

Abstract
STriatal Enriched protein tyrosine Phosphatase (STEP) is a brain-specific protein that is thought to play a role in synaptic plasticity. This hypothesis is based on previous findings demonstrating a role for STEP in the regulation of the extracellular signal-regulated kinase1/2 (ERK1/2). We have now generated a STEP knockout mouse and investigated the effect of knocking out STEP in the regulation of ERK1/2 activity. Here, we show that the STEP knockout mice are viable and fertile and have no detectable cytoarchitectural abnormalities in the brain. The homozygous knockout mice lack the expression of all STEP isoforms, whereas the heterozygous mice have reduced STEP protein levels when compared with the wild-type mice. The STEP knockout mice show enhanced phosphorylation of ERK1/2 in the striatum, CA2 region of the hippocampus, as well as central and lateral nuclei of the amygdala. In addition, the cultured neurons from KO mice showed significantly higher levels of pERK1/2 following synaptic stimulation when compared with wild-type controls. These data demonstrate more conclusively the role of STEP in the regulation of ERK1/2 activity.
Keywords: STEP, MAPK, ERK1/2, tyrosine phosphatase, synaptic plasticity, knockout
INTRODUCTION
STriatal Enriched protein tyrosine Phosphatase (STEP) has emerged as an important regulator of neuronal signal transduction. It is a neuron-specific tyrosine phosphatase that is enriched in the striatum. It is also expressed in the hippocampus, amygdala, cortex, and related regions of the brain that are involved in motor control and cognitive activities (Braithwaite et al., 2006). The striatum, where STEP is most abundant, receives two major sources of synaptic input: glutamatergic afferents from the cerebral cortex and dopaminergic afferents from the midbrain (Freund et al., 1984; Kotter, 1994). Both neurotransmitters have been linked to the regulation of STEP activity through phosphorylation of a serine residue within the regulatory domain in STEP, termed the kinase interacting motif (KIM). Dopamine/D1 receptor-mediated phosphorylation of this serine residue inactivates STEP, whereas glutamate/NMDA receptor-mediated dephosphorylation of the same serine residue activates STEP (Paul et al., 2000, 2003). Active STEP, in turn, modulates synaptic plasticity by regulating the activity of extracellular signal-regulated kinase1/2 (ERK1/2), a key signaling protein involved in memory formation. We have demonstrated that STEP binds to ERK1/2 and limits the duration of its activity and subsequent downstream signaling cascades (Paul et al., 2003). Administration of a substrate-trapping mutant of STEP fusion protein into the lateral amygdala disrupted fear memory consolidation, while addition to acute amygdala slices blocked long-term potentiation (Paul et al., 2007). This occurred, in part, through the blockade of ERK1/2 signaling, further confirming the importance of ERK1/2 in the consolidation of fear memories (Schafe et al., 2000).
The tyrosine kinase Fyn and the NR2B subunit of the N-methyl-d-aspartate receptor (NMDAR) are two additional substrates of STEP that are also involved in the regulation of synaptic plasticity (Braithwaite et al., 2006; Nguyen et al., 2002). The latter two substrates link STEP to NMDAR trafficking. Fyn normally phosphorylates NR2B at tyr1472, and phosphorylation at that site leads to rapid movement of NMDARs to membrane compartments (Hallett et al., 2006). STEP dephosphorylates Fyn at a regulatory tyrosine residue (tyr420), thereby inactivating Fyn (Nguyen et al., 2002). β-amyloid was recently shown to activate STEP, which in turn led to the dephosphorylation of NR2B at tyr1472 and internalization of the NMDAR complex (Snyder et al., 2005; Venkitaramani et al., 2007). The aforementioned findings indicate that activation of STEP leads to alterations in synaptic transmission, plasticity, and memory formation. However, to demonstrate the involvement of STEP more specifically in the regulation of synaptic plasticity, it is necessary to generate and characterize the STEP knockout mouse. In this study, we describe the generation of the STEP knockout mice and determine its effect on the regulation of ERK1/2 activity.
MATERIALS AND METHODS
Cloning of the mouse STEP gene and construction of the mutant targeting vector
Genomic clones of STEP gene were isolated from a λgt11 mouse genomic library (Clontech Mountain View, CA) by screening with a 2.6 kb mouse STEP cDNA (Lombroso et al., 1991). We isolated a clone that contained the 3′ region of the STEP genomic sequence including part of the phosphatase domain with the catalytic site (VHCSAGIGRTGCF) and the 3′ untranslated region (Fig. 1). We generated a targeting vector in which a 1284 bp genomic fragment containing the catalytic site was deleted and replaced with a 1913 bp neomycin gene (Fig. 1). To generate the targeting vector, we used pBluescript (pBS KS) as the backbone vector. The multiple cloning site in pBS KS was replaced by a 78-mer multiple cloning site containing seven unique restriction sites (KpnI, MluI, NheI, HindIII, SacII, NotI, and SacI). A 4447 bp genomic fragment immediately upstream of the deleted region was used as the 5′ homology domain and inserted between NheI and HindIII sites. The neo cassette was placed between HindIII and SacII restriction sites. A 1495 bp genomic fragment immediately downstream of the deleted region was used as the 3′ homology domain and placed between SacII and NotI within the multiple cloning site. Finally, the 2274 bp diptheria toxin (DTA) gene was inserted 3′ to the downstream homology domain between NotI and SacI, resulting in the 13.1 kb targeting plasmid. The plasmid was then sequenced to confirm all sequence changes.
Fig. 1.
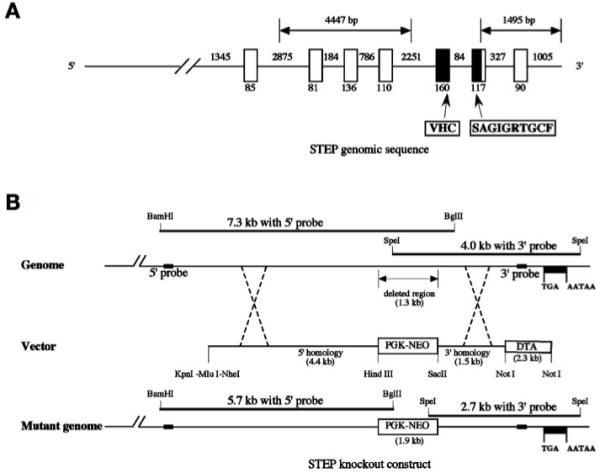
Genomic construct for the generation of STEP knockout mice. (A) Schematic diagram representing the genomic sequence of STEP along with the exons and introns. The exonic sequence deleted in the KO are shaded in black. (B) A neomycin cassette (PGK-NEO) was used to replace a 1.3 kb genomic sequence, disrupting the open reading frame. The insertion was confirmed by Southern blotting using 5′ (BamHI/BglII double digest) and 3′ (SpeI single digest) probes generated from the wild-type STEP genomic sequence, and the construct was then used to generate the STEP knockout mice (data not shown).
