

Abstract
Ribonuclease P (RNase P) complexed with an external guide sequence (EGS) represents a novel nucleic acid-based gene interference approach to modulate gene expression. This enzyme is a ribonucleoprotein complex for tRNA processing. In E. coli, RNase P contains a catalytic RNA subunit (M1 ribozyme) and a protein subunit (C5 cofactor). EGSs, which are RNAs derived from natural tRNAs, bind to a target mRNA and render the mRNA susceptible to hydrolysis by RNase P and M1 ribozyme. When covalently linked with a guide sequence, M1 can be engineered into a sequence-specific endonuclease, M1GS ribozyme, which cleaves any target RNAs that base pair with the guide sequence. Studies have demonstrated efficient cleavage of mRNAs by M1GS and RNase P complexed with EGSs in vitro. Moreover, highly active M1GS and EGSs were successfully engineered using in vitro selection procedures. EGSs and M1GS ribozymes are effective in blocking gene expression in both bacteria and human cells, and exhibit promising activity for antimicrobial, antiviral, and anticancer applications. In this review, we highlight some recent results using the RNase P-based technology, and offer new insights into the future of using EGS and M1GS RNA as tools for basic research and as gene-targeting agents for clinical applications.
Keywords: RNase P, External guide sequence, ribozyme, gene targeting, antisense, gene therapy
1. Introduction
1.1. Nucleic acid-based gene targeting approaches for modulating gene expression
Since their discovery, the idea of using catalytic nucleic acids for the purpose of gene inactivation has been highly touted [1, 2]. Derived from the originally discovered function of self-cleaving and catalytic RNAs including group I introns and ribonuclease P (RNase P) RNAs [3, 4], a wide variety of useful applications have been proposed and tested. Correcting genetic defects and targeted downregulation of pathogenic genes have been attempted, some with moderate success and other still at work. Of these strategies that range from commonly-used antisense oligonucleotides to over-expressing competitive RNA sequences to more recently discovered RNA interference technology, ribozymes stand out among the most extensively studied nucleic acid-based gene targeting approaches [1, 2]. In fact, the abilities of these agents have been well characterized and engineered in a way that they can block expression of a wide variety of genes (any given targets) in a sequence-specific and highly potent manner, bringing them even closer to the clinic [1].
Ribozymes exist as naturally occurring catalytic RNAs, expressed in a wide range of living organisms [5, 6]. In this review, we focus on RNase P and its derivatives and highlight some of the most recent studies in utilizing RNase P for the development of nucleic acid-based gene therapy, with a special focus on antiviral applications.
1.2. Ribonuclease P
RNase P, an essential cellular enzyme discovered about 35 years ago [7], is a key modulator in tRNA biogenesis; it is involved in the multistep processing of tRNA primary transcripts, being responsible for the removal of 5′ leader sequence from precursor tRNA transcripts (Figure 1) [8–10]. Other reported substrates of RNase P includes transfer-messenger RNA [11], bacterial operon RNAs [12, 13], riboswitches [14], phage regulatory RNAs [15], and signal recognition particle RNAs [16]. Recently, RNase P has been shown to play an important role in RNA polymerase III transcription, suggesting that transcription and early processing of tRNA may be coordinated [17]. The fundamental nature of RNase P’s presence is reflected by the fact that this enzyme has been found in every living cells across all kingdoms of life. The enzyme consists of a single RNA subunit providing the catalytic core and one protein subunit in bacteria, typically 4 in archaea and up to 10 protein subunits in eukarya [8, 18]. The presence of ubiquitous homologues of RNase P RNA subunits found across the most primitive phylogenetic domains suggests that the RNA existed in some of the earliest forms of life, even before the differentiation of the phylogenetic kingdoms.
Figure 1.
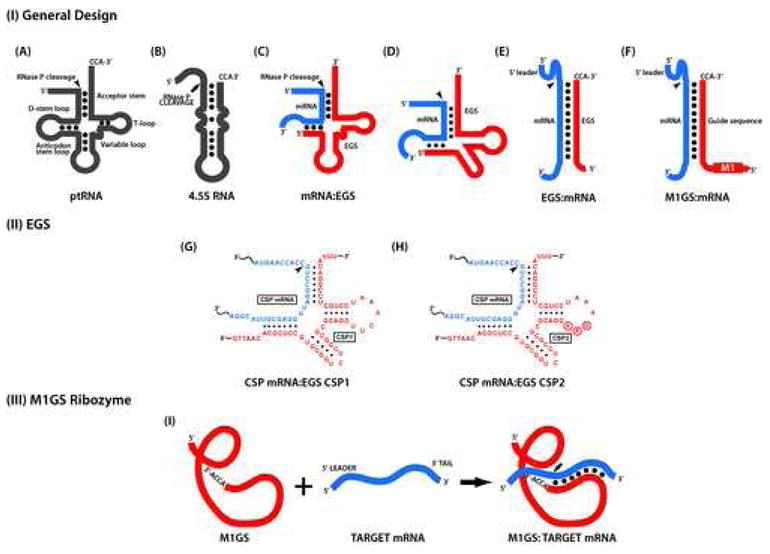
(A–B) Representation of natural substrates (pre-tRNA (A) and 4.5S RNA (B)). (C–E) A hybridized complex of a target RNA (e.g. mRNA) and an EGS that resembles a part of structure of a tRNA and can be cleaved by RNase P. (D) results from (C) by deleting the anticodon domain of the EGS, which is dispensable for EGS targeting activity, while (E) results from (D) by further deleting the D stem/loop and variable regions. Substrates in (C) and (D) can be cleaved by human RNase P and M1 ribozyme. In contrast, the stem structure in (E) can only serve as a substrate for M1 RNA and can not be cleaved by human RNase P. (F) A complex formed between an M1GS ribozyme and a target mRNA substrate. (G, H) Complexes between the HCMV capsid scaffolding protein (CSP) mRNA and EGS CSP1 and CSP2, respectively [82]. (I) Representation of an M1GS RNA construct to which a target RNA has hybridized. The arrow shows the site of the cleavage by RNase P and M1 RHA.
All bacterial RNase P enzymes consist of a single RNA and a single protein subunit. RNase P from Escherichia coli is composed of M1 RNA subunit of 377 nucleotides in length and a basic, 14 kDa C5 protein subunit [8, 9]. While both RNA and protein subunits are required for in vivo activity, increased ionic concentration can activate catalysis by the RNA subunit alone in vitro [8]. Such phenomenon can be explained by the idea that a high concentration of salt facilitates screening of electrostatic repulsion in the RNA subunit, where the active structure is otherwise maintained by the presence of the protein subunit. The role of the protein subunit has been extensively investigated. Early experimental evidence indicated that the protein helped stabilization of tertiary structures, while later studies indicated that it could be involved in enhancing pre-tRNA specificity over that of mature tRNAs or pre-organizing metal ion binding sites relevant to catalysis and E-S formation [3, 19–26].
Eurakyal RNase P enzymes are much more complex than their bacterial counterparts. The H1 RNA of human RNase P associates with at least ten different protein subunits [10, 27]. While the specific functions of the protein subunits have not been completely elucidated yet, some of the potential roles include RNA stabilization, localization [28], and contribution to the active site of the enzyme [29].
