

Abstract
Alpha-dystroglycanopathies such as Walker Warburg syndrome represent an important subgroup of the muscular dystrophies that have been related to defective O-mannosylation of alpha-dystroglycan. In many patients, the underlying genetic etiology remains unsolved. Isolated muscular dystrophy has not been described in the congenital disorders of glycosylation (CDG) caused by N-linked protein glycosylation defects. Here, we present a genetic N-glycosylation disorder with muscular dystrophy in the group of CDG type I. Extensive biochemical investigations revealed a strongly reduced dolichol-phosphate-mannose (Dol-P-Man) synthase activity. Sequencing of the three DPM subunits and complementation of DPM3-deficient CHO2.38 cells showed a pathogenic p.L85S missense mutation in the strongly conserved coiled-coil domain of DPM3 that tethers catalytic DPM1 to the ER membrane. Cotransfection experiments in CHO cells showed a reduced binding capacity of DPM3(L85S) for DPM1. Investigation of the four Dol-P-Man-dependent glycosylation pathways in the ER revealed strongly reduced O-mannosylation of alpha-dystroglycan in a muscle biopsy, thereby explaining the clinical phenotype of muscular dystrophy. This mild Dol-P-Man biosynthesis defect due to DPM3 mutations is a cause for alpha-dystroglycanopathy, thereby bridging the congenital disorders of glycosylation with the dystroglycanopathies.
Introduction
Congenital disorders of glycosylation (CDG) are inborn errors of metabolism and various defects affect the N-glycosylation of proteins. The complex N-glycan biosynthesis follows a sequential and highly ordered pathway in the cytoplasm, ER, and Golgi apparatus. Because N-glycosylation is a very common cotranslational modification, genetic defects in this pathway generally lead to a multisystem disease. Defects in the ER during the assembly of the lipid-linked oligosaccharide and transfer to nascent protein chains affect all N-linked proteins and typically lead to such a multisystem presentation in CDG-I patients.
Alpha-dystroglycanopathies represent a separate group of genetic glycosylation disorders, in which deficiency of tissue-specific alpha-dystroglycan O-mannosylation has been reported. These disorders present with a more organ-restricted clinical phenotype, including muscular dystrophy and a variable degree of structural brain and eye anomalies. At the severe end, serious muscular dystrophy, a complex brain development disorder, and eye abnormalities can be found in the congenital muscular dystrophies such as Walker-Warburg syndrome (WWS [MIM 236670]). Within the same spectrum, subtle eye abnormalities, clinically less significant brain malformation, and a milder muscular dystrophy can be observed in limb girdle muscular dystrophy (e.g., LGMD2I due to FKRP mutations1,2 [MIM 607155]). Several genes have been discovered underlying the phenotypic spectrum of the dystroglycanopathy syndromes (POMGnT1, POMT1, POMT2, LARGE, FKTN, and FKRP). In a large number of dystroglycanopathy patients, the underlying genetic etiology remains unsolved.3–5
Dolichol-phosphate-mannose (Dol-P-Man) synthase or GDP-Man:Dol-P mannosyltransferase (EC 2.4.1.83), plays an important role in four different glycosylation routes in the ER. N-glycosylation,6 C-mannosylation,7 O-mannosylation,8 and GPI-anchor formation9 depend on Dol-P-Man availability (Figure 1). In S. cerevisiae, the single-membrane-spanning DPM1 protein comprises the Dol-P-Man synthase activity, whereas in other yeasts and higher eukaryotes, a Dol-P-Man synthase complex exists that is composed of three subunits.10 The catalytic subunit DPM1 is located in the cytoplasm and transfers mannose from the nucleotide sugar GDP-Man to membrane-embedded Dol-P, resulting in the formation of Dol-P-Man. DPM1 is anchored to the ER membrane via the coiled-coil domain of DPM3 (Figure 1). DPM2 is located in the ER membrane as well and is required for stabilization of the complex. In addition, a role for DPM2 has been reported in GPI-anchor biosynthesis as the regulator of GPI-N-acetylglucosaminyltransferase (GPI-GnT).11
Figure 1.
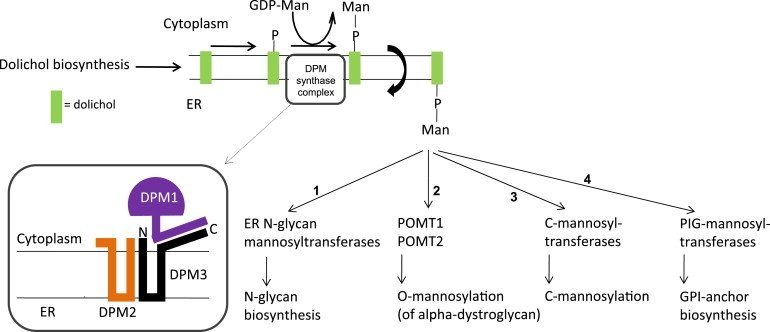
Architecture of the Dol-P-Man Synthase Complex
The enzymatically active DPM1 subunit in the cytoplasm is anchored to subunits DPM2 and DPM3 in the ER membrane via the C-terminal peptide of DPM3. Four biosynthetic pathways depend on Dol-P-Man: N-glycosylation (1), O-mannosylation (2), C-Mannosylation (3), and GPI-anchor biosynthesis (4).
In the group of CDG-I, many subtypes have been found with deficient glycosyltransferases and identification of new disease variants with aberrant LLO flipping from the cytosol to the ER lumen,12 and disturbed Dol-P biosynthesis13 has provided important insights in the complex glycosylation processes in human disease. Here, we identify a defect in an accessory protein (DPM3) of a glycosyltransferase in the ER. The unique clinical phenotype of a mild muscular dystrophy with dilated cardiomyopathy may relate to aberrant protein N-glycosylation and O-mannosylation of alpha-dystroglycan.
Material and Methods
Subjects
The use of CDG-Ie14 fibroblasts and serum was approved by informed consent of parents and treating physician (M. Garcia-Silva). DNA samples of Greek controls were obtained from A. Evangeliou. The use of fibroblasts and serum of the current patient has been approved by the parents and treating physician (Spilioti).
CDG Diagnostics
Transferrin isoelectric focusing was carried out as described before.15 The clinical history did not show any indication for the presence of alcohol abuse, fructosemia, or galactosemia as a possible secondary cause for CDG-I transferrin isoelectric-focusing profiles. A protein polymorphism was excluded by neuraminidase digestion of the samples and by the normal profiles of both parents. Analyses of lipid-linked oligosaccharides, formation of Dol-PP-GlcNAc1 and Dol-PP-GlcNAc2,16 elongation of Dol-PP-GlcNAc2 to Dol-PP-GlcNAc2Man5 by cytosolic mannosyltransferases,17 and oligosaccharyltransferase18 were performed in fibroblasts as described.
Mass Spectrometry
N-glycan analysis was carried out essentially as described.19 In brief, patient serum was denatured and N-glycans were released by overnight incubation with PNGaseF (New England Biolabs). After purification (PGC-SPE, Supelco), the glycan mixture was permethylated and purified on a SepPak C18 cartridge (Waters). Permethylated N-glycans were analyzed by MALDI-LTQ mass spectrometry (Thermo Finnigan). For molecular weight determination of serum transferrin, immunopurification and online desalting were followed by ESI-MS detection as described.20
Dol-P-Man Synthase in Fibroblasts and CHO Cells
Skin fibroblasts were cultured in M199 medium (Life Technologies) supplemented with 10% fetal calf serum and 1% penicillin + streptomycin in a humified atmosphere containing 5% CO2 at 37°C.