Generation of knockout mice and genotyping by PCR
After linearization with KpnI, the DNA was electroporated into 129 embryonic stem (ES) cells. Clones that survived G418 selection were identified by Southern blot analysis using two probes located outside the targeting vector. For Southern blot analyses, 5-10 μg of ES or mouse tail DNA were digested either with BamHI/BglII (double digest for 5′ probe) or with SpeI (for 3′ probe), separated on 0.7% agarose gel, transferred to ζ-Probe (Bio-Rad, Hercules, CA), and denatured with 0.4 N NaOH. Probes were labeled by random priming, and hybridization and washes performed as described previously (Coetzee et al., 1996).
The primers used for PCR genotyping were as follows: 5′ wild-type-CCCTACTCTCATTCC-TCCCTTCCC, 5′ neo primer-GGCGCGAAGGGGCCACC, and a 3′ common primer-GGCAGCAGATGCTGGTGGC. The multiprimer PCR reaction was performed using the following conditions: 94°C for 10 min, followed by 35 cycles of 94°C for 45 s, 59°C for 1 min, 72°C for 1 min, and a final extension at 72°C for 10 min. The PCR products were resolved on 1.2% agarose gels.
Western blots
Brains were rapidly dissected out and frozen on dry ice. Brain samples without the cerebellum as well as the cerebellum alone (where STEP is absent) were homogenized in TEVP buffer containing 10 mM Tris-HCl, pH 7.4, 5 mM NaF, 1 mM Na3VO4, 1 mM EDTA, 1 mM EGTA, and 320 mM sucrose and centrifuged at 3000 rpm to remove nuclear fraction and large debris (P1). The resulting supernatant (S1) was spun at 10,000 rpm to obtain the crude synaptosomal fraction (P2) and supernatant (S2) as described (Hu et al., 2007). For region-specific westerns, striatum and hippocampus were rapidly dissected out and processed as mentioned earlier. The levels of STEP and pERK1/2 were detected in both S2 and P2 fractions. Briefly, equal amount (30-50 μg) of proteins were separated on 10% SDS-PAGE gels and electroblotted onto nitrocellulose membrane. The membranes were blocked with 5% nonfat dry milk in Tris-buffered saline containing 0.1% Tween-20 (TBS-T), incubated overnight with primary antibody. Antibodies used in this study included STEP [23E5, 1:1000; (Boulanger et al., 1995)]; pT202pY204ERK1/2 [E10, 1:1000, Cell Signaling, Danvers, MA]; ERK2 [1:5000, Santa Cruz Biotechnology, Santa Cruz, CA], and followed by incubation with appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (sheep anti-mouse HRP-1:5000 or donkey anti-rabbit HRP-1:5000 [Amersham Biosciences, Piscataway, NJ]). The HRP signal was visualized using SuperSignal Western blot kit (Pierce Biotechnology, Rockford, IL) and detected using Chemi-HR16 gel imaging system (Syngene, Frederick, MD). The bands were quantified using ImageJ (Version 1.33, NIH, Bethesda, MD) and normalized to total ERK2 protein levels from the same blots. The normalized percentages are expressed as mean ± SE. The differences between groups were evaluated using one-way ANOVA and posthoc Tukey HSD.
Immunohistochemistry
Mice were anesthetized with an intraperitoneal injection of Nembutal (50 mg/kg body weight) and sacrificed by transcardial perfusion with ice-cold 4% paraformaldehyde (PFA) in 0.1 M phosphate-buffered saline (PBS). The brains were postfixed in 4% PFA overnight at 4°C and cryoprotected sequentially in 10, 20, and 30% sucrose in PBS over 48 h. The brains were embedded in optimal cutting temperature [O.C.T.] compound and quick-frozen in an ethanol-dry ice bath. For HRP staining, the brains were serially sectioned at 50 μM thickness, and either mounted onto VWR-coated slides as free floating sections for cresyl violet staining or stored at -20°C in cryoprotective buffer until antibody staining. Sections were washed in PBS, incubated with 0.3% H2O2 to block endogenous HRP activity, and then blocked with 10% normal goat serum (NGS) + 3% bovine serum albumin (BSA) in PBS containing 0.2% Triton-X-100 (PBS-T). The sections were incubated overnight with primary antibody (pERK1/2-1:400; STEP-1:5000; ERK2-1:5000) diluted in 1% NGS + 1% BSA in PBS-T. The sections were washed, incubated with biotinylated secondary antibody for 1 h, followed by avidin-biotin peroxidase complex reagent (ABC kit from Vector Laboratories, Burlingame, CA) for 1 h. The sections were washed extensively in PBS-T and developed using diamino benzidine (DAB)-Nickel chloride kit (Vector Laboratories, Burlingame, CA). Sections were mounted onto VWR precleaned coated slides, dehydrated through a graded-ethanol series, cleared using xylene, and coverslipped for light microscopy. Adjacent sections were Nissl-stained with cresyl violet and processed for light microscopy as described earlier. Images were acquired using Zeiss axiovision software, from a light microscope fitted with digital camera, and images were processed using Adobe Photoshop. For immunofluorescence staining, 20-μM-thick coronal sections were mounted directly onto precleaned/coated VWR slides and stored at -80°C. The staining was performed as described in Paul et al., 2007. Briefly, the sections were blocked with 10% NGS + 3% BSA in 1× PBS-T for an hour. The sections were incubated with primary antibodies (pERK1/2-1:400 and ERK2-1:800) in 1% NGS + 1% BSA overnight at 4°C. The sections were washed and incubated with secondary antibodies (goat anti-mouse Alexa Fluor 488 and goat anti-rabbit Alexa Fluor 594-1:400) at room temperature for 2 h. The sections were washed extensively with 1× PBS-T and finally with 1× PBS. The slides were coverslipped using VectaShield HardMount with DAPI and sealed with nail polish.
Cell culture
Primary hippocampal neuronal cultures were prepared from timed-pregnant E15-E16 WT and KO mice. Pregnant dams were sacrificed by cervical dislocation and embryos removed by Caesarean section. The brains were removed and the hippocampi dissected out under a dissecting microscope. Tissue was trypsinized with 0.025% trypsin-EDTA and dissociated in Hanks Balanced Salt Solution by tituration to isolate cells. Hippocampal cells were resuspended in Neurobasal medium containing B27 supplement and 5% fetal bovine serum (FBS) and plated in 12-well culture dishes containing poly-d-lysine (Sigma)-coated 18-mm German-glass coverslips. The cells were maintained in Neurobasal medium without FBS for 14-15 DIV at 37°C in a humidified 5% CO2 incubator. The Yale animal use and care committee approved all procedures.
