

Abstract
The present review discusses recent studies that have identified genetic differences in inflammatory proteins associated with different phenotypic presentations of systemic inflammation. Basic genetic terminology is defined. Implications of genetic influences on the inflammatory response are discussed. The published associations of specific polymorphisms in antigen recognition pathways, proinflammatory cytokines, anti-inflammatory cytokines, and effector molecules are reviewed. The strongest and most consistent associations thus far have been with the tumor necrosis factor, lymphotoxin-α, and IL-1 receptor antagonist polymorphisms. However, large, phenotypically detailed studies are required to address all of the other potential polymorphisms in inflammatory molecule genes and their interactions.
Keywords: gene polymorphisms, genetics, inflammation, pneumonia, sepsis
Introduction
The fact that individual genetic differences impact on the risk for developing or dying from various diseases has long been accepted. Typical examples include sickle cell trait and malaria, BRCA2 mutations and breast carcinoma, and trinucleotide repeats and a variety of neurologic diseases, including Huntington disease.
Physicians have also long been aware of the markedly different responses of seemingly similar individuals to the same inflammatory or infectious agents. The role of individual genetic differences as an explanation for these observations has been the subject of much speculation. The strongest observational evidence of a genetic influence comes from a study conducted by Sorenson and colleagues [1]. In their study of adoptees, the death of a biologic parent from infection was associated with a five times greater risk for death from infection. During the past half-decade, advances in knowledge of the human genome, greater understanding of the inflammatory response, and the development of genotyping technologies have allowed us to start the process of identifying specific genetic mutations associated with different inflammatory phenotypes.
In the present review, we discuss recent studies that have identified genetic differences in inflammatory proteins that are associated with different phenotypic presentations of system inflammation.
Basic genetic terminology
A region of DNA that encodes a protein product is called an exon. Introns are the noncoding regions of DNA that separate exons. Most genes consist of several exons and introns. The rate at which genes are transcribed is controlled by a variety of nuclear proteins that bind to different areas of DNA in the 5' (upstream) region from the first exon. The segment of DNA that controls the regulation of transcription of a gene is known as the promoter region.
A variety of mutations can occur in DNA, some of which lead to a change in function or production of a gene product. The simplest change is the substitution of one nucleotide for another, which is known as a single nucleotide polymorphism (SNP). A number of other mutations occur, including deletion or insertion of one or more nucleotides, and insertion of multiple repeating sequences (e.g. the trinucleotide repeats mentioned above). Mutations that occur in an exon may lead to a change in the protein structure encoded by the gene. Changes that occur in a promoter region may alter the binding of a transcription activating or suppressing factor, altering the rate of transcription of the gene. Although introns were considered to be 'junk DNA', and consequently mutations in introns were believed to be unimportant biologically, it is now appreciated that polymorphisms within introns can affect gene regulation, particularly when near the intron–exon boundary [2,3].
SNPs are referred to by their distance (i.e. the number of bp away) from the transcription activating site. Therefore, gene X -505 indicates that the SNP is 505 bp upstream of the X gene, potentially in the promoter region. Gene X +505 indicates the SNP is 505 bp downstream of the transcription activating site, potentially in an exon or intronic region.
Variations in a gene that arise due to mutations are referred to as alleles. An individual's genotype is often referred to by the nucleotide carried at the polymorphic site in question (i.e. tumor necrosis factor [TNF]-α-308A or TNF-α-308G). An alternative nomenclature is sometimes used when a mutation causes an amino acid change in the protein (i.e. Toll-ilke receptor [TLR]-4 Thr399Ile indicates an isoleucine is substituted for a threonine at amino acid position 399). Finally, the most common allele can be referred to as allele 1 (or A), the second most common as allele 2 (or B), and so on (i.e. TNF-α-308 allele 1). Because allele frequencies can vary significantly between populations, the latter convention for naming has the potential to lead to considerable confusion, and we therefore avoid that convention wherever possible in the present review.
The inflammatory response
Following the recognition of foreign antigens, a variety of proinflammatory cytokines are released, along with counter-regulatory or anti-inflammatory cytokines. Genetic polymorphisms with potential influences on the inflammatory response have been identified in a variety of antigen recognition pathways, proinflammatory cytokines, and anti-inflammatory cytokines (Table 1).
Table 1.
An enormous number of genes have been identified as having potentially important polymorphic sites
| Sites of specific polymorphism | Gene |
| Antigen recognition pathways | CD14 |
| TLR-4 | |
| TLR-2 | |
| HSP-70-1 | |
| HSP-70-2 | |
| HSP-70-HOM | |
| Proinflammatory cytokines | TNF-α + receptors |
| IL-1α + IL-1β + receptors | |
| IL-6, IL-2, IL-3, | |
| IL-8 + receptors | |
| IFN-γ | |
| IL-12 | |
| IL-18 | |
| GM-CSF | |
| IFN-α | |
| LT-α | |
| Anti-inflammatory cytokines | IL-10 |
| IL-1Ra | |
| IL-13 | |
| TGF-β1 and TGF-β2 | |
| IL-4 | |
| CTLA-4 |
CTLA, cytotoxic T-lymphocyte-associated antigen; GM-CSF, granulocyte/macrophage colony-stimulating factor; HSP, heat shock protein; IFN, interferon; IL-1Ra, interleukin 1 receptor antagonist; LT, lymphotoxin; TGF, transforming growth factor; TLR, Toll-like receptor; TNF, tumor necrosis factor.
