

Abstract
A gene causing autosomal-recessive, nonsyndromic hearing loss, DFNB39, was previously mapped to an 18 Mb interval on chromosome 7q11.22-q21.12. We mapped an additional 40 consanguineous families segregating nonsyndromic hearing loss to the DFNB39 locus and refined the obligate interval to 1.2 Mb. The coding regions of all genes in this interval were sequenced, and no missense, nonsense, or frameshift mutations were found. We sequenced the noncoding sequences of genes, as well as noncoding genes, and found three mutations clustered in intron 4 and exon 5 in the hepatocyte growth factor gene (HGF). Two intron 4 deletions occur in a highly conserved sequence that is part of the 3′ untranslated region of a previously undescribed short isoform of HGF. The third mutation is a silent substitution, and we demonstrate that it affects splicing in vitro. HGF is involved in a wide variety of signaling pathways in many different tissues, yet these putative regulatory mutations cause a surprisingly specific phenotype, which is nonsydromic hearing loss. Two mouse models of Hgf dysregulation, one in which an Hgf transgene is ubiquitously overexpressed and the other a conditional knockout that deletes Hgf from a limited number of tissues, including the cochlea, result in deafness. Overexpression of HGF is associated with progressive degeneration of outer hair cells in the cochlea, whereas cochlear deletion of Hgf is associated with more general dysplasia.
Introduction
There are hundreds of genes that, when mutated, cause hearing impairment.1–6 The completion of the Human Genome Project has greatly facilitated the identification of the causative mutations in these genes, especially those that have been positionally mapped by genetic linkage studies. When hearing impairment occurs as part of a genetic syndrome, candidate genes can usually be identified on the basis of a hypothesized function consistent with the physiological effects of the syndrome or by the correlation of expression pattern of the candidate gene with the tissues affected in the syndrome.2,7 But in the case of nonsyndromic hearing impairment (NSHI), the genes identified so far have shown such wide diversity of function and expression patterns that it is difficult to predict which gene in a linkage interval would be the best candidate.8,9 Of the ∼105 NSHI loci that have been mapped thus far, the causative mutation has been identified in 47 genes (Hereditary Hearing Loss homepage). These vary from cochlear-specific genes with functions specialized to the inner ear (e.g., OTOA [MIM 607038]10) to nearly ubiquitously expressed genes with presumed housekeeping functions (e.g., ACTG1 [MIM 102560]11,12). Given these precedents, researchers are left little alternative but to systematically sequence every gene in a linkage interval for a DFNA (dominantly inherited) or DFNB (recessively inherited) NSHI locus.
It is inevitable that for some DFNA or DFNB loci, no mutation in the coding sequences of any gene in the linked interval would be found. This follows by extrapolation from the few cases of well-defined monogenic disorders in which comprehensive analyses have revealed mutations in the noncoding regions of the gene. For example, there are 536 mutant alleles of the HBB (β-globin) gene (MIM 141900), and over 11% of them (61) are in noncoding sequences. These include mutations of 5′ untranslated regions (UTRs), 3′ UTRs, and introns and are hypothesized to affect regulation of HBB. But it is the occurrence of these mutations that defines these sequences as candidates for regulation of HBB, because regulatory sequences are difficult to predict from sequence information alone. Almost all regulatory mutations are identified only after the exclusion of mutation in the coding sequences of a gene with a well-established correlation to a specific phenotype.13,14 The identification of a disease gene through mutations in its putative regulatory sequences is unusual.
Here, we report the results of a mutation screen for the autosomal-recessive NSHI locus DFNB39 (MIM 608265).15 The linkage interval, with boundaries defined by recombinations in families that support highly significant LOD scores, was refined. We examined the coding sequences of every gene in the interval, as well as many noncoding sequences. We found three mutations in HGF, the gene encoding hepatocyte growth factor (MIM 142409). None of the mutations cause amino acid replacements. Two of the mutations occur in a region that we demonstrate to be contained in the 3′ UTR of an alternate splice form of HGF that to our knowledge has not been previously discovered. Additionally, evidence is provided that the third mutation, which is a third-position nucleotide change predicted to make a synonymous amino acid substitution, alters splicing by affecting the relative strengths of the splice acceptor sites in two known alternate splice forms of HGF. The NSHI phenotype of this widely expressed, and widely studied, gene will offer an opportunity for gaining insight on HGF regulation in general and its regulation and function in the cochlea in particular.
Subjects and Methods
Family Enrollment and Diagnosis
Consanguineous families with probands enrolled in schools for the deaf were ascertained in Pakistan and India. Written informed consent was collected from all participants after approval was obtained from the Combined Neuroscience Institutional Review Board (IRB) at the National Institutes of Health (NIH), Bethesda, MD, USA; the IRB at Baylor College of Medicine, Houston, TX, USA; the IRB at the National Centre of Excellence in Molecular Biology, Lahore, Pakistan; the IRB at Quaid-I-Azam University, Islamabad, Pakistan; and the IRB at the All India Institute of Medical Sciences, New Delhi, India. Participating hearing-impaired individuals were evaluated by medical-history interviews and a general physical examination. At least two hearing impaired individuals from each family underwent pure-tone audiological assessment. Some individuals were evaluated by funduscopy, serum chemistry, CBC, and urine analysis for ruling out features of a syndrome. Peripheral blood samples were obtained from each participating individual, and genomic DNA was extracted.16
Linkage Analysis
The DFNB39 locus was defined at 7q11.22-q21.12 by a genome-wide linkage screen of family DEM4011.15 Seventeen additional families were found to segregate recessive deafness linked to DFNB39 in genome-wide linkage scans. Eleven of the families were mapped with the use of ∼400 microsatellite markers with an average resolution of 10 cM from the Marshfield map,17 and six were mapped with the use of Illumina genome scan SNP marker loci panels with an average resolution of 1.5 cM (Table 1). An additional 23 families were found to be segregating the DFNB39 gene either by screening for linkage to the known DFNB loci with the use of a panel of STR markers (Hereditary Hearing Loss homepage) or by screening for mutations in HGF. Once linkage to the DFNB39 locus was established for these families, no further genetic linkage analyses were performed on them.
Table 1.
Summary of DFNB39 Families
| Family IDa | Genome Scan Method | LOD Scoreb | Mutationc |
|---|---|---|---|
| PKDF002de | STR | 4.27 | Δ3 |
| PKDF084de | SNP | 6.11 | Δ3 |
| PKDF121de | - | 4.00 | Δ3 |
| PKDF157de | - | 7.03 | Δ3 |
| PKDF204de | - | 4.80 | Δ3 |
| PKDF239ef | - | 7.41 | Δ3 |
| PKDF351ef | - | 5.34 | Δ3 |
| PKDF352e | - | 3.35 | Δ3 |
| PKDF402e | - | 4.16 | Δ3 |
| PKDF711e | - | 2.20 | Δ3 |
| PKDF841e | - | 2.89 | Δ3 |
| PKDF847 | - | 2.15 | Δ3 |
| PKDF879 | - | 3.24 | Δ3 |
| PKDF1113 | - | 12.00 | Δ3 |
| PKSR36ade | - | 1.84 | Δ3 |
| PKSR53ade | - | 3.22 | Δ3 |
| PKSR2bde | - | 3.52 | Δ3 |
| IDM13de | - | 3.15 | Δ3 |
| Kla2de | - | 2.33 | Δ3 |
| DEM4011e | STR | 3.7 | Δ3 |
| DEM4017A | STR | 3.1 | Δ3 |
| DEM4018e | STR | 6.03 | Δ3 |
| DEM4048 | STR | 3.02 | Δ3 |
| DEM4050 | STR | 1.93 | Δ3 |
| DEM4058 | STR | 2.37 | Δ3 |
| DEM4071 | STR | 5.75 | Δ3 |
| DEM4142e | STR | 2.53 | Δ3 |
| DEM4174e | STR | 3.54 | Δ3 |
| DEM4199e | STR | 3.73 | Δ3 |
| DEM4201 | SNP | 2.3 | Δ3 |
| DEM4212 | STR | 1.35 | Δ3 |
| DEM4320 | SNP | 1.93 | Δ3 |
| DEM4332 | SNP | 1.93 | Δ3 |
| DEM4333A | SNP | 1.33 | Δ3 |
| DEM4333B | SNP | 1.86 | Δ3 |
| DEM4434 | - | 6.57 | Δ3 |
| DEM4443 | - | 3.14 | Δ3 |
| DEM4467 | - | 2.53 | Δ3 |
| DEM4472 | - | 3.61 | Δ10 |
| PKDF601e | - | 4.22 | Δ10 |
| PKDF210de | - | 2.38 | c.495G→A |
All families except Indian families IDM13 and Kla2 were ascertained in Pakistan.
Maximum two-point LOD score obtained at θ = 0.
Δ3 = c.482+1986_1988delTGA; Δ10 = c.482+1991_2000delGATGATGAAA.
DNA from families sequenced for 374 amplicons in the DFNB39 linkage region.
DNA from families sequenced for all coding and conserved noncoding sequences of HGF.
Pedigrees that define the proximal and distal breakpoints for the minimal DFNB39 interval.
LOD scores for the families labeled with a PK, IDM, or Kla prefix were calculated in MLINK of the FASTLINK computer package.18 LOD scores for the families labeled with the DEM prefix were calculated in MLINK, used for two-point analyses, and with ALLEGRO19 and SIMWALK2, used for multipoint analyses.20,21 Marker allele frequencies were estimated with genotypes from founders and reconstructed founders from the DEM pedigrees. Haplotypes were reconstructed in ALLEGRO and SIMWALK2.
Candidate Gene Screening
Candidate genes within the DFNB39 interval were screened for mutations by direct sequencing of PCR products generated from genomic DNA from one affected individual of some DFNB39-linked families. Regions of conservation were identified with the Vertebrate Multiz Alignment and PhastCons Conservation (28 species) track of the UCSC Human Genome browser. Primers were designed with Primer3 software for amplification of all exons of each candidate gene, including the intron-exon boundaries. Primer pairs for HGF are listed in Table S1, available online. Methods for direct sequencing of PCR products were described previously.22 BigDye terminator reaction products were resolved on ABI377 or ABI3730 instruments. Sequencing traces were analyzed with the Phred, Phrap, and Consed software suite, and mutations were identified with the aid of Polyphred.23–25
Cosegregation of the mutation with deafness in each family was confirmed by direct sequencing of PCR products amplified from genomic DNA of all participating family members. Control DNA samples from Pakistan and India were analyzed for putative mutations, as were Caucasian control samples (HD200CAU) and panethnic control samples (HD01-09, HD27) from the Human Diversity Panel (Coriell Cell Repositories, NJ, USA) (Table S2).
