

Abstract
Absolute pitch (AP) is the rare ability to instantaneously recognize and label tones with their musical note names without using a reference pitch for comparison. The etiology of AP is complex. Prior studies have implicated both genetic and environmental factors in its genesis, yet the molecular basis for AP remains unknown. To locate regions of the human genome that may harbor AP-predisposing genetic variants, we performed a genome-wide linkage study on 73 multiplex AP families by genotyping them with 6090 SNP markers. Nonparametric multipoint linkage analyses were conducted, and the strongest evidence for linkage was observed on chromosome 8q24.21 in the subset of 45 families with European ancestry (exponential LOD score = 3.464, empirical genome-wide p = 0.03). Other regions with suggestive LOD scores included chromosomes 7q22.3, 8q21.11, and 9p21.3. Of these four regions, only the 7q22.3 linkage peak was also evident when 19 families with East Asian ancestry were analyzed separately. Though only one of these regions has yet reached statistical significance individually, we detected a larger number of independent linkage peaks than expected by chance overall, indicating that AP is genetically heterogeneous.
Main Text
Absolute pitch (AP [MIM 159300]), commonly referred to as perfect pitch, is the rare ability to identify tones with their corresponding musical note names without the aid of a reference tone. Identification is instantaneous and quite effortless for those who possess AP, much as the assignment of visible light frequencies with color names is for most people. It is distinguishable from relative pitch, a learned ability common in trained musicians, in which the pitch of a tone is inferred by mental comparison to an external reference tone.
AP is a complex trait, and both environmental and genetic factors may play a role in its genesis. Musical training during a critical period of childhood development1–4 probably contributes to the acquisition of AP, but this training alone is insufficient; many people receive early musical training but do not develop AP. Other environmental factors have been suggested to influence whether an individual develops AP, including the type of musical training that the individual received5 and the individual's tone language fluency.6 We and others hypothesize that the genetic makeup of the individual also contributes to the development of this ability.4,7–9 Familial aggregation studies have estimated the sibling recurrence-risk ratio (λs) for absolute pitch to be between 7.8 and 15.1 after controlling for early musical training.9,10 Twin observations, although limited, give further support to this hypothesis. Three pairs of monozygotic twins concordant for AP and one pair of dizygotic twins discordant for AP have been reported in the literature;11 in our study, seven of ten monozygotic twin pairs were confirmed as concordant for AP, whereas only nine of 20 dizygotic twin pairs were confirmed or reported to be concordant (E.T., unpublished data). Together, these data suggest that it is likely that a combination of environmental and genetic factors promote the genesis of AP.
Interestingly, one study showed that infants preferentially use AP cues over relative pitch cues in certain situations, suggesting that all people might be born with AP but that the majority lose their AP abilities with age.12 Thus, an attractive hypothesis is that genetic factors might extend this neurodevelopmental window to a duration sufficient to intersect with the onset of musical training.
In this study, as a first step toward identifying these genetic factors, we conducted whole-genome, nonparametric linkage analyses on multiplex AP families and successfully identified a region of significant linkage. Moreover, we found evidence for genetic heterogeneity both within and between populations of different ancestry.
To facilitate the recruitment of individuals with AP, we employed an online pitch-naming test and survey, as described previously.4,13 Subjects were advised that by filling in the survey they were providing implicit consent to participate in this aspect of the study. Participants who exceeded our threshold for AP on our pitch-naming test and who reported at least one relative with AP were asked to invite their relative(s) to also enter the study via the website. Study participants from families in which AP ability was documented in at least two family members who were not simply a parent-child relative pair and who resided in the United States or Canada were invited to contribute DNA samples to our linkage study. Participating family members were also encouraged to invite other family members who may be informative for our genetic analysis to contact us and provide a DNA sample, even if they did not possess AP. Participants who chose to donate mouthwash or saliva samples were given kits for self-collection of these samples. Blood samples were collected by a mobile phlebotomy service (ExamOne), and many of these were immortalized by Epstein Barr Virus (EBV) transformation.14 DNA was extracted from mouthwash samples, whole blood, and lymphoblastoid cell lines with Gentra Puregene DNA purification kits (QIAGEN). Saliva samples were collected in Oragene DNA self-collection kits and purified according to the manufacturer's instructions (DNA Genotek). Written informed consent was obtained from all participants who contributed DNA samples to our study. This study was approved by the Committee on Human Research at the University of California, San Francisco (UCSF).
Overall, DNA samples from 73 families with at least one non-parent-child AP relative pair were collected for linkage analysis (Figure S1, available online). Nineteen families reported predominantly East Asian ancestry (E Asian), eight families reported being Ashkenazi Jewish (AJ), one family was Indian (I), and the remaining 45 families were predominantly of mixed European ancestry (Eu) (Table S1). The distribution of AP relative pairs in these families is summarized in Table 1.