Immunofluorescence
Hippocampal cultures were treated for 5 min with increasing concentration of (RS)-3,5-dihydroxyphenylglycine (DHPG) at 37°C in a humidified 5% CO2 chamber. Following the DHPG treatment, the cells were sequentially treated with 4% paraformaldehyde, 4% sucrose in 1× PBS for 10 min at 4°C, and 100% ice-cold methanol for 5 min at 4°C. The neurons were permeabilized with 1× PBS-T (0.2% Triton-X-100) for 10 min, washed with 1× PBS, and blocked for 1 h at room temperature with 1× PBS containing 10% NGS and 1% BSA. After a brief wash with 1× PBS, the cells were incubated with primary antibodies against STEP (23E5, 1:2000) and/or pERK1/2 (Cell Signaling, 1:750), in 1% BSA/1× PBS overnight at 4°C. Following three washes in 1× PBS, the cells were incubated with goat anti-mouse Alexa Fluor 594 and/or goat anti-rabbit Alexa Fluor 488 secondary antibodies (1:600, Molecular Probes). The coverslips were washed extensively and mounted onto slides using Vectashield Hardmount with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Labs). Immunofluorescence was visualized using a Zeiss Axiovert 2000 microscope with an apotome.
Synaptoneurosome preparation
Hippocampi were dissected from 3- to 4-week-old WT and KO mice and processed using membrane filters to obtain synaptoneurosomes. The tissue was homogenized in ice-cold homogenization buffer containing 20 mM HEPES, pH 7.3, 124 mM NaCl, 3.2 mM KCl, 1 mM KH2PO4, 26 mM NaHCO3, 1.3 mM MgCl2, 2.5 mM CaCl2, 10 mM glucose, and complete protease inhibitor cocktail (Roche). The homogenates were filtered sequentially through two 100-μm nylon mesh filters (Sefar America, Richfield, MN) and a 5-μm nitrocellulose filter (Millipore). The filtrate was centrifuged at 1000g for 10 min at 4°C. The pelleted synaptoneurosomes were washed, resuspended in homogenization buffer, and preincubated at 37°C for 10 min prior to DHPG stimulation. The synaptoneurosomes were stimulated with increasing dose of DHPG for 5 min, and the samples were lysed by the addition of SDS to 1%. The lysed synaptoneurosomes were processed for Western blotting. The purity of the synaptoneurosomal prep was ascertained by probing the Western blots with histone H1 (only in homogenate), CaMKII (homogenate, cytosol and synaptoneurosomes), and PSD95 (synaptoneurosomes alone) antibodies.
RESULTS
Generation of STEP knockout mice
The mouse STEP (PTPN5) gene is located on chromosome 7, spans ∼55 kb and consists of 14 exons. The single STEP gene is alternatively spliced to produce transcripts that encode STEP61, STEP46, STEP38, and STEP20 isoforms (Bult et al., 1996, 1997; Sharma et al., 1995). STEP genomic clones encompassing the 3′ region were isolated and sequenced. One of these was selected for further analysis and contained the coding sequence including a part of the phosphatase domain, as well as the 3′ untranslated region (Fig. 1A). The STEP genomic sequence was later confirmed by comparison to NCBI (MGI: 97,807) and the Mouse Genome Sequencing Consortium.
To generate the STEP KO mice, using homologous recombination, a targeting vector was made in which a 1.3 kb genomic DNA fragment containing a portion of the phosphatase domain with the catalytic site was replaced by the neomycin gene (1.9 kb) (Fig. 1B). The targeting construct was electroporated into ES cells from mice of the 129 background, and clones that were resistant to the antibiotic G418 were isolated and used for screening by Southern blot. Homologous recombination of the targeting vector at the endogenous locus was confirmed by a shift of the genomic fragment from 7.3 to 5.7 kb when hybridized with a STEP genomic DNA probe generated from a region 5′ to the neomycin cassette. A second probe obtained from the STEP gene 3′ of the neomycin cassette showed the expected shift of the genomic fragment from 4.0 to 2.7 kb (data not shown).
The selected G418 resistant clones were injected into C57BL/6 blastocysts, which were implanted into pseudopregnant females to generate chimeric off-springs. The chimeras were crossed with C57BL/6 mice to produce mice heterozygous (HT) for STEP. The STEP HT mice were crossed to obtain STEP null mutants. The genotype of the STEP knockout mice was confirmed by PCR amplification of genomic DNA purified from tail biopsies. A 400 bp band was observed in the genomic DNA obtained from the WT mice, whereas the DNA obtained from KO mice had a 200 bp band, and both bands were present in the STEP HT mice (Fig. 2A). The progeny obtained from STEP HT crosses were used for all experiments. The table in Figure 2B indicates the number of WT (358, 26%), HT (648, 48%), and KO (353, 26%) animals generated to date. The observed percentages for each genotype are not significantly different from the expected Mendelian values of 25, 50, and 25%, respectively. STEP KO mice are viable, fertile, and the three genotypes are visibly indistinguishable. These results indicate that knocking down STEP expression did not affect embryonic or postnatal survival.
Fig. 2.
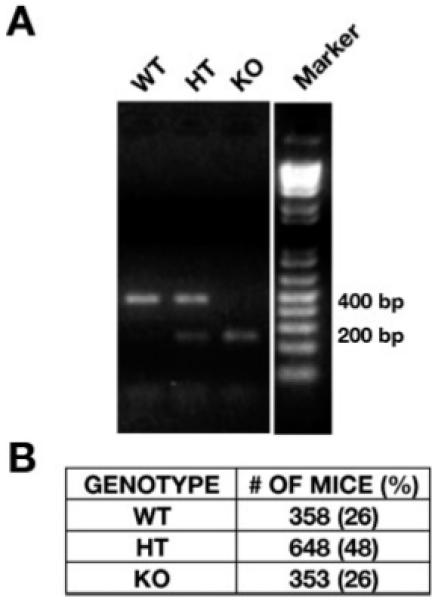
PCR genotyping of STEP genomic DNA from WT, HT, and KO mice. (A) The genotype of the KO mice was determined by PCR amplification of the genomic DNA using primers from the STEP genomic sequence. The genomic DNA from WT mice show an expected 400 base pair (bp) band and that from the KO mice show a smaller 200 bp product. Both the bands are present in the HT mice. The table in (B) shows the number of WT, HT, and KO animals generated and the percentage of each genotype. The observed percentages are not significantly different from the expected values of 25, 50, and 25%, respectively.