The outcome of an inflammatory response is dictated by a variety of factors, including the pathogenicity and duration of the stimulus, and the balance between the proinflammatory and anti-inflammatory response. An excessive proinflammatory (or deficient anti-inflammatory) response is thought to be important in the pathogenesis of septic shock [4]. Equally, a deficient proinflammatory (or excessive anti-inflammatory) response could result in failure to clear an invading pathogen, with equally deleterious effects. A further adverse result of the anti-inflammatory response is the period of relative immunosuppression (also known as immunoparalysis or compensatory anti-inflammatory response syndrome [4]) after an inflammatory insult. Prolonged compensatory anti-inflammatory response syndrome may be associated with excess mortality and morbidity because of increased risk for nosocomial infections [5].
With substantial overlap between the functions of many cytokines, and frequently multiple antagonists for any given agonist, the ability to compensate for a certain amount of divergence in production of individual cytokines is significant. Therefore, for a single mutation to influence the outcome of an inflammatory response, the mutation must markedly alter the production or function of a critical inflammatory protein. Although possible, a more likely scenario is the inheritance of multiple mutations in multiple proteins, each leading to small changes in production or function, but with a net serious deleterious effect.
Finally, for seemingly adverse mutations to be preserved in the human genome despite predisposition to a deleterious outcome in one disease, a survival advantage in another (i.e. different infection, malignancy, etc.) is quite possible. The sickle cell–malaria relationship is the most obvious example. Many of the polymorphisms described in this review were first studied in noninfectious or non-critical-care populations. A consistent relationship in these other inflammatory disorders adds to the validity of findings in the critically ill. Therefore, although identifying polymorphisms associated with adverse outcomes can provide useful insights into the inflammatory response, much more study will be needed before the full implications of carriage of specific polymorphisms in specific individuals can be determined.
The specific mutations in the systemic inflammatory response can be roughly grouped for discussion into three categories: antigen recognition, proinflammatory cytokines, and anti-inflammatory cytokines.
Antigen recognition pathways
CD14
CD14 is a glycosylphosphatidylinositol membrane-anchored protein that is expressed on the surface of macrophages, monocytes, and polymorphonuclear cells. Complexed with two other proteins, namely MD-2 and TLR-4, CD14 has been identified as a key endotoxin, or lippopolysaccharide (LPS) recognition pathway [6]. A second, CD14-independent LPS recognition pathway has also been identified that involves a complex of heat shock protein-70, heat shock protein-90, chemokine receptor-4, and growth differentiation factor-5 [7].
A polymorphism at -159 involving a cytosine to thymidine transition has been identified in the CD14 gene [8], with those who carry the T allele having greater circulating levels of soluble CD14. Because transgenic mice that over-express CD14 are highly susceptible to septic shock [9], increased expression of CD14 may be an important risk factor for septic shock. Gibot and colleagues [10] recently found that carriage of the CD14 -159 TT genotype was more common in 90 French patients with septic shock than in 122 age- and sex-matched, healthy control individuals (71% versus 48%; P = 0.008). Currently clinical trials of CD14 blocking agents are underway. However, increased mortality from Gram-negative infections in animals treated with anti-CD14 antibodies [11] suggests that therapeutic intervention may potentially be limited.
Toll-like receptors
One of the most important recent discoveries of mechanisms of foreign antigen recognition is the identification of the group of proteins known as the TLRs. Toll protein was initially recognized in Drosophila as an important signaling molecule in innate immunity against bacteria and fungi. Thus far, 10 TLRs have been identified in humans [12]. TLR-4 appears to be essential for signal recognition of LPS, whereas TLR-2 appears to be vital for recognition of peptidoglycans from Gram-positive bacteria [12].
Mice with a mutation in TLR-4 are highly resistant to LPS challenge [13], and several coding region variations in human TLR-4 have been identified [14], although their functional importance is as yet undetermined. Lorenz and colleagues [15] studied the TLR-4 Asp299Gly and TLR-4 Thr399Ile mutations in a French cohort of 91 patients with septic shock and 73 healthy control individuals. Interestingly, carriage of TLR-4 Asp299Gly was found only in the shock cohort (P = 0.05), although a potential confounding factor was a significant difference in the age of the control individuals (mean 37 years) as compared with cases (mean 58 years; P < 0.001). Although further studies are needed, the authors speculated that the TLR-4 Asp299Gly mutation may interrupt LPS signaling.
Consistent with its important role in the recognition of Gram-positive bacteria, a TLR-2 mutation (Arg753Gln) was found to be associated with a reduced inflammatory response to Borrelia burgdorferi and Treponema pallidum [16]. In the same cohort as the TLR-4 study above [15], the TLR-2 Arg753Gln mutation was found in only two individuals. Both had staphylococcal sepsis, which is consistent with this mutation predisposing to Gram-positive sepsis.
