga K., Goldstein R. F., Jurkuvenaite A., Chen L., Matalon S., Sorscher E. J., Bebok Z., Collawn J. F. Enhanced cell-surface stability of rescued ΔF508 cystic fibrosis transmembrane conductance regulator (CFTR) by pharmacological chaperones. Biochem. J. 2008;410:555–564. doi: 10.1042/BJ20071420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varghese B., et al. Polyubiquitination of prolactin receptor stimulates its internalization, postinternalization sorting, and degradation via the lysosomal pathway. Mol. Cell. Biol. 2008;28:5275–5287. doi: 10.1128/MCB.00350-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisz O. A. Acidification and protein traffic. Int. Rev. Cytol. 2003a;226:259–319. doi: 10.1016/s0074-7696(03)01005-2. [DOI] [PubMed] [Google Scholar]
- Weisz O. A. Organelle acidification and disease. Traffic. 2003b;4:57–64. doi: 10.1034/j.1600-0854.2003.40201.x. [DOI] [PubMed] [Google Scholar]
- Wu M. M., Grabe M., Adams S., Tsien R. Y., Moore H. P., Machen T. E. Mechanisms of pH regulation in the regulated secretory pathway. J. Biol. Chem. 2001;276:33027–33035. doi: 10.1074/jbc.M103917200. [DOI] [PubMed] [Google Scholar]
- Yates R. M., Hermetter A., Russell D. G. The kinetics of phagosome maturation as a function of phagosome/lysosome fusion and acquisition of hydrolytic activity. Traffic. 2005;6:413–420. doi: 10.1111/j.1600-0854.2005.00284.x. [DOI] [PubMed] [Google Scholar]
- Zeitlin P. L., Lu L., Rhim J., Cutting G., Stetten G., Kieffer K. A., Craig R., Guggino W. B. A cystic fibrosis bronchial epithelial cell line: immortalization by adeno-12-SV40 infection. Am J. Respir. Cell. Mol. Biol. 1991;4:313–319. doi: 10.1165/ajrcmb/4.4.313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.