Expression profile of STEP in KO, HT, and WT mice
In the next series of experiments, we examined the level of expression of the STEP protein in WT, HT, and KO mice using immunoblotting and immunohistochemical techniques. Subcellular fractions obtained from total cerebral homogenates from age- and sex-matched WT, HT, and KO animals were analyzed by SDS-PAGE gel electrophoresis and immunoblotted with a STEP antibody that recognizes all isoforms of STEP (23E5; Boulanger et al., 1995). The results show that no STEP protein was detectable in the KO mice, whereas the HT mice expressed ∼50% of the STEP protein when compared with the expression level in WT littermates (Fig. 3A). Immunoblot analysis of subcellular fractions of cerebral homogenates demonstrated that STEP46 is the predominant isoform present in the cytosolic fraction (S2), whereas STEP61 is the predominant isoform expressed in the membrane fraction (P2) of the WT mice. STEP46 is also present in the P2 fraction, albeit at low level. In HT mice, the expression level of STEP46 in the S2 fraction was 53% ± 9% (P < 0.001) when compared with WT controls (Fig. 3B). In the P2 fraction, HT mice expressed 70% ± 6% (P < 0.002) of STEP61 when compared with WT, whereas the level of STEP46 in HT was 54% ± 10% (P < 0.003) (Fig. 3B). As expected, both STEP61 and STEP46 bands were absent in the S2 and P2 fractions of KO mice.
Fig. 3.
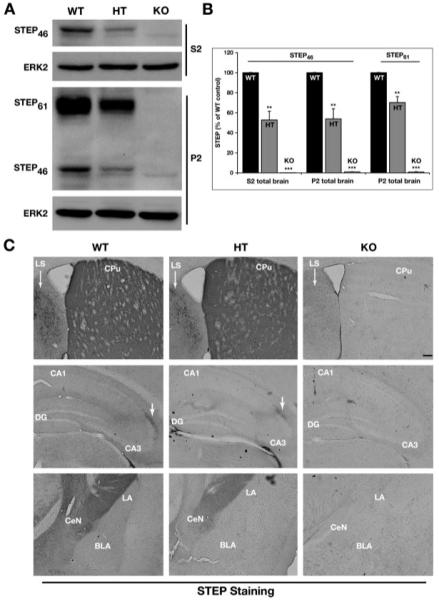
Expression profile of STEP protein in KO, HT, and WT mice. (A) The S2 and P2 fractions obtained from WT, HT, and KO mice total brain homogenates were analyzed by SDS-PAGE and immunoblotted with anti-STEP antibody (23E5). The levels of both STEP61 and STEP46 were significantly decreased in the samples obtained from HT mice, whereas both the isoforms of STEP were absent in the KO mice samples. The membranes were reprobed with anti-ERK2 antibody to ensure equal protein loading in each lane. (B) Quantification of immunoblots for STEP61 and STEP46 expressed in the three genotypes normalized with ERK2 loading control (n = 4). The normalized protein levels are expressed as a percentage of WT control (mean ± SE). Statistical analysis was performed using one-way ANOVA and posthoc Tukey HSD (**P < 0.01 and ***P < 0.001). (C) Coronal brain sections from the WT, HT, and KO mice were immunostained for STEP. The top panel shows representative sections through the striatum (CPu). The STEP staining was undetectable in the KO mice. STEP immunoreactivity was decreased in the lateral septum (LS) of HT when compared with WT. The middle and bottom panel show representative sections from the hippocampus and amygdala, respectively. The level of STEP expression in the HT was reduced and was not detected in the KO mice [Scale bar-200 μm].
The aforementioned findings were confirmed by immunohistochemical analysis of coronal brain sections obtained from 2- to 3-month-old WT, HT, and KO animals and stained with anti-STEP antibody. In the KO mice, there was no detectable STEP staining in any of the brain regions tested, whereas in the HT mice, STEP staining was observed in most of the regions examined, also confirming the specificity of STEP antibody. However, as is evident from Figure 3C (upper panel), the STEP staining in the lateral septum region (indicated by arrows) of the HT mice is substantially reduced when compared with the WT control animals. There is also a similar reduction in STEP immunoreactivity in the hippocampus and amygdala of HT mice when compared with WT littermates (Fig. 3C, middle and lower panel). These results demonstrate that the STEP KO mice do not express detectable levels of the STEP protein, whereas the HT mice have decreased expression patterns.
Anatomical characterization of the STEP KO mice brain
We next wanted to determine if knocking out the STEP gene has an effect on the cytoarchitecture of brain regions that normally express STEP. For these experiments, coronal brain sections (50 μm) from WT, HT, and KO mice were analyzed by cresyl violet staining. As is evident from the photomicrographs obtained from the striatal, hippocampal, and amygdala regions (Fig. 4), there are no gross architectural abnormalities in the KO and the HT brains when compared with the WT brain. Also, the cellular morphology and the cell population were similar between KO, HT, and WT mice. These results suggest that there are no major disruptions to brain development in the STEP KO mice at this level of analysis.
Fig. 4.
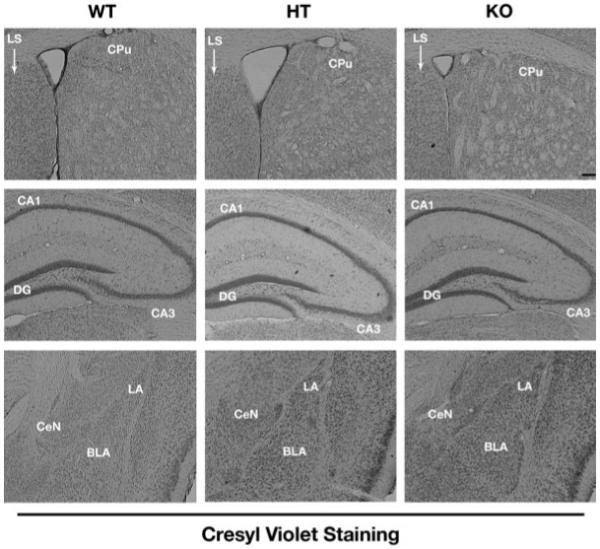
Morphological analysis of striatum, hippocampus, and amygdala from WT, HT, and KO mice. Coronal brain sections from the three genotypes were stained with cresyl violet to investigate anatomical changes in the striatum (upper panel), hippocampus (middle panel), and amygdala (lower panel). The staining did not detect any gross architectural abnormalities in the KO brain when compared with WT and HT sections at this level of analysis [Scale bar-200 μm].