Proinflammatory cytokines
Tumor necrosis factor-α
TNF-α is a critical cytokine in the inflammatory response to infection [17]. Accordingly, any genetic variability in the production of TNF-α after an infectious stimulus could have a significant impact on the degree of inflammatory response and therefore potentially influence the clinical outcome.
In vitro studies have consistently found marked individual variation in TNF-α production after a variety of inflammatory stimuli [18-21]. Early studies identified specific human leukocyte antigen (HLA) markers associated with variable TNF-α production (e.g. HLA A1B8DR3 haplotype in Caucasians [22]). Subsequently, the highly polymorphic nature of the TNF locus has become appreciated, with more than a dozen SNPs and several microsatellites (areas of multiple nucleotide repeat sequences) being described.
A significant amount of evidence supports the biologic importance of polymorphisms within the TNF-α promoter region. A guanine (G) to adenine (A) transition at TNF-α-308, associated with the ancestral haplotype mentioned above [22], is perhaps the best studied cytokine polymorphism and the one for which the best evidence of functional significance exists. Stimulation studies in healthy volunteers suggested that carriage of the TNF-α-308 A allele is associated with significantly greater TNF-α production [23] and TNF-α mRNA transcription [24], although the degree of difference may be depend on the cell type stimulated and the stimulus applied [25]. Carriage of the A allele of TNF-α-308 has been associated with an increased risk for many diseases, including septic shock [26], severe cerebral malaria [27], and death from meningococcal sepsis [28].
Additional polymorphisms within the TNF-α promoter may also influence the rate of transcription of TNF-α, including TNF-α-238 [29], TNF-α-376 [30], and TNF-α-1031 [31]. Clinical association studies suggest mutations at TNF-α-376 are risk factors for severe malaria [30] and mutations at TNF-α-238 are risk factors for death from community-acquired pneumonia [32], although not from meningococcal sepsis [33].
Complicating assessment of TNF-α polymorphisms is the high degree of linkage disequilibrium between TNF-α promoter polymorphisms and between other polymorphisms within other nearby genes, many of which also have significant inflammatory roles. Although not a comprehensive list, among the nearby genes (Fig. 1) are many with major inflammatory roles, including lymphotoxin-α (LT-α), lymphotoxin-β, the heat shock protein-70 complex, complement 4A and 4B, HLA B associated transcript 1, and the HLA A, B, C, DR, DP, and DQ loci.
Figure 1.
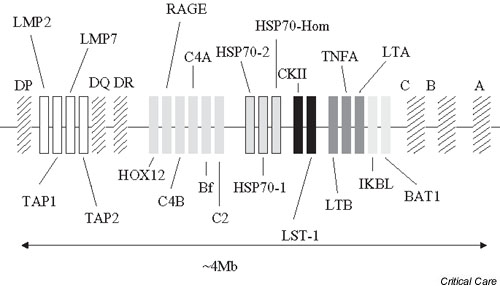
Area in short arm of chromosome 6, demonstrating the close proximity of many genes that are involved in inflammatory responses within the human leukocyte antigen (HLA) locus.
Lymphotoxin-α (also known as tumor necrosis factor-β)
LT-α +250, a G to A transition in the first intron of LT-α, has been identified as a potentially influential locus in many inflammatory conditions. This polymorphism is part of a complex haplotype including the nonsynonymous mutation LT-α +250 Asp26Thr. Linkage disequilibrium with the TNF-α-308G allele and the LT-α +250 A allele [34] requires that these polymorphisms not be assessed in isolation. Carriage of the A allele of LT-α +250 has been associated with increased TNF-α production both in vitro [35] and in vivo [36,37], providing a biologically plausible mechanism of effect, although the mechanism by which this mutation impacts on TNFα production is unknown.
In the landmark study of 40 patients with septic shock conducted by Stuber [36], carriage of the LT-α +250 AA genotype carried a substantially greater risk for death (P = 0.002). Subsequent studies by the same group suggested that carriage of LT-α +250 A is also a risk factor for the development of septic shock, at least in a population of 110 post-trauma patients [37]. We have also found LT-α +250 AA genotype to be a risk factor for septic shock in patients with community-acquired pneumonia [34]. Interestingly, respiratory failure in the absence of shock strongly correlated with TNF-α +250 GG genotype, again suggesting that polymorphisms may be 'good' or 'bad', depending on the outcome of interest.
Interleukin-1α and interleukin-1β
IL-1, a potent proinflammatory cytokine released by macrophages, also plays a key role in mediating endotoxin lethality [38]. The IL-1 family includes the agonists IL-1α and IL-1β, and the IL-1 receptor antagonist (IL-1Ra). Although both the IL-1β +3953 and -511 polymorphic sites may influence levels of IL-1β in stimulated peripheral blood mononuclear cells [39], no association was found with susceptibility to or outcome from septic shock by Fang and colleagues [40]. Kornman and coworkers [41] did find an association between IL-1β +3953 and risk for periodontitis, and so an effect of these polymorphisms on the inflammatory response in some conditions remains possible, but as yet not demonstrated.