2. Gene targeting strategy based on RNase P
2.1. Substrate recognition by RNase P and M1 RNA of E. coli
How RNase P recognizes its substrates has been a focus of intensive research, as its understanding is critical in revealing the mechanism of the enzyme’s activity. In the case of both RNase P holoenzyme and M1 RNA of E. coli, it has been shown that they primarily recognize the structure of the substrates, not their sequences (Figure 1A and B). In fact, early studies have shown that shortened model substrates that retain some structural elements of a pre-tRNA (natural substrate of RNase P) can be recognized and hydrolyzed by RNase P and M1 RNA (e.g. those similar structures in Figure 1C–D) [30, 31]. More specifically, deletion analysis of a tRNA substrate has shown that M1 RNA is capable of cleaving a “minimal model substrate” composed of the acceptor stem and T stem/loop, the 3′ CCA sequence, and the 5′ leader sequence of a pre-tRNA (e.g. the similar structure in Figure 1E) [30, 31].
2.2. External guide sequence (EGS)
The components of the model substrates can be simplified into two sequences: the 5′ proximal sequence (the 5′ leader and 5′ proximal acceptor stem sequence) and the 3′ proximal sequence (the 3′ proximal acceptor stem sequence and the rest tRNA-like sequence) (Figure 1C–E, G–H). Of these, the 3′ proximal sequence has been termed external guide sequence (EGS) [30]. The EGS guides either RNase P or M1 RNA to recognize the site of cleavage by base pairing to the targeted sequence (Figure 1C–E, G–H) [30]. Studies have shown that EGSs can be used for down-regulation of target genes in bacterial and mammalian cells [32–36]. Altman and co-workers first demonstrated highly specific targeting capability of EGS in their studies where the expression of β-galactosidase and alkaline phosphatase activity in E. coli was suppressed to 50–60% in bacterial strains that expressed EGSs targeting these genes [32]. No suppression has been observed in strains that carried non-specific EGSs, confirming high specificity of the EGS technology [32]. In another study, drug-resistant strains of E. coli have been converted to drug-sensitive strains by targeting the drug resistance gene with EGS and RNase P [33]. A different design of shorter EGS constructs has also been reported, in which the targeted mRNA resembles the 5′ leader sequence, 5′ acceptor stem, the variable region, and the 5′ T-stem/loop of a tRNA while the EGS of only 15–20 nucleotides looks like the 3′ acceptor stem and 3′ T-stem/loop regions [37].
Perhaps one of the biggest advantages of using RNase P is that the enzyme is endogenously expressed and active at all stages of the cell cycle. The enzyme is ubiquitous and essential as it is responsible for processing all tRNA molecules [8–10]. In addition, EGS-directed cleavage of target RNA by RNase P is highly specific and does not exhibit significant non-specific cleavage, which is commonly seen with RNase H-mediated cleavage induced by conventional antisense phosphorothioate oligonucleotides [8, 38]. Some of the fundamental issues regarding the general efficacy of EGS molecules and how to improve their inhibitory effects (e.g. by increasing catalytic efficiency or relieving rate-limiting steps) need to be addressed, and we believe further studies will reveal whether EGS technology is indeed practical for clinical applications.
2.3. M1GS RNA
Development of gene-targeting strategies based on M1 RNA and EGS has led to the development of a more efficient and easy-to-make agent, called M1GS RNA [34]. M1GS RNA is simply constructed through 3′ extension of M1 RNA by a guide sequence, which base pairs with the target and contains an unpaired 3′-NCCA end as present in natural tRNA substrates to allow efficient cleavage by the tethered M1 moiety (Figure 1F). This design is based on the idea that the guide sequence binds to its target mRNA and directs M1RNA, which is in close proximity due to covalent attachment to the guide sequence, to the site of cleavage (Figure 1I) [34, 39]. Subsequent studies in our as well as other laboratories have demonstrated that M1GS RNA is highly effective in cleaving its target mRNAs and blocking gene expression [34, 40, 41].
It will be interesting to determine how M1GS ribozymes efficiently function in human cells in the absence of C5 protein, which is the cofactor of M1 RNA from E. coli and is not found in human cells. It is generally believed that the regions of the catalytic M1 RNA domain in these M1GS RNAs, which may be homologous to those of the RNA subunit of human RNase P [42], interact with cellular proteins, including those associated with human RNase P [8, 34]. This hypothesis is consistent with previous observations that several “RNA chaperone” proteins have been shown to stimulate the activity of hammerhead and group I ribozymes by either facilitating folding of the ribozyme or dissociation of the product [43]. It is possible that these proteins may enhance M1GS activity in a similar manner as C5 protein by stabilizing the structure of the ribozymes, participating in substrate recognition, and contribution to the E-S formation. Further studies on potential interactions between M1GS RNAs and cellular proteins will provide insight into how a M1GS ribozyme functions in cultured cells.
3. General design of RNase P ribozymes and external guide sequences
In order to generate highly potent ribozymes, it is crucial to select targets that are appropriate for cleavage to occur. While M1 RNA is shown to be catalytically active in vitro against target RNAs, the situation may not be the same in cells or in vivo, as the RNA secondary structure and protein association can easily hamper the recognition and catalysis by M1GS RNA or RNase P. To address this issue, careful selection of a target region is necessary. Accessibility of the target mRNAs should be mapped in vitro and in vivo, using numerous methods. For example, we have utilized in vivo mapping of accessible regions in some target mRNAs with dimethyl sulfate (DMS) [41, 44]. DMS methylates N7 of guanine, N1 of adenine, and N3 of cytosine, of which the latter two can be examined by primer extension assay (transcription terminates at the base immediately before the modified one) [34, 45]. We postulated that the region that is exposed to DMS would also be accessible to binding by S RNA. Another method that has been commonly used is based on in vitro digestion of radiolabeled target RNA with RNase T1 [46] to probe regions that are exposed to enzymatic attack, based on the assumption that such regions will be accessible to M1GS RNA and RNase P as well [32, 47].
While some of the more empirical approaches are effective in determining site accessibility, it is also possible and often helpful to use in silico approximation [48, 49]. The substrate RNA, which would be the mRNA target, may be analyzed by RNA folding software programs that approximate secondary structures from sequence information. Using this approach, some of the more thermodynamically stable regions in the target mRNA could be identified [48, 49]. Target sites buried in these regions would not be suitable as potential targets of ribozymes. In silico prediction is limited by, however, the computational power and technique for such folding programs as the size of RNA increases. Also, gross approximation does not take into account the existence of tertiary structure and RNA-protein interactions, both of which play an important role for the accessibility of the RNA molecule.
Biological significance of the target mRNA could also provide useful information. If RNA-RNA interactions or RNA-protein interactions are known to be present at certain target sites, these interactions are predicted to reduce target accessibility. Indeed, considering such functional interactions contributed to the success of approaches screening for ribozyme targets in the HIV-1 genome. Most importantly, gag and tat genes have been used as targets, along with the 5′-leader region and Ψ packaging sites [50–56].
Site accessibility remains a very important issue that needs to be addressed in order to achieve effective ribozyme-mediated gene suppression. So far, it has been less straightforward and ambiguous at times. As mentioned above, in silico approaches are simple and direct methods to map accessible sites, while the results may not show high correlation with other in vitro or in vivo data. However, as technologies for in silico analysis become more mature, we believe that it will prove to be a valuable approach aside from in vivo mapping and in vitro screening with full length mRNA for the design of gene-targeting ribozymes and external guide sequences.