In Vitro Splicing Assay
The effect of each of the three mutations on splicing of HGF was evaluated by an exon trapping assay.26 A PCR fragment encompassing exons 4 and 5 of HGF was amplified from genomic DNA of a noncarrier from family PKDF402 (wild-type) and an affected individual from each of three families, PKDF402 (c.482+1986_1988delTGA), PKDF601 (c.482+1991_2000delGATGATGAAA), and PKDF210 (c.495G>A), with the primer pair 5′-CTGCAGGGTTGTCCTATCAGCAGTGGA-3′ and 5′-CTGCAGGAAGCAGGTGCTGGTTGAAT-3′. The HGF cDNA numbering is in reference to the “A” of the ATG start site of RefSeq No. NM_000601.
Each of the four 6.0 kb genomic DNA fragments were cloned into pSPL3 (GIBCOBRL), sequence verified, and transfected into COS-7 cells with Lipofectamine 2000 (Invitrogen). After 24 hr, total RNA was extracted from the transfected cells with the use of Trizol (Invitrogen) and cDNA was synthesized with Superscript III RT (Invitrogen) with the use of oligonucleotide SA2 5′-ATCTCAGTGGTATTTGTGAGC-3′. Primary and nested secondary PCR amplifications of the cDNA were performed with vector-specific primers, and products from the nested PCR were subcloned and sequence verified as recommended (Exon Trapping Kit, GIBCOBRL). The numbers given in Table 2 are the totals of four independent experiments for each allele. Contingency table probabilities were calculated with Prism 5 software (GraphPad).
Table 2.
In Vitro Exon Splicing Assay Results
| Allelea | Splice Isoform | No. of Clones | χ2 | p Value |
|---|---|---|---|---|
| Wild-Type | 5a | 33/63 | NA | NA |
| Wild-Type | 5b | 26/63 | NA | NA |
| Wild-Type | otherb | 4/63 | NA | NA |
| Δ3 | 5a | 23/47 | 0.673 | 0.71 |
| Δ3 | 5b | 19/47 | 0.673 | 0.71 |
| Δ3 | otherb | 5/47 | 0.673 | 0.71 |
| Δ10 | 5a | 20/37 | 0.184 | 0.912 |
| Δ10 | 5b | 14/37 | 0.184 | 0.912 |
| Δ10 | otherb | 3/37 | 0.184 | 0.912 |
| c.495G→A (p.S165S) | 5a | 0/39 | 31.69 | 0.0001 |
| c.495G→A (p.S165S) | 5b | 37/39 | 31.69 | 0.0001 |
| c.495G→A (p.S165S) | otherb | 2/39 | 31.69 | 0.0001 |
NA denotes “not applicable.”
Δ3 = c.482+1986_1988delTGA; Δ10 = c.482+1991_2000delGATGATGAAA.
A few aberrant splicing products were identified for each allele that skipped exon 4, exon 5, or both exons 4 and 5. These are presumed to be artifacts and are listed as “other.”
Hgf Conditional Knockout and Transgenic Mice
Mice with loxP sites flanking exon 5 of the Hgf gene (HGFex.5-flox)27 were used for the creation of an inner ear conditional knockout of Hgf. Homozygous Hgf floxed mice were bred to heterozygous Hgf floxed mice also segregating the cre recombinase gene under the control of the endogenous Foxg1 gene promoter [B6.129P2(Cg)-Foxg1tm1(cre)Skm/J] (The Jackson Laboratory).28 Wild-type (1.1 kb) and floxed (1.4 kb) exon 5 Hgf alleles were detected in genomic DNA from mouse tails by PCR amplification with the primer pair 5′-TGTGACCCTGGATCATCAGTGTAA-3′ and 5′-GCTGATTTAATCCCATTTCTTCA-3′. The presence of the cre gene was detected in genomic DNA from mouse tails by PCR amplification with the cre-specific primer pair (358 bp amplimer) 5′-ACGACCAAGTGACAGCAATG-3′ and 5′-CCATCGCTCGACCAGTTTAG-3′.
The generation of MH19 transgenic mice overexpressing full-length Hgf cDNA was previously described.29 The Hgf transgene was detected by PCR amplification of a 550 bp product from mouse tail genomic DNA with the primer pair 5′-TCTCGTAAACTCCAGAGC-3′ and 5′-GGGTCTTCCTTGGTAAG-3′. All animal procedures were approved and conducted according to the NIH Animal Care and Use Committee guidelines and Animal Protocols 1263-06 and 08-049 (to T.B.F. and G.M., respectively).
Phenotypic Evaluation of Mice
Mice were anesthetized with an intraperitoneal injection of a mixture of 100 mg/ml ketamine hydrochloride (42–56 mg/kg body weight) and 1 mg/ml medetomidine hydrochloride (0.56–0.75 mg/kg body weight) and placed on a heating pad in a sound-attenuated booth (Acoustic Systems). Auditory-evoked brainstem responses (ABRs) were measured with TDT System 3 hardware and software (Tucker-Davis Technologies) with free-field presentation of auditory stimuli through an ES1 electrostatic speaker. Clicks and pure-tone pips were presented as 3 msec bursts, with 1 msec rise and fall times at 21 stimuli/sec. The recorded ABR at each presentation level was a cumulative average from a minimum of 500 stimuli. After testing, the animals were administered 5 mg/ml Antisedan (2.75–3.67 mg/kg body weight) and allowed to recover from anesthesia in a warm cage. Effects of genotype and age on ABR thresholds were analyzed by two-way ANOVA with Prism 5 software (GraphPad). Distortion product otoacoustic emissions (DPOAEs) were recorded with DP2000 measurement software, version 3.0 (Starkey Laboratory), and an ER-10C acoustic probe (Etymotic Research). The DP (2f1−f2) amplitudes from both ears were averaged for each mouse.
Six conditional knockout mice (Hgf Fl/Fl; Foxg1cre/+) (three males, three females) and six littermate controls (Hgf Fl/+; Foxg1cre/+) (five males, one female) were evaluated at 7–8 wks of age by the Mouse Phenotyping Service, Division of Veterinary Resources, NIH. Mice were sacrificed with CO2 asphyxiation and necropsied. Mouse tissues were harvested and fixed in 10% formalin, processed with alcohols and xylenes, embedded in paraffin, sectioned at 5 microns, stained with hematoxylin and eosin, and visualized by light microscopy.
Fluorescence Confocal Microscopy
Inner ears of wild-type and mutant mice were fixed with 4% paraformaldehyde overnight at 4°C and rinsed with phosphate-buffered saline (PBS). The organs of Corti were dissected and incubated overnight at 4°C with rhodamine-phalloidin (Molecular Probes), mounted on glass slides in ProLong Gold antifade reagent (Invitrogen), and visualized on a Zeiss LSM710 laser confocal microscope.
Lymphoblastoid Cell Lines and cDNA Synthesis
Lymphoblastoid cell lines were established by the Coriell Cell Repository via Epstein-Barr-virus transformation of peripheral blood samples from one affected individual and one carrier individual from four DFNB39 families (PKDF002, PKDF084, PKDF121, and PKSR53a). Cells were grown in RPMI-1640 supplemented with L-glutamine (GIBCO) and 15% fetal bovine serum (FBS) (Atlanta Biologicals) at 37°C and 5% CO2.
Total RNA was extracted from lymphoblastoid cell cultures with Trizol (Invitrogen). Poly A+ RNA was isolated with an Oligotex mRNA Kit (QIAGEN), and cDNA was synthesized with Superscript III RT (Invitrogen) with the use of an Oligo(dT) SMART primer (Clontech). Human brain first-strand cDNA was synthesized from human brain polyA+ RNA (Clontech) with an Oligo(dT) primer and Powerscript Reverse Transcriptase (Clontech). The cDNAs were amplified with the primer pair 5′- ATGTGGGTGACCAAACTCCT-3′ and 5′- GGTGGCCAATGAAGGATACA-3′.
Results
Clinical Findings
Hearing was evaluated by pure-tone audiometry for at least two affected members (age range 3 to 45 yrs) from each of the 41 families (Figure 1 and Figure S1). The hearing loss was prelingual, bilateral, and severe to profound, with a downward-sloping audiometric configuration. Bone conduction threshold testing in some individuals confirmed a sensorineural etiology, but we cannot formally rule out a mixed hearing loss in others. Some affected individuals from families PKDF210 and PKDF601 segregate a slightly milder phenotype characterized by moderately severe hearing loss at low frequencies.
Figure 1.