The reaction contained in a final volume of 0.07 ml: 7 mM Tris-HCl (pH 7.2), 7 mM MgCl2, 0.025 μCi GDP-[14C] mannose (specific activity 303 mCi/ mmol), 2 μg dolichol-P, 0.1% Nonidet P-40, and 0.05 mg membrane protein. Incubation was at 24°C for the times indicated. The reaction was stopped by addition of chloroform-methanol (3:1, by volume) and processed by phase partitioning as described.21 Radioactive glycolipids were separated on silica gel 60 plates (Merck) developed in chloroform/methanol/water (65:25:4, by volume). Radioactivity was detected by Phospho-Imaging.
Genetic Analysis of the DPM1, DPM2, and DPM3 Genes
We designed intronic oligonucleotide primers in accordance with public sequences (GenBank accession number NM_153741) to flank the coding sequences and exon-intron junctions. Primer sequences are available on request. The exon fragments were amplified in a reaction volume of 25 μl containing 100 ng DNA template, 1 unit of Taq DNA polymerase (Invitrogen), 2.5 μl of 10× PCR buffer, 2.0 mM MgCl2, 50 ng of forward and reverse primers, 0.25 mM dNTPs, and 0.75 μl DMSO. After an initial step of 2 min at 94°C, PCR parameters were 35 cycles for 45 s denaturation at 94°C, 45 s annealing at Ta (between 53 and 58°C), and 1 min extension at 72°C. The cycles were followed by a final extension step of 10 min at 72°C. We ran the amplimers on a 1.0% agarose gel to check the specificity of the reaction. The same oligonucleotides were used for direct sequencing with the BigDye terminator cycle sequencing chemistry on an Applied Biosystems ABI PRISM 3130xl Genetic Analyzer. The SIFT website was used for intolerance prediction.
Culture and Transfection of CHO Cells
CHO cells were maintained at 37°C under 5% CO2 in Dulbecco's modified Eagle (DMEM; Invitrogen) containing 10% fetal calf serum (Pan Biotech), penicillin (6 mg/L), and streptomycin 100 mg/L. For transfection, FuGENE HD transfection reagent (Roche) was used in accordance with the manufacturer's instruction. Growing cells were inoculated at about 25% density and grown to ∼75% confluence, transfected with 18 μg DNA (plasmids as indicated in Figure 4D and prepared according to Ashida et al.22) and incubated for two days. Cells were harvested and resuspended in 30 mM Tris/HCl, pH 7.5, containing 35% (v/v) glycerol, 3 mM MgCl2, 1 mM dithiotreitol, 1 mM PMSF, and 1 mM benzamidine and centrifuged at 48 000 g for 20 min. The pellet was homogenized in 10 mM Tris/HCl, pH 7.5, containing 1 mM MgCl2, 1 mM dithiotreitol, 1 mM PMSF, and 1 mM benzamidine and centrifuged as above. The pellet was resuspended in 30 mM Tris/HCl, pH 7.5, containing 35% (by volume) glycerol, 3 mM MgCl2, and 1 mM dithiotreitol and was used as the enzyme source.
Figure 4.
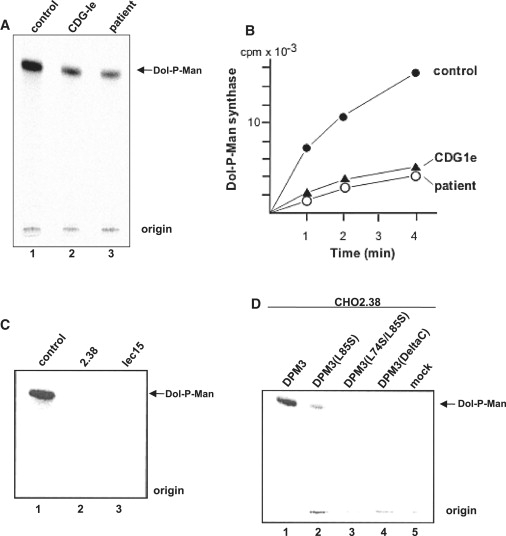
Dol-P-Man Synthase Activity in Fibroblasts and CHO Cells
Microsomal membranes from fibroblasts or CHO cells were incubated with dolichol-P and radioactive GDP-Man and then Dol-P-Man formation by TLC was analyzed. (A) shows Dol-P-Man synthase activity in human fibroblasts. (B) shows the time course of Dol-P-Man formation in patient and control fibroblasts. (C) shows Dol-P-Man synthase activity in CHO control, CHO2.38 (DPM3), and lec15 (DPM2) cells. (D) shows Dol-P-Man synthase activity in CHO2.38 cells after transient transfection with different DPM3 plasmids or an empty vector. DPM3 plasmids used are indicated in Figure 5C.
Interaction Analysis of DPM1-DPM3
CHO K1 (WT) cells were transiently cotransfected with pME/FLAG-DPM3 (WT, L85S, L74S/I78T/L85S or empty vector) and pME/3HSV-DPM1. After 40 hr cultivation, cells were lysed with 0.5% digitonin, and the soluble fractions were subjected to immunoprecipitation with anti-FLAG M2-agarose beads (Sigma). From unbound fractions, 3HSV-DPM1 was recovered with anti-HSV (Novagen) and protein G-Sepharose beads (GE Healthcare). Proteins were detected by western blotting with anti-FLAG M2 (Sigma) (×1000) or anti-HSV (×1000), and second antibody goat anti-mouse IgG (H+L chain)-HRP (MBL) (×4000). Detection was carried out by LAS1000 (Fuji Film) with a West Pico Chemiluminescent reagent (Pierce).
Immunohistochemistry on Muscle Biopsy
Immunohistochemical examination was performed on 10 μm frozen sections of a biopsy from the m. rectus femoris, after 10 min fixation in ice-cold acetone. One slide was stained with hematoxylin-phloxin for morphological comparison. Anti-spectrin, anti-merosin (laminin-alpha2) and anti-β-dystroglycan (all from Novocastra Laboratories Ltd, UK), and IIH6 (gift from K. Campbell) were used as primary antibodies; Power Vision (Goat a-Mouse/Rb HRP one component, Immunologic, Duiven) as was used as a secondary antibody. Slides were stained with AEC (ScyTek Laboratories) for detection.
Analysis of C-Mannosylation of Serum Properdin
Properdin was immunopurified from 100 μl of plasma or serum from the DPM3-deficient patient, a CDG type Ie patient, as well as seven healthy individuals, as described.23 The C-mannosylation of peptides T7 (residues 80–100; amino acid numbering as in UniProtKB/Swiss-Prot entry P27918) was analyzed by tandem LC–MS in the multiple reaction monitoring mode.23 Transitions specific for the two major glycoforms of peptide T7 were measured, as well as those for the glycoforms expected when C-mannosylation would be defective. For comparison of the properdin from different individuals, the nonglycosylated peptide T28 was measured; the measurement result reflected the amount of injected protein digest.
CD59 Expression on Fibroblasts
Cultured fibroblast cells were incubated on ice for 15 min with directly fluorochrome-labeled antibodies CD45-ECD and with CD59-PE directed against a GPI-anchored protein. CD45 was purchased from Beckman Coulter (Miami, FL, USA), CD59 from IQP, Groningen, The Netherlands). The cells were washed in PBS with BSA. Fluorescence was determined on a FC500 flow cytometer (Beckman Coulter, Hileah, FL, USA). We used mouse IgG1 to detect nonspecific binding and used cells incubated without antibodies to determine autofluorescence. Immunofluorescence was compensated for both background stainings. Data (collected in list mode) were analyzed with the CXP software (Beckman Coulter).
