

Abstract
We present a method for testing overrepresentation of biological pathways, indexed by gene-ontology terms, in lists of significant SNPs from genome-wide association studies. This method corrects for linkage disequilibrium between SNPs, variable gene size, and multiple testing of nonindependent pathways. The method was applied to the Wellcome Trust Case-Control Consortium Crohn disease (CD) data set. At a general level, the biological basis of CD is relatively well known for a complex genetic trait, and it thus acted as a test of the method. The method, known as ALIGATOR (Association LIst Go AnnoTatOR), successfully detected biological pathways implicated in CD. The method was also applied to a meta-analysis of bipolar disorder, and it implicated the modulation of transcription and cellular activity, including that which occurs via hormonal action, as an important player in pathogenesis.
Introduction
Genome-wide association (GWA) analysis can be a powerful method for identifying genes involved in complex disorders, which often arise from the interplay of multiple genetic and environmental risk factors.1
The GWA approach has proven to be successful in identifying susceptibility genes for several complex disorders2–4 on the basis of identification and replication of associated SNPs. It seems intuitively likely that susceptibility alleles for any given disorder are not randomly distributed among genes but, instead, are distributed among one (or more) set(s) of genes whose functions are to some extent related. Under such a model, although a number of SNPs would be expected to show modest association when analyzed in isolation, one would expect to see an overall excess of SNPs with moderate p values for association on a list of SNPs representing a set of genes from relevant related biological pathways.
Several methods exist for prioritizing gene pathways for involvement in disease susceptibility, based on functional annotation,5 gene-expression data,6 sequence features,7 protein-protein interactions,8 or a combination of multiple types of data.9 Recently, pathway-based approaches have been developed for application to the results of genome-wide linkage10 and association11 studies.
This paper presents a novel method, called ALIGATOR (Association LIst Go AnnoTatOR), for studying groups of genes by testing for overrepresentation of members of those groups within lists of genes containing significantly associated SNPs from GWA studies. The aim is to identify whether certain groups of genes are potentially disease causing. To illustrate the application of the method, we defined groups on the basis of membership in Gene Ontology (GO) database categories, though the approach is applicable to any other gene-membership classification system. Compared with single-locus analysis, group or pathway analysis may yield more secure insights into disease biology, because an associated pathway is likely to implicate function better than a hit in a single gene that may have many functional possibilities. Additionally, genetic heterogeneity may cause any one causal variant to exhibit only modest disease risk in the sample as a whole, because different individuals may possess different disease-risk alleles at different loci in the same gene or in different genes. This will reduce the power to detect any one variant by traditional association methods. However, if the genes in question are members of the same biological pathway, then considering the pathway as the unit of analysis may increase the power to detect association between the genes and disease. For similar reasons, association of disease with biological pathways may be easier to replicate across different studies than association to individual SNPs or genes. This approach can be regarded as complementary to the studies that focus on the top hits.
In our method, we define a list of significant SNPs, applying an arbitrary threshold of significance to the GWA study, and test for overrepresentation of categories of genes, defined by GO terms (henceforth referred to as GO categories) on this list. Our analysis method corrects for the presence of linkage disequilibrium (LD) between SNPs, variable gene size, overlapping genes, and multiple nonindependent GO categories. It can be applied to data from any GWA platform.
We apply our approach to the Wellcome Trust Case-Control Consortium (WTCCC) Crohn disease (CD) GWA data set. At a general level, the biological basis of CD is relatively well known for a complex genetic trait, so it can be regarded as a proof-of-principle test of the method. In addition, we apply our method to a meta-analysis of bipolar disorder (BD) GWA study data sets (including the WTCCC data set), for which imputed data are available. This enables us to investigate the improvements provided by a larger sample size and the increased gene coverage given by the imputed data.
Material and Methods
GO Categories
The GO database12 assigns biological descriptors (GO terms) to genes on the basis of the properties of their encoded products. These terms fall into three types: cellular component, biological process, and molecular function. Genes assigned the same GO term can thus be regarded as members of a category (“GO category”) of genes that are more closely related in terms of some aspect of their biology than are random sets of genes. Rather than restricting analysis to categories at an arbitrarily defined level in the GO hierarchy, we chose to analyze all GO categories containing at least three genes.
SNP ID and Chromosomal Location
The “dbSNP chromosome report” file, based on human genome assembly build 36.2, was downloaded from the NCBI ftp site for chromosomes 1–22 and X. From this file, the following three data fields were extracted for reference sequence entries only: rs# (SNP rs number), chr (chromosome), and chr pos (chromosome position).
Assigning SNPs to Genes and Gene Regions
The “seq-gene” file was downloaded from the NCBI ftp website. First, for the exclusion of pseudogenes, records with a “feature_id” of “pseudo” were removed. All records with a “feature_type” of “gene,” “group_label” of “reference,” and “tax_id” of “9606” (i.e., human) were extracted. The following four fields were retained: chromosome, chr_start, chr_stop, and feature_id (NCBI gene ID).
The extracted SNP ID and chromosomal location file was compared to this file, first by chromosome and second by position. Output files were generated, containing SNP rs numbers and the gene region(s) in which those SNPs lie. We generated two such output files. The first comprised SNPs assigned to genes on the basis of being located within the genomic sequence corresponding to the start of the first and the end of the last exon. For the second, we added SNPs within 20 kb of the 5′ and 3′ ends of the first and last exons, respectively. The choice of 20 kb has been used by us elsewhere for candidate gene analysis13 to capture proximal regulatory and other functional regions that may lie outside, but close to, the gene. If a SNP was found to be located within more than one gene or gene region, all entries were included.
Assigning GO Terms to Genes and Gene Regions
Subsequently, the GO categories associated with these genes were obtained by linking the gene Locus ID to GO categories with the use of the gene2go file available as part of the NCBI Entrez Gene database. More information on the gene2go file can be obtained from the readme file (see Web Resources). The gene2go file was generated with data from the Gene Ontology Annotation (GOA) database.14
This file gives a list of genes (indexed by EntrezGene ID) and GO categories of which each gene is a member. For each GO category listed in gene2go, the complete set of categories of which that category is a subset was obtained by recursive examination of the ontology file from the AmiGO website.
This set of categories was added to those already present in gene2go, producing a complete list of GO category memberships for each gene.
Platform SNP Lists
We used NCBI SNP lists based upon build 36.2. The annotations of SNPs present on the genotyping platforms were updated as necessary. Newly assigned SNP rs ID numbers were obtained with the NCBI SNP batch entry list (see Web Resources).