Increased phosphorylation of ERK1/2 in STEP KO mice brain
Our previous studies have demonstrated that ERK1/2 is a substrate of STEP (Paul et al., 2003, 2007), and hence, we reasoned that the phosphorylation level of ERK1/2 might be elevated at baseline in the STEP deficient mice. To test this hypothesis, S2 and P2 fractions obtained from total brain homogenates from WT, HT, and KO mice were analyzed by immunoblotting with an antibody that recognizes ERK1/2 that is dually phosphorylated at the regulatory threonine and tyrosine residues (TPEYP-ERK1/2 or pERK1/2). An increase in the level of pERK1/2 was observed in both the S2 (127% ± 2%; P < 0.002) and P2 (154% ± 15%; P < 0.04) fractions obtained from the KO mice when compared with those obtained from the WT mice (Figs. 5A and 5B). However, there were no significant differences in pERK1/2 levels in the HT mice when compared with WT controls (S2: 110 ± 5%; P > 0.1; P2: 121% ± 14%; P > 0.4). Total ERK2 levels remained unaltered in all the samples. If disrupting STEP expression is indeed responsible for the increased phosphorylation of ERK1/2 in the cerebrum, the phosphorylation level of ERK1/2 should remain unaltered in the cerebellum where STEP is not expressed. To demonstrate this, S2 fractions obtained from cerebellar homogenates of WT, HT, and KO mice were analyzed by immunoblotting to determine pERK1/2 levels. There was no significant difference in the phosphorylation levels of ERK1/2 in the S2 fractions obtained from WT (100%), HT (93% ± 7%, P > 0.8), and KO (98% ± 16%, P > 0.9) mice cerebellar homogenates (Figs. 5A and 5B). The total ERK2 level remained unaltered in all the samples. The P2 fractions obtained from the cerebellum also did not show any differences in the pERK1/2 levels between the WT, HT, and the KO mice (data not shown).
Fig. 5.
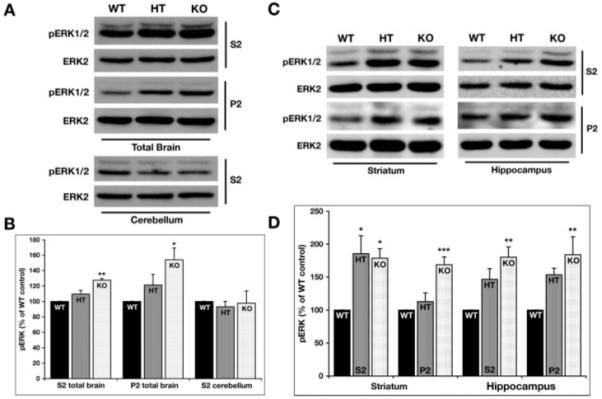
Increased baseline levels of phosphorylated ERK1/2 in STEP KO mice. (A) The levels of phosphorylated ERK1/2 were detected in the S2 and P2 fractions from total brain homogenates as well as the cerebellar S2 samples by probing with an antibody that recognizes ERK1/2 dually phosphorylated at the regulatory threonine and tyrosine residues (TPEYP-ERK1/2 or pERK1/2). The membranes were reprobed with anti-ERK2 antibody to ensure equal protein loading in each lane. STEP KO mice shows elevated levels of phosphorylated ERK1/2 when compared with WT controls in both the S2 and P2 fractions obtained from total brain homogenates. (B) Quantification of immunoblots for phosphorylated ERK2 expressed in the three genotypes normalized with total ERK2 protein loaded in the same blot (*P < 0.05 and **P < 0.01; n = 4). (C) pERK1/2 and ERK2 levels were detected in the S2 and P2 fractions from the striatum and hippocampus of WT, HT, and KO mice. The pERK1/2 levels were significantly elevated in both the fractions and regions tested. The HT showed significant increases only in the S2 fraction from the striatum. (D) The pERK1/2 levels from the immunoblots were quantified and normalized over the total ERK2 levels. The normalized pERK1/2 levels are plotted as a percentage of WT levels. The data were compared using one-way ANOVA, followed by posthoc Tukey HSD (*P < 0.05; **P < 0.01; ***P < 0.001; n = 4).
To further assess the basal pERK1/2 levels in specific brain regions, S2 and P2 fractions processed from striatum and hippocampus were analyzed by immunoblotting with pERK1/2 and ERK2 antibodies. The levels of pERK1/2 were significantly elevated in both subcellular fractions obtained from the striatum of the KO mice (S2: 179% ± 14%; P < 0.03 and P2: 169% ± 12%; P < 0.001) (Figs. 5C and 5D). In the striatal samples obtained from HT mice, there was a significant change in the S2 (185% ± 28%, P < 0.02), but not the P2 fraction (113% ± 13%; P > 0.6). Basal pERK1/2 levels were also found to be significantly elevated in both S2 and P2 fractions obtained from the hippocampus of the KO mice (S2: 180% ± 16%; P < 0.002 and P2: 183% ± 28%; P < 0.01) (Figs. 5C and 5D). The HT mice did not show significant changes in pERK1/2 levels in hippocampal S2 (147% ± 16%; P > 0.06) or P2 fractions (153% ± 10%; P > 0.1). Taken together, these findings show that the STEP KO mice have elevated phosphorylation of ERK1/2 in specific brain regions.
Immunohistochemical analyses of coronal brain sections extended these findings. There was a marked increase in pERK1/2 immunoreactivity in the striatum, hippocampus, and amygdala obtained from KO mice when compared with WT littermates (Fig. 6). The levels of phosphorylated ERK1/2 were slightly elevated in the lateral septum of KO mice when compared with WT and HT mice (data not shown). Although we did not observe major differences in the control ERK2 staining, there were slight variations in the level of cell body and neuropil staining between the genotypes (Fig. 6). Further, experiments are necessary to address these minor differences. These findings show that the STEP KO mice have elevated phosphorylation of ERK1/2 in specific brain regions.
Fig. 6.
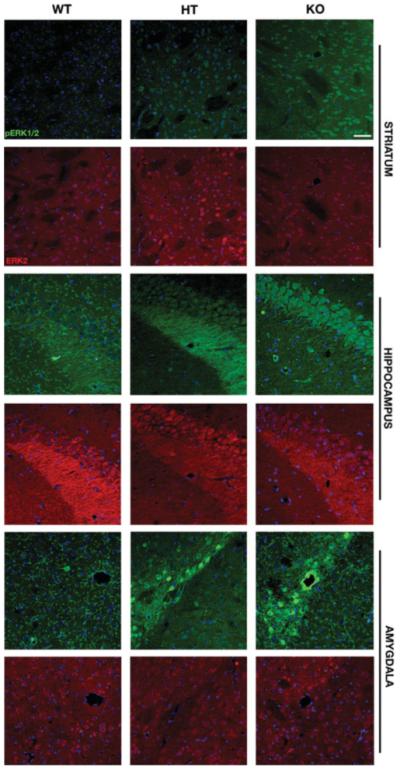
Immunohistochemical analysis of phosphorylated ERK1/2 and total ERK2 in the striatum, hippocampus, and amygdala of WT, HT, and KO mice. Coronal brain sections from the three genotypes were double-labeled with antibodies that recognize the dually phosphorylated ERK1/2 (green) and total ERK2 (red). The two upper panels illustrate staining in the striatum (CPu) from the three genotypes. pERK1/2 immunoreactivity was elevated in the KO mice when compared with WT and HT. The middle and bottom panels show representative sections from the hippocampus and amygdala, respectively. Increased pERK1/2 levels were noticed in the CA2 area of the hippocampus as well as central and lateral nuclei of the amygdala [Scale bar-50 μm].