Results
Discovery of a Congenital Disorder of Glycosylation Type I in a Patient with Muscular Dystrophy
This female patient, born to healthy Greek parents, was born at term after an uneventful pregnancy by spontaneous vaginal delivery. No consanguinity was known. Her birth weight, length, and head circumference were normal. She was nondysmorphic. Psychomotor development, growth, and pubertal development were normal and the patient was able to attend a regular primary school. A muscle biopsy was performed at 11 years because of mild muscle weakness and a waddling gait. Subtle structural changes were found suggesting a mild myopathy. After Achilles tenotomy, no further complaints were registered. At 20 years, dilated cardiomyopathy was diagnosed after episodes of precordial pain. No signs of cardiac muscle hypertrophy or outflow obstruction were detected. Elevated CK was detected (1500 to 3000, normal < 280 IU/L) with mildly elevated transaminases (ASAT 80, ALAT 98, normal < 50 IU/L). A heart muscle biopsy showed no structural or histological abnormalities. A second open biopsy of the m. rectus femoris revealed moderate muscular dystrophy (Figure 2) with fiber-size variation, multiple internal nuclei, necrotic fibers, rimmed vacuoles, fiber splitting, and interstitial fibrosis. At 21 years, the patient experienced a stroke-like episode involving the right temporo-parietal region. Cerebral angiography and ophthalmologic evaluation were normal. Metabolic investigations were normal, but results of transferrin isoelectric focusing showed an abnormal profile suggesting a CDG type I pattern (Figure 3A). In addition, isoelectric focusing of serum thyroxine-binding globulin was slightly abnormal. Baseline endocrine investigations, APTT, PTT, clotting factors, and protein C and S were normal. However, on preventive anticoagulant therapy the coagulation was difficult to control.
Figure 2.
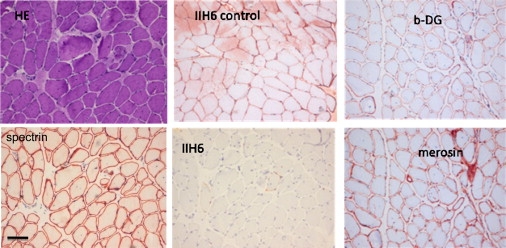
Muscle Biopsy of m. rectus femoris at 20 Years
Histochemistry with hematoxylin-phloxin (HE) and immunohistochemistry with IIH6 antibody against glycosylated alpha-dystroglycan and with antibodies against spectrin, merosin, and β-dystroglycan (b-DG). A staining of a control muscle biopsy with IIH6 is shown for comparison. The scale bar represents 100 μm.
Figure 3.
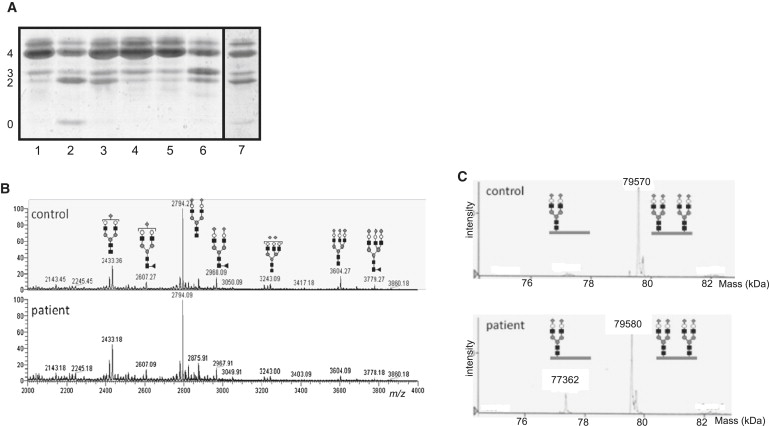
Determination of CDG Subtype
(A) Isoelectric focusing of serum transferrin. Profiles of patient and both parents are shown in lanes 3 and 4 + 5, respectively. Controls are presented in lane 1 (healthy control), lane 2 (CDG-Ia), lane 6 (CDG-II patient), and lane 7 (CDG-Ie). Numbers 0, 2, 3, and 4 indicate the sialotransferrin subfractions.
(B) Maldi mass spectrometry of permethylated N-glycans from serum expressed as relative percentage of the biantennary glycan at m/z 2794. Structures of the most abundant glycan isoforms are indicated.
(C) ESI-MS of immunopurified transferrin from serum. The increase of mass 77362 in the patient corresponds to an increase of monoglycosylated transferrin (GlcNAc, black square; Fuc, gray triangle; Man, gray circle; Gal, open circle; and NeuNAc, gray diamond).
After recovery from the stroke-like episode and on longterm antidiuretic therapy, the patient developed no further progression of her cardiac disease. Currently, at the age of 27 years, the patient has a low normal IQ, and normal head circumference, whereas her body length is below the tenth centile and weight for height is at the 85th centile. The patient can walk without assistance, but needs some support when climbing stairs. She has a mild proximal muscle weakness, more prominent on the left, and bilateral pes planus. There is no significant alteration in muscle mass, except for a mild pseudohypertrophy of the calf musculature. The deep tendon reflexes are decreased, except for the left extremity. The Babinski sign is positive on the left; the Gower's sign is negative. There is no sign of central or peripheral ataxia. Brain imaging showed no abnormalities that could be related to either CDG or alpha-dystroglycanopathy.
In a repeat serum sample, transferrin isoelectric focusing again showed a mildly abnormal profile with increased disialo- but also trisialotransferrin. Further mass spectrometric investigations of permethylated total serum N-glycans of the second sample did not indicate marked abnormalities apart from a very mild increase of m/z 2433 indicating a slight loss of sialic acid (Figure 3B). ESI-MS of immunopurified transferrin showed an increase of monoglycosylated transferrin with a mass of 77362 Da and an increased ratio of mono- versus diglycosylated transferrin (0.21, normal < 0.07). No nonglycosylated transferrin could be observed (Figure 3C). These results clearly indicated the partial absence of complete biantennary N-glycans, which is in agreement with a diagnosis CDG type I and defective N-glycosylation in the ER.
Identification of Reduced Dol-P-Man Synthase Activity in Patient Fibroblasts
After exclusion of CDG-Ia and CDG-Ib by enzyme analysis in fibroblasts, analysis of lipid-linked oligosaccharides in fibroblast from the patient showed a normal profile, excluding additional CDG-Ic, -Id, -Ig, -Ih, and -Il. Formation of Dol-PP-GlcNAc1 and Dol-PP-GlcNAc2, as well as the elongation of Dol-PP-GlcNAc2 to Dol-PP-GlcNAc2Man5, were unremarkable, thereby also ruling out subtypes CDG-Ii, -Ij, and -Ik. Oligosaccharyltransferase was normal as well. Because the mass spectrometry studies were clearly indicative for a CDG type I defect in this patient, more extended investigations were performed, including Dol-P-Man synthase activity measurement. Transfer of radioactive labeled mannose from GDP-Man to Dol-P in patient fibroblasts clearly showed a lowered enzymatic activity to ∼30% of controls (Figures 4A and 4B). Using this in vitro assay system, we found the residual Dol-P-Man synthase activity in fibroblasts of a known CDG-Ie patient14 with DPM1 mutations to be similar.