Statistical Analysis Method
The presence of LD between SNPs complicates analysis, as does variable gene size and number of SNPs per gene. We chose to preselect a p value criterion to define a list of significantly associated SNPs. These SNPs define a list of significantly associated genes, each gene counted only once regardless of the number of significantly associated SNPs that it contains. The number of significantly associated genes in each GO category can be counted. Analysis was restricted to categories containing at least two significant genes, in order to prevent small categories appearing to be significantly overrepresented on the basis of one (possibly chance) hit. This approach is simple, and it can be applied regardless of the LD relationships between the SNPs. However, the number of significantly associated genes in each category will not follow a standard distribution (such as the hypergeometric distribution), because the probability that a gene appears on the list will depend on the number of (effectively independent) SNPs that it contains. The more (effectively independent) SNPs that a gene contains, the more likely that at least one SNP, and therefore that gene, will be considered “significant.” Therefore, the significance of the number of significantly associated genes in each category was assessed by simulations, as follows: SNPs were drawn successively at random from the set of all SNPs used in the study, and the genes that contained that particular SNP were added to the list of significant genes. The process was repeated until the list of significant genes was the same length as that in the original study. Five thousand replicate gene lists were generated in this way, enabling empirical p values to be calculated for the number of significantly associated genes in each GO category (i.e., the proportion of replicate gene lists containing at least as many genes from that GO category as the original list). This procedure implicitly assumes that the level of LD between SNPs is approximately equal across GO categories; violations of that assumption will lead to test statistics that are overconservative for categories in which the average LD between SNPs within member genes is higher than the genome-wide average. This is because in contrast to the real data set, under the simulation procedure, the probability of selecting a gene to the list by chance is proportional to the number of SNPs in that gene, not the effective number of independent SNPs15 that the gene contains. For genes with several SNPs in high LD, the effective number of independent SNPs is much smaller than the total number of SNPs, so the probability that they are selected to the simulated gene lists is inflated relative to the actual data set, thereby reducing the significance of observing them on the original list. Ideally, one would like to perform replicates of the GWA study by permuting disease status, rank the SNPs from each replicate study in order of significance, and use these to generate gene lists. However, as noted,11 this is computationally intensive. Furthermore, permutation requires access to the individual genotype data, which may not always be available.
Correction for Multiple Testing
In an experiment of this nature, several GO categories will generally be tested simultaneously. It is therefore desirable to correct the individual category-specific p values for the number of categories being tested. Because the categories are not independent, standard methods, such as the Bonferroni and Sidak corrections, are inappropriate, as is the use of false discovery rate (FDR) procedures. We corrected for multiple testing by using a bootstrap approach. One of the 5000 replicate gene lists was selected at random to be the “observed data.” A sample of 5000 gene lists for assessing significance was generated by random sampling with replacement (thus, lists could be counted once, more than once, or not at all) from the remaining gene lists. p values for the number of significantly associated genes in each GO category in the “observed data” were calculated as before. This procedure was repeated 1000 times. Each p value from the original data can thus be corrected for testing multiple categories, the corrected p value being the proportion of bootstrap replicates for which the minimum p value across all categories is less than or equal to the category-specific p value from the original data. The “expected number of hits” for each category can be calculated as the average number of categories per bootstrap replicate with p values less than or equal to the (uncorrected) p value from the original data. Finally, the number of categories with p values less than a given value (0.05, 0.01, 0.001) in the original data can be compared with the corresponding values from the bootstrap replicates. An excess of significantly overrepresented GO categories suggests a nonrandom distribution of associations and thus provides support for a biological basis for the disease. A flow diagram of the method used for assessing statistical significance is given in Figure 1.
Figure 1.
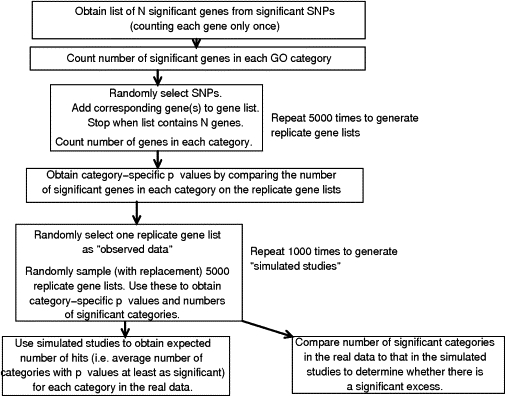
Flow Diagram Showing the Procedure for Estimating Statistical Significance
Application to Data
As a “proof of principle,” the method was applied to the results of the WTCCC CD case-control study, which, for a complex disease, is relatively well characterized and is thus a reasonable test of the effectiveness of the method. The method was also applied to the data from a meta-analysis of BD genome scans. BD is a disorder whose biological background is not well characterized, so the results of the analysis are of considerable interest.
The summary statistics for the CD study were downloaded from the WTCCC website. These data were produced from an analysis of 1748 cases versus 2953 controls, based upon the Affymetrix 500K Chip. After quality control procedures, the WTCCC retained genotypes on 469,557 SNPs (for more details, see the WTCCC article2), of which 181,961 lay within genes, covering 14,653 genes and 4685 GO categories. A total of 246,929 SNPs lay within 20 kb of a gene, covering 22,253 genes and 5177 GO categories. Extending the region within which a SNP is considered to map to a gene considerably increases the coverage of genes, although it is unknown a priori whether this increases or decreases the ratio of signal to noise. SNPs were defined as “significantly associated” if the Armitage Trend test had a p value of 1 × 10−4 or less. This is the criterion used by the WTCCC to define the SNPs of interest that were listed in their Supplemental Data. Less stringent criteria of p < 0.001 and p < 0.005 were also explored. These criteria were met by 308, 1226, and 3905 SNPs lying within 74, 253, and 833 genes, respectively. As mentioned previously, GO categories containing only one significant gene were not counted as overrepresented, because a single chance association in a small GO category could result in that category having false evidence of overrepresentation.
Data were also analyzed from a meta-analysis of the WTCCC BD sample together with BD samples from the United States (STEP-BD collection), University College London, and the Universities of Edinburgh and Dublin,16 consisting of a total of 4387 cases and 6209 controls. There were 325,690 SNPs genotyped in common between the samples and met quality control thresholds in each of the studies. Of these, 123,840 lay within genes, covering 13,204 genes and 4487 GO categories. Data were imputed for HapMap SNPs (for details, see Ferreira et al.16), giving a total set of 1,769,948 SNPs. Of these, 679,901 lay within a total of 17,249 genes and 4877 GO categories. Not surprisingly, imputed SNP data greatly enhanced gene coverage as compared to array genotypes alone, so these data were chosen for the main analysis presented in this paper. There were 593, 3759, and 15,979 SNPs that were significant at p < 1 × 10−4, p < 0.001, and p < 0.005, lying within 50 genes, 296 genes, and 1036 genes, respectively.
Analysis of CD with the LD-Pruned SNP Set
An LD-pruned SNP set was obtained for the WTCCC CD data set as follows: At each step, the SNP with the most significant p value was selected, and all SNPs within 1 Mb with r2 > 0.2 were removed (both criteria that we accept are arbitrary). Then, the most significant of the remaining SNPs was selected, and the process was repeated until no pair of SNPs within 1 Mb of each other and with r2 > 0.2 remained. This left 61,246 SNPs within genes, covering 12,899 genes (compared to 14,653 covered by the complete SNP list). The number of SNPs with p < 0.0001, p < 0.001, or p < 0.005 in each category was counted (counting multiple significant SNPs in the same gene separately), as was the total number of significant SNPs overall. Replicate lists of significant SNPs of the same length as the original list were generated by randomly sampling SNPs (assuming independence). The numbers of significant SNPs in each GO category in each replicate list were obtained, and these were used to obtain category-specific p values for overrepresentation. The same bootstrap technique as that described in the main manuscript was used for assessing whether there was an excess of significantly overrepresented categories.
This method counts multiple hits from the same gene, which may increase power. However, there is also the question of where to set the r2 cutoff for defining pairs of SNPs in LD—low values will result in a sparse map of SNPs, which may lose power as a result of reduced gene coverage, whereas if higher values are used, multiple signals in a gene may have considerable interdependence due to LD. This would result in false positives if the signals are analyzed under the assumption that the markers are independent.