Table 1.
Description of Families Used in Linkage Analysis
| Eu/AJ/I | Eua | E Asian | |
|---|---|---|---|
| No. of families | 54 | 45 | 19 |
| No. of individuals genotyped | 220 | 184 | 61 |
| No. of AP individuals genotyped | 128 | 108 | 40 |
| No. of AP sibling pairs | 73 | 65 | 16 |
| No. of AP avuncular pairs | 8 | 3 | 1 |
| No. of AP cousin and distant pairs | 5 | 5 | 4 |
| No. of AP relative pairsb | 86 | 73 | 21 |
The European descent (Eu) sample set is a subset of the Eu/AJ/I sample set, excluding one Indian and eight Ashkenazi Jewish families.
AP parent-child pairs were not included in the relative-pairs count.
DNA samples from 281 individuals (indicated by the + signs in Figure S1) were genotyped with 6090 SNPs on the Infinium HumanLinkage-12 BeadChip (Illumina) in the UCSF Genomics Core Facility. These SNPs are located at an average spacing of 0.58 cM (441 kb) throughout the human genome, and their genetic map positions have been estimated on the deCODE genetic map.15 The Genotyping Module of Illumina's BeadStudio software was used to manually inspect SNP genotype calls on intensity plots. Once obvious errors were resolved, the genotype data were analyzed with Pedcheck to locate Mendelian inconsistencies.16 These errors were corrected by the adjustment of genotype calls or by elimination of genotypes from the data set after reinspection of the intensity plots. Merlin was used for the detection and removal of unlikely genotype combinations that appeared to have arisen from excessive numbers of recombinations.17
Multipoint nonparametric linkage analyses were performed on the genotype data with the use of Merlin,17 which estimates identical-by-descent allele sharing among affected relatives. To anticipate potential locus heterogeneity within and between populations of different ancestry and potential allele frequency differences, we performed separate linkage analyses on the combined group of European, Ashkenazi Jewish, and Indian ancestry families (Eu/AJ/I) and the East Asian ancestry (E Asian) families, as well as the European ancestry (Eu) families alone. Because parental genotype data were lacking in some of our pedigrees, we used Merlin to form clusters18 of correlated markers that exhibited pairwise r2 values greater than 0.16, to ensure that marker-marker linkage disequilibrium was not inflating our multipoint linkage scores.19 HapMap marker allele frequencies were used for these analyses, though similar results were obtained when allele frequencies were estimated from the founders in our families. Multipoint Kong and Cox exponential nonparametric LOD scores20 obtained with Whittemore and Halpern's SALL statistic21 were then calculated for each marker or marker cluster.
We empirically estimated p values for our LOD scores by conducting 10,000 gene-dropping simulations under the null hypothesis of no linkage in Merlin17 and by retaining the LOD scores from the highest independent (separated by ≥ 40 cM) linkage peaks on the autosomes in each replicate. On average, there were about 78 independent linkage peaks per genome scan. These simulations used the same marker spacing, clustering, family structures, and informativeness of our study, and we conducted separate sets of simulations on the three subpopulations. The 500th highest LOD score from these simulations was taken to be the empirical threshold for statistical significance (expected to occur in one of every 20 genome scans by chance), and the 10,000th highest LOD score was the empirical threshold for suggestive linkage (expected to occur once in every genome scan by chance).
By conducting linkage analysis on the combined set of Eu/AJ/I families, we found that peak LOD scores for two regions of the genome exceeded our empirical threshold for suggestive linkage (LOD = 1.874): chromosome 8q24.21 at rs3057, with a LOD score of 2.330, and chromosome 8q21.11 at rs1007750, with a LOD score of 2.069 (Figure 1A and Table 2). These regions are shown in more detail in Figure 2, and the contributions of individual families to peak LOD scores is detailed in Table S1. In addition, regions on chromosomes 2, 6, 7, 8, 9, 11, and 14 achieved nominal LOD scores greater than 1.0 but did not meet the criteria for suggestive linkage (Table 3).
Figure 1.
