

Abstract
In humans, three genes—ADRB1, ADRB2 and ADRB3—encode β-adrenoreceptors (ADRB); these molecules mediate the action of catecholamines in multiple tissues and play pivotal roles in cardiovascular, respiratory, metabolic, and immunological functions. Genetic variants in ADRB genes have been associated with widespread diseases and conditions, but inconsistent results have often been obtained. Here, we addressed the recent evolutionary history of ADRB genes in human populations. Although ADRB1 is neutrally evolving, most tests rejected neutral evolution for ADRB2 in European, African, and Asian population samples. Analysis of inferred haplotypes for ADRB2 revealed three major clades with a coalescence time of 1–1.5 million years, suggesting that the gene is either subjected to balancing selection or undergoing a selective sweep. Haplotype analysis also revealed ethnicity-specific differences. Additionally, we observed significant deviations from Hardy-Weinberg equilibrium (HWE) for ADRB2 genotypes in distinct European cohorts; HWE deviation depends on sex (only females are in disequilibrium), and genotypes displaying maximum and minimum relative fitness differ across population samples, suggesting a complex situation possibly involving epistasis or maternal selection. Overall, our data indicate that future association studies involving ADRB2 will benefit from taking into account ethnicity-specific haplotype distributions and sex-based effects. With respect to ADRB3, our data indicate that the gene has been subjected to a selective sweep in African populations, the Trp64 variant possibly representing the selection target. Given the previous association of the ancestral ADRB3 Arg64 allele with obesity and type 2 diabetes, dietary adaptations might represent the underlying selective force.
Introduction
Adrenergic receptors are G protein-coupled molecules that mediate the action of catecholamines in multiple tissues. In particular, they are an integral part of the sympathetic nervous system and play pivotal roles in cardiovascular, respiratory, metabolic, and immunological functions.
Adrenergic receptors have been subdivided into two major types, α and β, on the basis of agonist-mediated responses, with subsequent classification into subtypes based on differential tissue localization (reviewed in 1). As far as members of the β type (ADRB) are concerned, three distinct receptors have been identified in humans: β1, β2 and β3, all encoded by small intronless or single-intron genes located on chromosomes 10, 5 and 8, respectively.
ADRBs have been the subjects of intensive investigations as a result of their possible role in the pathophysiology of widespread conditions such as insulin resistance, obesity (MIM 601665), asthma (MIM 600807), and cardiovascular disorders.2–6 Also, ADRBs are targets of many commonly used drugs; thus, the identification and analysis of functional variants is extremely relevant to pharamacogenetic studies. As a consequence, commonly occurring polymorphisms have been searched for and studied in ADRB genes and particular focus has been placed on nonsynonymous SNPs. A common R389G variant in ADRB1 (MIM 109630) has been shown7 to influence the receptor's coupling properties; the association of this SNP with obesity is controversial,8,9 but experiments in transgenic mice have indicated that the R389 allele predisposes to heart failure.10 Of four relatively common nonsynonymous SNPs in ADRB2 (MIM 109690), three are functional in vitro: Gly16 leads to enhanced agonist-mediated downregulation,11 Glu27 reduces such regulation,11 and Ile64 shows12 impaired agonist binding and decreased adenyl cyclase activity. Moreover, additional functional variants have been identified in the promoter region of ADRB2.13 The phenotypic effect of coding SNPs has been investigated in many studies; associations of Gly16 and Glu27 with asthma and obesity, respectively, have been reported, although not always validated in independent studies (reviewed in 14). Similarly, contradictory results have been obtained among studies aiming at correlating genetic variants in ADRB2 with hypertension (reviewed in 15). In addition, conditions such as autism (MIM 209850),16,17 preterm delivery,18–21 cerebral palsy,22 parasitic infection,23 rheumatoid arthritis (MIM 180300),24,25 and temporomandibular joint disorder26 have been associated with ADRB2 polymorphisms. With respect to ADRB3 (MIM 109691), a low-frequency ancestral allele has been associated with obesity and type 2 diabetes (MIM 125853) in some but not all populations (reviewed in 27).
Therefore, in analogy to many other cases, studies aiming at associating specific variants with common diseases have often been inconsistent with one another; in general, the lack of consistency among association studies is thought to be due to both false-positive and false-negative results rather than to variability in association for populations with different ethnic origins.28 Whatever the reason, a clear picture of whether ADRB variants influence disease susceptibility is still missing.
Further insight into the genetic basis of common diseases can be obtained through molecular evolutionary studies of candidate loci; indeed, inference about selection models operating on specific gene regions implicitly implies the presence of functional variants with some effect on fitness (at some time in human history). Moreover, it has recently been proposed (reviewed in 29,30) that the shift of environment and lifestyle that has been associated with two extremely relevant transitions, the out-of-Africa migration and the development of agricultural methods, might have rendered maladaptive genetic variants that had been selected for in earlier stages of human history. This hypothesis has been explicitly formulated for a few common diseases, such as obesity, hypertension, and asthma. On these bases, we set out to verify whether the neutral model of evolution applies to ADRB genes in humans.