Mutation Analysis Indicated a Homozygous Missense Mutation in DPM3
To identify the causative genetic defect, we subjected the coding sequences of the three genes encoding subunits of the Dol-P-Man synthase complex to DNA sequence analysis. No mutations could be identified in catalytic subunit DPM1 and accessory subunit DPM2, while sequencing of accessory subunit DPM3 displayed a homozygous missense mutation (c.254T>C [p.Leu85Ser]), present in heterozygous state in both parents (Figure 5A). The missense mutation was not detected in 240 control alleles from the same ethnic background.
Figure 5.
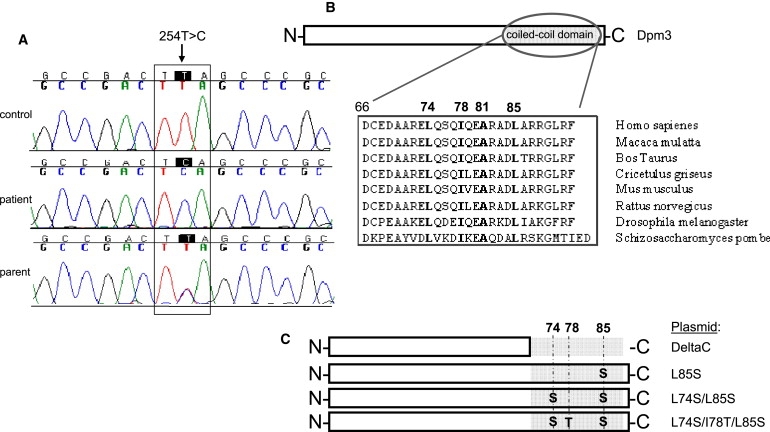
Mutation Analysis and DPM3 Plasmids Used
(A) Mutation analysis of the Dol-P-Man synthase genes DPM1-3 showed a c.254T>C (p.L85S) missense mutation in DPM3. Forward sequences are shown.
(B) Location of leucine 85 in the coiled-coil domain of the C-terminal peptide of DPM3 and its conservation. Hydrophobic amino acids important for DPM1 binding are shown in bold.
(C) Structure of the plasmids used for transient transfection of CHO2.38 cells as shown in Figure 3D and DPM1-DPM3-binding studies as shown in Figure 5.
The affected amino acid Leu85 is highly conserved during evolution (Figure 5B) and a change to Ser was predicted to be intolerant with the SIFT database, which uses conservation among species and amino acid characteristics for predicting the tolerance level of a certain mutation. Leucine 85 is present on the hydrophobic side of the coiled-coil domain in DPM3, thereby forming the DPM1-interacting region together with hydrophobic amino acids Leu74, Ile78, and Ala81 (Figure 5B).
Confirmation of the Pathogenicity of p.L85S Revealed by Analyses with CHO2.38 Cells
Given that S. cerevisiae, often used for identifying genetic defects by complementation of the corresponding orthologous mutant,24 does not contain DPM3, DPM3-deficient CHO2.38 cells were used as heterologous expression system. Because of the importance of the coiled-coil domain of DPM3 for DPM1 binding, we previously studied the effect of mutated amino acids in this region on GPI-anchor biosynthesis by FACS analysis of CD59 in CHO cells.22 Transient transfection of CHO2.38 cells with DPM3 plasmids containing DeltaC (deletion of C-terminal coiled-coil peptide), L74S/I78T/L85S triple, or L74S/L85S double mutations showed no or very low CD59 expression, whereas L85S transfection restored CD59 expression to wild-type levels. Protein expression was similar for all mutants studied. However, the enzymatic activity of Dol-P-Man synthase in these cells was not studied.22 In order to prove the pathogenicity of the p.L85S mutation in our patient, we measured Dol-P-Man synthase activity after transient transfection of CHO2.38 DPM3-deficient cells with different DPM3 plasmids (Figure 4D). No enzyme activity was found in CHO2.38 cells and in DPM2-deficient Lec15 cells (Figure 4C). Transfection of CHO2.38 cells with full-length DPM3 restored Dol-P-Man synthase activity. In contrast, transfection with plasmids DeltaC and the L74S/L85S double mutant did not show any complementation, whereas L85S transfection resulted in slight Dol-P-Man production. Previously,22 the binding capacity of DPM3 to DPM1 has been studied by cotransfection of CHO K1 cells with 3HSV-DPM1 and FLAG-DPM3, followed by immunoprecipitation with anti-FLAG beads. DeltaC and L74S/I78T/L85S triple mutants lost their capability of interacting with DPM1 resulting in cytoplasmic degradation of DPM1 via ubiquitination in the proteasome. In the present study, similar experiments were performed with the L85S single mutant (Figure 6). The amount of 3HSV-DPM1 bound to FLAG-DPM3(L85S) was significantly reduced compared to that bound to FLAG-DPM3(WT), under 0.5% digitonin solubilization (lane 2 versus lane 1). Furthermore, the total expressed amount of 3HSV-DPM1 (bound plus unbound) was equivalent after transfection with FLAG-DPM3(L85S) and FLAG-DPM3(L74S/I78T/L85S) (lane 3) or empty vector (lane 4), whereas total 3HSV-DPM1 was much higher after transfection with FLAG-DPM3(WT). These results suggest that the p.L85S mutation in DPM3 in our patient may cause a reduction in DPM1-binding capacity and DPM1 stability in vivo.
Figure 6.
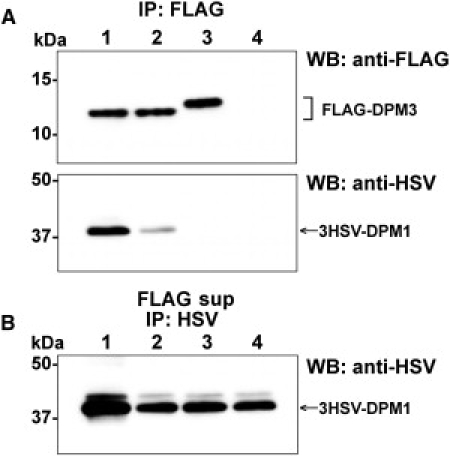
Binding of DPM1 and DPM3
CHO K1 cells were transiently cotransfected with pME/3HSV-DPM1 and either pME/FLAG-DPM3(WT) (lane 1), pME/FLAG-DPM3(L85S) (lane 2), pME/FLAG-DPM3(L74S/I78T/L85S) (lane 3), or an empty vector (lane 4). After 40 hr culture, the cells were lysed with 0.5% digitonin and the complex of FLAG-DPM3 and 3HSV-DPM1 was immunoprecipitated (IP) with anti-FLAG beads (A). Unbound 3HSV-DPM1 was recovered from the supernatant of FLAG IP (FLAG sup) with anti-HSV and protein G-beads (B). Proteins were detected by western blotting (WB) with indicated antibodies.