Results
The number of categories reaching uncorrected category-specific significance levels of 0.05, 0.01, and 0.001 for overrepresentation in the WTCCC CD data set, when analysis was restricted to SNPs lying within genes, is shown in Table 1. We also tested whether the total number of overrepresented categories was significantly in excess as compared with the null expectation. For CD, the significance of the excess of overrepresented categories increased with the stringency of the significance criterion defining overrepresentation. The significance of the number of overrepresented categories also increased as the p value cutoff for defining significant SNPs became more stringent. This suggests that associations for CD are concentrated in a relatively small number of categories, each of which shows strong evidence of overrepresentation.
Table 1.
Number of Significantly Overrepresented GO Categories: CD Data Set
| p < 0.05 |
p < 0.01 |
p < 0.001 |
||||||
|---|---|---|---|---|---|---|---|---|
| p Value Criterion for SNPs | No. of Top SNPs | No. of Genes | No. of Categories | p Value | No. of Categories | p Value | No. of Categories | p Value |
| 0.0001 | 308 | 74 | 55 | 0.091 | 32 | 0.009 | 17 | 0.001 |
| 0.001 | 1226 | 253 | 72 | 0.275 | 35 | 0.018 | 7 | 0.054 |
| 0.005 | 3905 | 833 | 124 | 0.151 | 35 | 0.088 | 9 | 0.032 |
Number of GO categories reaching various levels of significance for overrepresentation on the list of significant SNPs in the WTCCC CD data set and their corresponding genes, together with p values indicating whether this number is significantly greater than that expected by chance. Only categories containing two or more significant genes are counted. SNPs assigned to genes if they lie within that gene. Genotyped SNPs only.
The most significant individual categories, with a cutoff of p < 1 × 10−4 used for defining significant SNPs, are shown in Table 2, and complete category-specific results are presented in Table S1 (available online). We also present (as expected hits per study) the number of categories expected by chance to have a category-specific p value at least as significant as that of the test category, thus giving a measure of significance allowing for multiple testing of categories. Note that, for categories with a category-specific p value of 0, a smaller value of expected hits per study may be obtained by simulating more than 5000 replicate gene lists.
Table 2.
Top 30 Overrepresented GO Categories: CD Data Set
| GO Category | Type | Total Genes in Category | No. of Genes on List | Expected No. of Genes on List | p Value | Expected Hits per Study | Function |
|---|---|---|---|---|---|---|---|
| GO02504 | PROCESS | 11 | 2 | 0.02 | 0.0000 | 0.32 | antigen processing and presentation of peptide or polysaccharide antigen via MHC class II |
| GO32395 | FUNCTION | 9 | 2 | 0.01 | 0.0000 | 0.32 | MHC class II receptor activity |
| GO42613 | CELLULAR | 10 | 2 | 0.02 | 0.0000 | 0.32 | MHC class II protein complex |
| GO06955 | PROCESS | 365 | 7 | 1.21 | 0.0002 | 0.54 | immunological response |
| GO42611 | CELLULAR | 20 | 2 | 0.03 | 0.0002 | 0.54 | MHC protein complex |
| GO51184 | FUNCTION | 9 | 2 | 0.04 | 0.0004 | 0.77 | cofactor transporter activity |
| GO51181 | PROCESS | 8 | 2 | 0.02 | 0.0004 | 0.77 | cofactor transport |
| GO51183 | FUNCTION | 8 | 2 | 0.03 | 0.0004 | 0.77 | vitamin transporter activity |
| GO15226 | FUNCTION | 3 | 2 | 0.02 | 0.0004 | 0.77 | carnitine transporter activity |
| GO15879 | PROCESS | 3 | 2 | 0.02 | 0.0004 | 0.77 | carnitine transport |
| GO02376 | PROCESS | 492 | 8 | 1.95 | 0.0006 | 0.98 | immune system process |
| GO06511 | PROCESS | 143 | 5 | 0.59 | 0.0006 | 0.98 | ubiquitin-dependent protein catabolic process |
| GO19941 | PROCESS | 143 | 5 | 0.59 | 0.0006 | 0.98 | modification-dependent protein catabolic process |
| GO43632 | PROCESS | 143 | 5 | 0.59 | 0.0006 | 0.98 | modification-dependent macromolecule catabolic process |
| GO51603 | PROCESS | 144 | 5 | 0.59 | 0.0006 | 0.98 | proteolysis involved in cellular protein catabolic process |
| GO44257 | PROCESS | 146 | 5 | 0.6 | 0.0006 | 0.98 | cellular protein catabolic process |
| GO19882 | PROCESS | 34 | 2 | 0.07 | 0.0010 | 1.41 | antigen processing and presentation |
| GO30163 | PROCESS | 178 | 5 | 0.71 | 0.0012 | 1.6 | protein catabolic process |
| GO04221 | FUNCTION | 53 | 3 | 0.18 | 0.0012 | 1.6 | ubiquitin thiolesterase activity |
| GO04843 | FUNCTION | 56 | 3 | 0.19 | 0.0012 | 1.6 | ubiquitin-specific protease activity |
| GO19783 | FUNCTION | 57 | 3 | 0.19 | 0.0012 | 1.6 | small conjugating protein-specific protease activity |
| GO44265 | PROCESS | 244 | 5 | 0.91 | 0.0014 | 1.79 | cellular macromolecule catabolic process |
| GO51180 | PROCESS | 11 | 2 | 0.06 | 0.0014 | 1.79 | vitamin transport |
| GO16790 | FUNCTION | 66 | 3 | 0.22 | 0.0018 | 2.14 | thiolester hydrolase activity |
| GO43285 | PROCESS | 241 | 5 | 0.9 | 0.0024 | 2.73 | biopolymer catabolic process |
| GO07249 | PROCESS | 31 | 2 | 0.08 | 0.0028 | 3.11 | I-kappaB kinase/NF-kappaB cascade |
| GO09057 | PROCESS | 292 | 5 | 1.06 | 0.0038 | 3.95 | macromolecule catabolic process |
| GO06952 | PROCESS | 324 | 5 | 1.17 | 0.0044 | 4.42 | defense response |
| GO06512 | PROCESS | 384 | 6 | 1.6 | 0.0048 | 4.73 | ubiquitin cycle |
| GO44248 | PROCESS | 440 | 6 | 1.64 | 0.0054 | 5.19 | cellular catabolic process |
List of 30 most significantly overrepresented GO categories for CD (cutoff for significant SNPs: p < 1 × 10−4). The type of category and the expected number of categories with a category-specific overrepresentation p value at least as significant as that observed in the absence of any true overrepresentation are also shown.