Interleukin-6
IL-6 has been demonstrated to be a marker of the severity and outcome of sepsis by a number of groups, but whether this is an epiphenomenon or a more causative relationship is still undetermined. A haplotype involving at least four SNPs within the promoter has been identified and appears to influence the rate of transcription of IL-6 [42]. Schluter and colleagues [43] studied the IL-6 -174 C/G polymorphism – one of the SNPs in this haplotype – in 50 German patients with severe sepsis; they found that carriage of the IL6 -174 GG (low IL-6 secretor phenotype) genotype was associated with improved survival.
A common problem in interpreting the finding of an association between a polymorphism and a clinical outcome is demonstrated in the study conducted by Schluter and colleagues [43]. Despite the positive findings discussed above, no correlation was found between serum IL-6 levels and IL-6 -174 genotype. One interpretation of the lack of a phenotype–genotype correlation would be that the association is spurious. However, serum cytokine levels may have a poor correlation with tissue concentrations, and a single serum level taken at a variable time point after the onset of the inflammatory insult may tell us little about the amount of IL-6 produced in the early critical phases of the inflammatory response. Much greater research into genotype–phenotype relationships and how they are altered by external factors (such as comorbid diseases) is clearly required.
Anti-inflammatory cytokines
Interleukin-1 receptor antagonist
As already mentioned, IL-1Ra is part of the IL-1 family of proteins and, as its name suggests, is a naturally occurring antagonist of IL-1α and IL-1β. IL-1Ra knockout mice have increased susceptibility to endotoxin-induced lethality whereas mice that over-express IL-1Ra are protected [44], indicating that IL1-Ra is likely to play a key role in protection against the adverse effects of an inflammatory response.
Intron 2 of IL-1Ra contains a variable 86-bp tandem repeat containing at least three binding sites for DNA-binding proteins [45]. Alleles are named A1, A2, A3, A4, and A5, based on their relative frequency in healthy populations. In vitro studies suggest that fewer 86-bp repeats correlates with higher IL-1Ra protein production after LPS stimulation [46]. The relationship appears to be complex, with higher IL-1Ra found in the serum of healthy A2 allele carriers [46,47] but only in those who also carry the high IL-1β genotype of IL-1β-511 [47]. Carriage of the A2 allele has been associated with increased risk for a number of inflammatory disorders, including systemic lupus erythematosis, Sjögren's syndrome, myasthenia gravis, alopecia areata, Grave's disease, periodontitis, Henoch–Schonlein nephritis, multiple sclerolis, ulcerative colitis, tuberculous pleurisy and insulin-dependent diabetes mellitus.
In 93 patients with severe sepsis, Fang and colleagues [40] found carriage of the A2 allele to be associated with a significantly greater risk for septic shock, with carriage of both the LT-α +250 AA and IL-1Ra A2/A2 genotypes universally fatal. Ma and colleagues [48] recently confirmed these findings in a study of 60 Chinese patients with severe sepsis, and Arnalich and colleagues [49] also demonstrated a 6.47-fold increased risk for death in IL-1Ra A2 carriers in a cohort of 78 Spanish patients with severe sepsis. Taken together, the weight of evidence would certainly suggest that this IL-1Ra locus has a very influential effect on the outcome of an inflammatory response.
Interleukin-10
IL-10 is also a potent anti-inflammatory protein. In a study of LPS stimulated whole blood, Westendorp and colleagues [33] found that family members of children who died from meningococcemia had significantly greater IL-10 production than did family members of children who survived. This suggests that genetic differences in IL-10 production are likely to be important in the outcome of severe sepsis.
Several polymorphisms have been identified in IL-10, including a three-SNP promoter haplotype [50], and microsatellites in both the 3' and 5' regions [51,52]. The promoter haplotype influences IL-10 production, with stimulated lymphocytes from individuals carrying IL-10 -1082A/-819C/-592C haplotype producing less IL-10 after concanavalin A stimulation than those carrying the 1082G/-819C/-592C haplotype [50].
A variety of inflammatory diseases have been associated with IL-10 SNP promoter genotypes, including asthma severity, inflammatory bowel disease, rheumatoid arthritis, and systemic lupus erythematosus. The only published study of IL-10 genotype and severe sepsis to date found that the -1082 GG genotype was associated with a greater risk for meningococcal disease [53]. The -1082 GG genotype also appears to be a risk factor for chronic hepatitis C [54]. Further studies in severe sepsis are awaited.
Other proteins
Outside of the three broad groups of proteins detailed above are a large number of genes that may modify the outcome of a systemic inflammatory insult. Many of these genes encode proteins that are upregulated or downregulated by the inflammatory response, key examples of which include the coagulation system and tissue and wound repair. In some cases, gene mutations in the 'downstream' proteins may have a much greater impact than mutations in genes expressed early in the inflammatory response.
Studies of acute respiratory distress syndrome (ARDS), which is known to be associated with a marked inflammatory response [55,56], demonstrate the potential role of mutations within genes outside the inflammatory system. Homozygotes for the deletion allele of the angiotensin converting enzyme deletion/insertion polymorphism were markedly (P < 0.0001) over-represented in a cohort of patients with ARDS in the UK [57]. This angiotensin converting enzyme polymorphism has been associated with a wide arrange of predominantly vascular diseases, and the study conducted by Marshall and colleagues [57] suggests that the renin–angiotensin system may play a key role in the pathogenesis of ARDS.