Investigation of the Four Dol-P-Man-Dependent Glycosylation Pathways Reveals a Strong Effect on O-Mannosylation
As indicated in Figure 1, four different glycosylation routes are initiated by or depend on the transfer of mannose to protein or lipid-linked glycans in the ER. To investigate the O-mannosylation pathway, we performed immunohistochemical staining of a patient muscle biopsy by using the IIH6 antibody specific for O- mannosylation-dependent Laminin-binding glycans on alpha-dystroglycan. IIH6 staining was overall strongly reduced as compared to staining of the sarcolemma in a control muscle biopsy. Immunohistochemical analyses with antibodies against beta-dystroglycan, merosin (laminin-alpha2), and spectrin showed normal sarcolemmal staining of the patient biopsy (Figure 2). As described above, N-glycosylation of the serum protein transferrin was found to be mildly affected as studied by isoelectric focusing and mass spectrometry. C-mannosylation was investigated by mass spectrometric analysis of serum properdin that can be C-mannosylated and O-fucosylated.23 In controls, the most abundant forms of this peptide are MM00 (two C-mannosylated trypthophans, not O-fucosylated) and MMFG (two C-linked mannoses and an O-fucosylated threonine) as indicated in Figure 7A. The peptides with a single mannose residue (M0FG and M000) are present in much smaller amounts, whereas the nonmannosylated forms were not detected. In the DPM3-deficient patient (Figure 7B) and the CDG-Ie (MIM 608799) patient (data not shown), both doubly C-mannosylated species were found, but no increase was observed in the less-mannosylated species with no or a single C-linked mannose residue. If reduced Dol-P-Man levels would have affected the C-mannosylation of serum properdin, an increase of less-mannosylated species would have been expected. Interestingly, an increase in fucosylation state was observed in the mannosylated peptides in the CDG-Ie and DPM3-deficient patients. The results indicate that C-mannosylation of peptide T7 from serum properdin is unaltered in both patients as compared to the control sample and that, for an as yet unknown reason, O-fucosylation is significantly more pronounced (Figure 7B). The influence of DPM3 mutations on cell-surface CD59 expression as an indicator of GPI-anchor biosynthesis has been studied previously. Transfection of CHO2.38 cells with DPM3 plasmid containing the L85S mutation showed normal CD59 expression, whereas transfection with equal levels of the more severe DeltaC and L74S/I78T/L85S triple mutants clearly led to reduced CD59 expression.22 Analysis of CD59 expression on patient fibroblasts showed a similar result. DPM3-deficient fibroblasts showed an equal expression of CD59 as compared to control fibroblasts, whereas fibroblasts from a CDG-Ie patient showed a slight reduction in CD59 expression (Figure 7C).
Figure 7.
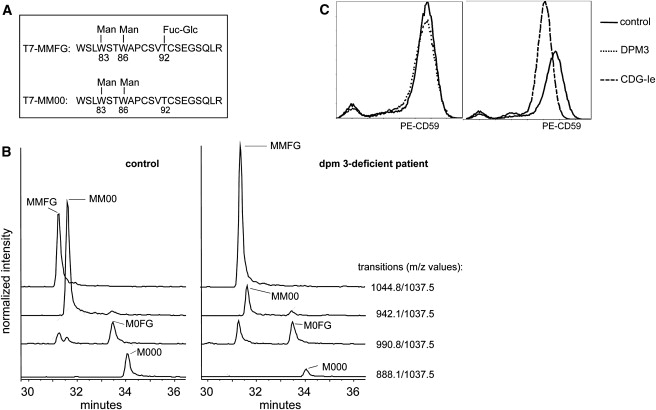
Analysis of C-Mannosylation and GPI-Anchored Protein CD59
(A) Structure of the two main glycoforms of peptide T7 from properdin.
(B) Properdin was immunopurified from serum or plasma of patients and healthy controls. C-mannosylated tryptic peptides were examined by tandem LC-MS in the multiple reaction monitoring (MRM) mode. Traces for the MRM transitions (for the precursor/y9 pair) are shown for a healthy control and patient. Peak identity was assigned on the basis of the presence of the other transitions specific for the different glycoforms. The signal intensities were normalized relative to the nonglycosylated peptide T28, thereby allowing comparison of samples.
(C) CD59 expression on control and patient fibroblasts by FACS analysis. A comparison is shown of a control fibroblast with DPM3-deficient fibroblasts (left panel) and CDG-Ie fibroblasts (right panel).
Discussion
In this report, we present a genetic glycosylation disorder in the group of alpha-dystroglycanopathies in a patient with mild muscular dystrophy and biochemical features of a congenital disorder of glycosylation. So far, CDG and dystroglycanopathies have been regarded as two separate groups of glycosylation disorders, each caused by deficiency of enzymes affecting a particular glycosylation pathway: N-glycosylation in CDG and O-mannosylation in the alpha-dystroglycanopathies. Here, we show that a mild defect in the accessory protein DPM3 of Dol-P-Man synthase, required for four different glycosylation routes, can lead to an isolated phenotype of muscular dystrophy most likely resulting from deficient O-mannosylation of alpha-dystroglycan.
Dol-P-Man is the glycosyl donor for all mannosylation reactions taking place on the luminal side of the ER. Cell models exist with a deficiency of the Dol-P-Man synthase subunits or MPDU1, a protein required for Dol-P-Man and Dol-P-Glucose utilization of which the specific biochemical function remains to be determined. In the mouse Thy-1 cells (DPM1),25,26 CHO Lec15 cells (DPM2),27 CHO2.38 cells (DPM3),22 and Lec35 cells (MPDU1),28 deficiency of Dol-P-Man leads to Dol-P-P-GlcNAc2Man5 accumulation in the ER and to reduced protein N-glycosylation. The absence of the expression of GPI-anchored proteins at the plasma membrane indicates defective GPI-anchor biosynthesis in the ER. C-mannosylation of RNase has been investigated in Lec35 cells28 and was shown to be strongly reduced. Because Dol-P-Man is required for C-mannosylation and O-mannosylation,8 these types of glycosylation can be expected to be abnormal in all Dol-P-Man synthase-deficient cell lines. The mild biochemical phenotype in this DPM3-deficient patient allowed us to investigate subtle effects of reduced Dol-P-Man availability on the different glycosylation pathways. O-mannosylation seemed to be most affected, as revealed by strongly reduced glycosylation-specific IIH6 staining of a muscle biopsy. It can be expected that CDG-Ie patients with DPM1 mutations suffer from a similar or even more severe deficiency of alpha-DG mannosylation. Analysis of O-mannosylation has so far not been reported in the few known CDG-Ie patients, although elevated CK was a common symptom in all reported cases. The isoelectric-focusing result with a mildly abnormal transferrin pattern indicates a slight effect on protein N-glycosylation. In the analysis of lipid-linked oligosaccharides, no abnormalities could be detected under low-glucose conditions in cultured fibroblasts of our patient, whereas in CDG-Ie fibroblasts, Dol-PP-GlcNAc2Man5 accumulates.29 Possibly, cell-culturing conditions for [3H]-Man incorporation for LLO analysis prevented the detection of mild abnormalities as present in the cell line of our patient. The abnormal N-glycosylation due to slightly reduced Dol-P-Man levels could be the result of two effects, i.e., reduced incorporation of mannose by the ER mannosyltransferases and also a reduced stimulation of UDP-GlcNAc:Dol-P GlcNAc-P-transferase by Dol-P-Man. This first enzyme in N-glycan biosynthesis is regulated by Dol-P-Man levels.30 Regulation of the other Dol-P-Man-dependent glycosylation pathways by Dol-P-Man concentrations is unknown. The normal GPI-anchor biosynthesis in DPM3(L85S) mutant cells suggests that this mild mutation is insufficient for affecting GPI-anchor formation. CD59 has been found reduced in cultured fibroblasts of a CDG-Ie patient,31 indicating an effect on GPI-anchor formation in the case of a more severe Dol-P-Man synthase deficiency. Finally, the normal results for serum properdin C-mannosylation indicate that the residual Dol-P-Man levels are sufficient for normal C-mannosylation in DPM3-deficient and CDG-Ie patients, whereas the severe Dol-P-Man deficiency in Lec35 cells is detrimental for C-mannosylation.28 The increased fucosylation of serum properdin is not readily explained by lowered Dol-P-Man levels. Possibly, an increased expression of protein O-fucosyltransferase as response to disease could explain this alteration. Similarly, increased expression of α1,6-fucosyltransferase has been postulated as cause for increased N-glycan core fucosylation in CDG-I patients.32 In addition, a potentially increased pool of GDP-Man due to lowered Dol-P-Man levels could increase the conversion of GDP-Man into GDP-Fucose.
