

Abstract
We present a theoretical framework that enables us to dissect out the parametric dependencies of the pathogenesis of prion diseases. We are able to determine the influence of both host-dependent factors (connectivity, cell density, protein synthesis rate, and cell death) and strain-dependent factors (cell tropism, virulence, and replication rate). We use a model based on a linked system of differential equations on a lattice to explore how the regional distribution of central nervous system pathology in Creutzfeldt-Jakob disease, Gerstmann-Sträussler-Scheinker syndrome, and fatal familial insomnia relates to each of these factors. The model then is used to make qualitative predictions about the pathology for two possible hypothetical triggers of neuronal loss in prion diseases. Pathological progression in overexpressing mouse models has been shown to depend on the site of initial infection. The model allows us to compare the pathologies resulting from different inoculation routes.
Keywords: strain, transmissible spongiform encephalopathies
Transmissible spongiform encephalopathies are neurodegenerative disorders characterized by an accumulation of a protease-resistant isoform (PrPSc) of the cellular prion protein (PrPc) (1, 2). The presence of PrPSc within nervous tissue results in the formation of amyloid plaques, spongiform changes, reactive astrocytosis, and neuronal loss (3–5). A characteristic feature of these diseases is the large degree of variation in the distribution and magnitude of pathological changes associated both with the host species and the PrPSc strain (6, 7). These variations have been attributed to several factors including the kinetics of prion propagation, cell tropism, differential toxicity, and host neuro-anatomy and genetic makeup. In experimental systems it is difficult to separate these diverse causes and thereby determine the impact of each variable independently. In the present study we develop a model for prion propagation within the central nervous system, which allows us to explore each of the parameters involved in prion pathogenesis.
All of the known prion diseases in animals and humans can be correlated with an accumulation of PrPSc in the central nervous system, suggesting that the information carrying pathogen is a protein (8). Pronounced differences in human pathology emerge in comparisons of Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome (GSS), and fatal familial insomnia (FFI) cases (3–5, 9). In familial CJD, loss of neurons and astrocytosis is prominent in gray matter, and PrP amyloid plaques sometimes are reported. These plaques tend to be found in the cerebellum adjacent to the Purkinje cells and sometimes are arranged in arrays. In new variant CJD numerous kuru-type PrP plaques are observed surrounded by a halo of spongiform degeneration. The number of plaques in CJD correlates positively with disease latency. In iatrogenic CJD, lesions and plaques are distributed more diffusely throughout the brain (3). In kuru around 70% of cases have plaques, and the number is greater in long latency cases. In GSS PrP plaques are common and have a unique multicentric configuration; in FFI no plaques have been observed. A similar spectrum of pathology is observed in the animal prion diseases (14). The genotype of the host has an enormous influence on disease susceptibility and progression. For example, mutations at codon 129 of the human prion gene have been shown to be as important in determining pathology as the PrP isotype (11).
Methods
This paper differs from previous theoretical work on prion infection (12–16) as it focuses on pathogenesis in the brain. This approach allows us to explore mechanisms giving rise to variations in the neuropathology. We start with a description of PrPSc replication kinetics. Throughout we assume that PrPSc is required for the transformation of PrPc, thus we subscribe to a protein-only transmission mechanism. This mechanism has been suggested to involve either some form of nucleation-dependent aggregation (15) or a conformational change of the PrP (1). In both cases one describes the metabolic pathways by which the diseased form of the protein is formed from the healthy PrPc and how it is then incorporated into β-amyloid. We model this process qualitatively and use a generic realization of the metabolic pathway:
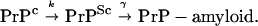 |
1 |
We describe the mathematical model of the disease kinetics in detail in Appendix 1. The within-cell dynamics consist of the PrPc → PrPSc conversion; PrPSc is free to move and can infect other cells (17). Strain parameters enter the dynamics in a straightforward way: for greater values of k, more PrPSc is produced more quickly, whereas greater values of γ increase the rate of PrP-plaque deposition. The parameter bij determines the rate of cell to cell diffusion and α/β control the rate of PrPc production/decay. The parameter K determines the average number of connections among cells (to capture close apposition and synaptic contact). Because of computational limitations we simulate values of K in series in the order of tens of connections rather than thousands.
Results
Effects of Varying PrPc Turnover.