The overrepresented categories for CD include those related to the major histocompatibility complex (MHC), immunological response, and antigen processing. The involvement of MHC in the genetic etiology of CD is well known.17,18 Likewise, immunological response and antigen processing are well-established features of this disease.19 Although these results are not novel, they provide a proof of principle of the effectiveness of the method. The list of categories for CD also contains several relating to ubiquitination, one of the two major intracellular protein-degradation systems—the other being autophagy (a catabolic process that involves delivery of cellular components to the lysosome for degradation). GWA studies have already implicated autophagy as an important functional pathway involved in CD,20 although we did not find exidence for overrepresentation of GO categories involving autophagy here. There are seven genes with SNPs significant at p < 1 × 10−4 in one or more of the four ubiquitination categories shown in Table 3 (GO: 6511, 6512, 4221, and 4383): CYLD (MIM 605018), USP4 (MIM 603486), RNF123, CUL2 (MIM 603135), KLHL20, USP7 (MIM 602519), and FAF1 (MIM 604460). CYLD is approximately 90 kb from NOD2 (MIM 605956), and its apparent association could be due to LD with NOD2. USP4 and RNF123 are in the previously published 3p21 locus.21 A nonsynonymous SNP in MST1 (MIM 142408) has been postulated as responsible for the association at this locus.22 However, neither USP4 nor RNF123 were genotyped, so their association with CD risk cannot be definitively excluded. CUL2 is in a “newly identified” locus on chromosome 10p11 showing convincing evidence for association in a large meta-analysis of GWA data.21 FAF1 (TNFRSF6 associated factor) is a particularly interesting candidate for involvement in CD susceptibility, because it shows evidence of association in both the WTCCC study2 (p = 1.6 × 10−5) and the meta-analysis21 (p = 1 × 10−4). Furthermore, it is well established as a negative regulator of NFkappaB, which is a key player in the pathogenesis of CD. There is also prior evidence linking ubiquitination with CD.23,24 Thus, the role of ubiquitination in CD is worthy of further study. Of further substantial interest is category 6955 (“immune response”). This category contains seven genes with SNPs significant at p < 1 × 10−4 (category-specific p = 0.0002 for overrepresentation). These genes include NOD2, IL23R (MIM 607562), and TNFSF15 (MIM 604052), all of which were identified as putative susceptibility genes for CD in a recent review20 and showed convincing evidence for association in the meta-analysis.21 The other four genes are SBNO2, CCL18 (MIM 603757), HLA-DQA2, and HLA-DQB2. These, and other genes in the category, may be interesting additional candidates for involvement in CD susceptibility.
Table 3.
Number of significantly Overrepresented GO Categories: BD Meta-Analysis Data Set
| p < 0.05 |
p < 0.01 |
p < 0.001 |
||||||
|---|---|---|---|---|---|---|---|---|
| p Value Criterion for SNPs | No. of Top SNPs | No. of Genes | No. of Categories | p Value | No. of Categories | p Value | No. of Categories | p Value |
| 0.0001 | 593 | 50 | 21 | 0.556 | 6 | 0.521 | 2 | 0.246 |
| 0.001 | 3759 | 296 | 61 | 0.546 | 17 | 0.363 | 3 | 0.301 |
| 0.005 | 15979 | 1036 | 133 | 0.112 | 46 | 0.017 | 13 | 0.009 |
| 0.01 | 29073 | 1698 | 232 | 0.001 | 74 | 0.001 | 22 | <0.001 |
Number of GO categories reaching various levels of significance for overrepresentation on the list of significant SNPs in the BD meta-analysis data set and their corresponding genes, together with p values indicating whether this number is significantly greater than that expected by chance. Only categories containing two or more significant genes are counted. SNPs assigned to genes if they lie within that gene. Genotyped and imputed SNPs.
The analysis method assumes that the probability that a gene is present on the list of significant genes is independent of the presence or absence of other genes in that list—in other words, that SNPs from different genes are not in LD. Although this is often a reasonable assumption, there are regions of the genome in which long-range LD is known to exist. One of these is the MHC region, located at chromosome 6p21.3, which is known to be implicated in autoimmune diseases such as CD. It is possible that the overrepresentation of significant categories in Table 1 could be due to hits in multiple genes from the MHC region and that these may be due to LD, rather than to several different genes being involved in disease etiology. To investigate this possibility, we reran the analysis, omitting all genes and SNPs in the MHC region, defined25 as the region between HLA-F (MIM 143110) and KIFC1 (MIM 603763). For a cutoff of p < 1 × 10−4 for defining significant SNPs, this resulted in 26 categories reaching a significance level of 0.01 for overrepresentation and 15 reaching a significance level of 0.001. These are both significantly higher than would be expected by chance (p = 0.032 and 0.003, respectively).
Thus, there is still a significant excess of overrepresented categories for CD, even after removal of the MHC region, suggesting that most of the overrepresented categories in Table 2 do not depend on multiple MHC genes. The most significantly overrepresented categories after removal of the MHC region are shown in Table S3. As expected, categories containing several genes from the MHC region, such as those involving immunological response or MHC activity, are no longer significant. Conversely, the prominence of categories related to ubiquitin (as noted earlier), as well as to carnitine transport, is enhanced. There is prior evidence from different sources that suggests that variants in the carnitine transporter genes SLC22A4 (MIM 604190) and SLC22A5 (MIM 603377) are associated with CD.26
Using a 20 kb window for assigning SNPs to genes resulted in similar categories being highlighted, although the significance of the number of overrepresented categories was reduced (see Table S4).
The number of categories reaching significance levels for overrepresentation of 0.05, 0.01, and 0.001 in the BD meta-analysis data set are shown in Table 3. Imputed SNPs were used, as well as SNPs that were assigned to genes if they lay within that gene (no window). The significance of the number of overrepresented categories increased as less stringent criteria were used in defining significant SNPs. In particular, the most significant results were obtained with a cutoff of p < 0.01. This suggests that, compared to CD, the genetic susceptibility to BD (at least as currently defined) may involve risk alleles with smaller individual effects.27
Given that this level of stringency resulted in the maximum enrichment for significant pathways, we used the threshold of p < 0.01 to examine specific associated pathways. The results are shown in Table 4, the full list of which is shown in Table S2. Many of the overrepresented GO categories implicate biological systems involved in the broad control of cellular activity, including the categories of hormone activity, RNA splicing, and macroautophagy. Autophagy is a catabolic process that is crucial to normal cell growth, development, and homeostasis, and it is known that lithium, the major prophylactic medication for BD, can induce autophagy.28 Within the category of hormone activity, both the genes encoding thyrotropin-releasing hormone (TRH [MIM 275120]) and those encoding thyroglobulin (TG [MIM 188450]) were identified. Both are involved in thyroid function, which modulates cellular activity, is known to influence mood in general29 and BD in particular,30 and is influenced by lithium.31 Also within the category of hormone activity is the gene encoding proopiomelanocortin preproprotein (POMC [MIM 176830]), whose protein product, adrenocorticotrophin, is essential for normal functioning of the hypothalamic-pituitary-adrenal (HPA) axis, which, like thyroid function, influences cellular activity. HPA dysfunction is known to be associated with mood disorders, including BD.32 Genes implicated within the category of RNA splicing include several members of the spliceosome C complex, including small nuclear ribonucleoprotein 200kDa (U5) (SNRNP2000 [MIM 601664]), heterogeneous nuclear ribonucleoprotein C (HNRPC [MIM 164020]), mago-nashi homolog (MAGOH [MIM 602603]), pre-mRNA processing factor 6 homolog (PRPF6), and small nuclear ribonucleoprotein 40kDa (U5) (SNRNP40 [MIM 607797]). This complex plays a major role in RNA splicing and, hence, regulation, of cellular activity.33 This category also includes A2BP1 (MIM 605104), a gene that encodes ataxin 2 binding protein, which showed a strong association signal in a recent GWA study of schizoaffective disorder, bipolar type,34 and QKI (MIM 605950), which encodes quaking homolog, a protein that is important for normal myelination and is implicated in human diseases, including schizophrenia.35
Table 4.