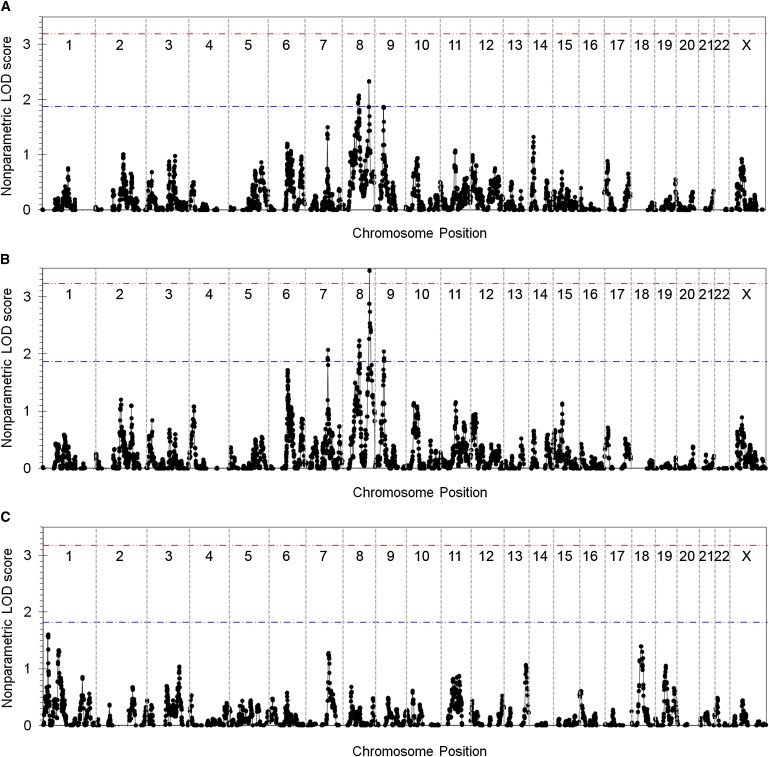
Results of Whole-Genome Linkage Analysis
Nonparametric multipoint exponential LOD scores were calculated for every marker or marker cluster position across the genome with the use Merlin for (A) the 54 families of European, Ashkenazi Jewish, and Indian (Eu/AJ/I) descent, (B) a subset of 45 families of mixed European (Eu) descent, and (C) 19 families of East Asian (E Asian) descent. Only nonnegative LOD scores are shown. Red and blue lines indicate empirical thresholds for significant and suggestive linkage, respectively, with 10,000 gene dropping simulations used.
Table 2.
Significant and Suggestive Chromosome Regions from Multipoint Nonparametric Linkage Analysis
| Sample Set | Region | Marker | deCODE cM | LODa | Empirical p Valueb | Interval Size (Mb)c | Flanking Markers |
|---|---|---|---|---|---|---|---|
| Eu/AJ/I | 8q21.11 | rs1007750 | 86.732 | 2.069 | 0.6490 | 22.86 | rs997493-rs10105219 |
| Eu/AJ/I | 8q24.21 | rs3057 | 139.741 | 2.330 | 0.3611 | 6.01 | rs1562435-rs2102861 |
| Eu | 7q22.3 | rs2028030 | 117.774 | 2.074 | 0.6402 | 4.04 | rs887882-rs1013920 |
| Eu | 8q21.11 | rs1007750 | 86.732 | 2.236 | 0.4500 | 11.75 | rs695167-rs716349 |
| Eu | 8q24.21 | rs3057 | 139.741 | 3.464 | 0.0300d | 5.54 | rs755520-rs2102861 |
| Eu | 9p21.3 | rs2169325 | 46.478 | 2.048 | 0.6786 | 7.91 | rs748530-rs9103 |
The top multipoint nonparametric exponential LOD scores from linkage analysis of the European, Ashkenazi Jewish, and Indian ancestry sample set (Eu/AJ/I) and the subset of families of European ancestry (Eu).
Empirical genome-wide p values were estimated for each sample set independently by calculating the average numbers of independent linkage peaks expected under the null hypothesis of no linkage per genome scan, with 10,000 autosomal simulations run.
Intervals are LOD − 1.
Bold italics denote significant results (p < 0.05).
Figure 2.