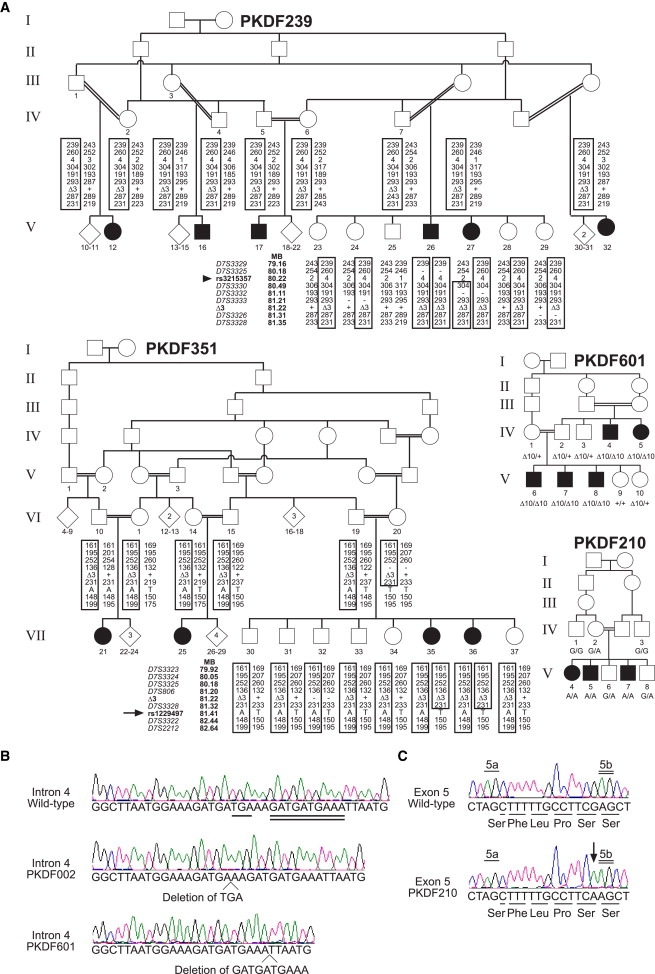
Four of 41 Families Segregating Mutant Alleles of HGF Associated with Deafness
(A) Filled symbols of pedigrees represent individuals with prelingual, severe to profound hearing loss, and a double horizontal line indicates a consanguineous marriage. Numbers below individual symbols indicate that the person was enrolled in the study and that their DNA sample was genotyped. Haplotype data for families PKDF239 and PKDF351 indicate the smallest linkage interval. The deafness in PKDF239 and PKDF351 cosegregates with c.482+1986_1988delTGA (Δ3). Only informative markers are shown for the sibships that define the genetic breakpoints. The proximal breakpoint (arrowhead) is defined by individual 27 of family PKDF239 at SNP rs3215357. The distal breakpoint (arrow) is defined by individuals 35 and 36 of family PKDF351 at SNP rs1229497, located between HGF and CACNA2D1. The deafness phenotype of family PKDF601 cosegregates with c.482+1991_2000delGATGATGAAA (Δ10) in intron 4 of HGF. The deafness phenotype of family PKDF210 cosegregates with c.495G>A (p.S165S) in HGF.
(B) Representative nucleotide sequence chromatograms of intron 4 of HGF showing the wild-type, homozygosity for the 3 bp deletion, and homozygosity for the 10 bp deletion.
(C) Wild-type and homozygous mutant nucleotide sequence chromatograms of exon 5 of HGF, illustrating homozygosity for the c.495G>A (p.S165S) mutation (arrow) and the 5a (underlined) and 5b (double underlined) splice acceptor sites.
General physical examination of affected individuals and retinal fundus examination of two affected individuals from family PKDF121 (24 and 28 years old) and two from PKDF879 (28 and 45 years old) ruled out Usher syndrome and obvious abnormalities in other organ systems. Serum chemistry, CBC, and urine analysis of two affected and two unaffected individuals from each of three families, PKDF121 (age range 24–40 years), PKDF841 (age range 8–15 years), and PKDF847 (age range 10–42 years), did not identify any abnormalities consistent among affected individuals. The clinical data for these families suggests a nonsyndromic, severe to profound hearing loss.
Genetic and Physical Map
DFNB39 was originally mapped to 7q11.22-q21.12 by genome-wide linkage analysis of family DEM4011.15 We report an additional 40 families with severe to profound deafness that cosegregates with markers at the DFNB39 locus. Additional Pakistani and Indian families segregating profound, prelingual, recessive deafness were screened for linkage to markers at all of the currently known DFNB loci (Hereditary Hearing Loss homepage). Deafness segregating in 19 families was linked to DFNB39 with LOD scores greater than the 3.3 threshold for significance.30,31 In 22 families, deafness cosegregates with the DFNB39 locus, but these families are too small to support LOD scores greater than 3.3 (Table 1, Figure 1, and Figure S1). With the assumption of genetic homogeneity at this locus, a total of 41 DFNB39 families were ascertained.
The minimal linkage interval of DFNB39 was defined by families that independently support highly significant evidence for linkage: PKDF239 (LOD 7.41, D7S3329) and PKDF351 (LOD 5.34, D7S3328) (Figure 1). Inspection of the haplotypes segregating in these families reveals a proximal meiotic breakpoint between SNP rs3215357 and STR marker D7S3330 in PKDF239 (arrowhead, Figure 1A) and a distal meiotic breakpoint between STR marker D7S3328 and SNP rs1229497 in PKDF351 (arrow, Figure 1A). The interval between rs3215357 and rs1229497 comprises 1.187 Mb at 7q21.11 (NCBI build 36.1) and is entirely within the original 18 Mb DFNB39 locus.15
Identification of DFNB39
Genes in or just outside the smallest linkage interval include CLDN3, CLDN4, MAGI2, GNAI1, GNAT3, CD36, SEMA3C, HGF, CACNA2D1, PCLO, SEMA3E, SEMA3A, and CLDN12 (Figure 2A). We sequenced all of the coding exons, including splice sites and ∼50 bp of flanking intronic sequence, in each of these genes in affected individuals from 11 DFNB39-linked families. In addition, conserved intronic sequences and DNA corresponding to transcripts of unknown function were sequenced, including mRNAs AY927633 and AY927634 (Unigene Hs.571267) because their small open reading frames have similarity to cadherin ectodomains.32 A total of 374 amplicons were sequenced in a subset of 11 DFNB39-linked families (families indicated in Table 1). Because the 41 families were enrolled in our respective studies over a period of several years, some of the DFNB39 families were evaluated for only a subset of the 374 amplicons. No missense, nonsense, or frameshift mutations were identified in the coding sequences of any of the genes in this interval. However, we did find three potential regulatory mutations in the HGF gene.
Figure 2.
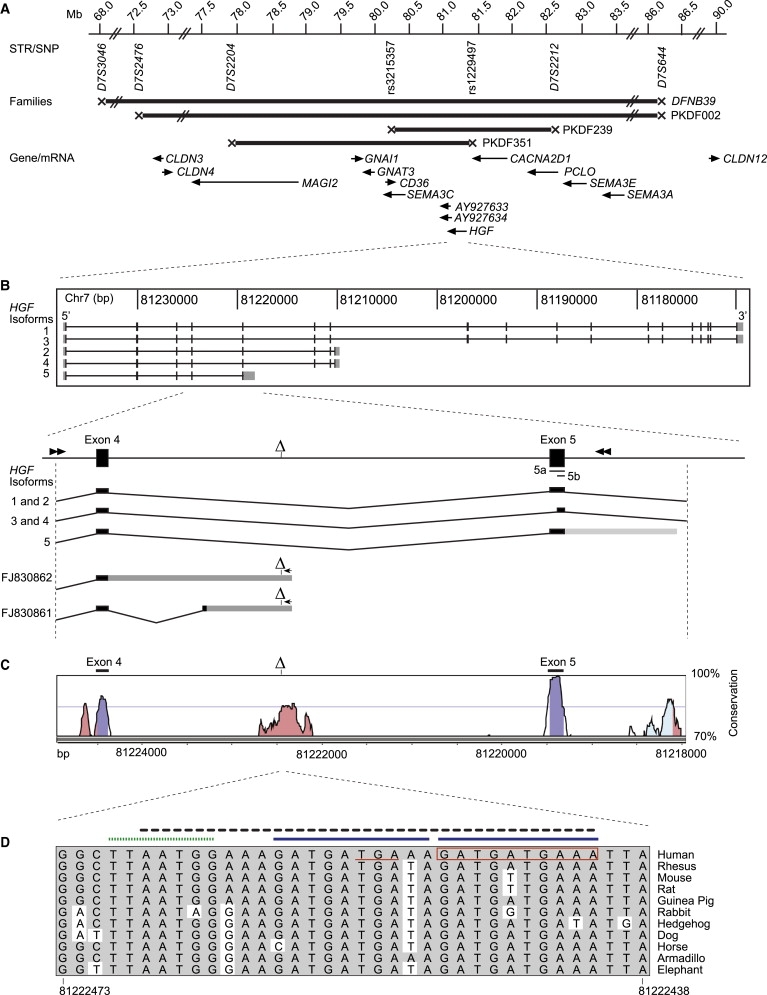
DFNB39 Locus, HGF Genomic Structure, Transcripts of HGF, and Intron 4 Sequence Conservation
(A) The linkage intervals for families DEM4011 and PKDF002, which defined the DFNB39 locus, and families PKDF239 and PKDF351, which define the smallest linkage interval, are shown as thick horizontal lines with Xs at the proximal and distal recombination sites. Relative locations and orientations of the genes and mRNAs are indicated with arrows.
(B) Exon structure of the Refseq splice isoforms of HGF and a magnification of the mutation region. Coding sequences are shaded black, 5′ and 3′ UTRs are shaded gray. Two previously unpublished transcripts (FJ830861 and FJ830862) are depicted with arrows indicating the location of the reverse PCR primer site. The forward primer (not shown) was located at the ATG site within exon 1. The 3 bp and 10 bp deletions are shown as a Δ within intron 4 and in the 3′ UTRs of these two isoforms. Double arrowheads indicate the primer locations for amplification of genomic DNA for in vitro splicing assays.
(C) Nucleotide conservation greater than 75% (100 bp window) between human and mouse genomic DNA viewed with the Vista Genome Browser. Conserved nucleotide sequences (pink); coding sequences (blue); UTRs of RefSeq isoforms (light blue).
(D) Nucleotide identity (gray shading) in intron 4 of HGF among placental mammals. The 3 bp (c.482+1986_1988delTGA) and 10 bp (c.482+1991_2000delGATGATGAAA) deletions are underlined and boxed in red, respectively. Solid blue bars represent tandemly repeated 10 bp sequences, one of which is deleted in family PKDF601. Note that the 3 bp and 10 bp deletions can be described as overlapping, but are depicted as adjacent to each other in order to conform to the nomenclature guidelines requiring that the nucleotide number designation begin at the first unambiguous occurrence of the deletion. The dotted green bar represents an SRP40 binding site (predicted by ESE Finder v3, score = 4.19). A black dashed line represents a region of 22 overlapping predicted exonic splice enhancer hexamer sites identified by Rescue-ESE.