Our results suggest that from all four glycosylation routes, O-mannosylation of alpha-dystroglycan catalyzed by POMT1 and POMT2 is most severely affected by decreased Dol-P-Man synthase activity. A possible explanation could be that the Km for this enzymatic reaction is higher than that for the other glycosylation routes. A reduction in Dol-P-Man levels would first affect O-mannosylation with a smaller effect on the other glycosylation routes. In HIBM33 (MIM 600737) and Sialuria patients34 (MIM 269921), decreased or increased levels of the sialic acid donor CMP-NANA mainly affected O-linked glycans and to a lesser extent N-glycans. At least in the case of Sialuria, this difference has been attributed to a higher Km value of the mucin core 1 O-glycan α2,6-sialyltransferase compared to other sialyltransferases. Although no experimental evidence exists, a regulatory role of DPM3 for alpha-dystroglycan O-mannosylation could also explain the more severe effect on O-mannosylation in comparison with the other glycosylation pathways. For example, the regulatory role of DPM2 in GPI-anchor biosynthesis27 could potentially lead to different biochemical abnormalities and a different clinical phenotype in DPM2-deficient patients. In view of the considerable residual enzymatic activity of Dol-P-Man synthase in our patient, it may be anticipated that patients with more severe mutations in DPM3 will present with a more severe multisystem presentation already in childhood. The full phenotypic spectrum will evolve when more patients with a DPM3 defect are identified.
The disease course in our patient with a slowly progressive muscle disease and elevated CK, dilated cardiomyopathy, and stroke-like episode, but no congenital brain or eye malformations, is significantly milder than in WWS or MEB (MIM 253280) disease and mimics dystroglycanopathy patients at the milder end of the spectrum similar to LGMD2I caused by loss of fukutin-related protein. Direct DNA sequencing of all coding exons and splice sites of FKRP in our patient did not show any mutations, whereas no homozygosity was identified for any of the six known dystroglycanopathy genes with coupled microsatellite markers (data not shown). In CDG-I patients, muscular dystrophy has not been reported explicitly, although severe muscle weakness was observed in dolichol kinase-deficient patients13 (CDG-Im [MIM 610768]). Progressive muscle disease with elevated CK was found in a few cases of CDG-II.35,36 Dilated cardiomyopathy has been described in milder cases of alpha-dystroglycan-deficient patients with a phenotype of limb-girdle muscular dystrophy, demonstrating an early-onset dilated cardiomyopathy already in their teens.37 Interestingly, cardiomyopathy has been reported in several children diagnosed with CDG, in most reported cases with a congenital hypertrophic or obstructive type, mostly associated with edema collection or hydrops.38 The few CDG cases described so far with a dilated cardiomyopathy included the dolichol-kinase-deficient patients.13 The occurrence of a stroke-like episode has been observed in CDG.39 In our patient, a stroke-like episode was rather peculiar; no cardiac thrombus or vascular anomaly was observed, cardiac function and blood pressure were within the normal range, and coagulation studies were normal as well. Still, the challenge to sustain an adequate level of anticoagulation after the vascular incident suggested an imbalance between different coagulants and anticoagulants. The same phenomenon has been observed in CDG-Ia (MIM 212065) patients who have minor protein C and S abnormalities and normal APTT/PT values, and who are receiving preventive anticoagulation therapy.40
Recently, it has been discussed that the main clinical features in CDG-Id (ALG3 [MIM 601110]),41 CDG-Ie (DPM1), and CDG-In (RFT1 [MIM 612015])12 could be explained by deficient N-glycosylation because of considerable overlap in the severity of symptoms. However, children with DPM1 mutations had severe congenital visual loss, optic atrophy, and seizures,29,31 and one of the children was diagnosed with congenital frontal cortical hypoplasia.14 These observations support a significant overlap with the dystroglycanopathies caused by POMGnT1, POMT1, and Fukutin mutations and are not common in N-glycosylation disorders. Children diagnosed with mutations in the MPDU1 gene (CDG-If [MIM 609180])42,43 also have reduced Dol-P-Man levels. The phenotype in two MPDU1-defective patients was reported as early-onset visual loss and severe epilepsy and is comparable to the presentation observed in children diagnosed with CDG-Ie. So far, no muscular dystrophy, cardiomyopathy, or CK elevation was described in CDG-If. The presence of skin abnormalities (erythroderma, hyperkeratosis, or ichtiosis) was absent in the so far described cases of Dol-P-Man deficiency. A recently discovered defect in GPI-anchor biosynthesis caused by mutations in the PIGM gene (MIM 610293), a mannosyltransferase, presents with refractory absence seizures and recurrent venous thrombosis.44 Interestingly, in DPM1-deficient patients, epilepsy is a prominent clinical feature, even though epilepsy occurs in other CDG subtypes, such as CDG-Ic. Our mild DPM3-deficient case did not present with epilepsy and we could not detect a GPI deficiency. Although no genetic disorders are currently associated with defective C-mannosylation, the normal results of serum properdin suggest that abnormal C-mannosylation does not contribute to the clinical phenotype in CDG-Ie and our patient.
In conclusion, it seems that in the very mild end of the clinical spectrum of Dol-P-Man-deficient patients, deficient O-mannosylation of alpha-dystroglycan is the most dominant factor in determining the clinical outcome, whereas in the more severely affected patients, other symptoms are present such as clotting abnormalities and epilepsy that could be related to, e.g., N-glycosylation and GPI synthesis defects. We have identified a genetic deficiency that we propose to name45 CDG-I(DPM3) in a patient with muscle dystrophy leading to a new cause for the mild spectrum of alpha-dystroglycanopathy patients, thereby bridging the congenital disorders of glycosylation with the dystroglycanopathies.
Recently, it has been suggested to leave the alphabetical numbers for assigning CDG subtype and include gene names to indicate function.45 We propose to keep CDG-I because this indicates an ER glycosylation defect and to include the gene name for adding function: CDG-I(DPM3). According to current CDG nomenclature, this new defect should be described as CDG-Io.
Web Resources
The URLs for data presented herein are as follows:
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
SIFT mutation prediction database, http://blocks.fhcrc.org/sift/SIFT.html
Acknowledgments
We would like to acknowledge the parents of the CDG-Ie patient and M. Garcia-Silva for providing the CDG-Ie fibroblast reference cell line. We gratefully acknowledge K. Campbell for the kind gift of IIH6 antibody. LC-MS experiments of immunopurified transferrin were performed by J. O'Brien. K. Huyben, K. Verrijp, and E. van Kaauwen are acknowledged for technical assistance. This work was supported by grants from the Deutsche Forschungsgemeinschaft and the Körber-Stiftung to L.L. and Euroglycanet (LSHM-CT2005-512131) and Metakids to D.J.L. and R.A.W. The work at the Friedrich Miescher Institute was supported by the Novartis Research Foundation.