Top 30 Overrepresented GO Categories: BD Meta-Analysis Data Set
| GO Category | Type | Total Genes in Category | No. of Genes on List | Expected No. of Genes on List | p Value | Expected Hits per Study | Function |
|---|---|---|---|---|---|---|---|
| GO05179 | FUNCTION | 75 | 10 | 2.83 | 0.0000 | 0.57 | hormone activity |
| GO03700 | FUNCTION | 692 | 88 | 64.03 | 0.0000 | 0.57 | transcription factor activity |
| GO16236 | PROCESS | 7 | 3 | 0.24 | 0.0000 | 0.57 | macroautophagy |
| GO30212 | PROCESS | 6 | 5 | 0.43 | 0.0000 | 0.57 | hyaluronan metabolic process |
| GO00045 | PROCESS | 6 | 3 | 0.24 | 0.0000 | 0.57 | autophagic vacuole formation |
| GO03677 | FUNCTION | 1776 | 189 | 148.04 | 0.0002 | 0.9 | DNA binding |
| GO03676 | FUNCTION | 2550 | 259 | 215.15 | 0.0002 | 0.9 | nucleic acid binding |
| GO33077 | PROCESS | 6 | 3 | 0.25 | 0.0002 | 0.9 | T cell differentiation in the thymus |
| GO08380 | PROCESS | 181 | 23 | 10.74 | 0.0002 | 0.9 | RNA splicing |
| GO06323 | PROCESS | 75 | 12 | 3.68 | 0.0002 | 0.9 | DNA packaging |
| GO42301 | FUNCTION | 6 | 3 | 0.3 | 0.0002 | 0.9 | phosphate binding |
| GO06465 | PROCESS | 7 | 4 | 0.6 | 0.0002 | 0.9 | signal peptide processing |
| GO04867 | FUNCTION | 81 | 17 | 7.24 | 0.0004 | 1.26 | serine-type endopeptidase inhibitor activity |
| GO05102 | FUNCTION | 572 | 74 | 53.92 | 0.0004 | 1.26 | receptor binding |
| GO33151 | PROCESS | 5 | 3 | 0.26 | 0.0004 | 1.26 | V(D)J recombination |
| GO06623 | PROCESS | 3 | 2 | 0.21 | 0.0004 | 1.26 | protein targeting to vacuole |
| GO07034 | PROCESS | 18 | 7 | 1.95 | 0.0006 | 1.63 | vacuolar transport |
| GO00398 | PROCESS | 57 | 9 | 2.58 | 0.0008 | 2.01 | nuclear mRNA splicing, via spliceosome |
| GO00377 | PROCESS | 57 | 9 | 2.58 | 0.0008 | 2.01 | RNA splicing, via transesterification reactions with bulged adenosine as nucleophile |
| GO00375 | PROCESS | 57 | 9 | 2.58 | 0.0008 | 2.01 | RNA splicing, via transesterification reactions |
| GO30528 | FUNCTION | 1066 | 128 | 101.76 | 0.0010 | 2.41 | transcription regulator activity |
| GO18345 | PROCESS | 4 | 3 | 0.3 | 0.0010 | 2.41 | protein palmitoylation |
| GO05634 | CELLULAR | 3617 | 372 | 328.82 | 0.0014 | 3.22 | nucleus |
| GO44249 | PROCESS | 602 | 70 | 51.32 | 0.0014 | 3.22 | cellular biosynthetic process |
| GO02521 | PROCESS | 51 | 10 | 4.06 | 0.0016 | 3.6 | leukocyte differentiation |
| GO30098 | PROCESS | 39 | 9 | 3.2 | 0.0018 | 4 | lymphocyte differentiation |
| GO05853 | CELLULAR | 5 | 2 | 0.07 | 0.0018 | 4 | eukaryotic translation elongation factor 1 complex |
| GO04364 | FUNCTION | 16 | 4 | 0.5 | 0.0018 | 4 | glutathione transferase activity |
| GO01958 | PROCESS | 3 | 3 | 0.46 | 0.0024 | 5.22 | endochondral ossification |
| GO07076 | PROCESS | 15 | 5 | 1.12 | 0.0028 | 6.02 | mitotic chromosome condensation |
List of 30 most significant GO categories for BD in the BD meta-analysis data set (cutoff for significant SNPs: p < 0.01). Genotyped and imputed SNPs used, with SNPs assigned to genes if they lie within that gene. The type of category and the expected number of categories with a category-specific overrepresentation p value at least as significant as that observed in the absence of any true overrepresentation are also shown.
The significance of the number of overrepresented categories ascertained on the basis of the analysis of the LD-pruned data set, with multiple independent hits per gene allowed, is given in Table S8, and the 30 most significant categories, with a cutoff of p < 0.0001 used for defining significant SNPs, are given in Table S9.
If Table S8 is compared to Table 1, it can be seen that using a cutoff of p < 0.0001 to define significant SNPs still gives the greatest excess of significantly overrepresented categories and that this excess has similar significance to that obtained when all SNPs are used but each gene is counted only once. The less-stringent cutoffs give less significant excess of overrepresented categories, and these are less significant than the corresponding values in Table 1. Thus, allowing multiple independent hits per gene does not increase the significance of the results here. The most significant categories, shown in Table S9, are also very similar to those in Table 2.
Discussion
We present a novel method for testing for overrepresentation of biological pathways among significant SNPs from GWA study data sets. Unlike previous approaches, our method corrects for varying numbers of SNPs per gene and multiple overlapping pathways. In addition to providing a measure of significance (corrected for multiple-testing) for individual pathways, the method also assesses whether the number of overrepresented pathways is significantly higher than expected (given the overlap between pathways), thus giving a measure of the overall significance of the list of associated genes. When applied to GWA data of CD, a disease with a relatively well-characterized biological background, the method identified several biological pathways known to be implicated in the disease etiology, thus demonstrating its validity for providing insights into the biological basis of complex diseases. Moreover, when we applied the method to GWA data from the BD meta-analysis, we identified a number of processes consistent with previous hypotheses concerning the etiology of this disorder, although none of those has the degree of prior empirical support equivalent to that of the involvement of the immunological system in CD. Analyses of additional data sets will be required for confirmation of which of these specific pathways are genuinely involved in disease.