The flanking sequence of the mRNA region to be targeted by RNase P or M1GS RNA should also exhibit several sequence features that need to be present in order to interact with an EGS and RNase P to achieve efficient cleavage. For M1GS targeting, these features include that the nucleotides 3′ and 5′ adjacent to the site of cleavage are a guanosine and a pyrimidine, respectively [57]. An additional sequence feature, in which a U is 8 nucleotides downstream from this cleavage site, is needed for human RNase P targeting [58]. These sequence elements interact with the EGS to facilitate the formation of the mRNA-EGS complex into a tRNA-like structure. Furthermore, the interactions of these elements with RNase P are critical for recognition and cleavage by the enzyme [58]. In their studies of the unique design of EGS constructs, Werner et al. have also investigated the effects of different sequences of the EGSs on their targeting activity [37].
4. In vitro selection of ribozymes
In vitro selection refers to a process where enrichment of molecules with desired properties is achieved through iterative cycles of isolation and amplification [59–61]. Functional RNA molecules such as ribozymes and EGS RNAs are great candidates for the technique as it is relatively easy to generate a large pool of mutagenized or randomized template DNA library and transcribe them in vitro to make the RNA sequence library. In vitro selection has been originally used in isolating RNA aptamers that bind to specific targets [59–61]. Since its introduction, the technology found itself in many different laboratories, used for different purposes from enhancing catalytic activity to enzyme engineering to isolating nucleic acids with novel functions.[58, 62–64]
We have used the in vitro selection technique to successfully isolate highly active M1GS RNA variants targeting the mRNA sequence encoding the thymidine kinase (TK) of herpes simplex virus type 1 (HSV-1) [65]. Our initial library of ribozymes contained random mutations in regions that are known to be conserved across all species of RNase P catalytic RNAs and are important for catalysis and substrate binding [66–68]. The in vitro evolution process was carried out in a series of steps that included (1) annealing of M1GS RNA pool with a 5′ biotinylated substrate, (2) binding the complex to streptavidin-agarose column, (3) M1GS RNA-mediated cleavage of the substrate in the presence of divalent ion-containing buffer, (4) recovering M1 RNA with denaturing gel electrophoresis, (5) synthesis of cDNA copies of RNA molecules with RT-PCR, followed by (6) in vitro transcription of the generated cDNAs with T7 RNA polymerase (Figure 2) [65, 69, 70]. The sequences isolated after several rounds of selection were cloned and determined. We selected representative sequences from each round of selection and assayed their catalytic efficiency to assess the progress of the selection process. Using this system, we were able to isolate RNase P ribozyme variants that are highly efficient in cleaving target mRNA in vitro and potently inhibiting the expression of target mRNA in cultured cells.
Figure 2.
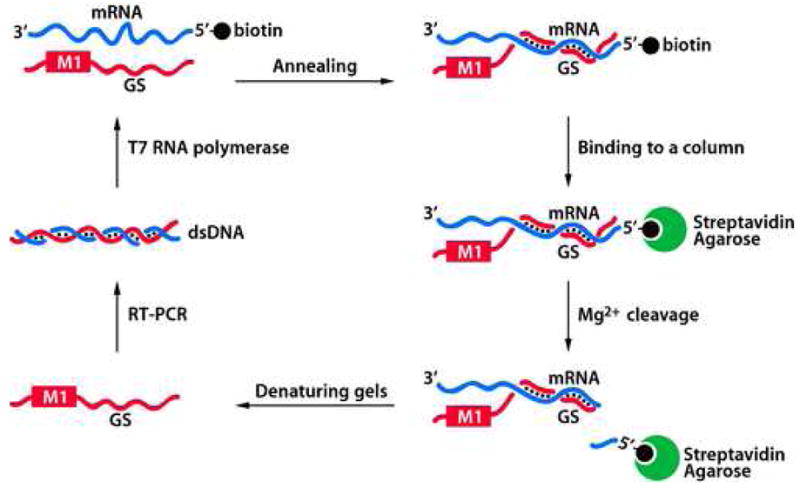
Schematic representation of the in vitro evolution procedure for the generation of highly active M1GS RNA ribozyme variants that specifically cleave a target mRNA.
5. Applications
Recent developments in synthetic nucleotide chemistry and gene expression technologies have augmented the feasibility of nucleic acid-based therapeutic applications [1, 2]. Blocking gene expression at the RNA level in a highly specific and potent fashion opens up many possibilities in molecular biology and pharmacology. Here, we summarize the recent progress on using RNase P and M1GS RNA for gene targeting applications.
5.1. Antibacterial agents
An elegant study by Altman and colleagues applied EGS technology to selectively inhibit expression of several essential bacterial genes, successfully achieving an antimicrobial effect [35]. More notably, carefully designed EGSs were shown to be highly specific and were capable of species-specific targeting. Sequences of mRNA differing between E. coli and S. typhimurium, but encoding identical proteins were both targeted. Results from this study demonstrated that the expression of EGSs complementary to E. coli mRNAs resulted in reduction of the expression of the target mRNAs in E. coli, while no effects were shown against S. typhimurium mRNAs and vice versa. If EGS technology can be used in antimicrobial therapy, its ability to achieve species-specific inhibition of bacterial viability could become very useful in circumventing the current limitation of narrow spectrum antimicrobials in inhibiting commensal nonpathogenic bacteria.
More recently, McKinney, Altman, and co-workers further attempted to use the EGS technology to disrupt S. typhimurium type III secretion system involved in invasion of host cells [71]. Using inducibly expressed EGSs targeting invB and invC resulted in directed cleavage of the target mRNAs by endogenous RNase P, leading to effective depletion of the corresponding proteins. Type III secretion was reduced and host cell invasion was blocked, strongly implicating the possibility of using EGS technology in antibacterial strategies [71].
5.2. Antiviral agents
Utilization of RNase P and M1 RNA for the development of antiviral agents has been extensively pursued in several laboratories, including ours, for blocking infection of human immunodeficiency virus (HIV) [72, 73], human influenza virus [74], and three human herpesviruses such as human cytomegalovirus (HCMV) [41, 69], herpes simplex virus 1 (HSV-1) [65, 70, 75], and Kaposi’s sarcoma-associated herpesvirus (KSHV) [44]. Herpes simplex virus 1 (HSV-1) is a causative agent of cold sores and encephalitis in newborns [76]. HCMV is the leading viral cause of birth defects in the United States and accounts for a significant portion of mortality associated with organ transplant patients [77]. KSHV is believed to be the causative agent of the leading AIDS-associated neoplasms such as Kaposi’s sarcoma and primary effusion lymphoma [78].
Our early attempts at using ribozymes for antiviral applications focused on targeting the mRNA encoding ICP4 of HSV-1, the major transcriptional activator [41]. ICP4 is one of the immediate-early genes expressed by HSV-1 and is responsible for expression of many viral early and late genes [76]. We showed that M1GS ribozyme targeting ICP4 is highly specific and potent in downregulating the expression of the target gene, as 80% of ICP4 expression could be suppressed, resulting in nearly 1000-fold reduction in viral growth [41]. The specificity of M1GS in targeting the mRNA of choice was verified as it did not affect the expression of other immediate-early genes. More recently, we showed that a single ribozyme targeting multiple viral mRNAs can be even more effective in inhibition of viral replication in cultured cells. We designed a M1GS ribozyme that targets the overlapping region of two HCMV mRNAs, coding for the viral protease (PR) and assembly protein (AP), both of which are essential for viral replication [79]. We observed specific and potent inhibition of viral gene expression, leading to a reduction of more than 2000-fold in viral titer in cells expressing the ribozymes.