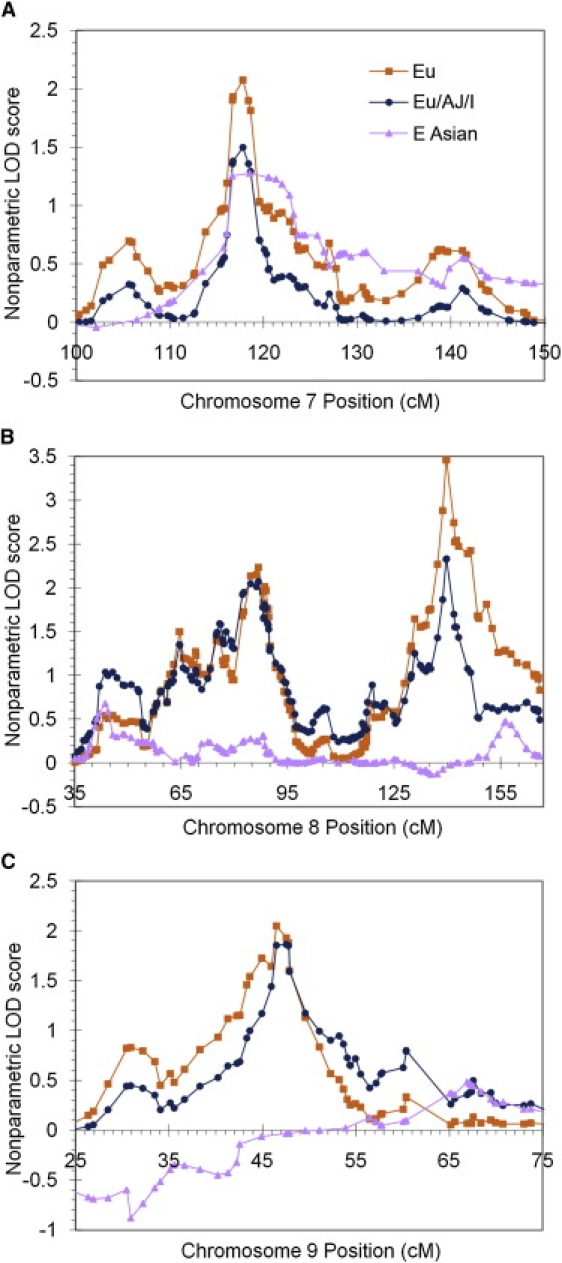
Multipoint Nonparametric Exponential LOD Scores at Each Marker or Marker Cluster Position
LOD scores were calculated with the use of data from 54 families of European, Ashkenazi Jewish, and Indian descent (Eu/AJ/I), the subset of 45 families of European descent (Eu), and 19 East Asian families (E Asian). Genetic distance is measured in deCODE cM. Results are shown for (A) chromosome 7, (B) chromosome 8, and (C) chromosome 9.
Table 3.
Evaluation of Nonparametric Peak LOD Scores > 1.0 by Locus Counting
| Sample Seta | Chr | deCODE cM | Observed LOD (Z) | Rank (r) | No. of LODs ≥ Z Generated Per Simulated Scanb | Proportion of Simulations with r LODs ≥ Zc |
|---|---|---|---|---|---|---|
| Eu/AJ/I | 8 | 139.741 | 2.330 | 1 | 0.3611 | 0.3034 |
| Eu/AJ/I | 8 | 86.732 | 2.069 | 2 | 0.6490 | 0.1372 |
| Eu/AJ/I | 9 | 47.565 | 1.864 | 3 | 1.0240 | 0.0878 |
| Eu/AJ/I | 7 | 117.774 | 1.499 | 4 | 2.2296 | 0.1852 |
| Eu/AJ/I | 14 | 27.385 | 1.322 | 5 | 3.2538 | 0.2299 |
| Eu/AJ/I | 6 | 99.679 | 1.201 | 6 | 4.2168 | 0.2462 |
| Eu/AJ/I | 11 | 78.450 | 1.080 | 7 | 5.4576 | 0.3032 |
| Eu/AJ/I | 8 | 43.577 | 1.038 | 8 | 5.9693 | 0.2498 |
| Eu/AJ/I | 2 | 145.803 | 1.007 | 9 | 6.3761 | 0.1898 |
| Eu | 8 | 139.741 | 3.464 | 1 | 0.0300d | 0.0293d |
| Eu | 8 | 86.732 | 2.236 | 2 | 0.4500 | 0.0750 |
| Eu | 7 | 117.774 | 2.074 | 3 | 0.6402 | 0.0262d |
| Eu | 9 | 46.478 | 2.048 | 4 | 0.6786 | 0.0042d |
| Eu | 6 | 99.679 | 1.723 | 5 | 1.3904 | 0.0128d |
| Eu | 2 | 130.661 | 1.205 | 6 | 4.2078 | 0.2494 |
| Eu | 11 | 78.450 | 1.160 | 7 | 4.6294 | 0.1839 |
| Eu | 10 | 41.725 | 1.147 | 8 | 4.7538 | 0.1072 |
| Eu | 15 | 47.975 | 1.133 | 9 | 4.8958 | 0.0590 |
| Eu | 2 | 185.422 | 1.100 | 10 | 5.2524 | 0.0378d |
| Eu | 4 | 25.557 | 1.082 | 11 | 5.4560 | 0.0209d |
| E Asian | 1 | 25.394 | 1.606 | 1 | 1.6028 | 0.7996 |
| E Asian | 18 | 49.984 | 1.399 | 2 | 2.5007 | 0.7123 |
| E Asian | 1 | 81.597 | 1.326 | 3 | 2.9287 | 0.5621 |
| E Asian | 7 | 118.517 | 1.277 | 4 | 3.2562 | 0.4078 |
| E Asian | 13 | 114.683 | 1.064 | 5 | 5.1098 | 0.5854 |
| E Asian | 19 | 55.795 | 1.051 | 6 | 5.2597 | 0.4296 |
| E Asian | 3 | 168.852 | 1.040 | 7 | 5.3825 | 0.2912 |
Both b and c were calculated for all observed independent linkage peaks with LOD scores exceeding 1.0.