An entirely different set of polymorphisms within the surfactant protein (SP)-A, SP-B, and SP-D genes has also been studied in patients with ARDS. In a case–control study, Lin and colleagues [58] found carriage of the SP-B +1580 C allele (which is associated with a change of isoleucine to threonine at amino acid 131) was associated with a 2.4 times increased risk for ARDS. Because SPs are known to be important in a number of pulmonary processes, including local host defense and modulating pulmonary inflammation, the association is biologically plausible.
Conclusion
An increasing array of polymorphisms in diverse inflammatory genes have been identified as candidates to explain part of the enormous phenotypic variability in the systemic inflammatory response. Currently published studies, although illuminating, are already inadequate to assess the relative influence of and interactions between the currently identified loci. Large, phenotypically detailed studies, with adequate statistical power to address these issues are now required. Clearly, the size of these studies will be beyond a single institution and will require large, multicenter, collaborative efforts. Despite the size and complexity of the task, enormous potential to develop new therapeutic interventions is clearly possible once we understand and can predict individual inflammatory responses.
Competing interests
None declared.
Abbreviations
ARDS = acute respiratory distress syndrome; bp = base pair; HLA = human leukocyte antigen; IL = interleukin; IL-1Ra = interleukin-1 receptor antagonist; LPS = lipopolysaccharide; LT-α = lymphotoxin-α; SNP = single nucleotide polymorphism; SP = surfactant protein; TLR = Toll-like receptor; TNF = tumor necrosis factor.
References
- Sorensen TI, Nielsen GG, Andersen PK, Teasdale TW. Genetic and environmental influences on premature death in adult adoptees. N Engl J Med. 1988;318:727–732. doi: 10.1056/NEJM198803243181202. [DOI] [PubMed] [Google Scholar]
- Li M, Pritchard PH. Characterization of the effects of mutations in the putative branchpoint sequence of intron 4 on the splicing within the human lecithin:cholesterol acyltransferase gene. J Biol Chem. 2000;275:18079–18084. doi: 10.1074/jbc.M910197199. [DOI] [PubMed] [Google Scholar]
- Artiga MJ, Saez AI, Romero C, Sanchez-Beato M, Mateo MS, Navas C, Mollejo M, Piris MA. A short mutational hot spot in the first intron of BCL-6 is associated with increased BCL-6 expression and with longer overall survival in large B-cell lymphomas. Am J Pathol. 2002;160:1371–1380. doi: 10.1016/S0002-9440(10)62564-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. 1996;24:1125–1128. doi: 10.1097/00003246-199607000-00010. [DOI] [PubMed] [Google Scholar]
- Peters M, Petros A, Dixon G, Inwald D, Klein N. Acquired immunoparalysis in paediatric intensive care: prospective observational study. BMJ. 1999;319:609–610. doi: 10.1136/bmj.319.7210.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantafilou M, Triantafilou K. Lipopolysaccharide recognition: CD14, TLRs and the LPS-activation cluster. Trends Immunol. 2002;23:301–304. doi: 10.1016/S1471-4906(02)02233-0. [DOI] [PubMed] [Google Scholar]
- Triantafilou K, Triantafilou M, Dedrick RL. A CD14-independent LPS receptor cluster. Nat Immunol. 2001;2:338–345. doi: 10.1038/86342. [DOI] [PubMed] [Google Scholar]
- Baldini M, Lohman IC, Halonen M, Erickson RP, Holt PG, Martinez FD. A polymorphism* in the 5' flanking region of the CD14 gene is associated with circulating soluble CD14 levels and with total serum immunoglobulin E. Am J Respir Cell Mol Biol. 1999;20:976–983. doi: 10.1165/ajrcmb.20.5.3494. [DOI] [PubMed] [Google Scholar]
- Ferrero E, Jiao D, Tsuberi BZ, Tesio L, Rong GW, Haziot A, Goyert SM. Transgenic mice expressing human CD14 are hypersensitive to lipopolysaccharide. Proc Natl Acad Sci USA. 1993;90:2380–2384. doi: 10.1073/pnas.90.6.2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibot S, Cariou A, Drouet L, Rossignol M, Ripoll L. Association between a genomic polymorphism within the CD14 locus and septic shock susceptibility and mortality rate. Crit Care Med. 2002;30:969–973. doi: 10.1097/00003246-200205000-00003. [DOI] [PubMed] [Google Scholar]