It has been observed that increased production of PrPc reduces the latency of prion disease onset (18). In Fig. 1 a and b we follow the accumulation of PrPc in a 25 by 25 array of simulated nerve cells for different rates of PrPc production. Infection is initiated by inoculation of PrPSc into the cell in the center of the array, assuming a baseline level of PrPc production (Fig. 1a) and a higher rate of production (Fig. 1b). After a short period (T2) infection has spread to a number of cells through bidirectional connections in Fig. 1 a and b. In Fig. 1a the concentration of PrPSc in each cell other than at the site of the inoculum remains negligible. By contrast, the high production series (Fig. 1b) shows that each of the strongly connected cells already has accumulated considerable PrPSc. By T3 the low turnover series (Fig. 1a) is building up PrPSc concentration, whereas the high production series is already nearing saturation. Thus turnover significantly influences the spatial and temporal distribution of PrPSc.
Figure 1.

PrPSc accumulation pattern over time. (a) α = 200 and no inhibition for K = 4. (b) Effect of increased PrPc production, α = 250 (no inhibition, K = 4). (c) Effect of axonal inhibition as PrPSc is accumulated (α = 250, K = 4). (d) Effect of higher local connectivity, K = 8 (α = 250, no inhibition). In all cases six long-range axons have randomly been placed between cells in addition to the K local connection per neuron.
The same pattern is true for the distribution of PrP-amyloid (Fig. 2). The high turnover series (Fig. 2b) shows a much faster rate of deposition and a more abundant distribution of amyloid than the baseline infection (Fig. 2a).
Figure 2.

PrP-amyloid deposition patterns for the same parameters as in Fig. 1. (a) α = 200, no inhibition, K = 4. (b) Effect of increased PrPc production. (c) Effect of inhibition of axonal transport. (d) Effect of increased local connectivity.
Inhibitory Effects of PrPSc.
If the strength of the interneuronal connections is weakened by PrPSc-induced killing of target cells, the spread of the infectious agent will be reduced when it is at high concentrations within a cell. Thus infectious propagation can depend on the concentration gradients of PrPSc and the inhibitory influence of the protein on axonal/dendritic connections. We assume a simple linear relation between PrPSc concentration in the cells and strength of the inter neuronal connection (see Appendix 2). In Fig. 1c we follow the influence of inhibition on the accumulation of PrPSc through time. In the early stages of infection inhibition has little influence on the propagation of infection, because intracellular PrPSc has not yet reached a concentration that induces the axons to regress. By the final epoch, the inhibition series is distinguished by a more patchy distribution of PrPSc than in the absence of inhibition. The amyloid deposition series (Fig. 2c) reflects this pattern only very weakly and the distribution of plaques is effectively indistinguishable from the baseline case (Fig. 2a).
Effects of Local Neuronal Connectivity.
Different regions in the central nervous system possess different numbers of connections per neuron. Within the cerebellum the average number of connections a neuron can form varies from a few dozen for granular cells to 150,000 for Purkinje cells. Therefore we will next investigate the influence of structural variation in different brain regions on the patterns of PrPSc and amyloid deposition.
In Fig. 1d we have doubled the mean connectivity in our model while keeping all other parameters fixed. Early in the infectious process we notice that more cells are involved than in the baseline infection (Fig. 1a). By the final stages of infection PrPSc is abundant in each cell. Thus PrP deposition is increased significantly in highly connected regions, and progression to clinical disease is expected to be more rapid in these regions.
Unlike high PrPc turnover (Fig. 2b) where there are many regions without amyloid-free cells, in Fig. 2d there are few regions in which groups of contiguous cells all have deposited plaques. In the figure this is suggested by the clusters of amyloid-free cells within infected regions.
PrPSc Accumulation Vs. PrPc Depletion and Neuronal Loss.
There is mounting evidence that apoptosis is responsible for cell death in prion diseases (19, 20). We explore two possible mechanisms for the induction of cell loss. In one, PrPSc accumulation directly induces cell death by invoking the apoptotic pathway. In the second mechanism, PrPc is assumed to play a (as yet unknown) crucial role in the neuron; PrPc depletion therefore leads to cell death through loss of function. These two mechanisms are implemented, assuming either that (i) a 50% reduction of the PrPc concentration leads to cell death or (ii) a PrPSc concentration exceeding the normal cellular concentration of PrPc in the healthy cell by 50% produces cell death.