To further enhance the efficiency of ribozymes, we employed an in vitro selection procedure (Figure 2) and selected for RNase P ribozyme variants that efficiently cleaved a different target, the mRNA encoding thymidine kinase (TK) of HSV-1 [65, 80]. Our method of selecting catalytically superior variants proved to be successful, as we were able to generate mutant ribozymes that exhibited at least 20-fold higher cleavage efficiency (represented as kcat/Km) than the wild type M1 RNA. Accordingly, expressing one of these ribozyme sequences in cells infected by HSV-1 resulted in highly reduced levels of TK mRNA and protein products, where inhibition of up to 99% has been observed [65, 80]. Another M1GS ribozyme variant showed a comparably potent activity in inhibiting TK expression [70].
EGS found its application in antiviral therapeutics as well. Recent studies in our laboratory and others have shown that using EGS to recruit endogenous RNase P for targeted degradation of viral mRNA is highly effective. For example, Altman and colleagues constructed EGSs that targeted the mRNAs coding for the polymerase and nucleocapsid protein of human influenza virus, which are essential for viral replication [74]. The EGS RNAs are efficient in inducing RNase P to cleave the target viral mRNAs in vitro. The expression of these EGSs in human cells led to inhibition of the expression of the target genes and blockage of viral replication. This study further provided the direct evidence that targeting two different mRNAs simultaneously by the EGS technology inhibits viral growth more effectively than targeting of one mRNA only [74].
By constructing EGS RNAs against the sequence of the HIV RNA genome, Hnatyzyn and co-workers have elegantly demonstrated that RNase P is highly effective in inhibiting HIV gene expression and replication in cultured cells [72, 73]. Expression of the constructed EGS RNAs in human heterogeneous T cell cultures upon HIV challenge was able to maintain CD4 levels, devoid of cytopathology, and did not produce significant level of HIV p24 through 30 days infection. Impressively, the cells that expressed the EGSs were resistant to HIV clinical isolates from clades A, B, C, and F [72, 73]. These results provide the first direct evidence that RNase P-associated EGSs may represent a new class of potential therapeutic agents for anti-HIV therapy.
EGS RNAs were also highly active in targeting RNase P to cleave HSV-1 TK mRNA in vitro. Reduction in both TK mRNA and protein levels was observed when EGSs were expressed in human cells infected with HSV-1 [81]. When EGS RNAs targeting HCMV mRNAs were expressed in human cells infected with HCMV, they were also effective in inducing RNase P to inhibit the expression of the target mRNAs and block HCMV growth [82]. More recently, EGS-based gene interference strategy has been attempted in blocking gene expression and growth of yet another member of the herpesvirus family, namely Kaposi’s sarcoma-associated herpesvirus (KSHV). Chemically modified EGS molecules were constructed to target the mRNA encoding KSHV immediate-early transactivator called Rta [44]. Exogenous administration of 2′-O-methyl-modified EGS to KSHV-infected human primary-effusion lymphoma cells significantly inhibited Rta expression and subsequently resulted in 150-fold reduction in viral growth [44].
In vitro selection has been utilized in EGS technology as well. EGS RNA molecules targeting HSV-1 TK mRNA have been subjected to many rounds of selection and highly active EGS variants that exhibited up to 35 times higher activity than the natural tRNA substrate have been isolated [75]. One of the selected EGS RNAs was also highly effective in human RNase P-mediated inhibition of TK expression in HSV-1 infected cells [75]. Moreover, the selected molecules were also used to construct EGSs that target the overlapping region of the mRNAs coding for HCMV essential transcription regulatory factors IE1 and IE2. These constructed EGSs exhibited a substantially higher activity than the EGS derived from a natural tRNA in down-regulating gene expression [83]. In cultured cells infected with HCMV, the expression of these EGS molecules resulted in a reduction of about 93% in IE1/IE2 gene expression, and a reduction of 3000 fold in viral growth. These results demonstrated the feasibility of developing effective EGS RNA variants for antiviral applications by using in vitro selection procedures.
To harness the existing system even further, multiple ribozymes can be constructed to target several mRNAs that encode viral proteins as well as host proteins involved in establishment of viral infection [84, 85]. Complete suppression of viral replication and subsequent disease progression may be achievable with this approach.
5.3 Anticancer agents
The RNase P ribozyme has also found its way into anti-cancer strategies. In a series of elegant experiments, Sánchez-García and colleagues constructed M1GS ribozymes to specifically cleave chimeric RNA molecules generated due to chromosomal abnormalities [40]. They used a well-characterized model of BCR and ABL genes where aberrant translocation results in BCR-ABL oncogenes that cause chronic myelogenous leukemia and acute lymphoblastic leukemias. M1 RNA with a guide sequence that recognized the oncogenic mRNA at the fusion site appeared to be highly effective and specific in cleaving the target mRNA in vitro and blocking the effect of BCR-ABL function in cultured mammalian cells [40].
Using the EGS technology, Ma, Stein, and colleagues have successfully induced RNase P-mediated cleavage of the mRNA that encodes protein kinase C-α (PKC-α) [38]. In their study, EGSs equipped with 2′-O-methyl modification for enhanced stability were exogenously administered into T24 bladder carcinoma cells for specific downregulation of PKC-α expression. Modified EGSs that targeted the 3′ untranslated region of PKC-α were highly potent. No non-specific cleavage, which is usually associated with RNase H-induced reactions, was found, providing direct evidence that RNase P-mediated cleavage induced by EGS is highly specific in targeting its mRNA [38]. In addition, potent downregulation of antiapoptotic protein bcl-xL was observed, further suggesting a general applicability of the EGS technology for anticancer applications [38].
5.4 A research tool for studies of gene function
Extensive studies have also been carried out to develop M1GS RNA and RNase P as a research tool for studies of gene function. Recently, M1GS RNA was used to study the mechanism of HCMV capsid maturation and the role of HCMV protease (PR) in viral replication [86]. A M1GS ribozyme variant that targeted the HCMV PR mRNA was generated and expressed in human cells to produce viral capsids that lack PR. Expression and processing of scaffolding proteins and viral encapsidation were significantly reduced as a consequence of the inhibition of the PR expression. High-resolution electron cryomicroscopy of these PR-minus capsids showed that the PR is required for DNA encapsidation and subsequent maturation steps for production of infectious virion particles [86].
6. Advantage and disadvantage of M1GS and RNase P-EGS technology
Traditional antisense technology relies on cellular RNase H to cleave an RNA-DNA hybrid in order to degrade the mRNA target [1, 2]. However, non-specific cleavage at non-targeted sites is a potential problem, as RNase H does not require a 100% complementary duplex to degrade hybridized mRNA and only six or seven contiguous base pairs with the target RNA are required to direct cleavage [1, 2]. Compared to conventional antisense DNA and RNA, ribozymes such as M1GS RNA can be designed to be highly specific in cleaving its targeted mRNA [8, 40]. In an elegant study, M1GS ribozyme has been shown to be specific in cleaving one substrate over another even though the two substrates share the first nine contiguous base pairs complementary to the guide sequence [40].