Simulations were conducted independently for the European, Ashkenazi Jewish, and Indian sample set (Eu/AJ/I), for the European ancestry subset (Eu), and for the East Asian (E Asian) sample set.
Average numbers of independent linkage peaks per genome scan observed under the null hypothesis of no linkage in 10,000 autosomal simulations.
The proportion of 10,000 autosomal simulations that had at least r linkage regions with LOD scores greater than or equal to the observed LOD score.
Bold italics denote significant results (p < 0.05).
Examining data for the Eu families alone, we detected one region, with a maximum LOD score at rs3057 on chromosome 8q24.21 (Figure 1B), that showed strong evidence for linkage, having a nonparametric multipoint exponential LOD score of 3.464 (empirical genome-wide p = .0300). This value exceeded the empirical threshold for significant linkage (LOD = 3.231) obtained from 10,000 autosomal gene-dropping simulations. We then used the method of Camp and Farnham22 to correct for multiple testing. A linear regression of the Eu/AJ/I nonparametric LOD scores versus the corresponding Eu nonparametric LOD scores had an r2 value of 0.7874, indicating that these two analyses represented 1.213 independent tests. After this correction, the 8q24.21 linkage peak remained significant, with a p value of 0.0364. Three additional regions, on chromosomes 8q21.11 (LOD = 2.236 at rs1007750), 7q22.3 (LOD = 2.074 at rs2028030), and 9p21.3 (LOD = 2.048 at rs2169325), exceeded the empirical threshold for suggestive linkage (LOD = 1.869). Figure 2 shows these regions in more detail. In addition to these significant and suggestive hits (Table 2), several other regions of the genome on chromosomes 2, 4, 6, 10, 11, and 15 had peak LOD scores greater than 1.0 (Table 3). We did not conduct a similar analysis on the eight Ashkenazi Jewish families, because we felt that the sample size was too small to allow linkage detection with any certainty, but it appears that the Ashkenazi Jewish families do not show linkage to the top Eu linkage regions (Table S1). Overall, this linkage analysis indicates that there is a genetic basis for AP in the Eu population.
Using the E Asian families, we observed that no linkage peak exceeded the empirical threshold for suggestive linkage (LOD = 1.822), but regions on chromosomes 1, 3, 7, 13, 18 and 19 had linkage peaks with LOD scores greater than 1.0 (Figure 1C and Table 3). Notably, there was no evidence in the E Asian population for linkage in the region of significant linkage (8q24.21) from the Eu sample set. In fact, the chromosome 7 region was the only E Asian region with a LOD score over 1.0 that showed overlap with linkage peaks observed in the Eu data set (Figure 2A).
Because AP is a complex trait and many loci could potentially be involved in its genesis, we also used locus-counting methods23,24 to evaluate the significance of our linkage results. Again, as with our linkage analyses, we considered the two main sample sets (Eu/AJ/I and E Asian) and the Eu subset separately. First, the top observed linkage regions with LOD scores > 1.0 were arranged in order by rank (r). For each of these observed LOD scores (Z), the number of independent linkage regions (separated by a genetic distance of at least 40 cM) that had LOD scores at least as large as Z in 10,000 autosomal gene-dropping simulations were tallied and divided by 10,000 to determine the average number of times that a LOD score of Z's magnitude was seen in a simulation scan. The final step was to determine the proportion of simulations that had at least as many independent linkage peaks at or above Z as we observed in our linkage analysis (r). If 5% or more of the 10,000 simulations did not contain at least as many independent linkage peaks as our linkage scan did at a LOD score threshold of Z, the excess of linkage peaks at that threshold was considered significant.