In Fig. 3 we plot the time course of neuronal loss. In the top series (Fig. 3a) we assume direct activation of apoptosis whereas in the bottom series we assume death through loss of function (Fig. 3b). Over the first four epochs of infection degeneration is indistinguishable. This is because we are seeing the early phase of infection, during which there is transport and transformation of susceptible cells rather than cell death. By the time T5 the PrPSc neurotoxicity series (Fig. 3a) is beginning to show signs of cell death as more of the brain degenerates. At T6 the differences between the two treatments have become very pronounced. PrPSc accumulates rapidly within the infected cells in a density-dependent fashion, whereas PrPc depletes slowly as cells are constantly synthesizing new PrP, resulting in a delayed onset of cell loss.
Figure 3.

Neurotoxicity of PrPSc (a) vs. loss of function through PrPc depletion (b) as triggers for neuronal loss. The distribution of uninfected cells (gray), infected (black), and absence of neurons (white) for the two possible simple mechanisms are shown at 10 equidistant time intervals until all cells are dead. The arbitrary threshold was set such that the cell dies when c(PrPSc) > 1.5 × c0(PrPc) (a) and c(PrPc) < 0.5 × c0(PrPc) (b), respectively.
The differences in PrP-amyloid deposition are more revealing. In Fig. 4 we plot the absolute amyloid concentrations for epochs T1, T5, and T10 corresponding to Fig. 3. Assuming a toxic PrP we see very little amyloid deposition as the rapid accumulation of PrPSc kills cells before they can deposit large numbers of plaques. Whereas with loss of function, cells die at a lower rate and thereby allow a greater amount of amyloid to be deposited over the protracted lifetime of the cells.
Figure 4.

Spatial PrP-amyloid deposition characteristics for the case of PrPSc neurotoxicity (a) and PrPc depletion (b) as triggers for neuronal loss. The time labels refer to the corresponding times in Fig. 3. Depletion results in significantly higher (by a factor of ≈10) concentrations of PrP-amyloid than neurotoxicity.
Effects of Cellular Tropism.
Tropism of PrPSc for different neuronal cell types has been proposed as an explanation for the diversity of prion-associated pathologies upon infection with different PrPSc strains (21, 22). We model tropism as strain-specific rates k of transformational change of PrPc into PrPSc. Thus a strain has a higher rate of transformation within cells for which it has a preferential tropism.
In Fig. 5a we have assumed that k = k0 in the front half of the lattice and k = 2 × k0 in the back half (k0 = 0.0001). In Fig. 5b we have moved the site of initial inoculation. Independent of the site of inoculation, PrPSc deposition and disease progression are more pronounced in the region with the higher value of k. If different forms of PrPc do occur in the same brain, then it is possible for small variations in the value of k associated with different strains to lead to radically different PrPSc accumulation patterns.
Figure 5.

Neurotropism as a cause for characteristic PrPSc accumulation patterns. In our model neurotropism is attributed to a local difference in the PrPSc replication rate, k. The initial infection sites are clearly visible in the T1 frames. Because of the higher value of k in the neurons in the rear part of the spatial model, PrPSc deposition is increased in that region whereas comparatively little PrPSc accumulates in the front half.
Discussion
We have modeled neuropathogenesis by a spatial model incorporating the reaction kinetics of PrPc conversion to PrPSc. Using the modeling approach we have attempted to separate out strain- and host-dependent factors. In the following qualitative discussion we relate our results to empirical findings. We were only able to show characteristic cases but our study has shown remarkable robustness in all of the parameters including k, γ, and K. Moreover we found that the results for the cell loss triggers are qualitatively the same for a wide range of threshold values. Simulations were run with up to 5,000 cells, and no qualitative difference was found for the smaller numbers presented here. The documented C-source code used for the simulations can be downloaded on our web site: http://mathbiol.zoo.ox.ac.uk/michael/prions.html.
The model shows the huge influence PrPc turnover and neuronal connectivity exerts on disease progression. Axonal regression, which is observed in prion diseases (23), on the other hand, was not found to alter the spatial dynamics of the infectious agent significantly. Loss of connectivity is, however, functionally equivalent to loss of neurons, and we therefore expect to find neurological change as axons regress.
The model shows that neuronal connectivity profoundly influences pathogenesis; it is probably the high density of neuronal connections in the cerebellum (which holds about 30% of the neurons in 10% of the total volume of the brain) that gives rise to the very similar early clinical progressions observed in the human prion diseases. This is also confirmed by neuropathology: in kuru and GSS PrP-amyloid is found at relatively high concentrations in the cerebellum. Neuronal loss is also most severe in GSS (3–5).