A transition mutation in HGF resulting in a synonymous substitution (c.495G>A [p.S165S]) cosegregates with deafness in family PKDF210 (maximum LOD score of 2.38) (Figures 1A and 1C). This G-to-A transition mutation occurs 13 nucleotides into exon 5 relative to the exon 5a splice acceptor site of HGF isoform 1 (RefSeq no. NM_000601), but at the minus 3 position (c.483-3G>A) relative to the exon 5b splice acceptor site of HGF isoform 3 (RefSeq no. NM_001010932) (Figures 1C and 2B). This nucleotide substitution is predicted to alter the relative strengths of these two splice acceptor sites. For example, Genscan33 predicts exon 5 with the 5a acceptor site and a probability (P) of 0.682 when given the c.495G wild-type sequence as input, but prefers the 5b acceptor site with p = 0.592 when the c.495A mutation sequence is given as input. The c.495G>A transition was not found among 1040 chromosomes from the Pakistani controls, Caucasian controls, and Human Diversity Panel controls (Table S2), nor was it found in the dbSNP database.
We sequenced intragenic and intergenic regions of the DFNB39 interval that were highly conserved among species, including a 500 bp region of intron 4 of HGF that is 79% identical to the corresponding mouse sequence (Figure 2C). By comparison, the coding sequences of human HGF and mouse Hgf are 88% identical. This intronic region is transcribed in mice as part of the 3′ UTR of a short isoform of Hgf (RefSeq no. AK082461). We subsequently determined that this region is also transcribed in humans as part of the 3′ UTR of a previously uncharacterized short HGF isoform (Figure 2B). We identified a 3 nt deletion in intron 4 (c.482+1986_1988delTGA) that cosegregates with deafness in 36 Pakistani and two Indian families (Figures 1A and 1B, Figure S1). A 10 bp deletion (c.482+1991_2000delGATGATGAAA), located in intron 4 of HGF, adjacent to the 3 bp deletion, cosegregates with deafness in families PKDF601 and DEM4472 (LOD 4.22 and 3.61, respectively) (Figure 1A and Figure S1). This 10 bp sequence is tandemly duplicated in humans, the first duplication encompassing the 3 bp deletion as well (solid blue lines, Figure 2D). These duplications are also part of a stretch of 21 overlapping 6 bp exon splice enhancer sites predicted by the Rescue-ESE program (black dashed line, Figure 2D).34
Heterozygosity for the 3 bp deletion, c.482+1986_1988delTGA, was identified in 2 of 429 representative samples from unaffected Pakistani individuals (858 control chromosomes), but was not found in a total of 830 Caucasian, Indian, or Human Diversity Panel control chromosomes (Table S2). No carriers of the 10 bp deletion, c.482+1991_2000delGATGATGAAA, were found among 1688 control chromosomes.
We genotyped seven SNPs in 35 DFNB39 families that share the same c.482+1986_1988delTGA deletion in order to evaluate the haplotype of the 221,150 bp region (Table S3). All of the families with the 3 bp deletion share the same homozygous haplotype, which consisted of SNPs rs5745752, rs2286194, and rs1558001. HapMap data show that this haplotype is found among 15% of Europeans and 11% of East Asians. Although the frequency of this haplotype in the Pakistani and Indian populations is not known, the 3 bp deletion is not on a rare haplotype found only in the Indian subcontinent.
c.495G>A Alters Splicing of HGF Exon 5
Given the predicted effect of the c.495G>A mutation on splicing at the exon 5a and 5b acceptor sites, as well as the proximity of the intron 4 deletions to exon 5 of HGF, we performed in vitro splicing assays to determine whether any of the three mutant alleles affected splicing of exons 4, 5a, or 5b of HGF (Table 2). With the wild-type construct, we recovered cDNA clones using the 5a or 5b splice acceptor sites in roughly equal amounts: 52% and 41%, respectively. Seven percent of the clones skipped exon 5 or represented other splicing events. The proportion of these other splicing events did not change significantly among experiments and therefore were considered aberrant. There was no significant alteration of the proportion of 5a and 5b cDNA clones recovered in experiments using a construct with the 3 bp deletion (49% 5a and 40% 5b) (χ2 = 0.673, p value = 0.71) or a construct with the 10 bp deletion (54% 5a and 38% 5b) (χ2 = 0.184, p value = 0.91). However, the in vitro splicing assay using a construct with the c.495G>A transition produced clones with the exon 5b splice acceptor site exclusively (0% 5a and 95% 5b), thus altering the ratio of isoforms of HGF containing exons 5a and 5b (χ2 = 31.69, p value = 0.0001).
HGF and Hgf Isoforms
There are multiple isoforms transcribed at the HGF locus (Figure 2B). Full-length HGF isoform 1 (NM_000601) encodes a preprotein that is cleaved into alpha and beta chains.35 The alpha chain is composed of a hairpin loop homologous to the plasminogen-activation peptide, followed by four N-terminal kringle domains, which are cysteine-rich motifs involved in protein-protein interactions. The beta chain has homology to trypsin-like serine proteases but has no catalytic function. After cleavage, the alpha and beta chains form a heterodimer via disulfide bonds.36 Isoform 2 (NM_001010931) encodes only two kringle domains (NK2). Isoforms 3 (NM_001010932) and 4 (NM_001010933) are similar to isoforms 1 and 2, respectively, differing only in the usage of the alternate exon 5b splice acceptor site. A fifth isoform (NM_001010934), encoding a single kringle domain (NK1), utilizes the 5a acceptor site (Figure 2B).
The UCSC Genome Browser annotation of the mouse Hgf locus (mm9, NCBI build 37) shows orthologs of isoforms 1, 3, and 5, as well an mRNA (AK082461) that ends with a 3′ UTR located in intron 4. The open reading frame of this isoform is predicted to encounter a stop codon at the beginning of intron 4 and encode no additional amino acids beyond exon 4. The 3′ UTR of this short isoform includes the region conserved with humans encompassing the 3 bp and 10 bp deletions. By RT-PCR, we were able to demonstrate that mRNAs homologous to this short mouse isoform are transcribed in human brain and lymphoblastoid cell lines from individuals who are homozygous for the 3 bp deletion, as well as from heterozygous and wild-type individuals (FJ830862) (Figure 2B). We also recovered cDNA clones that show utilization of the splice donor site for exon 4 and a splice acceptor site in intron 4 that results in a 3′ UTR that encompasses the region of the 3 bp and 10 bp deletions (FJ830861). This transcript is predicted to encode 24 additional amino acids before encountering a stop codon.
Evaluation of Hgf Mutant Mice
In the mouse, homozygosity for mutant alleles of Hgf causes embryonic lethality in two different knockout-mouse models.37,38 An allele of Hgf in which exon 5 is floxed (Hgf Fl/Fl) was created and used in making a conditional knockout of Hgf in liver tissue, resulting in a viable mouse with impaired liver-regeneration ability.27 We used the Hgf Fl/Fl mouse to generate a conditional knockout Hgf in the inner ear39 so that we could determine whether there would be a hearing deficit in the absence of HGF. We created conditional knockouts by crossing exon 5-floxed mice (Hgf Fl/Fl)27 to mice carrying cre driven by the endogenous Foxg1 gene promoter (Foxg1cre/+),28 with subsequent backcrossing for production of (Hgf Fl/Fl; Foxg1cre/+) mice. In the Foxg1cre/+ mouse, cre recombinase expression corresponds to wild-type Foxg1 mRNA transcription in the telencephelon, otic vesicle, optic vesicle, pharyngeal region, and foregut.28 The Foxg1cre/+ mouse has been used in generating inner ear knockouts of Bmp4,40 Sox9,41 Tbx1,42 and Fgfr1.43
ABR thresholds were obtained for Hgf conditional knockout mice at approximately 4 wks and 7 wks of age; some mice were tested at both ages. There was no difference in ABR thresholds between the two age groups, and mice tested at both time points showed no evidence of progression in their hearing thresholds (data not shown). The conditional knockout mice (Hgf Fl/Fl; Foxg1cre/+) displayed no response at maximum stimulus levels for 8 kHz, 16 kHz, 32 kHz, and click stimuli, whereas their heterozygous littermate controls with or without cre recombinase (Hgf Fl/+; Foxg1cre/+ and Hgf Fl/+; Foxg1+/+) displayed normal ABR thresholds (Figure 3A).
Figure 3.