The understanding of BD is much less advanced than that of CD. There are no laboratory tests for BD as of yet, and diagnoses are based on clinical features.36 To date, the strongest signals that have emerged from a meta-analysis of published GWA studies of BD16 have implicated genes whose products are involved in ion channel activity (and, hence, control of neuronal excitability), including ANK3 (MIM 600465) and CACNA1C (MIM 114205). The findings from our current analysis (which uses the same combined data set) implicates several GO categories that suggest that some aspects of the broad control of cellular activity may also be important players in the pathogenesis of BD, including the categories of hormone activity, RNA splicing, and macroautophagy. At an intuitive level, there is plausible face validity to the possibility that some aspects of the broad control of cellular activity could influence the BD phenotype: BD is an episodic disorder that is affected by environmental changes, and, at least at a simplistic explanatory level, both hyper- and hypoenergized states can occur with reversion to periods of normal function between acute episodes. It is well known that episodes of BD can be precipitated by stressors, natural hormonal changes, and administration of steroid medication,36 and changes in transcriptional activity are a key mechanism by which such stimuli interact with genotype to influence phenotype. If this finding is replicated in other data sets, it will be important to refine observations to identify (1) the most important biological systems affected by the general transcriptional changes and (2) the extent to which the findings may contribute to the genetic overlap between schizophrenia and BD.37,38
A limitation of the method is the requirement for specification of a p value cutoff in defining the list of significantly associated SNPs, an approach similar to that taken by programs, such as GO-stat,39 that analyze lists of genes (e.g., from microarray expression studies) directly. Clearly, the choice of this threshold could be arbitrary. Depending upon the sample size and the distribution of genetic effect sizes, a relatively stringent cutoff such as p < 0.0001 will focus attention on SNPs most likely to be genuine associations; empirically, this worked well for CD. However, for BD, less stringent cutoffs gave more significant results, in terms of overrepresented categories, the best results being obtained with a cutoff of p < 0.01. It is likely that the genetic basis of complex traits will show considerable heterogeneity, and this is particularly true for phenotypes such as BD, of which the diagnosis is entirely clinical and there are currently no diagnostic tests for validation of classification.37 Most associations with individual SNPs will have a small effect size in the sample as a whole. Thus, one of the major aims of pathway-based analyses is detection of pathways in which several genes show moderate association individually. This rationale would argue for the use of a less stringent p value criterion for selection of the list of significant SNPs. Clearly, there is a balance to be struck between being confident that the associations tested are genuine, which is greatest when a stringent cutoff is used in defining significant SNPs, and ensuring that genuine associations of small magnitude are not missed. It is likely that the optimal cutoff will depend on the disease. A pragmatic solution to the problem of choosing an arbitrary threshold, a solution that we adopt here, is to apply a range of cutoffs, determine which gives the most significant increase in overrepresented categories, and examine the individual categories highlighted by this cutoff.
The results for the BD meta-analysis were much more significant than those that would have been obtained from analyzing the WTCCC BD sample alone (results shown in Table S5), highlighting the importance of using large sample sizes to give high power to detect associations of small magnitude.
Several promising methods have recently been developed for imputing genotypes at untyped SNPs with the use of the genotyped SNPs and the Hapmap data.40 We expected that the use of imputed SNPs might increase power of the method, because more genes would become informative. Our analysis of the BD meta-analysis data confirmed that this hypothesis was correct (results obtained when genotype data was used alone are in Table S6).
Another issue that is applicable to any method based upon genes concerns how the boundaries of genes are defined. We used two options. First, SNPs were assigned to genes only if they lay within the start of the first and the end of the last known exon. In the second approach, SNPs mapping within 20 kb of a gene (5′ or 3′) were assigned to that gene. When the optimal cutoff for selecting significant SNPs (p < 0.01) was used, we observed no apparent improvement in the results by using the latter approach (see Table S7), which was designed to capture the proximal functional elements of most genes. The choice of 20 kb is not entirely arbitrary—a recent study of gene expression41 found that the majority of eQTLs lay within 20 kb of genes. However, other window sizes are also justifiable; for example, a 500 kb window has been used.11 Again, there is a balance to be struck between narrow windows (running the risk of missing regulatory regions) and wide regions (increasing the chance that an associated SNP has no functional relationship with the gene to which it is assigned).
It is possible to reduce the multiple-testing burden by restricting analysis to a subset of GO categories; for example, GO level 4 categories containing between 20 and 200 genes.11 Alternatively, a partitioning method42 could be used for selection of informative subsets of categories for analysis. However, for a disease such as BD, for which previous biological information is limited, there is no a priori indication of which size or level of category will best reflect the underlying biological processes. Thus, selecting any subset of categories for analysis risks the loss of information. Because the aim of our study was to investigate what the GWA results could tell us about the biological basis of BD, while making as few assumptions as possible, we chose to analyze all GO categories. Although our results must necessarily be regarded as exploratory and do require replication in other studies, it should be noted that we did observe an experiment-wide excess of significantly overrepresented categories even after correcting for multiple categories.
A limitation of our method is that it counts each gene only once. If a gene contains multiple independent hits, it is possible that counting each of these separately could increase the power of the study. We investigated this in the CD data by selecting a subset of 61,246 SNPs in low LD (r2 < 0.2) lying within genes. Each significant SNP in a GO category was counted separately, and significance of overrepresentation was tested by generating random lists of “significant” SNPs of the same length as the original. This did not increase the significance of the results, nor did it alter the most significant GO categories (see Tables S8 and S9). One reason for this might be that the subset of SNPs covered fewer genes than the original SNP set (12,899 to 14,653), so potentially important genes might have been “lost.” It is possible that counting significant SNPs in low LD separately may increase power for some diseases, particularly if a relatively lax cutoff is used in defining significance (because this makes multiple independent signals more likely). However, such SNPs may not be truly independent (dependent on the r2 cutoff), so analyzing them as independent may cause false-positive results. Using a stricter r2 cutoff will reduce this possibility, but it may result in a loss of power due to decreased gene coverage. SNP selection strategies that maximize power merit further work.
An alternative method for testing for overrepresentation of pathways among significant SNPs from GWA studies is given by Wang et al.11 Their method involves ranking all genes in order of significance (based on association-test statistics for individual SNPs), then comparing the distribution of ranks of genes in a particular pathway to the remaining genes via a Kolmogorov-Smirnov test. This test can be modified to take into account the actual test statistics associated with the genes, with higher weights assigned to more significant genes. This is a modification of the method used by the program GSEA.43 This approach has the advantage of not requiring a criterion for significant SNPs, and genes, to be specified, because it is based on the distribution of ranks in the whole set of genes. However, the Kolmogorov-Smirnov test does not take into account where in the overall distribution the differences in ranks lie. Thus, a significant result could be based on differences in rank occurring among genes some way down the list, with nonsignificant p values for association. Such a result would be of limited interest. Conversely, the weighting scheme used by Wang et al.11 favors pathways in which a few genes have very large test statistics in comparison to the others over pathways with several genes of approximately similar significance. As noted above, the aim of pathway analysis is, at least arguably, to detect the latter sort of pathway, given that genes with very large test statistics will be apparent from inspection of the individual SNP p values. Perhaps a useful compromise between these extremes would be to define a list of SNPs, and thus genes, of interest, as in the method proposed here, but to assign each gene a score based on its rank within the list. For example, if the list has n genes, assign the most significant a score of n, the next a score of n − 1, and so on. Genes not on the list would be assigned a score of zero. A score for each pathway could then be obtained by summing the scores of its genes, and the significance of the pathway score could be tested in a manner similar to that described here.
As noted by Wang et al.,11 ranking genes on the basis of the most significant SNP within each gene favors large genes with several SNPs, and any analysis of the significance must allow for this. Ideally, one would reanalyze the entire genome, permuting case and control status, to obtain sets of ranked genes to which the observed results can be compared. Such a method allows for varying sizes and LD structure among genes, but it is computationally demanding, particularly if imputed data are used. Furthermore, permutation requires individual genotype data to be available. Therefore, a single p value is assigned to each gene by Wang et al.,11 and these are permuted randomly among all genes. Two methods are proposed for obtaining a single p value for each gene: (1) using the most significant p value and (2) applying a Simes correction to the p values from that gene and using the corrected p value in the analysis. Using the most significant p value will increase the chance that large genes with several SNPs rank highly by chance, and thus it may falsely inflate the significance of pathways that contain such genes. Conversely, the Simes correction will be very conservative for large genes containing only a few highly significant SNPs, particularly if the analysis method for overrepresentation uses the actual p value (as does the method used by Wang et al.11) rather than just presence on a list of significant genes. Furthermore, applying the Simes correction may alter the order of genes from that in the original data, which may be undesirable when rank-based methods are being used. Our method allows for the varying sizes of genes by a random selection of SNPs for generation of the replicate gene lists against which the observed data are tested. Thus, the probability that a gene is added to the list is proportional to the number of SNPs that it contains. This assumes that SNPs in different genes are not in LD, so the position of a gene on the list does not depend on that of nearby genes. Regions of the genome that are known to show long-range LD, such as the MHC region, will violate this assumption. Reanalyzing the data, omitting SNPs and genes within these regions, provides a means of checking that significant overrepresentation of GO categories is not solely a result of a few genes being in LD with each other. When the MHC region was removed, the significance of GO categories containing several MHC genes (such as those involving immunological response or MHC activity) was reduced, removing them from the list of significant categories in Table S3. This does not necessarily mean that the significantly overrepresented MHC-related categories observed in Table 2 are false positives, since multiple hits from the MHC region could still be independent. Indeed, the fact that the MHC-activity- and immunological-response-related categories were the most significant when the LD-pruned SNP set was used (see Table S9) suggests that multiple hits from the MHC region are not in strong LD with each other. Careful examination of the LD patterns between significant SNPs would be required for assessment of whether the multiple significant genes in the MHC region could result from the same association signal or represent multiple distinct signals.