- Frevert CW, Matute-Bello G, Skerrett SJ, Goodman RB, Kajikawa O, Sittipunt C, Martin TR. Effect of CD14 blockade in rabbits with Escherichia coli pneumonia and sepsis. J Immunol. 2000;164:5439–5445. doi: 10.4049/jimmunol.164.10.5439. [DOI] [PubMed] [Google Scholar]
- Lien E, Ingalls RR. Toll-like receptors. Crit Care Med. 2002;30(suppl):S1–S11. [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, VanHuffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Beutler B, Du X, Poltorak A. Identification of Toll-like receptor 4 (Tlr4) as the sole conduit for LPS signal transduction: genetic and evolutionary studies. J Endotoxin Res. 2001;7:277–280. [PubMed] [Google Scholar]
- Lorenz E, Mira JP, Frees KL, Schwartz DA. Relevance of mutations in the TLR4 receptor in patients with gram-negative septic shock. Arch Intern Med. 2002;162:1028–1032. doi: 10.1001/archinte.162.9.1028. [DOI] [PubMed] [Google Scholar]
- Lorenz E, Mira JP, Cornish KL, Arbour NC, Schwartz DA. A novel polymorphism in the toll-like receptor 2 gene and its potential association with staphylococcal infection. Infect Immun. 2000;68:6398–6401. doi: 10.1128/IAI.68.11.6398-6401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler B, Grau GE. Tumor necrosis factor in the pathogenesis of infectious diseases. Crit Care Med. 1993;21(suppl):S423–S435. [PubMed] [Google Scholar]
- Jacob CO, Fronek Z, Lewis GD, Koo M, Hansen JA, McDevitt HO. Heritable major histocompatibility complex class II-associated differences in production of tumor necrosis factor alpha: relevance to genetic predisposition to systemic lupus erythematosus. Proc Natl Acad Sci USA. 1990;87:1233–1237. doi: 10.1073/pnas.87.3.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molvig J, Baek L, Christensen P, Manogue KR, Vlassara H, Platz P, Nielsen LS, Svejgaard A, Nerup J. Endotoxin-stimulated human monocyte secretion of interleukin 1, tumour necrosis factor alpha, and prostaglandin E2 shows stable interindividual differences. Scand J Immunol. 1988;27:705–716. doi: 10.1111/j.1365-3083.1988.tb02404.x. [DOI] [PubMed] [Google Scholar]
- Aguillon JC, Escobar A, Ferreira V, Aguirre A, Ferreira L, Molina MC, Ferreira A. Daily production of human tumor necrosis factor in lipopolysaccharide (LPS)-stimulated ex vivo blood culture assays. Eur Cytokine Netw. 2001;12:105–110. [PubMed] [Google Scholar]
- Yaqoob P, Newsholme EA, Calder PC. Comparison of cytokine production in cultures of whole human blood and purified mononuclear cells. Cytokine. 1999;11:600–605. doi: 10.1006/cyto.1998.0471. [DOI] [PubMed] [Google Scholar]
- Price P, Witt C, Allcock R, Sayer D, Garlepp M, Kok CC, French M, Mallal S, Christiansen F. The genetic basis for the association of the 8.1 ancestral haplotype (A1, B8, DR3) with multiple immunopathological diseases. Immunol Rev. 1999;167:257–274. doi: 10.1111/j.1600-065x.1999.tb01398.x. [DOI] [PubMed] [Google Scholar]
- Louis E, Franchimont D, Piron A, Gevaert Y, Schaaf-Lafontaine N, Roland S, Mahieu P, Malaise M, De Groote D, Louis R, Belaiche J. Tumour necrosis factor (TNF) gene polymorphism influences TNF-alpha production in lipopolysaccharide (LPS)-stimulated whole blood cell culture in healthy humans. Clin Exp Immunol. 1998;113:401–406. doi: 10.1046/j.1365-2249.1998.00662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeger KM, Carville KS, Abraham LJ. The -308 tumor necrosis factor-alpha promoter polymorphism effects transcription. Mol Immunol. 1997;34:391–399. doi: 10.1016/S0161-5890(97)00052-7. [DOI] [PubMed] [Google Scholar]
- Kroeger KM, Steer JH, Joyce DA, Abraham LJ. Effects of stimulus and cell type on the expression of the -308 tumour necrosis factor promoter polymorphism. Cytokine. 2000;12:110–119. doi: 10.1006/cyto.1999.0529. [DOI] [PubMed] [Google Scholar]
- Mira JP, Cariou A, Grall F, Delclaux C, Losser MR, Heshmati F, Cheval C, Monchi M, Teboul JL, Riche F, Leleu G, Arbibe L, Mignon A, Delpech M, Dhainaut JF. Association of TNF2, a TNF-alpha promoter polymorphism, with septic shock susceptibility and mortality: a multicenter study. JAMA. 1999;282:561–568. doi: 10.1001/jama.282.6.561. [DOI] [PubMed] [Google Scholar]
- McGuire W, Hill AV, Allsopp CE, Greenwood BM, Kwiatkowski D. Variation in the TNF-alpha promoter region associated with susceptibility to cerebral malaria. Nature. 1994;371:508–510. doi: 10.1038/371508a0. [DOI] [PubMed] [Google Scholar]