Table 3 summarizes the results of this locus-counting analysis for each sample set. In the Eu subset, we observed four independent linkage peaks at or above a LOD score of 2.048; however, on the basis of 10,000 simulations, only 0.68 independent linkage peaks would be expected under the null hypothesis of no linkage at that threshold. The difference between the observed number of linkage peaks and the number expected under the null hypothesis of no linkage based on the simulations was significant (p = 0.0042). Similarly, a significant (p < 0.05) excess of linkage peaks was observed for the 1st-, 3rd-, 5th-, 10th-, and 11th-ranked independent linkage regions at LOD score thresholds of 3.464, 2.074, 1.723, 1.1, and 1.082, respectively (Table 3). These results indicate that the genetic basis for AP exhibits locus heterogeneity, at least in the Eu population. Though an excess of linkage peaks was also observed when the Eu/AJ/I sample set was used, this difference was not significant. No obvious excess of linkage peaks was found in the E Asian linkage scan with this analysis.
Together, the findings discussed above provide strong evidence that at least one gene promotes the genesis of AP in individuals of European ancestry and that AP probably results from genetic factors that vary both within and between different populations, conclusions that are supported by evidence for linkage in more regions than expected by chance.
The top linkage peak on chromosome 8q24.21 in the Eu subset had a maximum multipoint nonparametric LOD score of 3.464 at the SNP rs3057, which lies at approximately 140 cM on the chromosome 8 deCODE genetic map. In the UCSC genome browser, it appears that four genes lie closest to this peak: GSDMC (gasdermin C [MIM 608384]), FAM49B (a hypothetical protein-coding gene), ASAP1 (ArfGAP with SH3 domain, ankyrin repeat and PH domain 1 [MIM 605953]), and ADCY8 (adenylate cyclase 8 (brain) [MIM 103070]). ASAP1 is expressed in a variety of tissues, including the brain,25 and ADCY8 is expressed almost exclusively in the brain26 and is thought to play a role in learning and memory.27,28 Given that the linkage peak is observed in a single, although broad, population, linkage disequilibrium analysis may help to narrow the interval in the search for genetic variants that lead to AP.
The genetic basis for musical abilities is a subject of growing curiosity. A recent linkage study on Finnish families with musical aptitude, as determined by several different tests, reported significant evidence for linkage at 4q22 and suggestive evidence for linkage at 8q13-21.29 Though we saw no evidence for linkage of AP to 4q22, we did observe suggestive evidence for linkage to 8q21.11 at 86.7 cM, close to the peak (chromosome 8 at 92 cM) reported in the musical-aptitude study. AP and musical aptitude may share linkage to this genomic region, though it was not the top linkage peak in either study. Another recent study suggested that polymorphisms in the arginine vasopressin receptor 1A gene may be associated with musical memory,30 but none of our top linkage peaks fall near the AVPR1A gene on chromosome 12.
This study bears extension by further recruitment within our own laboratory and replication by other groups interested in this question. Our LOD scores were modest in comparison to the theoretical maximum nonparametric multipoint exponential LOD scores predicted for our samples (maximum exponential LOD scores were 40.03, 34.31, and 12.34 for the Eu/AJ/I, Eu, and E Asian analyses, respectively), and our study was probably underpowered, especially in the case of the E Asian and AJ families. Theoretically, a study of 100 affected sibling pairs could have greater than 90% power to detect linkage, assuming that the λs = 10, the recombination fraction between the marker and trait locus (θ) < .05, and the markers are fully informative, and a similar study of only 40 affected sibling pairs would have 20%–70% power, depending on θ.31 Though the SNPs in our study were closely spaced (θ < 0.27 on average), they were not completely informative (average polymorphism information content [PIC] = 0.35).32 Moreover, we were unable to acquire DNA from informative relatives, such as parents, in some of the families, so the probabilities that AP relatives share alleles identically by descent were difficult to determine with certainty for these families, thus reducing power further. Despite these considerations, our study was able to detect significant linkage at one locus in the Eu subset, though it was probably underpowered to detect loci that make smaller contributions to predispose individuals to develop AP.
AP is a complex trait, and the discovery of genes responsible for AP will provide the first step in unraveling the interplay between genetic predisposition and environmental influences. For some of our families, incomplete penetrance can be partially explained by a lack of early musical training in some family members. Overall, however, gene-environment interactions may not be the sole complexity. This degree of complexity prompted us to use nonparametric linkage analysis to search for susceptibility loci for AP, relying on the calculation of likelihoods that alleles are shared identically by descent among AP relatives. Though parametric linkage analysis is theoretically a more powerful approach, it would have required us to estimate the prevalence, penetrance, and mode of inheritance of AP, which are uncertain. Our study shows that not one but a number of different loci may foster AP, despite the fact that it is a dichotomous trait.13 Although one genetic variant in any of a variety of genes could predispose individuals to developing AP by itself, it is also possible that gene-gene interactions might be required for the development of AP. The existence of more than one gene involved in AP could also be beneficial in elucidating the molecular pathways that give rise to it.