An additional assumption of our method is that LD within genes is approximately constant (so that the effective number of tests per gene is roughly proportional to the number of SNPs), but when that assumption is violated, such as in regions of high LD, our analysis will be conservative. It is difficult to fully allow for variable LD levels without resorting to simulation-based methods (for which the full genotype data are required). The SNP-pruning approach mentioned above is a possibility, but it needs further evaluation. It should be noted, however, that our method is only an initial stage in highlighting interesting genes and pathways for further study and that issues of inter-SNP LD will need to be resolved by more detailed analyses of individual genotype data.
In summary, we detail a method for implicating biological pathways likely to be involved in disease susceptibility. In a proof-of-principle application, we correctly identified pathways known or suspected to be involved in CD. When applied to BD, a disorder whose pathophysiology is almost entirely unknown, the results suggest that biological systems involved in modulation of transcription and cellular activity are implicated, as is hormonal function, including thyroid hormone. These observations suggest that a core feature of pathogenesis of BD may be a disturbance in regulation of transcriptional activity. Although intriguing, these results need to be replicated in additional large studies.
Supplemental Data
Supplemental Data include a complete list of WTCCC members and nine tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
List of category-specific p values for overrepresentation for all GO categories for Crohn disease (cutoff for significant SNPs: p < 1 × 10−4). The type of category and the expected number of categories with a category-specific overrepresentation p value at least as significant as that observed in the absence of any true overrepresentation are also shown.
List of category-specific p values for overrepresentation for all GO categories for the bipolar disorder meta-analysis data set (cutoff for significant SNPs: p < 0.01). The type of category and the expected number of categories with a category-specific overrepresentation p value at least as significant as that observed in the absence of any true over-representation are also shown.
Web Resources
The URLs for data presented herein are as follows:
AmiGO database, http://www.geneontology.org/GO.downloads.ontology.shtml
Computer programs and files for carrying out the analyses described in this manuscript, http://x004.psycm.uwcm.ac.uk/∼peter
Gene Ontology Annotation (GOA) database, http://www.ebi.ac.uk/GOA
NCBI ftp site, http://www.ncbi.nlm.nih.gov/ftp
NCBI SNP batch entry list, http://www.ncbi.nlm.nih.gov/SNP/dbSNP.cgi?list=rslist
NCBI chr_rpts download site, ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606/chr_rpts/
NCBI detailed information on chr_rpts file, ftp://ftp.ncbi.nih.gov/snp/00readme.txt
NCBI seq_gene download site, ftp://ftp.ncbi.nih.gov/genomes/H_sapiens/mapview/
NCBI gene2go file download site, ftp://ftp.ncbi.nih.gov/gene/DATA
NCBI Detailed information on gene2go file, ftp://ftp.ncbi.nih.gov/gene/README
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/
Wellcome Trust Case-Control Consortium website, https://www.wtccc.org.uk/
Acknowledgments
The authors would like to thank Chris Mathew for helpful comments and advice on the relevance of the results to Crohn disease biology. We are indebted to all individuals who have participated in our bipolar disorder research. We thank MDF the Bipolar Organisation for the help of its staff and members. Funding for recruitment and phenotype assessment has been provided by the Wellcome Trust and the Medical Research Council. The genotype analyses were funded by the Wellcome Trust and undertaken within the context of the Wellcome Trust Case Control Consortium (WTCCC). The members of the WTCCC are listed in the Supplemental Data.
References
- 1.Freimer N.B., Sabatti C.S. Human genetics: variants in common diseases. Nature. 2007;445:828–830. doi: 10.1038/nature05568. [DOI] [PubMed] [Google Scholar]
- 2.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeggini E., Weedon M.N., Lindgren C.M., Frayling T.M., Elliott K.S., Lango H., Timpson N.J., Perry J.R., Rayner N.W., Freathy R.M. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manolio T.A., Brooks L.D., Collins F.S. A HapMap harvest of insights into the genetics of common disease. J. Clin. Invest. 2008;118:1590–1605. doi: 10.1172/JCI34772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tiffin N., Kelso J.F., Powell A.R., Pan H., Bajic V.B., Hide W.A. Integration of text- and data-mining using ontologies successfully selects disease gene candidates. Nucleic Acids Res. 2005;33:1544–1552. doi: 10.1093/nar/gki296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Curtis R.K., Oresic M., Vidal-Puig A. Pathways to the analysis of microarray data. Trends Biotechnol. 2005;23:429–435. doi: 10.1016/j.tibtech.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 7.Adie E.A., Adams R.R., Evans K.L., Porteous D.J., Pickard B.S. Speeding disease gene discovery by sequence based candidate prioritization. BMC Bioinformatics. 2005;6:55. doi: 10.1186/1471-2105-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kohler S., Bauer S., Horn D., Robinson P.N. Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet. 2008;82:949–958. doi: 10.1016/j.ajhg.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franke L., van Bakel H., Fokkens L., de Jong E.D., Egmont-Petersen M., Wijmenga C. Reconstruction of a functional human gene network, with an application for prioritizing positional candidate genes. Am. J. Hum. Genet. 2006;78:1011–1025. doi: 10.1086/504300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iossifov I., Zheng T., Baron M., Gilliam T.C., Rzhetsky A. Genetic-linkage mapping of complex hereditary disorders to a whole-genome molecular-interaction network. Genome Research. 2008;18:1150–1162. doi: 10.1101/gr.075622.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang K., Li M., Bucan M. Pathway-based approaches for analysis of genomewide association studies. Am. J. Hum. Genet. 2007;81:1278–1283. doi: 10.1086/522374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris M.A., Clark J., Ireland A., Lomax J., Ashburner M., Foulger R., Eilbeck K., Lewis S., Marshall B., Mungall C. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res Database. 2004;32:D258–D261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buxbaum J.D., Georgieva L., Young J.J., Plescia C., Kajiwara Y., Jiang Y., Moskvina V., Norton N., Peirce T., Williams H. Molecular dissection of NRG1-ERBB4 signaling implicates PTPRZ1 as a potential schizophrenia susceptibility gene. Mol. Psychiatry. 2008;13:162–172. doi: 10.1038/sj.mp.4001991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Camon E., Barrell D., Lee V., Dimmer E., Apweiler R. The Gene Ontology Annotation (GOA) Database - an integrated resource of GO annotations to the UniProt Knowledgebase. In Silico Biol. 2004;4:5–6. [PubMed] [Google Scholar]