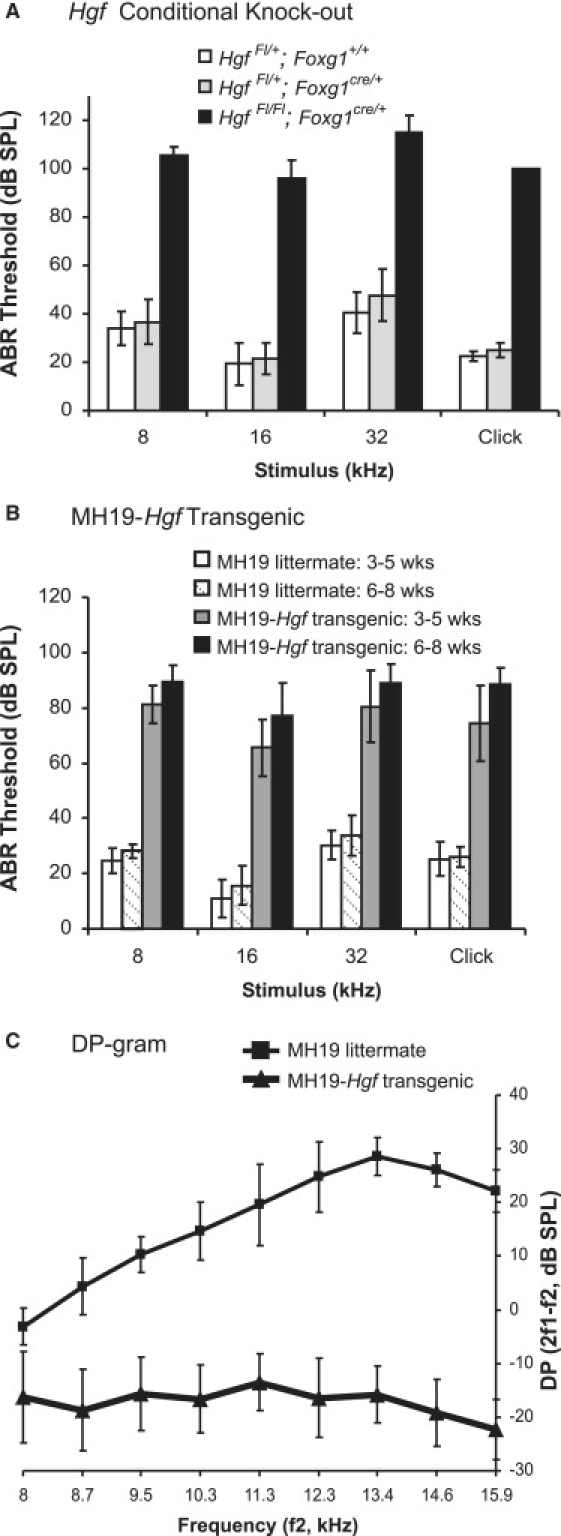
Average ABR Thresholds for Hgf Conditional Knockout and MH19-Hgf Transgenic Mice and DPOAE Thresholds for MH19-Hgf Transgenic Mice
(A and B) ABR thresholds in decibels (dB), sound pressure level (SPL) at 8 kHz, 16 kHz, 32 kHz pure-tone pips, and click stimuli were measured in Hgf conditional knockout mice and their littermate controls (A) and in MH19-Hgf transgenic mice and their littermate controls (B). For Hgf Fl/+; Foxg1+/+: n = 21 for pure-tone pips and n = 13 for clicks. For Hgf Fl/+; Foxg1cre/+, n = 11 for pure-tone pips and n = 8 for clicks. For Hgf Fl/Fl; Foxg1cre/+, n = 10 for pure-tone pips and n = 8 for clicks. Thresholds at all frequencies were significantly different between conditional knockout mice and littermate controls (p < 0.0001). MH19-Hgf transgenic mice (n = 4 for 3–5 wks, n = 3 for 6–8 wks) and littermate controls (n = 4 for 3–5 wks; n = 3 for 6–8 wks). Vertical bars indicate standard deviations. Average ABR thresholds of MH19-Hgf transgenic mice are significantly different from those of their littermate controls (p < 0.0001).
(C) Mean DPOAEs in MH19-Hgf transgenic mice (n = 13) and littermate controls (n = 9) at 4–6 wks of age. The mean response when f2 = 55 dB SPL and f2/f1 = 1.25 is plotted. Vertical bars indicate standard deviations.
Six conditional knockout mice (Hgf Fl/Fl; Foxg1cre/+) and six littermate controls (Hgf Fl/+; Foxg1cre/+) were evaluated by the NIH phenotyping service. There was no significant difference in serum chemistries or CBC between the mutant mice and littermate controls. The littermate controls had significantly larger body weights and brain weights than the homozygous mutant mice. Five of the six mutant mice had dilated and thinned muscular walls of the esophagus, which may be a consequence of Foxg1cre/+ expression in the foregut.28 All six of the mutant mice had evidence of esophageal reflux, leading to aspiration pneumonia in all six and rhinitis in two. Five of the six mutant mice and one littermate control had otitis media.
Light microscopy of tissue sections through the inner ears of Hgf Fl/Fl; Foxg1cre/+ mice revealed morphological defects not found in littermate controls. The tectorial membrane appears disorganized in mutant mice as compared to littermates, and Reissner's membrane is collapsed onto the tectorial membrane (Figure 4B). The stria vascularis appears thin and flattened in mutant mice (Figure 4C), with occasional clumps of cellular proliferation (Figure 4B), and the spiral ganglion appears hypoplastic. Confocal microscopy shows outer hair cell (OHC) degeneration throughout the organ of Corti, from base to apex (Figure 4E).
Figure 4.
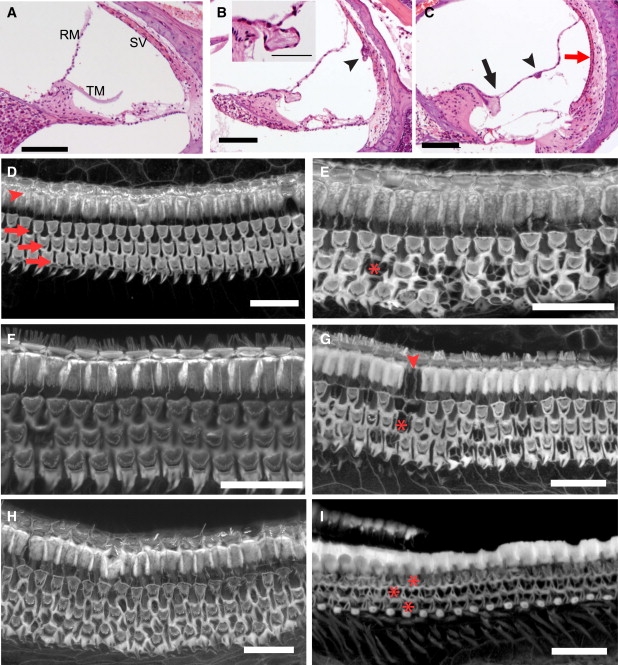
Inner Ear Defects in Adult Hgf Conditional Knockout Mice and MH19-Hgf Transgenic Mice
(A) Cross section through the middle turn of the cochlea of an adult Hgf Fl/+; Foxg1 cre/+ mouse shows normal morphology of the organ of Corti, Reissner's membrane (RM), the stria vascularis (SV), and the acellular tectorial membrane (TM).
(B) Cross section of an adult Hgf Fl/Fl; Foxg1 cre/+ mutant mouse at the middle turn of the cochlea. Note the hypoplasia of supporting cells and the hyperplastic clump of cells in the stria vascularis (arrowhead), which otherwise appears thin. The inset shows a higher magnification of the tectorial membrane, which appears splayed and covered in epithelial cells, presumably derived from Reissner's membrane, which is attached to the tectorial membrane.
(C) Another cross section from an adult Hgf Fl/Fl; Foxg1 cre/+ mutant mouse (different than in B), showing the attachment of Reissner's membrane to the tectorial membrane (arrow), a clump of hyperproliferative RM cells (arrowhead), and a thin stria vascularis (red arrow).
(D and E) Confocal images of organ of Corti wholemounts stained with phalloidin from a normal Hgf Fl/+; Foxg1 cre/+ mouse (D) and his Hgf Fl/Fl; Foxg1 cre/+ mutant littermate (E). Arrowhead in D indicates the row of normal inner hair cells (IHCs), and arrows mark three rows of outer hair cells (OHCs). In E, the IHCs are normal but there are numerous degenerating OHCs, one of which is indicated by an asterisk. The x-shaped processes that replace OHCs are the borders of neighboring Deiter's cells.
(F–I) Confocal images of phalloidin-stained wholemounts from MH19 mice. F shows a region from the middle turn of a wild-type littermate of the animal from which images (G–I) were taken. G is the middle turn of an MH19-Hgf transgenic mouse, showing numerous degenerating OHCs (example marked by an asterisk), normal IHCs, and a missing pillar cell (arrowhead). Panel H shows a section of the apex of the cochlea, with essentially normal complement of OHCs, whereas I is a section of the base where virtually every OHC has degenerated. (Missing IHCs in I are a dissection artifact.)
Scale bars represent 100 μm (A–C), 5 μm (inset, B), and 20 μm (D–I).
Having demonstrated that conditional knockout of Hgf can result in cochlear pathology, we examined an existing Hgf transgenic mouse model29 to determine whether overexpression of HGF could affect cochlear function or development. MH19-Hgf transgenic mice express ectopic full-length HGF driven by a metallothionein promoter, with expression 3- to 50-fold higher than endogenous levels in all adult tissues tested.29 MH19-Hgf transgenic mice exhibit ectopic skeletal muscle development and pigment cells in the central nervous system, patterned hyperpigmentation of the dermis and epidermis, tumorigenesis of mesenchymal and epithelial origin, and frequent kidney failure and gastrointestinal obstructions.29,44,45 Hearing loss was not reported in the MH19-Hgf transgenic mice. We tested the hearing of MH19-Hgf transgenic mice and their littermate controls by ABR and DPOAE and evaluated dissections of the inner ears for evidence of pathology.
Average ABR thresholds of the MH19-Hgf transgenic mice were 60 dB greater at 8 kHz, 16 kHz, 32 kHz, and click stimuli than those of their littermate controls (Figure 3B). Hearing loss progressed with age in all mice tested, but most of the hearing loss was attributable to genotype (Two-way ANOVA results: p < 0.0001 for genotype, p = 0.09 for age, and p = 0.45 for interaction of genotype and age).
A 60 dB threshold shift is consistent with the loss of cochlear amplification as a result of OHC degeneration.46 OHC function can be assessed by measurement of DPOAEs, which were significantly lower in MH19-Hgf transgenic mice (Figure 3C). Inspection of organ of Corti whole mounts via confocal microscopy confirms that MH19-Hgf transgenic mice display an OHC degeneration in a spatial gradient, from complete OHC loss at the base to near-normal complement of OHCs at the apex at ∼5 wks of age (Figures 4F–4I). The inner hair cells appear normal. Cells other than the OHCs appear normal in inspection of whole mounts via confocal laser microscopy or of cross sections with light microscopy. No obvious defects of the tectorial membrane, stria vascularis, Reissner's membrane, or supporting cells were observed (data not shown).
Discussion
HGF was initially identified as a factor that was secreted from cultured hepatocytes and acted as a potent mitogen.36 It was later shown to be identical to “scatter factor,” which is secreted from fibroblasts and causes epithelial cells to disperse in culture.47 Thus, HGF is often referred to in the literature as HGF/SF. In addition to its properties as both a mitogen and a motogen, HGF has also been implicated in branching morphogenesis and invasion,48 tumorigenesis,49 erythroid differentiation,50 neuronal survival,51–53 and wound healing.54
The full open reading frame of HGF encodes (from amino termini to carboxy termini) a signal peptide, an alpha chain containing four kringle domains, and a beta chain.55 This inactive pro-HGF polypeptide is converted to the active form by proteases that cleave between the alpha and beta chains. The two chains are joined by a disulfide bond to complete the active heterodimer form of HGF. There are several proteases that can convert pro-HGF to HGF. One of them, HGF activator (HGFAC [MIM 604552]), is expressed exclusively in damaged tissue.56 Both the inactive pro-HGF and HGF can bind the tyrosine kinase MET receptor (MET [MIM 164860]), but only the active form can stimulate downstream pathways through the MET receptor.57,58 The multiplicity of the pathways mediated by MET59 is one likely reason for the pleiotropic effects influenced by HGF. Another, less well characterized, reason for the sometimes paradoxical effects of HGF is the variant forms that arise as a result of alternative splicing at the HGF locus.