Enhanced ERK1/2 activation in STEP KO cultures following DHPG stimulation
Previous studies indicate that the activation of the ERK1/2 pathway in the hippocampus is essential for metabotropic glutamate receptor (mGluR1) dependent long-term depression (Gallagher et al., 2004). To evaluate a putative role of STEP in mGluR1-mediated activation of ERK1/2, hippocampal neuronal cultures (14 DIV) obtained from WT and STEP KO mice were stimulated with increasing concentrations of an mGluR1 agonist, DHPG (10, 50, and 100 μM) for 5 min. Immunocytochemical studies showed that under basal conditions the immunoreactivity of pERK1/2 in the neurons obtained from the KO mice was significantly higher when compared with that in the corresponding control (Fig. 7A). DHPG stimulation led to a dose-dependent increase in the accumulation of pERK1/2 in the nucleus of both the experimental (KO) and control (WT) neuronal cultures. However, the increase in immunoreactivity of pERK1/2 was much more prominent in the hippocampal cultures obtained from the KO mice.
Fig. 7.
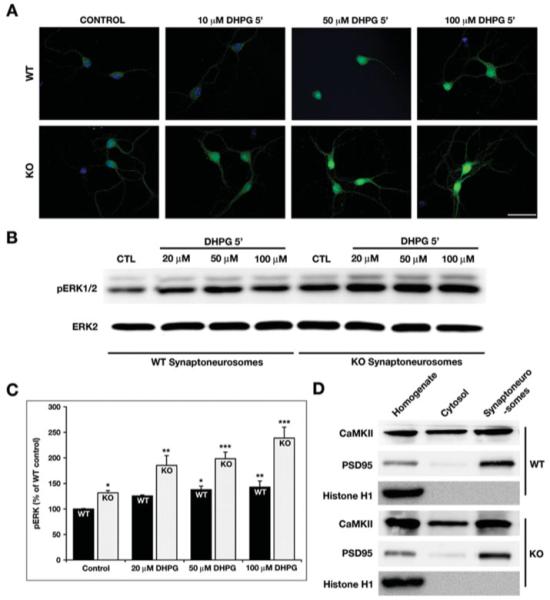
Enhanced phosphorylation of ERK1/2 in the KO hippocampal cultures and synaptoneurosomes. (A) The STEP WT and KO cultures were stimulated with increasing concentration of DHPG for 5 min and stained with pERK1/2 antibody. DHPG leads to activation of pERK1/2 in both the WT and KO cultures. However, the phosphorylation of ERK1/2 is more pronounced in the KO cells when compared with WT. This suggests that in the absence of STEP, there is a higher level of pERK1/2 activation [Scale bar-50 μm]. (B) Synaptoneurosomes prepared from WT and KO mice hippocampi were stimulated with increasing concentration of DHPG and processed for Western blotting. The membranes were probed with pERK1/2 and ERK2 antibodies sequentially. (C) The pERK1/2 levels from WT and KO synaptoneurosomes were measured using ImageJ and normalized to total ERK2 levels. The normalized pERK1/2 levels are plotted as a percentage of WT control levels. The data were compared using one-way ANOVA, followed by posthoc Tukey HSD (WT 20 μm: 126% ± 2%, P > 0.1; WT 50 μm: 138% ± 7%, P < 0.02; WT 100 μm: 143% ± 12%, P < 0.01; KO Ctl: 132% ± 5%, P < 0.02; KO 20 μm: 185% ± 19%, P < 0.01; KO 50 μm: 198% ± 13%, P < 0.005; and KO 100 μm: 237% ± 37%, P < 0.001; *P < 0.05; **P < 0.01; ***P < 0.001; n = 3). (D) The integrity of the synaptoneurosomes was determined by probing the input, cytosolic and synaptosomal fractions with histone H1 (present only in the input), CaMKII (detected in all fractions), and PSD95 (present mainly in the input and synaptosomal fraction).
To quantify the magnitude of ERK activation in the hippocampus of WT and KO mice, we prepared synaptoneurosomes from hippocampal tissue. The synaptoneurosomes were stimulated with increasing concentration of DHPG for 5 min and processed by Western blotting. The synaptoneurosomes obtained from WT as well as KO hippocampal tissue showed a dose-dependent increased in pERK1/2 activation. However, the extent of DHPG-mediated ERK activation was more significant in the KO samples than in the WT controls (Figs. 7B and 7C; WT 20 μM: 126% ± 2%, P > 0.1; WT 50 μM: 138% ± 7%, P < 0.02; WT 100 μM: 143% ± 12%, P < 0.01; KO Ctl: 132% ± 5%, P < 0.02; KO 20 μM: 185% ± 19%, P < 0.01; KO 50 μM: 198% ± 13%, P < 0.005; and KO 100 μM: 237% ± 37%, P < 0.001). The integrity of the preparation was confirmed by probing with PSD95, a specific marker for synaptoneurosomes (Fig. 7D), and the purity was established by the absence of histone H1 and the presence of CaMKII in cytosolic and synaptoneurosomal fractions.
DISCUSSION
Considerable information regarding the biology of STEP has accumulated since its discovery (Lombroso et al., 1991). STEP was the first member of the family of nonreceptor tyrosine phosphatases to show specific expression within the CNS (Boulanger et al., 1995; Lombroso et al., 1993). Alternative splicing produces a cytosolic isoform STEP46 and a membrane-associated isoform, STEP61 (Bult et al., 1996, 1997). They differ from one another by the presence of specific amino acid domains known to be involved in signal transduction. These domains localize STEP isoforms to different compartments, determine substrate specificity, and regulate phosphatase activity (Nguyen et al., 2002; Paul et al., 2000, 2003). Ultrastructural analysis using electron microscopy has localized STEP61 to the endoplasmic reticulum and postsynaptic densities (Boulanger et al., 1995; Oyama et al., 1995).