- 15.Nyholt D.R. A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am. J. Hum. Genet. 2004;74:765–769. doi: 10.1086/383251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferreira M.A., O'Donovan M.C., Meng Y.A., Jones I.R., Ruderfer D.M., Jones L., Fan J., Kirov G., Perlis R.H., Green E.K. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet. 2008;40:1056–1058. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cariappa A., Sands B., Forcione D., Finkelstein D., Podolsky D.K., Pillai S. Analysis of MHC class II DP, DQ and DR alleles in Crohn's disease. Gut. 1998;43:210–215. doi: 10.1136/gut.43.2.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forcione D.G., Sands B., Isselbacher K.J., Rustgi A., Podolsky D.K., Pillai S. An increased risk of Crohn's disease in individuals who inherit the HLA class II DRB3∗0301 allele. Proc. Natl. Acad. Sci. USA. 1996;93:5094–5098. doi: 10.1073/pnas.93.10.5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown S.J., Mayer L. The immune response in inflammatory bowel disease. Am. J. Gastroenterol. 2007;102:2058–2069. doi: 10.1111/j.1572-0241.2007.01343.x. [DOI] [PubMed] [Google Scholar]
- 20.Mathew C.G. New links to the pathogenesis of Crohn disease provided by genome-wide association scans. Nat. Rev. Genet. 2008;9:9–14. doi: 10.1038/nrg2203. [DOI] [PubMed] [Google Scholar]
- 21.Barrett J.C., Hansoul S., Nicolae D.L., Cho J.H., Duerr R.H., Rioux J.D., Brant S.R., Silverberg M.S., Taylor K.D., Barmada M.M. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat. Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goyette P., Lefebvre C., Ng A., Brant S.R., Cho J.H., Duerr R.H., Silverberg M.S., Taylor K.D., Latioano A., Aumais G. Gene-centric association mapping of chromosome 3p implicates MST1 in IBD pathogenesis. Mucosal Immunology. 2008;1:131–138. doi: 10.1038/mi.2007.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayor A., Martinon F., De Smedt T., Petrilli V., Tschopp J. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat. Immunol. 2007;8:497–503. doi: 10.1038/ni1459. [DOI] [PubMed] [Google Scholar]
- 24.Sakiyama T., Fujita H., Tsubouchi H. Autoantibodies against ubiquitination factor E4A (UBE4A) are associated with severity of Crohn's disease. Inflamm. Bowel Dis. 2008;14:310–317. doi: 10.1002/ibd.20328. [DOI] [PubMed] [Google Scholar]
- 25.MHC Sequencing Consortium Complete sequence and gene map of a human major histocompatibility locus. Nature. 1999;401:921–923. doi: 10.1038/44853. [DOI] [PubMed] [Google Scholar]
- 26.Peltekova V.D., Wintle R.F., Rubin L.A., Amos C.I., Huang Q., Gu X., Newman B., Van Oene M., Cescon D., Greenberg G. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat. Genet. 2004;36:471–475. doi: 10.1038/ng1339. [DOI] [PubMed] [Google Scholar]
- 27.Craddock N., O'Donovan M.C., Owen M.J. Genome-wide association studies in psychiatry: lessons from early studies of non-psychiatric and psychiatric phenotypes. Mol. Psychiatry. 2008;13:649–653. doi: 10.1038/mp.2008.45. [DOI] [PubMed] [Google Scholar]
- 28.Heiseke A., Aguib Y., Riemer C., Baier M., Schätzl H.M. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J. Neurochem. 2009;109:25–34. doi: 10.1111/j.1471-4159.2009.05906.x. [DOI] [PubMed] [Google Scholar]
- 29.Young E.A., Korszun A. The hypothalamic-pituitary-gonadal axis in mood disorders. Endocrinol. Metab. Clin. North Am. 2002;31:63–78. doi: 10.1016/s0889-8529(01)00002-0. [DOI] [PubMed] [Google Scholar]
- 30.Hendrick V., Altshuler L., Whybrow P. Psychoneuroendocrinology of mood disorders. The hypothalamic-pituitary-thyroid axis. Psychiatr. Clin. North Am. 1998;21:277–292. doi: 10.1016/s0193-953x(05)70005-8. [DOI] [PubMed] [Google Scholar]
- 31.Lazarus J.H. The effects of lithium therapy on thyroid and thyrotropin-releasing hormone. Thyroid. 1998;8:909–913. doi: 10.1089/thy.1998.8.909. [DOI] [PubMed] [Google Scholar]
- 32.Daban C., Vieta E., Mackin P., Young A.H. Hypothalamic-pituitary-adrenal axis and bipolar disorder. Psychiatr. Clin. North Am. 2005;28:469–480. doi: 10.1016/j.psc.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 33.Kornblihtt A.R., de la Mata M., Fededa J.P., Munoz M.J., Nogues G. Multiple links between transccription and splicing. RNA. 2004;10:1489–1498. doi: 10.1261/rna.7100104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamshere M.L., Green E.K., Jones I.R. Strong genetic support for broadly defined bipolar schizoaffective disorder as a useful diagnostic concept. Br. J. Psychiatry. 2009 doi: 10.1192/bjp.bp.108.061424. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chénard C.A., Richard S. New implications for the QUAKING RNA binding protein in human disease. J. Neurosci. Res. 2008;86:233–242. doi: 10.1002/jnr.21485. [DOI] [PubMed] [Google Scholar]
- 36.Goodwin J. Oxford University Press; Oxford: 2007. Manic-depressive illness. [Google Scholar]
- 37.Craddock N., Owen M.J. Rethinking psychosis: the disadvantages of a dichotomous classification now outweigh the advantages. World Psychiatry. 2007;6:84–91. [PMC free article] [PubMed] [Google Scholar]
- 38.Craddock N., O'Donovan M.C., Owen M.J. Psychosis genetics: modeling the relationship between schizophrenia, bipolar disorder and mixed (or “schizoaffective”) psychoses. Schizophr. Bull. 2009;35:482–490. doi: 10.1093/schbul/sbp020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beissbarth T., Speed T.P. GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics. 2004;20:1464–1465. doi: 10.1093/bioinformatics/bth088. [DOI] [PubMed] [Google Scholar]
- 40.Marchini J., Howie B., Myers S., McVean G., Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 41.Veyrieras J.B., Kudaravalli S., Kim S.Y., Dermitzakis E.T., Gilad Y., Stephens M., Pritchard J.K. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet. 2008;4:e1000214. doi: 10.1371/journal.pgen.1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alterovitz G., Xiang M., Mohan M., Ramoni M.F. GO PaD: the Gene Ontology Partition Database. Nucleic Acids Res. 2007;35:D322–D327. doi: 10.1093/nar/gkl799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of category-specific p values for overrepresentation for all GO categories for Crohn disease (cutoff for significant SNPs: p < 1 × 10−4). The type of category and the expected number of categories with a category-specific overrepresentation p value at least as significant as that observed in the absence of any true overrepresentation are also shown.
List of category-specific p values for overrepresentation for all GO categories for the bipolar disorder meta-analysis data set (cutoff for significant SNPs: p < 0.01). The type of category and the expected number of categories with a category-specific overrepresentation p value at least as significant as that observed in the absence of any true over-representation are also shown.