When comparing M1GS ribozyme to other ribozymes, such as hammerhead and hairpin, M1GS ribozyme possesses several unique features as a gene targeting tool. First, M1GS ribozyme can fold into a defined active conformation in the absence of its substrates. Second, while M1GS can cleave any designed sequence, hammerhead and hairpin ribozyme are limited by the requirement for the presence of specific nucleotide sequence (-GUX-) in the target mRNA for the cleavage to occur [1, 5]. Furthermore, a single point mutation in the required GUX sequence could render the ribozymes ineffective for target mRNA cleavage. The low sequence requirements at the cleavage site provide M1GS ribozyme strategies with the flexibility to be used against almost any target, including positionally fixed target sites such as the fusion junction of two chromosomes resulting in an oncogenic chimeric mRNA [40]. Third, the small ribozymes may have the disadvantage to be either rather inefficient under physiological conditions (e.g. in the case of the minimal hammerhead ribozymes) or to catalyze ligation quite efficiently (e.g. in the case of natural hammerhead ribozymes or the hairpin ribozymes); ligation will limit the efficiency of target cleavage, a clear disadvantage relative to RNase P and M1GS RNAs which do not catalyze the reverse reaction.
In recent years, the use of the RNA interference (RNAi) approach to degrade mRNA associated with human diseases has been the focus for nucleic acids-based gene interference studies [1]. RNAi has the advantage of utilizing the cellular machinery in its process to knockdown mRNA and can be effective in small concentration. However, the siRNA technology may “sequester or misguide” a cellular machinery which may have consequences for cell function not foreseeable at present. It will be interesting to compare the activity and effectiveness of M1GS RNA and RNAi approaches for knocking down gene expression in human cells.
Compared to other nucleic acid-based gene interference approaches, the EGS technology with the use of endogenous human RNase P exhibits several unique and attractive features as a gene-targeting tool. First, the mechanism of the EGS technology for degradation of a specific mRNA is different from other RNA- or DNA-based gene-targeting approaches. It uses the endogenous RNase P, which is one of the most ubiquitous, abundant, stable and efficient enzymes in all type of cells [6, 8, 85]. This essential enzyme is highly expressed (5×104 copies per cell) and is responsible for the processing of all tRNA precursors that account for approximately 2% of total cellular RNA [6]. The action of RNase P with the EGS will result in irreversible cleavage of the target mRNA in a highly efficient catalytic fashion. Second, the sequence specificity of the EGS technology is governed by two different types of interactions between the EGS and the target mRNA: (1) the base-pairing interactions in which the sequence of 12 nucleotides in the EGS hybridizes with the target mRNA, and (2) the interactions between the target mRNA and the other part of the EGS sequence (equivalent to the T-stem and T-loop, and variable regions of a tRNA) which are required for folding of the RNase P-recognizable tertiary structure. Thus, the EGS-based technology is highly specific and does not generate nonspecific “irrelevant cleavage” that is observed in RNase H-mediated cleavage induced by conventional antisense phosphorothioate molecules [36, 38]. Third, EGSs exhibit little sign of cytotoxicity because cells expressing these molecules for more than 40 days appear to be normal [36, 38, 44, 81].
As with any gene therapy design, stability and delivery of the agents remain a big concern. The delivery problem affects the siRNA technology to the same extent as the EGS technology. For stability, the ribozymes and EGSs could be chemically synthesized with 2′ hydroxyl modification and/or phosphorothioates to resist cellular endonucleases [87]. As an alternative to the viral vector approach, smaller ribozymes can be delivered ex vivo by encapsulating them in liposomes or other biodegradable polymeric matrix [37, 38, 44]. In the case of M1GS, chemical synthesis of a functional active ribozyme is at present technically difficult and economically impractical due to its large size (~400 nucleotides). The amount of M1GS ribozyme required for ex vivo delivery is similarly unattractive. Thus, endogenous and stable expression of M1GS ribozyme by viral vectors remains one of the most practical choices for M1GS expression and delivery. EGSs are small molecules of 25–60 nucleotides. Therefore, the EGSs can be easily synthesized and modified chemically [37, 38, 44]. Thus, an EGS can be delivered directly (in naked form or with the aid of liposomes) to cells as well as delivered by expression vectors such as retroviral vectors.
7. Future perspectives
Recent studies have demonstrated that M1GS ribozymes and RNase P complexed with EGS RNAs are efficient and specific in cleaving an mRNA sequence in vitro, and are effective in down-regulating the expression of both cellular and viral genes in cultured cells. Moreover, ribozyme and EGS variants that are more active and effective in inhibiting gene expression can be generated using in vitro selection procedures. Detailed biochemical characterization of these variants has provided significant insights into the mechanism of how M1GS ribozymes and RNase P complexed with EGSs cleave a mRNA substrate. These results should generate guidelines for constructing highly effective M1GS ribozymes and EGS RNAs for gene-targeting applications.
To develop M1GS and EGS RNAs for gene therapy applications, the efficacy of the ribozyme and RNase P complexed with EGSs needs to be evaluated in animal models and ultimately, in human clinical trials. Several issues like ribozyme/EGS delivery, stability, and colocalization may need to be addressed in order to achieve successful ribozyme/EGS-based gene therapy against viral infections and other human diseases. Meanwhile, the M1GS and RNase P-EGS approaches represent two different promising gene-targeting strategies for studying gene functions. If the sequence of the target is known, ribozymes and EGSs can be readily designed to shut down the expression of the desired gene. M1GS ribozymes/EGSs could also be employed to elucidate the roles of genetic messages (e.g. expressed sequence tags) that currently have unknown functions. Further studies on the biochemistry of M1GS ribozyme and RNase P complexed with EGS in vitro and on its activity in cultured cells and in animal models should facilitate the development of these agents as novel gene-targeting agents for both in vitro and in vivo applications.
8. Conclusions
Among the wide variety of methods to manipulate gene expression, which have been introduced in recent years for various purposes ranging from simple gene inactivation to gene therapy, RNase P-based technology is unique. In the case of EGS technology, gene inactivation is mediated by cleavage of the target mRNA by endogenous RNase P, which is a highly active enzyme ubiquitously present inside a living cell. The EGS-based technology is highly specific and potent in accomplishing its task, without the risk of non-specific cleavage reactions that have been associated with some of the other technologies such as RNase H-mediated antisense DNA/RNA strategies.
Equally promising is the potential use of M1GS in gene targeting applications, especially in antiviral application. Similar to the EGS technology, M1GS possesses high specificity and efficacy to suppress expression of target genes. While delivery of these antiviral agents in vivo remains an important question to be addressed, several strategies from modification of nucleotides and stable transduction using viral gene therapy vectors for ex vivo delivery can be considered. Further studies of these issues, as well as investigation aimed to understand the biochemistry of RNase P and M1GS and increase their cleavage efficiency and sequence specificity, will greatly facilitate the development of these two unique gene-targeting agents to be used for both basic research and clinical applications.