Activation of ERK1/2 MAP kinase in the hippocampus and amygdala is required for the consolidation of declarative, spatial, and emotional memories (Giovannini, 2006; Sweatt, 2004). Previous studies showed that STEP regulates the duration of ERK1/2 signaling by dephosphorylating a regulatory tyrosine residue in its activation domain (Paul et al., 2003). In addition, STEP also dephosphorylates the tyrosine kinase, Fyn and NR2B subunit of NMDAR (Nguyen et al., 2002; Snyder et al., 2005). Dephosphorylation by STEP leads to inactivation of Fyn, while in the case of NR2B subunit, it leads to internalization of the NMDAR complex. These studies led to the hypothesis that STEP normally opposes the development of synaptic plasticity by dephosphorylating key signaling molecules (Braithwaite et al., 2006).
To study the role of STEP in regulating synaptic plasticity, we made a substrate-trapping inactive variant of STEP carrying a point mutation (Cys to Ser) within the catalytic domain (Paul et al., 2003). This protein was made cell-permeable by the addition of a TAT-peptide to the N-terminus. TAT-STEP (C to S) binds endogenous substrates, but does not dephosphorylate them, and blocks endogenous STEP from interacting with these substrates. TAT-STEP thus acts as a substrate-trapping protein (Flint et al., 1997). The construct can be added to primary cell culture media, brain slices, or the brain itself, and gains entry to virtually 100% of cells within minutes (Paul et al., 2007). Under these conditions, TAT-STEP (C to S) binds to ERK, sequesters it in the cytoplasm, and prevents ERK-mediated transcriptional activation (Paul et al., 2003, 2007). We were able to block the consolidation of fear memories by infusing this fusion protein into the lateral amygdala (Paul et al., 2007). However, these experiments tell us more about proteins that STEP binds to than about the function of STEP itself. The importance of pERK1/2 in the consolidation of memories is well established and the experiments did not directly address the role of STEP. To that end, we generated STEP KO mice.
Here, we present our initial characterization of the STEP KO mice in an effort to specifically address the regulation of ERK1/2 by STEP. The KO mice are viable, fertile, and appear healthy. The frequency of each genotype in the litters born thus far is as expected for a gene that does not result in early lethality (WT = 26%, KO = 26%, HT = 48%). We have confirmed the absence of all STEP isoforms in the KO mice using Western blotting and immunohistochemistry. In the HT mice, STEP expression was reduced to about 50% of WT control. Cresyl violet staining was used to establish that the KO mice did not show gross anatomical changes in the different brain regions, although additional studies will be necessary to determine whether more subtle changes are present, such as changes in spine density.
Consistent with the role of STEP in regulating ERK1/2 activity, we found elevated levels of pERK in total brain homogenates obtained from KO mice. Similar findings were obtained when we determined pERK1/2 levels in specific brain regions. The increase of pERK1/2 in striatal and hippocampal S2 and P2 fractions was higher than the overall increase in total brain homogenates. The immunohistochemical data also showed that pERK1/2 is selectively elevated in certain brain regions including the striatum, hippocampus, and amygdala. These results suggest that the enhanced ERK1/2 activation may be limited to brain regions that normally express STEP. STEP is not expressed in the cerebellum, and the absence of similar increases in pERK1/2 levels in cerebellar homogenates serves as an internal control. Interestingly, the cerebellum expresses a closely related tyrosine phosphatase termed PTP-SL (PTP-STEP-like), although whether it serves similar regulatory function in the cerebellum has not yet been determined (Van Den Maagdenberg et al., 1999).
These results demonstrated that baseline levels of pERK1/2 were elevated in the STEP KO mice. We next asked whether synaptic stimulation would lead to a further increase in pERK1/2 levels. To this end, we stimulated hippocampal cultures with DHPG, as previous studies have shown that DHPG stimulation leads to the phosphorylation of ERK1/2 (Gallagher et al., 2004). Although we detected a dose-dependent increase in ERK1/2 activation in both the WT and KO hippocampal cultures and synaptoneurosomes following DHPG stimulation, the magnitude of change was more prominent in the KO. These data indicate that pERK1/2 levels are elevated both at baseline and after DHPG stimulation in the KO compared with WT controls.
STEP regulates the basal levels of pERK1/2 in the KO mice, although the magnitude of pERK1/2 elevation at baseline varies between different regions. This variation may due, in part, to STEPs activity under baseline conditions as well as differential distribution of the STEP isoforms. Thus, STEP61 is the only isoform found in the hippocampus and lateral amygdala, whereas both STEP46 and STEP61 are found in the striatum (Paul et al., 2007; Pelkey et al., 2002). STEP also plays a role in modulating the degree of pERK1/2 elevation following synaptic stimulation. These findings support the hypothesis that one of the important functions of STEP is to regulate ERK signaling.
Activation of ERK1/2 MAP kinase in the hippocampus and amygdala is required for the consolidation of declarative, spatial, and emotional memories (Giovannini, 2006, Sweatt, 2004). STEP is also known to play a role in the modulation of long-term potentiation and memory formation (Paul et al., 2007; Pelkey et al., 2002). Our findings suggest that this important function of STEP may also be because of its ability to regulate ERK-dependent synaptic plasticity and support the hypothesis that the STEP KO mice will develop synaptic plasticity more easily than WT control mice. Recent data suggest that the tyrosine phosphorylation of the other STEP substrates is elevated in the KO mice. Also, preliminary electrophysiological and behavioral experiments support our hypothesis that the STEP KO will show increased LTP and enhanced learning and memory. We are currently performing a battery of biochemical, electrophysiological, and behavioral experiments to further confirm these results.
ACKNOWLEDGMENTS
The authors thank Keith Haskell and Mary Bauchmann for their help in the production of STEP KO mice. They also thank members of the laboratory for their suggestions and critical reading of this manuscript.
Contract grant sponsor: NIH; Contract grant numbers: MH01527, MH52711, DA017360; Contract grant sponsors: The National Association of Research on Schizophrenia and Depression (NARSAD) to PJL, Brown-Coxe fellowship to DVV.