As a result of the use of mice overexpressing PrP in experimental inoculation studies (9, 24) pathology often differs from naturally occurring diseases (where oral intake is presumably more common). PrPc overexpression accelerates intracellular replication dynamics and loss of neurons. This has the result that early clinical symptoms stem from neuronal degradation close to the inoculation site and not from damage to regions toward which PrPSc is “naturally” attracted. To gain a better understanding of human prion disease pathogenesis it therefore seems important to use transgenic models that (i) are inoculated orally and (ii) do not overexpress human PrPc. The problem is that overexpression is required to ensure infection over a manageable time scale; but, using the modeling approach outlined above, predictions about the pathology in naturally occurring disease also can be derived from experiments with overexpressing mice.
Although the precise mechanism by which neuro-degeneration occurs remains uncertain, evidence suggests that apoptosis is responsible for neuronal death (19, 20). The accumulation of PrPSc could induce neurons to enter into apoptosis either through direct activation of cell death (25, 26) or through the competitive elimination of essential PrPc molecules (27). The former explanation often has been favored as prion knockout mice exhibit few deleterious phenotypes (40). In the absence of positive, conditional knockout mice models the nature of the cell loss trigger remains currently undecided by experiments (2).
Our model has allowed us to explore the neuropathological implications of both of these mechanisms. In the case where PrPSc is neurotoxic, a large number of cells are induced into cell death before an appreciable quantity of PrP-amyloid has been deposited. Whereas when cell death results from PrPc depletion, cells take a longer time to die and thereby produce a much larger quantity of PrP-amyloid. Thus abundant PrP-amyloid may be symptomatic of loss of function, whereas cell loss may be symptomatic of direct activation of apoptosis. With this result in mind we can contrast the pathologies of CJD, GSS, and FFI. In CJD there is a large amount of cell death and relatively few plaques. In GSS (and also in kuru) there are numerous plaques and less cell death. We hypothesize that in CJD and in particular in FFI, PrPSc is able to directly induce cell loss. By contrast, PrPSc/GSS is less toxic and hence cell death is primarily a consequence of PrPc depletion. Of course, amyloid frequency also reflects the strain and potentially the body's capability of partially digesting that particular strain.
Numerous studies have demonstrated that PrPSc accumulation is highly strain-dependent (22). The existence of differentially glycosylated PrPc forms in the same host brain or of different molecular chaperones in the PrPc→ PrPSc structural change can lead to radically different PrPSc and PrP-amyloid accumulation patterns and lesion profiles. A recent finding is that in scrapie-infected mice, cortical parvalbumin neurons are severely reduced whereas calbindin and calretinin neurons remain unaffected (29). In CJD-inoculated mice, loss of parvalbumin neurons is severe and occurs very early after inoculation (30).
Although we cannot explain the mechanisms behind cellular tropism of PrPSc with our model, we can approximate them, assuming differences in the replication rate of PrPSc with different forms of PrPc. Thus an important result of our model has been to show how partial tropism can lead to strain-specific PrPSc accumulation patterns. In fact, at least in our model, in a protein-only mechanism of transmissible spongiform encephalopathies cellular tropism is both necessary and sufficient to generate different pathological phenotypes.
The present work suggests a path toward an understanding of prion-related pathogenesis. We believe that by adding structural detail to the model predictions for empirically derived PrPSc accumulation patterns after inoculation are in reach. Furthermore, the model provides a conceptual assay of the neurotoxicity of different PrPSc isoforms in prion diseases.
Acknowledgments
We thank Lynn Enquist for his insightful comments on the manuscript. M.P.H.S. gratefully acknowledges support through fellowships from the Wellcome Trust and Linacre College, Oxford. D.C.K. thanks the Leon Levy and Shelby White Initiatives Fund, the Florence Gould Foundation, the J. Seward Johnson, Sr. Charitable Trusts, the Ambrose Monell Foundation, and the Alfred P. Sloan Foundation.