Isoform 1 is the canonical, full-length isoform (Figure 2B). It is encoded by transcripts that are either 2.8 kb or 6.0 kb in length, depending on the use of alternate polyadenylation sites in the 3′ UTR.36 Transcripts utilizing an alternate exon and polyadenylation site in intron 7 encode isoform 2, which contains only the amino terminal through the first two kringle domains, and the protein product is thus referred to as NK2.55,60 Isoform 5, which contains the amino terminal through only the first kringle domain (NK1), is encoded by transcripts ending in exon 5 with an alternate 3′ UTR and polyadenylation signal in intron 5.61,62 Isoforms 3 and 4 are identical to isoforms 1 and 2, respectively, except in the usage of an alternate splice acceptor site in exon 5, which results in the exclusion of five amino acids from the first kringle domain.63,64 In culture, the NK1 and NK2 isoforms can exhibit antagonist activity, depending on the cell type expressing the MET receptor, and so have been proposed as therapeutic agents to counteract HGF in tumors.60,61 However, transgenic mice expressing NK1 cDNA show that it can act as a potent agonist,62 and NK2 transgene expression promotes metastasis, despite inhibiting tumor growth.65
Almost all of the studies characterizing the differential effects of HGF, NK1, and NK2 focus exclusively on those isoforms encoded by the transcripts utilizing the 5a splice acceptor site. Yet, a functional difference has been demonstrated for the 5a versus 5b splice acceptor usage. The exclusion of five amino acids from the first kringle domain apparently does not affect binding of HGF to MET, but is thought to alter its ability to bind heparin sulfate proteoglycans in the extracellular matrix.66,67 Interaction with heparin induces NK1, at least, to dimerize, and in turn causes the MET receptor to dimerize on binding.68,69 The interaction with heparin and subsequent dimerization may underlie some of the paradoxical agonist versus antagonist properties of NK1.70–72
Other HGF transcripts have been identified as well, including those that encode only the beta subunit,73 which possibly forms a hybrid cytokine with IL-7.74 We report here two additional alternatively spliced transcripts in humans that include only exons 1–4. The first, FJ830861, utilizes the exon 4 splice donor site and a splice acceptor site within intron 4. Twenty-four additional amino acids are predicted to be encoded before reaching a stop codon. The second, FJ830862, does not splice after exon 4 but reads directly into intron 4, similar to the short mouse Hgf isoform AK082461. The open reading frame ends immediately after the last codon of exon 4. A highly conserved region of the 3′ UTRs of both of these alternate splice forms is partially deleted in deaf individuals from 40 of our 41 DFNB39 families. The deletions occur in a region of predicted exonic splice enhancer sites and adjacent to a predicted splice factor binding site (Figure 2D, black dashed line and green dotted line). This suggests that the deletions might cause dysregulation of one or more HGF isoforms.
Dysregulation of HGF isoforms is also suggested by the transition mutation c.495G>A (p.S165S). In an in vitro splicing assay, c.495G>A significantly alters the ratio of HGF splice isoforms that utilize either exon 5a or exon 5b splice acceptor sites. A change in the ratio of 5a versus 5b protein isoforms may decrease heparin binding, which could influence whether the isoform acts as a dimer or a monomer, as mentioned earlier. It could also affect local HGF sequestration in the extracellular matrix adjacent to the mesenchymal cells secreting HGF.
The possible effects of HGF dysregulation on HGF-MET-mediated pathways are myriad and difficult to predict, especially because the mutations described here most likely affect smaller isoforms of HGF and the differential activities of the various isoforms of HGF are not well understood. The primary pathological effect of these regulatory mutations may be on HGF's function as a neurotrophic factor.51,53 In rats exposed to kanamycin, ectopic expression of human HGF in spiral ganglion cells protected them against apoptosis and helped maintain normal auditory function.75 Whether endogenous HGF expression plays a role in protecting the cochlea from ototoxic insult is unknown. HGF has also been implicated in wound-healing processes, which may be important in regulating responses to noise and chemical trauma in the cochlea.76,77
Another possible mechanism for DFNB39-induced pathology is through the morphogenetic pathways mediated by HGF. In cell culture, one of the genes that is upregulated in response to HGF treatment is SPRY2 (MIM 602466), which in turn acts as a negative feedback regulator of HGF and MET signaling.78 SPRY2 has been implicated in pathways that determine supporting cell fate in the organ of Corti. A knockout mouse model (Spry2−/−) is deaf and exhibits supernumary pillar cells at the expense of Deiter's cells.79
These hypotheses of DFNB39-mediated pathology, as well as other hypotheses, will require an additional mouse model to test. It is interesting to note that two mouse models of Hgf dysregulation result in deafness. The conditional deletion of exon 5 of Hgf in the cochlea results in a profound, nonprogressive hearing loss associated with extensive morphogenetic pathology, and apparently no other phenotype. Conversely, the ubiquitous overexpression of Hgf in a transgenic mouse results in progressive hearing loss associated with degeneration of the outer hair cells. The degeneration of hair cells in both types of Hgf mutant mice is consistent with the general base-to-apex gradient of development of the cochlea80 and hair cell degeneration in the mammalian cochlea.81 Although neither of these mouse models proves HGF is the DFNB39 gene, they do corroborate the hypothesis that dysregulation of Hgf isoforms can cause surprisingly specific cochlear pathology. A knockin mouse model of the DFNB39 3′ UTR regulatory changes will confirm our hypothesis. A full evaluation of the relative amounts of all Hgf isoforms transcribed in the presence and absence of the mutation can be made only in a mouse knockin model. Hypotheses concerning which HGF-MET-mediated pathways are affected will then be tested directly.
Supplemental Data
Supplemental Data include three figures and three tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
dbSNP homepage, http://www.ncbi.nlm.nih.gov/SNP/
ESE Finder, http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi
Genscan, http://genes.mit.edu/GENSCAN.html
Hereditary Hearing Loss homepage, http://webh01.ua.ac.be/hhh/
National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Primer3, http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi
Rescue-ESE Web Server, http://genes.mit.edu/burgelab/rescue-ese/
UCSC Genome Browser, http://genome.ucsc.edu/
Vista Genome Browser, http://pipeline.lbl.gov/cgi-bin/gateway2?bg=hg16
Accession Numbers
The GenBank accession numbers for the two transcripts reported in this paper are FJ830861 and FJ830862.
Acknowledgments
We thank all of the families whose participation made this project possible. We thank Gregory Frolenkov, Ayala Lagziel, Inna Belyantseva, Jonathan Bird, Linda Peters, Erich Boger, and Marissa Reuveni for technical assistance, Sadaf Naz for helpful suggestions on the project, Penelope Friedman and Andrew Griffith for evaluation of clinical data, James McGehee for veterinary services, and Doris Wu and Andrew Griffith for critique of the manuscript. J.M.S. received a National Research Council Senior Research Associateship Award. Genotyping services were provided to S.M.L. by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the NIH to Johns Hopkins University, contract number N01-HG-65403. This work was supported in part by the Intramural Research Program of the NIH, Office of Research Services. This study was supported by the Higher Education Commission and Ministry of Science and Technology, Islamabad, Pakistan, to Sh.R. and W.A.; National Institute of Deafness and Other Communication Disorders (NIDCD) grant R01 DC03594 to S.M.L.; and intramural funds from NIDCD 1 Z01 DC000039-11 to T.B.F.