Acknowledgments
We apologize to the many authors whose publications were, due to space constraints, left unreferenced in this review. Special thanks go to Phong Trang and Paul Rider for invaluable discussions and comments on the manuscript. K.K acknowledges support from University of California Predoctoral Block Grant. The authors acknowledge the National Institutes of Health for generous support (AI041927 and DE014145).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Scherer LJ, Rossi JJ. Approaches for the sequence-specific knockdown of mRNA. Nat Biotechnol. 2003;21:1457–65. doi: 10.1038/nbt915. [DOI] [PubMed] [Google Scholar]
- 2.Stein CA, Cheng YC. Antisense oligonucleotides as therapeutic agents--is the bullet really magical? Science. 1993;261:1004–12. doi: 10.1126/science.8351515. [DOI] [PubMed] [Google Scholar]
- 3.Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell. 1983;35:849–57. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- 4.Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell. 1982;31:147–57. doi: 10.1016/0092-8674(82)90414-7. [DOI] [PubMed] [Google Scholar]
- 5.Doudna JA, Cech TR. The chemical repertoire of natural ribozymes. Nature. 2002;418:222–8. doi: 10.1038/418222a. [DOI] [PubMed] [Google Scholar]
- 6.Gopalan V, Vioque A, Altman S. RNase P: variations and uses. J Biol Chem. 2002;277:6759–62. doi: 10.1074/jbc.R100067200. [DOI] [PubMed] [Google Scholar]
- 7.Robertson HD, Altman S, Smith JD. Purification and properties of a specific Escherichia coli ribonuclease which cleaves a tyrosine transfer ribonucleic acid presursor. J Biol Chem. 1972;247:5243–51. [PubMed] [Google Scholar]
- 8.Altman S, Kirsebom L. In: The RNA World. Gesteland R, Cech T, Atkins J, editors. Cold Spring Harbor Laboratory Press; 1999. pp. 351–380. [Google Scholar]
- 9.Frank DN, Pace NR. Ribonuclease P: unity and diversity in a tRNA processing ribozyme. Annu Rev Biochem. 1998;67:153–80. doi: 10.1146/annurev.biochem.67.1.153. [DOI] [PubMed] [Google Scholar]
- 10.Xiao S, Scott F, Fierke CA, Engelke DR. Eukaryotic ribonuclease P: a plurality of ribonucleoprotein enzymes. Annu Rev Biochem. 2002;71:165–89. doi: 10.1146/annurev.biochem.71.110601.135352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Komine Y, Kitabatake M, Yokogawa T, Nishikawa K, Inokuchi H. A tRNA-like structure is present in 10Sa RNA, a small stable RNA from Escherichia coli. Proc Natl Acad Sci U S A. 1994;91:9223–7. doi: 10.1073/pnas.91.20.9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Altman S. Polarity effects in the lactose operon of Escherichia coli. J Mol Biol. 2004;339:31–9. doi: 10.1016/j.jmb.2004.03.041. [DOI] [PubMed] [Google Scholar]
- 13.Alifano P, Rivellini F, Piscitelli C, Arraiano CM, Bruni CB, Carlomagno MS. Ribonuclease E provides substrates for ribonuclease P-dependent processing of a polycistronic mRNA. Genes Dev. 1994;8:3021–31. doi: 10.1101/gad.8.24.3021. [DOI] [PubMed] [Google Scholar]
- 14.Altman S, Wesolowski D, Guerrier-Takada C, Li Y. RNase P cleaves transient structures in some riboswitches. Proc Natl Acad Sci U S A. 2005;102:11284–9. doi: 10.1073/pnas.0505271102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartmann RK, Heinrich J, Schlegl J, Schuster H. Precursor of C4 antisense RNA of bacteriophages P1 and P7 is a substrate for RNase P of Escherichia coli. Proc Natl Acad Sci U S A. 1995;92:5822–6. doi: 10.1073/pnas.92.13.5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peck-Miller KA, Altman S. Kinetics of the processing of the precursor to 4.5 S RNA, a naturally occurring substrate for RNase P from Escherichia coli. J Mol Biol. 1991;221:1–5. doi: 10.1016/0022-2836(91)80194-y. [DOI] [PubMed] [Google Scholar]
- 17.Reiner R, Ben-Asouli Y, Krilovetzky I, Jarrous N. A role for the catalytic ribonucleoprotein RNase P in RNA polymerase III transcription. Genes Dev. 2006;20:1621–35. doi: 10.1101/gad.386706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hartmann E, Hartmann RK. The enigma of ribonuclease P evolution. Trends Genet. 2003;19:561–9. doi: 10.1016/j.tig.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 19.Crary SM, Niranjanakumari S, Fierke CA. The protein component of Bacillus subtilis ribonuclease P increases catalytic efficiency by enhancing interactions with the 5′ leader sequence of pre-tRNAAsp. Biochemistry. 1998;37:9409–16. doi: 10.1021/bi980613c. [DOI] [PubMed] [Google Scholar]
- 20.Gopalan V, Baxevanis AD, Landsman D, Altman S. Analysis of the functional role of conserved residues in the protein subunit of ribonuclease P from Escherichia coli. J Mol Biol. 1997;267:818–29. doi: 10.1006/jmbi.1997.0906. [DOI] [PubMed] [Google Scholar]
- 21.Hsieh J, Andrews AJ, Fierke CA. Roles of protein subunits in RNA-protein complexes: lessons from ribonuclease P. Biopolymers. 2004;73:79–89. doi: 10.1002/bip.10521. [DOI] [PubMed] [Google Scholar]
- 22.Kim JJ, Kilani AF, Zhan X, Altman S, Liu F. The protein cofactor allows the sequence of an RNase P ribozyme to diversify by maintaining the catalytically active structure of the enzyme. Rna. 1997;3:613–23. [PMC free article] [PubMed] [Google Scholar]
- 23.Kurz JC, Niranjanakumari S, Fierke CA. Protein component of Bacillus subtilis RNase P specifically enhances the affinity for precursor-tRNAAsp. Biochemistry. 1998;37:2393–400. doi: 10.1021/bi972530m. [DOI] [PubMed] [Google Scholar]
- 24.Niranjanakumari S, Stams T, Crary SM, Christianson DW, Fierke CA. Protein component of the ribozyme ribonuclease P alters substrate recognition by directly contacting precursor tRNA. Proc Natl Acad Sci U S A. 1998;95:15212–7. doi: 10.1073/pnas.95.26.15212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Westhof E, Wesolowski D, Altman S. Mapping in three dimensions of regions in a catalytic RNA protected from attack by an Fe(II)-EDTA reagent. J Mol Biol. 1996;258:600–13. doi: 10.1006/jmbi.1996.0272. [DOI] [PubMed] [Google Scholar]
- 26.Kurz JC, Fierke CA. The affinity of magnesium binding sites in the Bacillus subtilis RNase P x pre-tRNA complex is enhanced by the protein subunit. Biochemistry. 2002;41:9545–58. doi: 10.1021/bi025553w. [DOI] [PubMed] [Google Scholar]
- 27.Jarrous N, Reiner R, Wesolowski D, Mann H, Guerrier-Takada C, Altman S. Function and subnuclear distribution of Rpp21, a protein subunit of the human ribonucleoprotein ribonuclease P. Rna. 2001;7:1153–64. doi: 10.1017/s1355838201010469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duarte CM, Wadley LM, Pyle AM. RNA structure comparison, motif search and discovery using a reduced representation of RNA conformational space. Nucleic Acids Res. 2003;31:4755–61. doi: 10.1093/nar/gkg682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leontis NB, Westhof E. Analysis of RNA motifs. Curr Opin Struct Biol. 2003;13:300–8. doi: 10.1016/s0959-440x(03)00076-9. [DOI] [PubMed] [Google Scholar]