Abbreviations
- PrP
prion protein
- PrpSc
protease-resistant isoform of PrP
- Prpc
cellular PrP
- CJD
Creutzfeldt-Jakob disease
- GSS
Gerstmann-Sträussler-Scheinker syndrome
- FII
fatal familial insomnia
The Kinetic Model
We model prion propagation with the following system of ordinary differential equations:
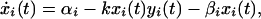 |
2 |
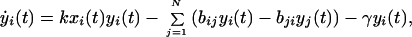 |
3 |
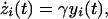 |
4 |
where x, y, z represent the concentrations of PrPc, PrPSc, and extracellular PrP plaques, respectively, and the index i labels the cells. PrPc is assumed to be created at a cell specific rate αi and destroyed within cells at a rate βi (in the absence of tropism we have αi = α and βi = β ∀i). PrPc is converted into PrPSc upon contact at a rate kxi(t)yi(t) (15). We assume that PrPSc spreads predominantly along axons and dendrites by slow axonal transport (17). The rate of PrPSc spread from one cell to another is a function of the connection strength, bji, and the difference in concentration of the diffusing PrPSc, |yi(t) − yj(t)|. Because PrPc is uniformly distributed throughout the mammalian brain we believe that PrPc diffusion is negligible. Assuming symmetrical transport we have for the elements of the connectivity matrix (bij)
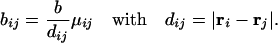 |
5 |
The parameter dij is the metric distance between cells i and j; μij embodies the local structure of the brain and is defined by
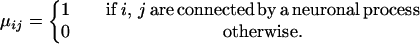 |
6 |
The PrP bound in plaques, z, remains attached to the cell where it is produced, thereby assuming negligible rates of transport of (amyloid) plaques.
The cellular dynamics of the PrPc/PrPSc system (Eqs. 2 and 3) show two equilibria,
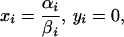 |
7 |
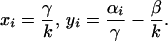 |
8 |
Eq. 7 represents the case of no infection where the PrPc abundance is determined by the ratio of its production and decay rates. Eq. 8 is the infection equilibrium and hence the PrPc abundance is given by the ratio of transformation rate and PrPSc decay rate. It is important to note that PrPSc >> PrPc at the infectious equilibrium. The equilibria do not depend on the connective strengths among cells (bij).
Axonal Regression
We define the function whereby axons and dendrites degenerate according to the death of anterograde and retrograde nerve cells. Assuming that the cells provide connections with some form of trophic support we write
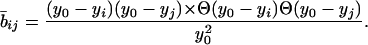 |
9 |
Here y0 is the threshold level of PrPSc at which the connection breaks down and yi, yj are the concentrations of PrPSc in cells i and j. The step functions [Θ(y0 − yi/j) Θ(X) = 0 for x < 0 and Θ(x) = 1 for x > 0] ensure that (i) the axon “dies” if the PrPSc concentration exceeds the critical level y0 and (ii) 1 b̄ij ≥ 0. Eq. 9 is the simplest representation of a function describing inhibition of axonal transport with increased PrPSc deposition, and we can multiply the right-hand side of Eq. 5 by b̄ij to arrive at a inhibitory connectivity matrix. In general we expect that the axons regress before the neurons die, i.e., y0 < yc.
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.180317097.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.180317097
References
- 1.Prusiner S. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 2.Aguzzi A, Weissmann C. Nature (London) 1997;389:795–798. doi: 10.1038/39758. [DOI] [PubMed] [Google Scholar]
- 3.Bell J, Ironside J. Br Med Bull. 1993;49:738–777. doi: 10.1093/oxfordjournals.bmb.a072645. [DOI] [PubMed] [Google Scholar]
- 4.Ironside J, Bell J. In: Prion Diseases. Collinge J, Palmer M, editors. Oxford: Oxford Univ. Press; 1997. pp. 57–88. [Google Scholar]
- 5.DeArmond S, Ironside J. In: Prion Biology and Diseases. Prusiner S, editor. Plainview, NY: Cold Spring Harbor Lab. Press; 1999. pp. 585–652. [Google Scholar]
- 6.Bruce M. Br Med Bull. 1993;49:822–838. doi: 10.1093/oxfordjournals.bmb.a072649. [DOI] [PubMed] [Google Scholar]