In summary, we found several regions of the human genome that show evidence of linkage to AP in this study, including one region of significant linkage on chromosome 8q24.21. These findings suggest that multiple regions of the genome harbor AP-predisposing variants that could be discovered upon further research. These results provide strong support for the role of genetics in the etiology of AP and will provide the basis for future studies dissecting the interplay between nature and nurture in the development of AP.
Acknowledgments
We thank the many research participants and their families whose desire to understand the genesis of their talent prompted them to enter our study. We gratefully acknowledge Siamak Baharloo, Barbara Levinson, E. Alexandra Athos, Amy Kistler, and Jason Zemansky for their work in recruiting participants for this project and Hernan Consengco for cell line immortalization. We thank Jon Woo, Elaine Carlson, and the UCSF Genomics Core Facility for their assistance with genotyping and their careful attention to this project. We thank Nelson Freimer and Victoria Carlton, who helped to formulate our thinking during the design of this project, as well as Neil Risch, Robert Nussbaum, and Steve Hamilton for many stimulating discussions. We acknowledge Joshua Melcon for writing software programs to assist with computational aspects of the project, and we acknowledge Aaron Calhoun for website development. This material is based upon work supported under a National Science Foundation (NSF) Graduate Research Fellowship (E.T.). This work was also supported by a Guggenheim Fellowship, a REAC grant from UCSF, and an NSF grant to J.G. J.G. was previously an Investigator with HHMI, who funded part of this research.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Merlin, http://www.sph.umich.edu/csg/abecasis/Merlin/index.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Pedcheck, http://watson.hgen.pitt.edu/register/docs/pedcheck.html
UCSC Genome Browser, http://genome.ucsc.edu/
UCSF Absolute Pitch Study website, http://perfectpitch.ucsf.edu
References
- 1.Sergeant D. Experimental investigation of absolute pitch. J. Res. Music Educ. 1969;17:135–143. [Google Scholar]
- 2.Miyazaki K. Musical pitch identification by absolute pitch possessors. Percept. Psychophys. 1988;44:501–512. doi: 10.3758/bf03207484. [DOI] [PubMed] [Google Scholar]
- 3.Takeuchi A.H., Hulse S.H. Absolute pitch. Psychol. Bull. 1993;113:345–361. doi: 10.1037/0033-2909.113.2.345. [DOI] [PubMed] [Google Scholar]
- 4.Baharloo S., Johnston P.A., Service S.K., Gitschier J., Freimer N.B. Absolute pitch: an approach for identification of genetic and nongenetic components. Am. J. Hum. Genet. 1998;62:224–231. doi: 10.1086/301704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gregersen P.K., Kowalsky E., Kohn N., Marvin E.W. Early childhood music education and predisposition to absolute pitch: teasing apart genes and environment. Am. J. Med. Genet. 2001;98:280–282. doi: 10.1002/1096-8628(20010122)98:3<280::aid-ajmg1083>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 6.Deutsch D., Dooley K., Henthorn T., Head B. Absolute pitch among students in an American music conservatory: association with tone language fluency. J. Acoust. Soc. Am. 2009;125:2398–2403. doi: 10.1121/1.3081389. [DOI] [PubMed] [Google Scholar]
- 7.Bachem A. The genesis of absolute pitch. J. Acoust. Soc. Am. 1940;11:434–439. [Google Scholar]
- 8.Profita J., Bidder T.G. Perfect pitch. Am. J. Med. Genet. 1988;29:763–771. doi: 10.1002/ajmg.1320290405. [DOI] [PubMed] [Google Scholar]
- 9.Gregersen P.K., Kowalsky E., Kohn N., Marvin E.W. Absolute pitch: prevalence, ethnic variation, and estimation of the genetic component. Am. J. Hum. Genet. 1999;65:911–913. doi: 10.1086/302541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baharloo S., Service S.K., Risch N., Gitschier J., Freimer N.B. Familial aggregation of absolute pitch. Am. J. Hum. Genet. 2000;67:755–758. doi: 10.1086/303057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gregersen P.K. Instant recognition: The genetics of pitch perception. Am. J. Hum. Genet. 1998;62:221–223. doi: 10.1086/301734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saffran J.R., Griepentrog G.J. Absolute pitch in infant auditory learning: evidence for developmental reorganization. Dev. Psychol. 2001;37:74–85. [PubMed] [Google Scholar]