- 30.Forster AC, Altman S. External guide sequences for an RNA enzyme. Science. 1990;249:783–6. doi: 10.1126/science.1697102. [DOI] [PubMed] [Google Scholar]
- 31.McClain WH, Guerrier-Takada C, Altman S. Model substrates for an RNA enzyme. Science. 1987;238:527–30. doi: 10.1126/science.2443980. [DOI] [PubMed] [Google Scholar]
- 32.Guerrier-Takada C, Li Y, Altman S. Artificial regulation of gene expression in Escherichia coli by RNase P. Proc Natl Acad Sci U S A. 1995;92:11115–9. doi: 10.1073/pnas.92.24.11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guerrier-Takada C, Salavati R, Altman S. Phenotypic conversion of drug-resistant bacteria to drug sensitivity. Proc Natl Acad Sci U S A. 1997;94:8468–72. doi: 10.1073/pnas.94.16.8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu F, Altman S. Inhibition of viral gene expression by the catalytic RNA subunit of RNase P from Escherichia coli. Genes Dev. 1995;9:471–80. doi: 10.1101/gad.9.4.471. [DOI] [PubMed] [Google Scholar]
- 35.McKinney J, Guerrier-Takada C, Wesolowski D, Altman S. Inhibition of Escherichia coli viability by external guide sequences complementary to two essential genes. Proc Natl Acad Sci U S A. 2001;98:6605–10. doi: 10.1073/pnas.121180398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan Y, Hwang ES, Altman S. Targeted cleavage of mRNA by human RNase P. Proc Natl Acad Sci U S A. 1992;89:8006–10. doi: 10.1073/pnas.89.17.8006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Werner M, Rosa E, Nordstrom JL, Goldberg AR, George ST. Short oligonucleotides as external guide sequences for site-specific cleavage of RNA molecules with human RNase P. Rna. 1998;4:847–55. doi: 10.1017/s1355838298980323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma M, Benimetskaya L, Lebedeva I, Dignam J, Takle G, Stein CA. Intracellular mRNA cleavage induced through activation of RNase P by nuclease-resistant external guide sequences. Nat Biotechnol. 2000;18:58–61. doi: 10.1038/71924. [DOI] [PubMed] [Google Scholar]
- 39.Frank DN, Harris ME, Pace NR. Rational design of self-cleaving pre-tRNA-ribonuclease P RNA conjugates. Biochemistry. 1994;33:10800–8. doi: 10.1021/bi00201a030. [DOI] [PubMed] [Google Scholar]
- 40.Cobaleda C, Sanchez-Garcia I. In vivo inhibition by a site-specific catalytic RNA subunit of RNase P designed against the BCR-ABL oncogenic products: a novel approach for cancer treatment. Blood. 2000;95:731–7. [PubMed] [Google Scholar]
- 41.Trang P, Kilani A, Kim J, Liu F. A ribozyme derived from the catalytic subunit of RNase P from Escherichia coli is highly effective in inhibiting replication of herpes simplex virus 1. J Mol Biol. 2000;301:817–26. doi: 10.1006/jmbi.2000.4022. [DOI] [PubMed] [Google Scholar]
- 42.Kazantsev AV, Pace NR. Bacterial RNase P: a new view of an ancient enzyme. Nat Rev Microbiol. 2006;4:729–40. doi: 10.1038/nrmicro1491. [DOI] [PubMed] [Google Scholar]
- 43.Bertrand EL, Rossi JJ. Facilitation of hammerhead ribozyme catalysis by the nucleocapsid protein of HIV-1 and the heterogeneous nuclear ribonucleoprotein A1. Embo J. 1994;13:2904–12. doi: 10.1002/j.1460-2075.1994.tb06585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu J, Trang P, Kim K, Zhou T, Deng H, Liu F. Effective inhibition of Rta expression and lytic replication of Kaposi’s sarcoma-associated herpesvirus by human RNase P. Proc Natl Acad Sci U S A. 2004;101:9073–8. doi: 10.1073/pnas.0403164101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zaug AJ, Cech TR. Analysis of the structure of Tetrahymena nuclear RNAs in vivo: telomerase RNA, the self-splicing rRNA intron, and U2 snRNA. Rna. 1995;1:363–74. [PMC free article] [PubMed] [Google Scholar]
- 46.Knapp G. Enzymatic approaches to probing of RNA secondary and tertiary structure. Methods Enzymol. 1989;180:192–212. doi: 10.1016/0076-6879(89)80102-8. [DOI] [PubMed] [Google Scholar]
- 47.Trang P, Hsu AW, Liu F. Nuclease footprint analyses of the interactions between RNase P ribozyme and a model mRNA substrate. Nucleic Acids Res. 1999;27:4590–7. doi: 10.1093/nar/27.23.4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci U S A. 2004;101:7287–92. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–15. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lo KM, Biasolo MA, Dehni G, Palu G, Haseltine WA. Inhibition of replication of HIV-1 by retroviral vectors expressing tat-antisense and anti-tat ribozyme RNA. Virology. 1992;190:176–83. doi: 10.1016/0042-6822(92)91203-7. [DOI] [PubMed] [Google Scholar]
- 51.Ojwang JO, Hampel A, Looney DJ, Wong-Staal F, Rappaport J. Inhibition of human immunodeficiency virus type 1 expression by a hairpin ribozyme. Proc Natl Acad Sci U S A. 1992;89:10802–6. doi: 10.1073/pnas.89.22.10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sarver N, Cantin EM, Chang PS, Zaia JA, Ladne PA, Stephens DA, Rossi JJ. Ribozymes as potential anti-HIV-1 therapeutic agents. Science. 1990;247:1222–5. doi: 10.1126/science.2107573. [DOI] [PubMed] [Google Scholar]
- 53.Sun LQ, Warrilow D, Wang L, Witherington C, Macpherson J, Symonds G. Ribozyme-mediated suppression of Moloney murine leukemia virus and human immunodeficiency virus type I replication in permissive cell lines. Proc Natl Acad Sci U S A. 1994;91:9715–9. doi: 10.1073/pnas.91.21.9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun LQ, Pyati J, Smythe J, Wang L, Macpherson J, Gerlach W, Symonds G. Resistance to human immunodeficiency virus type 1 infection conferred by transduction of human peripheral blood lymphocytes with ribozyme, antisense, or polymeric trans-activation response element constructs. Proc Natl Acad Sci U S A. 1995;92:7272–6. doi: 10.1073/pnas.92.16.7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang L, Witherington C, King A, Gerlach WL, Carr A, Penny R, Cooper D, Symonds G, Sun LQ. Preclinical characterization of an anti-tat ribozyme for therapeutic application. Hum Gene Ther. 1998;9:1283–91. doi: 10.1089/hum.1998.9.9-1283. [DOI] [PubMed] [Google Scholar]
- 56.Weerasinghe M, Liem SE, Asad S, Read SE, Joshi S. Resistance to human immunodeficiency virus type 1 (HIV-1) infection in human CD4+ lymphocyte-derived cell lines conferred by using retroviral vectors expressing an HIV-1 RNA-specific ribozyme. J Virol. 1991;65:5531–4. doi: 10.1128/jvi.65.10.5531-5534.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu F, Altman S. Requirements for cleavage by a modified RNase P of a small model substrate. Nucleic Acids Res. 1996;24:2690–6. doi: 10.1093/nar/24.14.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yuan Y, Altman S. Selection of guide sequences that direct efficient cleavage of mRNA by human ribonuclease P. Science. 1994;263:1269–73. doi: 10.1126/science.8122108. [DOI] [PubMed] [Google Scholar]