References
- 1.Poppe M., Bourke J., Eagle M., Frosk P., Wrogemann K., Greenberg C., Muntoni F., Voit T., Straub V., Hilton-Jones D. Cardiac and respiratory failure in limb-girdle muscular dystrophy 2I. Ann. Neurol. 2004;56:738–741. doi: 10.1002/ana.20283. [DOI] [PubMed] [Google Scholar]
- 2.Brockington M., Yuva Y., Prandini P., Brown S.C., Torelli S., Benson M.A., Herrmann R., Anderson L.V., Bashir R., Burgunder J.M. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum. Mol. Genet. 2001;10:2851–2859. doi: 10.1093/hmg/10.25.2851. [DOI] [PubMed] [Google Scholar]
- 3.van Reeuwijk J., Brunner H.G., van Bokhoven H. Glyc-O-genetics of Walker-Warburg syndrome. Clin. Genet. 2005;67:281–289. doi: 10.1111/j.1399-0004.2004.00368.x. [DOI] [PubMed] [Google Scholar]
- 4.Mercuri E., Messina S., Bruno C., Mora M., Pegoraro E., Comi G.P., D'Amico A., Aiello C., Biancheri R., Berardinelli A. Congenital muscular dystrophies with defective glycosylation of dystroglycan. A population study. Neurology. 2009;72:1802–1809. doi: 10.1212/01.wnl.0000346518.68110.60. [DOI] [PubMed] [Google Scholar]
- 5.Godfrey C., Clement E., Mein R., Brockington M., Smith J., Talim B., Straub V., Robb S., Quinlivan R., Feng L. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. 2007;130:2725–2735. doi: 10.1093/brain/awm212. [DOI] [PubMed] [Google Scholar]
- 6.Burda P., Aebi M. The dolichol pathway of N-linked glycosylation. Biochim. Biophys. Acta. 1999;1426:239–257. doi: 10.1016/s0304-4165(98)00127-5. [DOI] [PubMed] [Google Scholar]
- 7.Doucey M.A., Hess D., Cacan R., Hofsteenge J. Protein C-mannosylation is enzyme-catalysed and uses dolichyl-phosphate-mannose as a precursor. Mol. Biol. Cell. 1998;9:291–300. doi: 10.1091/mbc.9.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manya H., Chiba A., Yoshida A., Wang X., Chiba Y., Jigami Y., Margolis R.U., Endo T. Demonstration of mammalian protein O-mannosyltransferase activity: Coexpression of POMT1 and POMT2 required for enzymatic activity. Proc. Natl. Acad. Sci. USA. 2004;101:500–505. doi: 10.1073/pnas.0307228101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menon A.K., Mayor S., Schwarz R.T. Biosynthesis of glycosyl-phosphatidylinositol lipids in Trypanosoma brucei: Involvement of mannosyl-phosphoryldolichol as the mannose donor. EMBO J. 1990;9:4249–4258. doi: 10.1002/j.1460-2075.1990.tb07873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maeda Y., Kinoshita T. Dolichol-phosphate mannose synthase: Structure, function and regulation. Biochim. Biophys. Acta. 2008;1780:861–868. doi: 10.1016/j.bbagen.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 11.Watanabe R., Murakami Y., Marmor M.D., Inoue N., Maeda Y., Hino J., Kangawa K., Julius M., Kinoshita T. Initial enzyme for glycosylphosphatidylinositol biosynthesis requires PIG-P and is regulated by DPM2. EMBO J. 2000;19:4402–4411. doi: 10.1093/emboj/19.16.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haeuptle M.A., Pujol F.M., Neupert C., Winchester B., Kastaniotis A.J., Aebi M., Hennet T. Human RFT1 deficiency leads to a disorder of N-linked glycosylation. Am. J. Hum. Genet. 2008;82:600–606. doi: 10.1016/j.ajhg.2007.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kranz C., Jungeblut C., Denecke J., Erlekotte A., Sohlbach C., Debus V., Kehl H.G., Harms E., Reith A., Reichel S. A defect in dolichol phosphate biosynthesis causes a new inherited disorder with death in early infancy. Am. J. Hum. Genet. 2007;80:433–440. doi: 10.1086/512130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia-Silva M.T., Matthijs G., Schollen E., Cabrera J.C., Del Pozo J.S., Herreros M.M., Simon R., Maties M., Hernandez E.M., Hennet T. Congenital disorder of glycosylation (CDG) type Ie. A new patient. J. Inherit. Metab. Dis. 2004;27:591–600. doi: 10.1023/b:boli.0000042984.42433.d8. [DOI] [PubMed] [Google Scholar]
- 15.de Jong G., van Noort W.L., van Eijk H.G. Optimized separation and quantitation of serum and cerebrospinal fluid transferrin subfractions defined by differences in iron saturation or glycan composition. Adv. Exp. Med. Biol. 1994;356:51–59. doi: 10.1007/978-1-4615-2554-7_6. [DOI] [PubMed] [Google Scholar]
- 16.Bickel T., Lehle L., Schwarz M., Aebi M., Jakob C.A. Biosynthesis of lipid-linked oligosaccharides in Saccharomyces cerevisiae: ALG13p and ALG14p form a complex required for the formation of GlcNAc2-PP-Dol. J. Biol. Chem. 2005;280:34500–34506. doi: 10.1074/jbc.M506358200. [DOI] [PubMed] [Google Scholar]
- 17.Schwarz M., Thiel C., Lubbehusen J., Dorland B., de Koning T., von Figura K., Lehle L., Korner C. Deficiency of GDP-Man:GlcNAc2-PP-dolichol mannosyltransferase causes congenital disorder of glycosylation type Ik. Am. J. Hum. Genet. 2004;74:472–481. doi: 10.1086/382492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knauer R., Lehle L., Hanefeld F., von Figura K. Normal N-oligosaccharyltransferase activity in fibroblasts from patients with carbohydrate-deficient glycoprotein syndrome. J. Inherit. Metab. Dis. 1994;17:541–544. doi: 10.1007/BF00711588. [DOI] [PubMed] [Google Scholar]
- 19.Jang-Lee J., North S.J., Sutton-Smith M., Goldberg D., Panico M., Morris H., Haslam S., Dell A. Glycomic profiling of cells and tissues by mass spectrometry: Fingerprinting and sequencing methodologies. Methods Enzymol. 2006;415:59–86. doi: 10.1016/S0076-6879(06)15005-3. [DOI] [PubMed] [Google Scholar]
- 20.Babovic-Vuksanovic D., O'Brien J.F. Laboratory diagnosis of congenital disorders of glycosylation type I by analysis of transferrin glycoforms. Mol. Diagn. Ther. 2007;11:303–311. doi: 10.1007/BF03256251. [DOI] [PubMed] [Google Scholar]
- 21.Lehle L. Biosynthesis of the core region of yeast mannoproteins. Formation of a glucosylated dolichol-bound oligosaccharide precursor, its transfer to protein and subsequent modification. Eur. J. Biochem. 1980;109:589–601. doi: 10.1111/j.1432-1033.1980.tb04832.x. [DOI] [PubMed] [Google Scholar]
- 22.Ashida H., Maeda Y., Kinoshita T. DPM1, the catalytic subunit of dolichol-phosphate mannose synthase, is tethered to and stabilized on the endoplasmic reticulum membrane by DPM3. J. Biol. Chem. 2006;281:896–904. doi: 10.1074/jbc.M511311200. [DOI] [PubMed] [Google Scholar]
- 23.Hess D., Keusch J.J., Oberstein S.A., Hennekam R.C., Hofsteenge J. Peters Plus syndrome is a new congenital disorder of glycosylation and involves defective Omicron-glycosylation of thrombospondin type 1 repeats. J. Biol. Chem. 2008;283:7354–7360. doi: 10.1074/jbc.M710251200. [DOI] [PubMed] [Google Scholar]