- 13.Athos E.A., Levinson B., Kistler A., Zemansky J., Bostrom A., Freimer N., Gitschier J. Dichotomy and perceptual distortions in absolute pitch ability. Proc. Natl. Acad. Sci. USA. 2007;104:14795–14800. doi: 10.1073/pnas.0703868104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neitzel H. A routine method for the establishment of permanent growing lymphoblastoid cell lines. Hum. Genet. 1986;73:320–326. doi: 10.1007/BF00279094. [DOI] [PubMed] [Google Scholar]
- 15.Kong A., Gudbjartsson D.F., Sainz J., Jonsdottir G.M., Gudjonsson S.A., Richardsson B., Sigurdardottir S., Barnard J., Hallbeck B., Masson G. A high-resolution recombination map of the human genome. Nat. Genet. 2002;31:241–247. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- 16.O'Connell J.R., Weeks D.E. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 18.Abecasis G.R., Wigginton J.E. Handling marker-marker linkage disequilibrium: pedigree analysis with clustered markers. Am. J. Hum. Genet. 2005;77:754–767. doi: 10.1086/497345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyles A.L., Scott W.K., Martin E.R., Schmidt S., Li Y.J., Ashley-Koch A., Bass M.P., Schmidt M., Pericak-Vance M.A., Speer M.C. Linkage disequilibrium inflates type I error rates in multipoint linkage analysis when parental genotypes are missing. Hum. Hered. 2005;59:220–227. doi: 10.1159/000087122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kong A., Cox N.J. Allele-sharing models: LOD scores and accurate linkage tests. Am. J. Hum. Genet. 1997;61:1179–1188. doi: 10.1086/301592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whittemore A.S., Halpern J. A class of tests for linkage using affected pedigree members. Biometrics. 1994;50:118–127. [PubMed] [Google Scholar]
- 22.Camp N.J., Farnham J.M. Correcting for multiple analyses in genomewide linkage studies. Ann. Hum. Genet. 2001;65:577–582. doi: 10.1017/S0003480001008922. [DOI] [PubMed] [Google Scholar]
- 23.Wiltshire S., Cardon L.R., McCarthy M.I. Evaluating the results of genomewide linkage scans of complex traits by locus counting. Am. J. Hum. Genet. 2002;71:1175–1182. doi: 10.1086/342976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abecasis G.R., Burt R.A., Hall D., Bochum S., Doheny K.F., Lundy S.L., Torrington M., Roos J.L., Gogos J.A., Karayiorgou M. Genomewide scan in families with schizophrenia from the founder population of Afrikaners reveals evidence for linkage and uniparental disomy on chromosome 1. Am. J. Hum. Genet. 2004;74:403–417. doi: 10.1086/381713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown M.T., Andrade J., Radhakrishna H., Donaldson J.G., Cooper J.A., Randazzo P.A. ASAP1, a phospholipid-dependent arf GTPase-activating protein that associates with and is phosphorylated by Src. Mol. Cell. Biol. 1998;18:7038–7051. doi: 10.1128/mcb.18.12.7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ludwig M.G., Seuwen K. Characterization of the human adenylyl cyclase gene family: cDNA, gene structure, and tissue distribution of the nine isoforms. J. Recept. Signal Transduct. Res. 2002;22:79–110. doi: 10.1081/rrs-120014589. [DOI] [PubMed] [Google Scholar]
- 27.Wong S.T., Athos J., Figueroa X.A., Pineda V.V., Schaefer M.L., Chavkin C.C., Muglia L.J., Storm D.R. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron. 1999;23:787–798. doi: 10.1016/s0896-6273(01)80036-2. [DOI] [PubMed] [Google Scholar]
- 28.de Quervain D.J., Papassotiropoulos A. Identification of a genetic cluster influencing memory performance and hippocampal activity in humans. Proc. Natl. Acad. Sci. USA. 2006;103:4270–4274. doi: 10.1073/pnas.0510212103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pulli K., Karma K., Norio R., Sistonen P., Goring H.H., Jarvela I. Genome-wide linkage scan for loci of musical aptitude in Finnish families: evidence for a major locus at 4q22. J. Med. Genet. 2008;45:451–456. doi: 10.1136/jmg.2007.056366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Granot R.Y., Frankel Y., Gritsenko V., Lerer E., Gritsenko I., Bachner-Melman R., Israel S., Ebstein R.P. Provisional evidence that the arginine vasopressin 1a receptor gene is associated with musical memory. Evol. Hum. Behav. 2007;28:313–318. [Google Scholar]
- 31.Risch N. Linkage strategies for genetically complex traits. II. The power of affected relative pairs. Am. J. Hum. Genet. 1990;46:229–241. [PMC free article] [PubMed] [Google Scholar]
- 32.Risch N. Linkage strategies for genetically complex traits. III. The effect of marker polymorphism on analysis of affected relative pairs. Am. J. Hum. Genet. 1990;46:242–253. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.