References
- 1.Friedman T.B., Griffith A.J. Human nonsyndromic sensorineural deafness. Annu. Rev. Genomics Hum. Genet. 2003;4:341–402. doi: 10.1146/annurev.genom.4.070802.110347. [DOI] [PubMed] [Google Scholar]
- 2.Friedman T.B., Schultz J.M., Ben-Yosef T., Pryor S.P., Lagziel A., Fisher R.A., Wilcox E.R., Riazuddin S., Ahmed Z.M., Belyantseva I.A. Recent advances in the understanding of syndromic forms of hearing loss. Ear Hear. 2003;24:289–302. doi: 10.1097/01.AUD.0000079804.00047.CE. [DOI] [PubMed] [Google Scholar]
- 3.Petersen M.B., Wang Q., Willems P.J. Sex-linked deafness. Clin. Genet. 2008;73:14–23. doi: 10.1111/j.1399-0004.2007.00913.x. [DOI] [PubMed] [Google Scholar]
- 4.Petersen M.B., Willems P.J. Non-syndromic, autosomal-recessive deafness. Clin. Genet. 2006;69:371–392. doi: 10.1111/j.1399-0004.2006.00613.x. [DOI] [PubMed] [Google Scholar]
- 5.Petit C., Levilliers J., Hardelin J.P. Molecular genetics of hearing loss. Annu. Rev. Genet. 2001;35:589–646. doi: 10.1146/annurev.genet.35.102401.091224. [DOI] [PubMed] [Google Scholar]
- 6.Schrijver I., Gardner P. Hereditary sensorineural hearing loss: advances in molecular genetics and mutation analysis. Expert Rev. Mol. Diagn. 2006;6:375–386. doi: 10.1586/14737159.6.3.375. [DOI] [PubMed] [Google Scholar]
- 7.Yan D., Liu X.Z. Cochlear molecules and hereditary deafness. Front. Biosci. 2008;13:4972–4983. doi: 10.2741/3056. [DOI] [PubMed] [Google Scholar]
- 8.Eisen M.D., Ryugo D.K. Hearing molecules: contributions from genetic deafness. Cell. Mol. Life Sci. 2007;64:566–580. doi: 10.1007/s00018-007-6417-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petit C. From deafness genes to hearing mechanisms: harmony and counterpoint. Trends Mol. Med. 2006;12:57–64. doi: 10.1016/j.molmed.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 10.Zwaenepoel I., Mustapha M., Leibovici M., Verpy E., Goodyear R., Liu X.Z., Nouaille S., Nance W.E., Kanaan M., Avraham K.B. Otoancorin, an inner ear protein restricted to the interface between the apical surface of sensory epithelia and their overlying acellular gels, is defective in autosomal recessive deafness DFNB22. Proc. Natl. Acad. Sci. USA. 2002;99:6240–6245. doi: 10.1073/pnas.082515999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Wijk E., Krieger E., Kemperman M.H., De Leenheer E.M., Huygen P.L., Cremers C.W., Cremers F.P., Kremer H. A mutation in the gamma actin 1 (ACTG1) gene causes autosomal dominant hearing loss (DFNA20/26) J. Med. Genet. 2003;40:879–884. doi: 10.1136/jmg.40.12.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu M., Yang T., Wei S., DeWan A.T., Morell R.J., Elfenbein J.L., Fisher R.A., Leal S.M., Smith R.J., Friderici K.H. Mutations in the gamma-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26) Am. J. Hum. Genet. 2003;73:1082–1091. doi: 10.1086/379286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beffagna G., Occhi G., Nava A., Vitiello L., Ditadi A., Basso C., Bauce B., Carraro G., Thiene G., Towbin J.A. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res. 2005;65:366–373. doi: 10.1016/j.cardiores.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 14.Smirnov D.A., Cheung V.G. ATM gene mutations result in both recessive and dominant expression phenotypes of genes and microRNAs. Am. J. Hum. Genet. 2008;83:243–253. doi: 10.1016/j.ajhg.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wajid M., Abbasi A.A., Ansar M., Pham T.L., Yan K., Haque S., Ahmad W., Leal S.M. DFNB39, a recessive form of sensorineural hearing impairment, maps to chromosome 7q11.22-q21.12. Eur. J. Hum. Genet. 2003;11:812–815. doi: 10.1038/sj.ejhg.5201041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grimberg J., Nawoschik S., Belluscio L., McKee R., Turck A., Eisenberg A. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res. 1989;17:8390. doi: 10.1093/nar/17.20.8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Broman K.W., Murray J.C., Sheffield V.C., White R.L., Weber J.L. Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am. J. Hum. Genet. 1998;63:861–869. doi: 10.1086/302011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schaffer A.A. Faster linkage analysis computations for pedigrees with loops or unused alleles. Hum. Hered. 1996;46:226–235. doi: 10.1159/000154358. [DOI] [PubMed] [Google Scholar]
- 19.Gudbjartsson D.F., Jonasson K., Frigge M.L., Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat. Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 20.Sobel E., Lange K. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am. J. Hum. Genet. 1996;58:1323–1337. [PMC free article] [PubMed] [Google Scholar]
- 21.Weeks D.E., Harby L.D. The affected-pedigree-member method: power to detect linkage. Hum. Hered. 1995;45:13–24. doi: 10.1159/000154250. [DOI] [PubMed] [Google Scholar]
- 22.Bork J.M., Peters L.M., Riazuddin S., Bernstein S.L., Ahmed Z.M., Ness S.L., Polomeno R., Ramesh A., Schloss M., Srisailpathy C.R. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am. J. Hum. Genet. 2001;68:26–37. doi: 10.1086/316954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ewing B., Hillier L., Wendl M.C., Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- 24.Gordon D., Abajian C., Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- 25.Nickerson D.A., Tobe V.O., Taylor S.L. PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res. 1997;25:2745–2751. doi: 10.1093/nar/25.14.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buckler A.J., Chang D.D., Graw S.L., Brook J.D., Haber D.A., Sharp P.A., Housman D.E. Exon amplification: a strategy to isolate mammalian genes based on RNA splicing. Proc. Natl. Acad. Sci. USA. 1991;88:4005–4009. doi: 10.1073/pnas.88.9.4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phaneuf D., Moscioni A.D., LeClair C., Raper S.E., Wilson J.M. Generation of a mouse expressing a conditional knockout of the hepatocyte growth factor gene: demonstration of impaired liver regeneration. DNA Cell Biol. 2004;23:592–603. doi: 10.1089/dna.2004.23.592. [DOI] [PubMed] [Google Scholar]
- 28.Hebert J.M., McConnell S.K. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev. Biol. 2000;222:296–306. doi: 10.1006/dbio.2000.9732. [DOI] [PubMed] [Google Scholar]
- 29.Takayama H., La Rochelle W.J., Anver M., Bockman D.E., Merlino G. Scatter factor/hepatocyte growth factor as a regulator of skeletal muscle and neural crest development. Proc. Natl. Acad. Sci. USA. 1996;93:5866–5871. doi: 10.1073/pnas.93.12.5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lander E., Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat. Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 31.Sawcer S., Jones H.B., Judge D., Visser F., Compston A., Goodfellow P.N., Clayton D. Empirical genomewide significance levels established by whole genome simulations. Genet. Epidemiol. 1997;14:223–229. doi: 10.1002/(SICI)1098-2272(1997)14:3<223::AID-GEPI1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 32.Cheng J., Kapranov P., Drenkow J., Dike S., Brubaker S., Patel S., Long J., Stern D., Tammana H., Helt G. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science. 2005;308:1149–1154. doi: 10.1126/science.1108625. [DOI] [PubMed] [Google Scholar]
- 33.Burge C., Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- 34.Fairbrother W.G., Yeo G.W., Yeh R., Goldstein P., Mawson M., Sharp P.A., Burge C.B. RESCUE-ESE identifies candidate exonic splicing enhancers in vertebrate exons. Nucleic Acids Res. 2004;32(Web Server issue):W187–W190. doi: 10.1093/nar/gkh393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakamura T., Nawa K., Ichihara A., Kaise N., Nishino T. Purification and subunit structure of hepatocyte growth factor from rat platelets. FEBS Lett. 1987;224:311–316. doi: 10.1016/0014-5793(87)80475-1. [DOI] [PubMed] [Google Scholar]
- 36.Nakamura T., Nishizawa T., Hagiya M., Seki T., Shimonishi M., Sugimura A., Tashiro K., Shimizu S. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989;342:440–443. doi: 10.1038/342440a0. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt C., Bladt F., Goedecke S., Brinkmann V., Zschiesche W., Sharpe M., Gherardi E., Birchmeier C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature. 1995;373:699–702. doi: 10.1038/373699a0. [DOI] [PubMed] [Google Scholar]
- 38.Uehara Y., Minowa O., Mori C., Shiota K., Kuno J., Noda T., Kitamura N. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature. 1995;373:702–705. doi: 10.1038/373702a0. [DOI] [PubMed] [Google Scholar]
- 39.Yu Y., Zuo J. The practical use of Cre and loxP technologies in mouse auditory research. Methods Mol. Biol. 2009;493:87–102. doi: 10.1007/978-1-59745-523-7_6. [DOI] [PubMed] [Google Scholar]
- 40.Chang W., Lin Z., Kulessa H., Hebert J., Hogan B.L., Wu D.K. Bmp4 is essential for the formation of the vestibular apparatus that detects angular head movements. PLoS Genet. 2008;4:e1000050. doi: 10.1371/journal.pgen.1000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barrionuevo F., Naumann A., Bagheri-Fam S., Speth V., Taketo M.M., Scherer G., Neubuser A. Sox9 is required for invagination of the otic placode in mice. Dev. Biol. 2008;317:213–224. doi: 10.1016/j.ydbio.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 42.Arnold J.S., Braunstein E.M., Ohyama T., Groves A.K., Adams J.C., Brown M.C., Morrow B.E. Tissue-specific roles of Tbx1 in the development of the outer, middle and inner ear, defective in 22q11DS patients. Hum. Mol. Genet. 2006;15:1629–1639. doi: 10.1093/hmg/ddl084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pirvola U., Ylikoski J., Trokovic R., Hebert J.M., McConnell S.K., Partanen J. FGFR1 is required for the development of the auditory sensory epithelium. Neuron. 2002;35:671–680. doi: 10.1016/s0896-6273(02)00824-3. [DOI] [PubMed] [Google Scholar]
- 44.Takayama H., LaRochelle W.J., Sharp R., Otsuka T., Kriebel P., Anver M., Aaronson S.A., Merlino G. Diverse tumorigenesis associated with aberrant development in mice overexpressing hepatocyte growth factor/scatter factor. Proc. Natl. Acad. Sci. USA. 1997;94:701–706. doi: 10.1073/pnas.94.2.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takayama H., Takagi H., Larochelle W.J., Kapur R.P., Merlino G. Ulcerative proctitis, rectal prolapse, and intestinal pseudo-obstruction in transgenic mice overexpressing hepatocyte growth factor/scatter factor. Lab. Invest. 2001;81:297–305. doi: 10.1038/labinvest.3780238. [DOI] [PubMed] [Google Scholar]