- 59.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–22. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 60.Joyce GF. Directed molecular evolution. Sci Am. 1992;267:90–7. doi: 10.1038/scientificamerican1292-90. [DOI] [PubMed] [Google Scholar]
- 61.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–10. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 62.Berzal-Herranz A, Joseph S, Burke JM. In vitro selection of active hairpin ribozymes by sequential RNA-catalyzed cleavage and ligation reactions. Genes Dev. 1992;6:129–34. doi: 10.1101/gad.6.1.129. [DOI] [PubMed] [Google Scholar]
- 63.Joseph S, Burke JM. Optimization of an anti-HIV hairpin ribozyme by in vitro selection. J Biol Chem. 1993;268:24515–8. [PubMed] [Google Scholar]
- 64.Lorsch JR, Szostak JW. In vitro evolution of new ribozymes with polynucleotide kinase activity. Nature. 1994;371:31–6. doi: 10.1038/371031a0. [DOI] [PubMed] [Google Scholar]
- 65.Kilani AF, Trang P, Jo S, Hsu A, Kim J, Nepomuceno E, Liou K, Liu F. RNase P ribozymes selected in vitro to cleave a viral mRNA effectively inhibit its expression in cell culture. J Biol Chem. 2000;275:10611–22. doi: 10.1074/jbc.275.14.10611. [DOI] [PubMed] [Google Scholar]
- 66.Chen JL, Nolan JM, Harris ME, Pace NR. Comparative photocross-linking analysis of the tertiary structures of Escherichia coli and Bacillus subtilis RNase P RNAs. Embo J. 1998;17:1515–25. doi: 10.1093/emboj/17.5.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haas ES, Brown JW, Pitulle C, Pace NR. Further perspective on the catalytic core and secondary structure of ribonuclease P RNA. Proc Natl Acad Sci U S A. 1994;91:2527–31. doi: 10.1073/pnas.91.7.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Massire C, Jaeger L, Westhof E. Derivation of the three-dimensional architecture of bacterial ribonuclease P RNAs from comparative sequence analysis. J Mol Biol. 1998;279:773–93. doi: 10.1006/jmbi.1998.1797. [DOI] [PubMed] [Google Scholar]
- 69.Trang P, Hsu A, Zhou T, Lee J, Kilani AF, Nepomuceno E, Liu F. Engineered RNase P ribozymes inhibit gene expression and growth of cytomegalovirus by increasing rate of cleavage and substrate binding. J Mol Biol. 2002;315:573–86. doi: 10.1006/jmbi.2001.5291. [DOI] [PubMed] [Google Scholar]
- 70.Zou H, Lee J, Kilani AF, Kim K, Trang P, Kim J, Liu F. Engineered RNase P ribozymes increase their cleavage activities and efficacies in inhibiting viral gene expression in cells by enhancing the rate of cleavage and binding of the target mRNA. J Biol Chem. 2004;279:32063–70. doi: 10.1074/jbc.M403059200. [DOI] [PubMed] [Google Scholar]
- 71.McKinney JS, Zhang H, Kubori T, Galan JE, Altman S. Disruption of type III secretion in Salmonella enterica serovar Typhimurium by external guide sequences. Nucleic Acids Res. 2004;32:848–54. doi: 10.1093/nar/gkh219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hnatyszyn H, Spruill G, Young A, Seivright R, Kraus G. Long-term RNase P-mediated inhibition of HIV-1 replication and pathogenesis. Gene Ther. 2001;8:1863–71. doi: 10.1038/sj.gt.3301606. [DOI] [PubMed] [Google Scholar]
- 73.Kraus G, Geffin R, Spruill G, Young AK, Seivright R, Cardona D, Burzawa J, Hnatyszyn HJ. Cross-clade inhibition of HIV-1 replication and cytopathology by using RNase P-associated external guide sequences. Proc Natl Acad Sci U S A. 2002;99:3406–11. doi: 10.1073/pnas.052651199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Plehn-Dujowich D, Altman S. Effective inhibition of influenza virus production in cultured cells by external guide sequences and ribonuclease P. Proc Natl Acad Sci U S A. 1998;95:7327–32. doi: 10.1073/pnas.95.13.7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou T, Kim J, Kilani AF, Kim K, Dunn W, Jo S, Nepomuceno E, Liu F. In vitro selection of external guide sequences for directing RNase P-mediated inhibition of viral gene expression. J Biol Chem. 2002;277:30112–20. doi: 10.1074/jbc.M200183200. [DOI] [PubMed] [Google Scholar]
- 76.Roizman B, Knipe DM. In: Fields Virology. Knipe DM, Howley PM, editors. Lippincott-William & Wilkins; Philadelphia, Pa: 2001. pp. 2399–2460. [Google Scholar]
- 77.Mocarski ES, Courcelle CT. In: Fields Virology. Knipe DM, Howley PM, editors. Lippincott-William & Wilkins; Philadelphia, Pa: 2001. pp. 2629–2673. [Google Scholar]
- 78.Moore PS, Chang Y. In: Fields Virology. Knipe DM, Howley PM, editors. Lippincott-William & Wilkins; Philadelphia, Pa: 2001. pp. 2803–2834. [Google Scholar]
- 79.Kim K, Umamoto S, Trang P, Hai R, Liu F. Intracellular expression of engineered RNase P ribozymes effectively blocks gene expression and replication of human cytomegalovirus. Rna. 2004;10:438–47. doi: 10.1261/rna.5178404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim K, Liu F. In vitro selection of RNase P ribozymes that efficiently cleave a target mRNA. Methods Mol Biol. 2004;252:399–412. doi: 10.1385/1-59259-746-7:399. [DOI] [PubMed] [Google Scholar]
- 81.Kawa D, Wang J, Yuan Y, Liu F. Inhibition of viral gene expression by human ribonuclease P. Rna. 1998;4:1397–406. doi: 10.1017/s1355838298980918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li H, Trang P, Kim K, Zhou T, Umamoto S, Liu F. Effective inhibition of human cytomegalovirus gene expression and growth by intracellular expression of external guide sequence RNA. Rna. 2006;12:63–72. doi: 10.1261/rna.2184706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang YH, Li H, Zhou T, Kim K, Liu F. Engineered external guide sequences are highly effective in inducing RNase P for inhibition of gene expression and replication of human cytomegalovirus. Nucleic Acids Res. 2006;34:575–83. doi: 10.1093/nar/gkj431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Raj SM, Liu F. Engineering of RNase P ribozyme for gene-targeting applications. Gene. 2003;313:59–69. doi: 10.1016/s0378-1119(03)00677-2. [DOI] [PubMed] [Google Scholar]
- 85.Trang P, Kim K, Liu F. Developing RNase P ribozymes for gene-targeting and antiviral therapy. Cell Microbiol. 2004;6:499–508. doi: 10.1111/j.1462-5822.2004.00398.x. [DOI] [PubMed] [Google Scholar]
- 86.Yu X, Trang P, Shah S, Atanasov I, Kim YH, Bai Y, Zhou ZH, Liu F. Dissecting human cytomegalovirus gene function and capsid maturation by ribozyme targeting and electron cryomicroscopy. Proc Natl Acad Sci U S A. 2005;102:7103–8. doi: 10.1073/pnas.0408826102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Verma S, Eckstein F. Modified oligonucleotides: synthesis and strategy for users. Annu Rev Biochem. 1998;67:99–134. doi: 10.1146/annurev.biochem.67.1.99. [DOI] [PubMed] [Google Scholar]