- 24.Lehle L., Strahl S., Tanner W. Protein glycosylation, conserved from yeast to man: A model organism helps elucidate congenital human diseases. Angew. Chem. Int. Ed. Engl. 2006;45:6802–6818. doi: 10.1002/anie.200601645. [DOI] [PubMed] [Google Scholar]
- 25.Tomita S., Inoue N., Maeda Y., Ohishi K., Takeda J., Kinoshita T. A homologue of Saccharomyces cerevisiae Dpm1p is not sufficient for synthesis of dolichol-phosphate-mannose in mammalian cells. J. Biol. Chem. 1998;273:9249–9254. doi: 10.1074/jbc.273.15.9249. [DOI] [PubMed] [Google Scholar]
- 26.Sugiyama E., DeGasperi R., Urakaze M., Chang H.M., Thomas L.J., Hyman R., Warren C.D., Yeh E.T. Identification of defects in glycosylphosphatidylinositol anchor biosynthesis in the Thy-1 expression mutants. J. Biol. Chem. 1991;266:12119–12122. [PubMed] [Google Scholar]
- 27.Maeda Y., Tomita S., Watanabe R., Ohishi K., Kinoshita T. DPM2 regulates biosynthesis of dolichol phosphate-mannose in mammalian cells: Correct subcellular localization and stabilization of DPM1, and binding of dolichol phosphate. EMBO J. 1998;17:4920–4929. doi: 10.1093/emboj/17.17.4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anand M., Rush J.S., Ray S., Doucey M.A., Weik J., Ware F.E., Hofsteenge J., Waechter C.J., Lehrman M.A. Requirement of the Lec35 gene for all known classes of monosaccharide-P-dolichol-dependent glycosyltransferase reactions in mammals. Mol. Biol. Cell. 2001;12:487–501. doi: 10.1091/mbc.12.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim S., Westphal V., Srikrishna G., Mehta D.P., Peterson S., Filiano J., Karnes P.S., Patterson M.C., Freeze H.H. Dolichol phosphate mannose synthase (DPM1) mutations define congenital disorder of glycosylation Ie (CDG-Ie) J. Clin. Invest. 2000;105:191–198. doi: 10.1172/JCI7302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kean E.L. Stimulation by dolichol phosphate-mannose and phospholipids of the biosynthesis of N-acetylglucosaminylpyrophosphoryl dolichol. J. Biol. Chem. 1985;260:12561–12571. [PubMed] [Google Scholar]
- 31.Imbach T., Schenk B., Schollen E., Burda P., Stutz A., Grunewald S., Bailie N.M., King M.D., Jaeken J., Matthijs G. Deficiency of dolichol-phosphate-mannose synthase-1 causes congenital disorder of glycosylation type Ie. J. Clin. Invest. 2000;105:233–239. doi: 10.1172/JCI8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Callewaert N., Schollen E., Vanhecke A., Jaeken J., Matthijs G., Contreras R. Increased fucosylation and reduced branching of serum glycoprotein N-glycans in all known subtypes of congenital disorder of glycosylation I. Glycobiology. 2003;13:367–375. doi: 10.1093/glycob/cwg040. [DOI] [PubMed] [Google Scholar]
- 33.Tajima Y., Uyama E., Go S., Sato C., Tao N., Kotani M., Hino H., Suzuki A., Sanai Y., Kitajima K. Distal myopathy with rimmed vacuoles: Impaired O-glycan formation in muscular glycoproteins. Am. J. Pathol. 2005;166:1121–1130. doi: 10.1016/S0002-9440(10)62332-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wopereis S., Abd Hamid U.M., Critchley A., Royle L., Dwek R.A., Morava E., Leroy J.G., Wilcken B., Lagerwerf A.J., Huijben K.M. Abnormal glycosylation with hypersialylated O-glycans in patients with Sialuria. Biochim. Biophys. Acta. 2006;1762:598–607. doi: 10.1016/j.bbadis.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 35.Peters V., Penzien J.M., Reiter G., Korner C., Hackler R., Assmann B., Fang J., Schaefer J.R., Hoffmann G.F., Heidemann P.H. Congenital disorder of glycosylation IId (CDG-IId)–a new entity: Clinical presentation with Dandy-Walker malformation and myopathy. Neuropediatrics. 2002;33:27–32. doi: 10.1055/s-2002-23597. [DOI] [PubMed] [Google Scholar]
- 36.Wu X., Steet R.A., Bohorov O., Bakker J., Newell J., Krieger M., Spaapen L., Kornfeld S., Freeze H.H. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat. Med. 2004;10:518–523. doi: 10.1038/nm1041. [DOI] [PubMed] [Google Scholar]
- 37.Murakami T., Hayashi Y.K., Noguchi S., Ogawa M., Nonaka I., Tanabe Y., Ogino M., Takada F., Eriguchi M., Kotooka N. Fukutin gene mutations cause dilated cardiomyopathy with minimal muscle weakness. Ann. Neurol. 2006;60:597–602. doi: 10.1002/ana.20973. [DOI] [PubMed] [Google Scholar]
- 38.Marquardt T., Hulskamp G., Gehrmann J., Debus V., Harms E., Kehl H.G. Severe transient myocardial ischaemia caused by hypertrophic cardiomyopathy in a patient with congenital disorder of glycosylation type Ia. Eur. J. Pediatr. 2002;161:524–527. doi: 10.1007/s00431-002-1029-2. [DOI] [PubMed] [Google Scholar]
- 39.Ishikawa N., Tajima G., Ono H., Kobayashi M. Different neuroradiological findings during two stroke-like episodes in a patient with a congenital disorder of glycosylation type Ia. Brain Dev. 2008;31:240–243. doi: 10.1016/j.braindev.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 40.van Geet C., Jaeken J., Freson K., Lenaerts T., Arnout J., Vermylen J., Hoylaerts M.F. Congenital disorders of glycosylation type Ia and IIa are associated with different primary haemostatic complications. J. Inherit. Metab. Dis. 2001;24:477–492. doi: 10.1023/a:1010581613821. [DOI] [PubMed] [Google Scholar]
- 41.Korner C., Knauer R., Stephani U., Marquardt T., Lehle L., von Figura K. Carbohydrate deficient glycoprotein syndrome type IV: Deficiency of dolichyl-P-Man:Man(5)GlcNAc(2)-PP-dolichyl mannosyltransferase. EMBO J. 1999;18:6816–6822. doi: 10.1093/emboj/18.23.6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kranz C., Denecke J., Lehrman M.A., Ray S., Kienz P., Kreissel G., Sagi D., Peter-Katalinic J., Freeze H.H., Schmid T. A mutation in the human MPDU1 gene causes congenital disorder of glycosylation type If (CDG-If) J. Clin. Invest. 2001;108:1613–1619. doi: 10.1172/JCI13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schenk B., Imbach T., Frank C.G., Grubenmann C.E., Raymond G.V., Hurvitz H., Korn-Lubetzki I., Revel-Vik S., Raas-Rotschild A., Luder A.S. MPDU1 mutations underlie a novel human congenital disorder of glycosylation, designated type If. J. Clin. Invest. 2001;108:1687–1695. doi: 10.1172/JCI13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Almeida A.M., Murakami Y., Layton D.M., Hillmen P., Sellick G.S., Maeda Y., Richards S., Patterson S., Kotsianidis I., Mollica L. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat. Med. 2006;12:846–851. doi: 10.1038/nm1410. [DOI] [PubMed] [Google Scholar]
- 45.Jaeken J., Hennet T., Freeze H.H., Matthijs G. On the nomenclature of congenital disorders of glycosylation (CDG) J. Inherit. Metab. Dis. 2008;31:669–672. doi: 10.1007/s10545-008-0983-x. [DOI] [PubMed] [Google Scholar]