- Nadel S, Newport MJ, Booy R, Levin M. Variation in the tumor necrosis factor-alpha gene promoter region may be associated with death from meningococcal disease. J Infect Dis. 1996;174:878–880. doi: 10.1093/infdis/174.4.878. [DOI] [PubMed] [Google Scholar]
- Grove J, Daly AK, Bassendine MF, Day CP. Association of a tumor necrosis factor promoter polymorphism with susceptibility to alcoholic steatohepatitis. Hepatology. 1997;26:143–146. doi: 10.1002/hep.510260119. [DOI] [PubMed] [Google Scholar]
- Knight JC, Udalova I, Hill AV, Greenwood BM, Peshu N, Marsh K, Kwiatkowski D. A polymorphism that affects OCT-1 binding to the TNF promoter region is associated with severe malaria. Nat Genet. 1999;22:145–150. doi: 10.1038/9649. [DOI] [PubMed] [Google Scholar]
- Higuchi T, Seki N, Kamizono S, Yamada A, Kimura A, Kato H, Itoh K. Polymorphism of the 5'-flanking region of the human tumor necrosis factor (TNF)-alpha gene in Japanese. Tissue Antigens. 1998;51:605–612. doi: 10.1111/j.1399-0039.1998.tb03002.x. [DOI] [PubMed] [Google Scholar]
- Waterer GW, Quasney MW, Cantor RA, Wunderink RG. Increased risk of death from community-acquired pneumonia (CAP) associated with the A allele of the TNFa-238 polymorphism [abstract] Am J Respir Crit Care Med. 2001;163:A380. doi: 10.1164/ajrccm.163.7.2011088. [DOI] [PubMed] [Google Scholar]
- Westendorp RG, Langermans JA, Huizinga TW, Elouali AH, Verweij CL, Boomsma DI, Vandenbroucke JP, Vandenbrouke JP. Genetic influence on cytokine production and fatal meningococcal disease. Lancet. 1997;349:170–173. doi: 10.1016/S0140-6736(96)06413-6. [DOI] [PubMed] [Google Scholar]
- Waterer GW, Quasney MW, Cantor RM, Wunderink RG. Septic shock and respiratory failure in community-acquired pneumonia have different TNF polymorphism associations. Am J Respir Crit Care Med. 2001;163:1599–1604. doi: 10.1164/ajrccm.163.7.2011088. [DOI] [PubMed] [Google Scholar]
- Messer G, Spengler U, Jung MC, Honold G, Blomer K, Pape GR, Riethmuller G, Weiss EH. Polymorphic structure of the tumor necrosis factor (TNF) locus: an NcoI polymorphism in the first intron of the human TNF-beta gene correlates with a variant amino acid in position 26 and a reduced level of TNF-beta production. J Exp Med. 1991;173:209–219. doi: 10.1084/jem.173.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuber F. A genomic polymorphism within the tumor necrosis factor locus influences plasma tumor necrosis factor-α concentrations and outcome of patients with severe sepsis. Crit Care Med. 1996;24:381–384. doi: 10.1097/00003246-199603000-00004. [DOI] [PubMed] [Google Scholar]
- Majetschak M, Flohe S, Obertacke U, Schroder J, Staubach K, Nast-Kolb D, Schade FU, Stuber F. Relation of a TNF gene polymorphism to severe sepsis in trauma patients. Ann Surg. 1999;230:207–214. doi: 10.1097/00000658-199908000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boermeester MA, van Leeuwen PA, Coyle SM, Wolbink GJ, Hack CE, Lowry SF. Interleukin-1 blockade attenuates mediator release and dysregulation of the hemostatic mechanism during human sepsis. Arch Surg. 1995;130:739–748. doi: 10.1001/archsurg.1995.01430070061012. [DOI] [PubMed] [Google Scholar]
- Pociot F, Molvig J, Wogensen L, Wosaae H, Nerup J. A TaqI polymorphism in the human interleukin-1 beta (IL-1 beta) gene correlates with IL-1 beta secretion in vitro. Eur J Clin Invest. 1992;22:396–402. doi: 10.1111/j.1365-2362.1992.tb01480.x. [DOI] [PubMed] [Google Scholar]
- Fang XM, Schroder S, Hoeft A, Stuber F. Comparison of two polymorphisms of the interleukin-1 gene family: interleukin-1 receptor antagonist polymorphism contributes to susceptibility to severe sepsis. Crit Care Med. 1999;27:1330–1334. doi: 10.1097/00003246-199907000-00024. [DOI] [PubMed] [Google Scholar]
- Kornman KS, Pankow J, Offenbacher S, Beck J, di Giovine F, Duff GW. Interleukin-1 genotypes and the association between periodontitis and cardiovascular disease. J Periodontal Res. 1999;34:353–357. doi: 10.1111/j.1600-0765.1999.tb02265.x. [DOI] [PubMed] [Google Scholar]
- Terry CF, Loukaci V, Green FR. Cooperative influence of genetic polymorphisms on interleukin 6 transcriptional regulation. J Biol Chem. 2000;275:18138–18144. doi: 10.1074/jbc.M000379200. [DOI] [PubMed] [Google Scholar]