Abbreviations
- AMPAR
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors
- BSA
bovine serum albumin
- CA
Cornu ammonis
- DHPG
(RS)-3,5-dihydroxyphenylglycine
- ERK
extracellular signal-regulated kinase
- HRP
horseradish peroxidase
- KIM
kinase interacting motif
- LTD
long-term depression
- NGS
normal goat serum
- NMDAR
N-methyl d-aspartate receptor
- PAGE
polyacrylamide gel electrophoresis
- PBS
phosphate buffered saline
- SDS
sodium dodecyl sulfate
- STEP
striatal enriched protein tyrosine phosphatase
REFERENCES
- Boulanger LM, Lombroso PJ, Raghunathan A, During MJ, Wahle P, Naegele JR. Cellular and molecular characterization of a brain-enriched protein tyrosine phosphatase. J Neurosci. 1995;15:1532–1544. doi: 10.1523/JNEUROSCI.15-02-01532.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braithwaite SP, Adkisson M, Leung J, Nava A, Masterson B, Urfer R, Oksenberg D, Nikolich K. Regulation of NMDA receptor trafficking and function by striatal-enriched tyrosine phosphatase (STEP) Eur J Neurosci. 2006;23:2847–2856. doi: 10.1111/j.1460-9568.2006.04837.x. [DOI] [PubMed] [Google Scholar]
- Braithwaite SP, Paul S, Nairn AC, Lombroso PJ. Learning and Memory: One STEP at time. Trends Neurosci. 2006;29:452–458. doi: 10.1016/j.tins.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bult A, Zhao F, Dirkx R, Sharma E, Lukacsi E, Solimena M, Naegele JR, Lombroso PJ. STEP61: A new member of a family of brain-enriched PTPs is localized to the ER. J Neurosci. 1996;16:7821–7831. doi: 10.1523/JNEUROSCI.16-24-07821.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bult A, Zhao F, Dirkx R, Raghunathan A, Solimena M, Lombroso PJ. STEP: A family of brain enriched PTPs: Alternative splicing produces transmembrane, cytosolic and truncated isoforms. Eur J Cell Biol. 1997;72:337–344. [PubMed] [Google Scholar]
- Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, Popko B. Myelination in the absence of galactocerebroside and sulfatide: Normal structure with abnormal function and regional instability. Cell. 1996;86:209–219. doi: 10.1016/s0092-8674(00)80093-8. [DOI] [PubMed] [Google Scholar]
- Flint AJ, Tiganis T, Barford D, Tonks NK. Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc Natl Acad Sci USA. 1997;13:1189–1215. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Powell JF, Smith AD. Tyrosine hydroxylase-immunoreactive boutons in synaptic contact with identified striatonigral neurons, with particular reference to dendritic spines. Neuroscience. 1984;13:1189–1215. doi: 10.1016/0306-4522(84)90294-x. [DOI] [PubMed] [Google Scholar]
- Gallagher SM, Daly CA, Bear MF, Huber KM. Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J Neurosci. 2004;24:4859–4864. doi: 10.1523/JNEUROSCI.5407-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannini MG. The role of the extracellular signal-regulated kinase pathway in memory encoding. Rev Neurosci. 2006;17:619–634. doi: 10.1515/revneuro.2006.17.6.619. [DOI] [PubMed] [Google Scholar]
- Hallett PJ, Spoelgen R, Standaert DG, Dunah AW. Dopamine D1 activation potentiates striatal NMDA receptors by tyrosine phosphorylation-dependent subunit trafficking. J Neurosci. 2006;26:4690–4700. doi: 10.1523/JNEUROSCI.0792-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Zhang Y, Venkitaramani DV, Lombroso PJ. Translation of striatal-enriched protein tyrosine phosphatase (STEP) after β1-adrenergic receptor stimulation. J Neurochem. 2007;103:531–541. doi: 10.1111/j.1471-4159.2007.04749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotter R. Postsynaptic integration of glutamatergic dopaminergic signals in the striatum. Prog Neurobiol. 1994;44:163–196. doi: 10.1016/0301-0082(94)90037-x. [DOI] [PubMed] [Google Scholar]
- Lombroso PJ, Murdoch G, Lerner M. Molecular characterization of a protein-tyrosine-phosphatase enriched in striatum. Proc Natl Acad Sci USA. 1991;88:7242–7246. doi: 10.1073/pnas.88.16.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombroso PJ, Naegele JR, Sharma E, Lerner M. A protein tyrosine phosphatase expressed within dopaminoceptive neurons of the basal ganglia and related structures. J Neurosci. 1993;13:3064–3074. doi: 10.1523/JNEUROSCI.13-07-03064.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TH, Liu J, Lombroso PJ. Striatal enriched phosphatase 61 (STEP61) dephosphorylates Fyn at phosphotyrosine 420. J Biol Chem. 2002;277:24274–24279. doi: 10.1074/jbc.M111683200. [DOI] [PubMed] [Google Scholar]
- Oyama T, Goto S, Nishi T, Sato K, Yamada K, Yoshikawa M, Ushio Y. Immunocytochemical localization of the striatal enriched protein tyrosine phosphatase in the rat striatum: A light and electron microscopic study with a complementary DNA-generated polyclonal antibody. Neuroscience. 1995;69:869–880. doi: 10.1016/0306-4522(95)00278-q. [DOI] [PubMed] [Google Scholar]
- Paul S, Hisayuki Y, Snyder G, Picciotto MR, Nairn AC, Lombroso PJ. Dopamine/D1 receptor mediates the phosphorylation and inactivation of the protein tyrosine phosphatase. STEP, through a PKA-mediated pathway. J Neurosci. 2000;20:5630–5638. doi: 10.1523/JNEUROSCI.20-15-05630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Nairn AC, Wang P, Lombroso PJ. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat Neurosci. 2003;6:34–42. doi: 10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- Paul S, Olausson P, Venkitaramani DV, Ruchkina I, Moran TD, Tronson N, Mills E, Hakim S, Salter MW, Taylor JR, Lombroso PJ. The protein tyrosine phosphatase STEP gates long-term potentiation and fear memory in the lateral amygdala. Biol Psychol. 2007;61:1049–1061. doi: 10.1016/j.biopsych.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey K, Askalan R, Paul S, Kalia LV, Nguyen TH, Pitcher GM, Salter MW, Lombroso PJ. Tyrosine phosphatase STEP is a tonic brake on induction of long-term potentiation. Neuron. 2002;34:127–138. doi: 10.1016/s0896-6273(02)00633-5. [DOI] [PubMed] [Google Scholar]
- Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE. Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of Pavlovian fear conditioning. J Neurosci. 2000;2:8177–8187. doi: 10.1523/JNEUROSCI.20-21-08177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma E, Zhao F, Bult A, Lombroso PJ. Identification of two alternatively spliced transcripts of STEP: A subfamily of brain-enriched protein tyrosine phosphatases. Mol Brain Res. 1995;32:87–93. doi: 10.1016/0169-328x(95)00066-2. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CM, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by β-amyloid. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Van Den Maagdenberg AM, Bachner D, Schepens JT, Peters W, Fransen JA, Wieringa B, Hendricks WJ. The mouse Ptprr gene encodes two protein tyrosine phosphatases. PTP-SL and PTPBR7 that display distinct patterns of expression during neural development. Eur J Neurosci. 1999;11:3832–3844. doi: 10.1046/j.1460-9568.1999.00802.x. [DOI] [PubMed] [Google Scholar]
- Venkitaramani DV, Chin J, Netzer WJ, Gouras GK, Lesne S, Malinow R, Lombroso PJ. β-amyloid modulation of synaptic transmission and plasticity. J Neurosci. 2007;27:11832–11837. doi: 10.1523/JNEUROSCI.3478-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]