- 46.Dallos P., Harris D. Properties of auditory nerve responses in absence of outer hair cells. J. Neurophysiol. 1978;41:365–383. doi: 10.1152/jn.1978.41.2.365. [DOI] [PubMed] [Google Scholar]
- 47.Stoker M., Gherardi E., Perryman M., Gray J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature. 1987;327:239–242. doi: 10.1038/327239a0. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y.W., Vande Woude G.F. HGF/SF-met signaling in the control of branching morphogenesis and invasion. J. Cell. Biochem. 2003;88:408–417. doi: 10.1002/jcb.10358. [DOI] [PubMed] [Google Scholar]
- 49.Sakata H., Takayama H., Sharp R., Rubin J.S., Merlino G., LaRochelle W.J. Hepatocyte growth factor/scatter factor overexpression induces growth, abnormal development, and tumor formation in transgenic mouse livers. Cell Growth Differ. 1996;7:1513–1523. [PubMed] [Google Scholar]
- 50.Kmiecik T.E., Keller J.R., Rosen E., Vande Woude G.F. Hepatocyte growth factor is a synergistic factor for the growth of hematopoietic progenitor cells. Blood. 1992;80:2454–2457. [PubMed] [Google Scholar]
- 51.Hayashi K., Morishita R., Nakagami H., Yoshimura S., Hara A., Matsumoto K., Nakamura T., Ogihara T., Kaneda Y., Sakai N. Gene therapy for preventing neuronal death using hepatocyte growth factor: in vivo gene transfer of HGF to subarachnoid space prevents delayed neuronal death in gerbil hippocampal CA1 neurons. Gene Ther. 2001;8:1167–1173. doi: 10.1038/sj.gt.3301498. [DOI] [PubMed] [Google Scholar]
- 52.Jung W., Castren E., Odenthal M., Vande Woude G.F., Ishii T., Dienes H.P., Lindholm D., Schirmacher P. Expression and functional interaction of hepatocyte growth factor-scatter factor and its receptor c-met in mammalian brain. J. Cell Biol. 1994;126:485–494. doi: 10.1083/jcb.126.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miyazawa T., Matsumoto K., Ohmichi H., Katoh H., Yamashima T., Nakamura T. Protection of hippocampal neurons from ischemia-induced delayed neuronal death by hepatocyte growth factor: a novel neurotrophic factor. J. Cereb. Blood Flow Metab. 1998;18:345–348. doi: 10.1097/00004647-199804000-00001. [DOI] [PubMed] [Google Scholar]
- 54.Bevan D., Gherardi E., Fan T.P., Edwards D., Warn R. Diverse and potent activities of HGF/SF in skin wound repair. J. Pathol. 2004;203:831–838. doi: 10.1002/path.1578. [DOI] [PubMed] [Google Scholar]
- 55.Miyazawa K., Kitamura A., Naka D., Kitamura N. An alternatively processed mRNA generated from human hepatocyte growth factor gene. Eur. J. Biochem. 1991;197:15–22. doi: 10.1111/j.1432-1033.1991.tb15876.x. [DOI] [PubMed] [Google Scholar]
- 56.Miyazawa K., Shimomura T., Kitamura N. Activation of hepatocyte growth factor in the injured tissues is mediated by hepatocyte growth factor activator. J. Biol. Chem. 1996;271:3615–3618. doi: 10.1074/jbc.271.7.3615. [DOI] [PubMed] [Google Scholar]
- 57.Bottaro D.P., Rubin J.S., Faletto D.L., Chan A.M., Kmiecik T.E., Vande Woude G.F., Aaronson S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802–804. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- 58.Naka D., Ishii T., Yoshiyama Y., Miyazawa K., Hara H., Hishida T., Kidamura N. Activation of hepatocyte growth factor by proteolytic conversion of a single chain form to a heterodimer. J. Biol. Chem. 1992;267:20114–20119. [PubMed] [Google Scholar]
- 59.Birchmeier C., Birchmeier W., Gherardi E., Vande Woude G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 60.Chan A.M., Rubin J.S., Bottaro D.P., Hirschfield D.W., Chedid M., Aaronson S.A. Identification of a competitive HGF antagonist encoded by an alternative transcript. Science. 1991;254:1382–1385. doi: 10.1126/science.1720571. [DOI] [PubMed] [Google Scholar]
- 61.Cioce V., Csaky K.G., Chan A.M., Bottaro D.P., Taylor W.G., Jensen R., Aaronson S.A., Rubin J.S. Hepatocyte growth factor (HGF)/NK1 is a naturally occurring HGF/scatter factor variant with partial agonist/antagonist activity. J. Biol. Chem. 1996;271:13110–13115. doi: 10.1074/jbc.271.22.13110. [DOI] [PubMed] [Google Scholar]
- 62.Jakubczak J.L., LaRochelle W.J., Merlino G. NK1, a natural splice variant of hepatocyte growth factor/scatter factor, is a partial agonist in vivo. Mol. Cell. Biol. 1998;18:1275–1283. doi: 10.1128/mcb.18.3.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rubin J.S., Chan A.M., Bottaro D.P., Burgess W.H., Taylor W.G., Cech A.C., Hirschfield D.W., Wong J., Miki T., Finch P.W. A broad-spectrum human lung fibroblast-derived mitogen is a variant of hepatocyte growth factor. Proc. Natl. Acad. Sci. USA. 1991;88:415–419. doi: 10.1073/pnas.88.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seki T., Hagiya M., Shimonishi M., Nakamura T., Shimizu S. Organization of the human hepatocyte growth factor-encoding gene. Gene. 1991;102:213–219. doi: 10.1016/0378-1119(91)90080-u. [DOI] [PubMed] [Google Scholar]
- 65.Otsuka T., Jakubczak J., Vieira W., Bottaro D.P., Breckenridge D., Larochelle W.J., Merlino G. Disassociation of met-mediated biological responses in vivo: the natural hepatocyte growth factor/scatter factor splice variant NK2 antagonizes growth but facilitates metastasis. Mol. Cell. Biol. 2000;20:2055–2065. doi: 10.1128/mcb.20.6.2055-2065.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shima N., Nagao M., Ogaki F., Tsuda E., Murakami A., Higashio K. Tumor cytotoxic factor/hepatocyte growth factor from human fibroblasts: cloning of its cDNA, purification and characterization of recombinant protein. Biochem. Biophys. Res. Commun. 1991;180:1151–1158. doi: 10.1016/s0006-291x(05)81187-8. [DOI] [PubMed] [Google Scholar]
- 67.Zhou H., Mazzulla M.J., Kaufman J.D., Stahl S.J., Wingfield P.T., Rubin J.S., Bottaro D.P., Byrd R.A. The solution structure of the N-terminal domain of hepatocyte growth factor reveals a potential heparin-binding site. Structure. 1998;6:109–116. doi: 10.1016/s0969-2126(98)00012-4. [DOI] [PubMed] [Google Scholar]
- 68.Chirgadze D.Y., Hepple J.P., Zhou H., Byrd R.A., Blundell T.L., Gherardi E. Crystal structure of the NK1 fragment of HGF/SF suggests a novel mode for growth factor dimerization and receptor binding. Nat. Struct. Biol. 1999;6:72–79. doi: 10.1038/4947. [DOI] [PubMed] [Google Scholar]
- 69.Zhou H., Casas-Finet J.R., Heath Coats R., Kaufman J.D., Stahl S.J., Wingfield P.T., Rubin J.S., Bottaro D.P., Byrd R.A. Identification and dynamics of a heparin-binding site in hepatocyte growth factor. Biochemistry. 1999;38:14793–14802. doi: 10.1021/bi9908641. [DOI] [PubMed] [Google Scholar]
- 70.Lyon M., Deakin J.A., Lietha D., Gherardi E., Gallagher J.T. The interactions of hepatocyte growth factor/scatter factor and its NK1 and NK2 variants with glycosaminoglycans using a modified gel mobility shift assay. Elucidation of the minimal size of binding and activatory oligosaccharides. J. Biol. Chem. 2004;279:43560–43567. doi: 10.1074/jbc.M408510200. [DOI] [PubMed] [Google Scholar]
- 71.Rubin J.S., Day R.M., Breckenridge D., Atabey N., Taylor W.G., Stahl S.J., Wingfield P.T., Kaufman J.D., Schwall R., Bottaro D.P. Dissociation of heparan sulfate and receptor binding domains of hepatocyte growth factor reveals that heparan sulfate-c-met interaction facilitates signaling. J. Biol. Chem. 2001;276:32977–32983. doi: 10.1074/jbc.M105486200. [DOI] [PubMed] [Google Scholar]
- 72.Tolbert W.D., Daugherty J., Gao C., Xie Q., Miranti C., Gherardi E., Woude G.V., Xu H.E. A mechanistic basis for converting a receptor tyrosine kinase agonist to an antagonist. Proc. Natl. Acad. Sci. USA. 2007;104:14592–14597. doi: 10.1073/pnas.0704290104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hartmann G., Naldini L., Weidner K.M., Sachs M., Vigna E., Comoglio P.M., Birchmeier W. A functional domain in the heavy chain of scatter factor/hepatocyte growth factor binds the c-Met receptor and induces cell dissociation but not mitogenesis. Proc. Natl. Acad. Sci. USA. 1992;89:11574–11578. doi: 10.1073/pnas.89.23.11574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lai L., Goldschneider I. Cutting edge: Identification of a hybrid cytokine consisting of IL-7 and the beta-chain of the hepatocyte growth factor/scatter factor. J. Immunol. 2001;167:3550–3554. doi: 10.4049/jimmunol.167.7.3550. [DOI] [PubMed] [Google Scholar]
- 75.Oshima K., Shimamura M., Mizuno S., Tamai K., Doi K., Morishita R., Nakamura T., Kubo T., Kaneda Y. Intrathecal injection of HVJ-E containing HGF gene to cerebrospinal fluid can prevent and ameliorate hearing impairment in rats. FASEB J. 2004;18:212–214. doi: 10.1096/fj.03-0567fje. [DOI] [PubMed] [Google Scholar]
- 76.Hordichok A.J., Steyger P.S. Closure of supporting cell scar formations requires dynamic actin mechanisms. Hear. Res. 2007;232:1–19. doi: 10.1016/j.heares.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tornabene S.V., Sato K., Pham L., Billings P., Keithley E.M. Immune cell recruitment following acoustic trauma. Hear. Res. 2006;222:115–124. doi: 10.1016/j.heares.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 78.Lee C.C., Putnam A.J., Miranti C.K., Gustafson M., Wang L.M., Vande Woude G.F., Gao C.F. Overexpression of sprouty 2 inhibits HGF/SF-mediated cell growth, invasion, migration, and cytokinesis. Oncogene. 2004;23:5193–5202. doi: 10.1038/sj.onc.1207646. [DOI] [PubMed] [Google Scholar]
- 79.Shim K., Minowada G., Coling D.E., Martin G.R. Sprouty2, a mouse deafness gene, regulates cell fate decisions in the auditory sensory epithelium by antagonizing FGF signaling. Dev. Cell. 2005;8:553–564. doi: 10.1016/j.devcel.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 80.Lim D.J., Anniko M. Developmental morphology of the mouse inner ear. A scanning electron microscopic observation. Acta Otolaryngol. Suppl. 1985;422:1–69. [PubMed] [Google Scholar]
- 81.Taylor R.R., Nevill G., Forge A. Rapid Hair Cell Loss: A Mouse Model for Cochlear Lesions. J. Assoc. Res. Otolaryngol. 2008;9:44–64. doi: 10.1007/s10162-007-0105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.