- Schluter B, Raufhake C, Erren M, Schotte H, Kipp F, Rust S, Van Aken H, Assmann G, Berendes E. Effect of the interleukin-6 promoter polymorphism (-174 G/C) on the incidence and outcome of sepsis. Crit Care Med. 2002;30:32–37. doi: 10.1097/00003246-200201000-00005. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Irikura VM, Paul SM, Hirsh D. Functions of interleukin 1 receptor antagonist in gene knockout and overproducing mice. Proc Natl Acad Sci USA. 1996;93:11008–11013. doi: 10.1073/pnas.93.20.11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarlow JK, Blakemore AI, Lennard A, Solari R, Hughes HN, Steinkasserer A, Duff GW. Polymorphism in human IL-1 receptor antagonist gene intron 2 is caused by variable numbers of an 86-bp tandem repeat. Hum Genet. 1993;91:403–404. doi: 10.1007/BF00217368. [DOI] [PubMed] [Google Scholar]
- Danis VA, Millington M, Hyland VJ, Grennan D. Cytokine production by normal human monocytes: inter-subject variation and relationship to an IL-1 receptor antagonist (IL-1Ra) gene polymorphism. Clin Exp Immunol. 1995;99:303–310. doi: 10.1111/j.1365-2249.1995.tb05549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurme M, Santtila S. IL-1 receptor antagonist (IL-1Ra) plasma levels are co-ordinately regulated by both IL-1Ra and IL-1beta genes. Eur J Immunol. 1998;28:2598–2602. doi: 10.1002/(SICI)1521-4141(199808)28:08<2598::AID-IMMU2598>3.3.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Ma P, Chen D, Pan J, Du B. Genomic polymorphism within interleukin-1 family cytokines influences the outcome of septic patients. Crit Care Med. 2002;30:1046–1050. doi: 10.1097/00003246-200205000-00015. [DOI] [PubMed] [Google Scholar]
- Arnalich F, Lopez-Maderuelo D, Codoceo R, Lopez J, Solis-Garrido LM, Capiscol C, Fernandez-Capitan C, Madero R, Montiel C. Interleukin-1 receptor antagonist gene polymorphism and mortality in patients with severe sepsis. Clin Exp Immunol. 2002;127:331–336. doi: 10.1046/j.1365-2249.2002.01743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DM, Williams DM, Sankaran D, Lazarus M, Sinnott PJ, Hutchinson IV. An investigation of polymorphism in the interleukin-10 gene promoter. Eur J Immunogenet. 1997;24:1–8. doi: 10.1111/j.1365-2370.1997.tb00001.x. [DOI] [PubMed] [Google Scholar]
- Eskdale J, Gallagher G. A polymorphic dinucleotide repeat in the human IL-10 promoter. Immunogenetics. 1995;42:444–445. doi: 10.1007/BF00179416. [DOI] [PubMed] [Google Scholar]
- Eskdale J, Kube D, Gallagher G. A second polymorphic dinucleotide repeat in the 5' flanking region of the human IL10 gene. Immunogenetics. 1996;45:82–83. doi: 10.1007/s002510050174. [DOI] [PubMed] [Google Scholar]
- Ahmed S, Ihara K, Bassuny WM, Kuromaru R, Kohno H, Miyako K, Matsuura N, Iwata I, Nagafuchi S, Hara T. Association study between CD30 and CD30 ligand genes and type 1 diabetes in the Japanese population. Genes Immun. 2002;3:96–101. doi: 10.1038/sj.gene.6363837. [DOI] [PubMed] [Google Scholar]
- Vidigal PG, Germer JJ, Zein NN. Polymorphisms in the interleukin-10, tumor necrosis factor-alpha, and transforming growth factor-beta1 genes in chronic hepatitis C patients treated with interferon and ribavirin. J Hepatol. 2002;36:271–277. doi: 10.1016/S0168-8278(01)00243-4. [DOI] [PubMed] [Google Scholar]
- Armstrong L. Relative production of tumour necrosis factor a and interleukin 10 in adult respiratory distress syndrome. Thorax. 1997;52:442–446. doi: 10.1136/thx.52.5.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller IJ, Cohen AB, Nagao S, Griffith D, Maunder RJ, Martin TR, Weiner-Kronish JP, Sticherling M, Christophers E, Matthay MA. Elevated levels of NAP-1/Interleukin-8 are present in the airspaces of patients with the adult respiratory distress syndrome and are associated with increased mortality. Am Rev Respir Dis. 1992;146:427–432. doi: 10.1164/ajrccm/146.2.427. [DOI] [PubMed] [Google Scholar]
- Marshall RP, Webb S, Bellingan GJ, Montgomery HE, Chaudhari B, McAnulty RJ, Humphries SE, Hill MR, Laurent GJ. Angiotensin converting enzyme insertion/deletion polymorphism is associated with susceptibility and outcome in acute respiratory distress syndrome. Am J Respir Crit Care. 2002;166:646–650. doi: 10.1164/rccm.2108086. [DOI] [PubMed] [Google Scholar]
- Lin Z, Pearson C, Chinchilli V, Pietschmann SM, Luo J, Pison U, Floros J. Polymorphisms of human SP-A, SP-B, and SP-D genes: association of SP-B Thr131Ile with ARDS. Clin Genet. 2000;58:181–191. doi: 10.1034/j.1399-0004.2000.580305.x. [DOI] [PubMed] [Google Scholar]