- 7.Lasmezas C, Deslys J, Demaimay R, Adjou K, Hauw J, Dormont D. J Gen Virol. 1996;77:1601–1609. doi: 10.1099/0022-1317-77-7-1601. [DOI] [PubMed] [Google Scholar]
- 8.Prusiner S, Fuzi M, Scott M, Serban D, Serban H, Taraboulos A, Gabriel J, Wells G, Wilesmith J, Bradley R, et al. J Infect Dis. 1993;167:602–613. doi: 10.1093/infdis/167.3.602. [DOI] [PubMed] [Google Scholar]
- 9.DeArmond S, Prusiner S. Brain Pathol. 1995;5:77–89. doi: 10.1111/j.1750-3639.1995.tb00579.x. [DOI] [PubMed] [Google Scholar]
- 10.Bradley R. In: Prion Diseases. Collinge J, Palmer M, editors. Oxford: Oxford Univ. Press; 1997. pp. 89–129. [Google Scholar]
- 11.Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr L, Budka H, Kopp N, Piccardo P, et al. Ann Neurol. 1999;46:224–233. [PubMed] [Google Scholar]
- 12.Griffiths J. Nature (London) 1967;215:1043–1044. doi: 10.1038/2151043a0. [DOI] [PubMed] [Google Scholar]
- 13.Eigen M. Biophys Chem. 1996;63:A1–A18. doi: 10.1016/s0301-4622(96)02250-8. [DOI] [PubMed] [Google Scholar]
- 14.Payne R, Krakauer D. Proc R Soc London Ser B. 1998;265:2341–2346. doi: 10.1098/rspb.1998.0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nowak M, Krakauer D, Klug A, May R. Integrative Biol. 1998;1:3–15. [Google Scholar]
- 16.Masel J, Jansen V, Nowak M. Biophys Chem. 1999;77:139–152. doi: 10.1016/s0301-4622(99)00016-2. [DOI] [PubMed] [Google Scholar]
- 17.Scott J, Fraser H. Brain Res. 1989;504:301–305. doi: 10.1016/0006-8993(89)91373-5. [DOI] [PubMed] [Google Scholar]
- 18.Prusiner S, Groth D, Serban A, Koehler R, Foster D, Torchia M, Burton D, Yand S, DeArmond S. Proc Natl Acad Sci USA. 1993;90:10608–10612. doi: 10.1073/pnas.90.22.10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giese A, Groschup M, Hess B, Kretzschmar H. Brain Pathol. 1995;5:213–221. doi: 10.1111/j.1750-3639.1995.tb00597.x. [DOI] [PubMed] [Google Scholar]
- 20.Schatzl H, Laszlo L, Holtzman D, Tzelt J, DeArmond S, Weiner R, Mobley W, Prusiner S. J Virol. 1997;71:8821–8831. doi: 10.1128/jvi.71.11.8821-8831.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeArmond S, Yang S, Cayetano-Canlas J, Groth D, Prusiner S. Philos Trans R Soc London B. 1994;343:415–423. doi: 10.1098/rstb.1994.0038. [DOI] [PubMed] [Google Scholar]
- 22.DeArmond S, Sanchez H, Yehiely F, Qiu Y, Ninchak-Casey A, Daggett V, Camerino A, Cayetano J, Rogers M, Groth D, et al. Neuron. 1997;19:1337–1348. doi: 10.1016/s0896-6273(00)80424-9. [DOI] [PubMed] [Google Scholar]
- 23.Clinton J, Forsyth C, Royston M, Roberts G. NeuroReport. 1993;4:65–68. doi: 10.1097/00001756-199301000-00017. [DOI] [PubMed] [Google Scholar]
- 24.Raeber A, Brander S, Klein M, Benninger Y, Musahl C, Frigg R, Roeckl C, Fischer M, Weissmann C, Aguzzi A. Brain Pathol. 1998;8:715–733. doi: 10.1111/j.1750-3639.1998.tb00197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin M, Mirzabekov T, Kagan B. J Biol Chem. 1997;272:44–47. doi: 10.1074/jbc.272.1.44. [DOI] [PubMed] [Google Scholar]
- 26.Florio T, Thellung S, Amico C, Robello M, Salmona M, Bugiani O, Tagliavini F, Forloni G, Schettini G. J Neurosci Res. 1998;54:341–352. doi: 10.1002/(SICI)1097-4547(19981101)54:3<341::AID-JNR5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 27.Kuwahara C, Takeuchi A, Nishimura T, Haraguchi K, Kubosaki A, Matsumoto Y, Sacki K, Matsumoto Y, Yokoyama T, Itohara S, et al. Nature (London) 1999;400:225–226. doi: 10.1038/22241. [DOI] [PubMed] [Google Scholar]
- 28.Katamine S, Nishida N, Sugimoto T, Noda T, Sakaguchi S, Shigematsu K, Kataoka Y, Hasegawa S, Moriuchi R, Miyamoto T. Cell Mol Neurobiol. 1998;18:731–742. doi: 10.1023/A:1020698305911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yun S, Choi E, Ju W, Ahn M, Carp R, Wisniewski H, Kim Y. Mol Chem Neuropathol. 1998;34:121–132. doi: 10.1007/BF02815074. [DOI] [PubMed] [Google Scholar]
- 30.Guentchev M, Groschup M, Kordek R, Liberski P, Budka H. Brain Pathol. 1998;8:615–623. doi: 10.1111/j.1750-3639.1998.tb00188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]