Material and Methods
DNA Samples, Sequencing, and Genotyping
Human genomic DNA for East Asians (EAS), Australian Aborigines (AUA), and native South Americans (NSA) was obtained from either the Coriell Institute for Medical Research or the European Collection of Cell Cultures. For population genetics analyses, the 6 kb genomic portion encompassing ADRB2 coding and promoter regions was PCR amplified in overlapping fragments. PCR products were treated with ExoSAP-IT (USB Corporation, Cleveland, OH, USA), directly sequenced on both strands with a Big Dye Terminator Sequencing Kit (v3.1 Applied Biosystems), and run on an Applied Biosystems ABI 3130 XL Genetic Analyzer (Applied Biosystems). Sequences were assembled with AutoAssembler version 1.4.0 (Applied Biosystems) and inspected manually by two distinct operators, and singletons were reamplified and resequenced. Primer sequences are available upon request. The number of subjects resequenced was as follows: AUA, 12; NSA, 24; EAS, 25.
For the study of haplotype and genotype frequencies in an Italian population sample, rs1042714 (Arg16Gly) and rs1042713 (Gln27Glu) were genotyped in 654 Italian individuals. Genomic DNA from these subjects was obtained from different sources; in particular, 263 subjects were collected and genotyped as previously described,31 and the remaining individuals were either provided by the Telethon Bank of DNA, Nerve and Muscle Tissues (no. GTF02008), located at the Department of Neurological Sciences, IRCCS Ospedale Maggiore Policlinico, Mangiagalli and Regina Elena Foundation, Milan, or recruited as volunteers at the Scientific Institute IRCCS E.Medea, after informed consent was obtained. Genotyping for these subjects was performed by direct sequencing of the gene region encompassing rs1042714 and rs1042713, with the use of the same procedures described above. The study was approved by the Ethical Committee of the Scientific Institute IRCCS E. Medea-Associazione La Nostra Famiglia, in agreement with Italian standards.
Data Retrieval and Haplotype Construction
Genotype data for two population samples, one of African and one of European ancestry, were retrieved from the SeattleSNPs website. In particular, ADRB2 and ADRB3 have been resequenced in 24 Yoruba (YRI) and 24 Utah residents with ancestry from northern and western Europe. Data for ADRB1 refer to 24 African Americans (AA) and 23 individuals with European ancestry (20 Utah residents with ancestry from northern and western Europe and three French individuals). Populations with European ancestry are hereafter referred to as EU. The nucleotide positions for all analyzed genes correspond to those of SeattleSNPs, which in turn derive from the following GenBank accession numbers: ADRB1, AY567837; ADRB2, DQ094845; ADRB3, DQ104441.
Genotype data for 238 resequenced human genes were derived from the National Institute of Environmental Health Sciences (NIEHS) SNPs Program website. In particular, we selected genes that had been resequenced in genotype samples of populations of defined ethnicity, including AA, EU, YRI, and EAS (NIEHS panel 2). Data from the AA and EU samples were used as a comparison for ADRB1, which has been resequenced in these same populations. Similarly, for ADRB2 and ADRB3, the comparison was performed with YRI and EU samples.
Haplotypes were inferred with PHASE version 2.1,32,33 a program for reconstructing haplotypes from unrelated genotype data through a Bayesian statistical method. Inferred haplotypes for all individuals used in this study are available in the Supplemental Data (Tables S1 and S2, available online).
Statistical Analysis
Tajima's D statistic,34 Fu and Li's D∗ and F∗35 statistics, as well as diversity parameters θW36 and π37 and Fay and Wu's H,38 were calculated with libsequence,39 a C++ class library providing an object-oriented framework for the analysis of molecular population-genetic data. The scaled per-nucleotide recombination parameter (R = 4Ner) was estimated with MaxDip.40
Calibrated coalescent simulations were performed with the cosi package41 and its best-fit parameters for YRI, AA, EU, and Asian populations with 10000 iterations.
A composite-likelihood-ratio test and coalescent simulations under a selective sweep regime were performed with the clsw and ssw programs, kindly provided by Yuseob Kim.
The FST statistic42 estimates genetic differentiation among populations and was calculated as previously proposed.43
The multilocus HKA test was performed with the HKA software distributed by Jody Hey. Sixteen reference loci were randomly selected among NIEHS loci that are shorter than 20 kb and have been resequenced in genotype samples of the three populations (YRI, EU, and EAS; panel 2). The only criterion was that Tajima's D did not suggest the action of natural selection (i.e., DT is higher than the 5th and lower than the 95th percentiles in the distribution of NIEHS genes). The reference set was accounted for by the following genes: VNN3 (MIM 606592), PLA2G2D (MIM 605630), MB (MIM 160000), MAD2L2 (MIM 604094), HRAS (MIM 190020), CYP17A1 (MIM 609300), ATOX1 (MIM 602270), BNIP3 (MIM 603293), CDC20 (MIM 603618), NGB (MIM 605304), TUBA1 (MIM 191110), MT3 (MIM 139255), NUDT1 (MIM 600312), PRDX5 (MIM 606583), RETN (MIM 605565), and JUND (MIM 165162). For each locus, 1000 coalescent simulations were performed with the cosi package41 and only neutrally evolving sites were considered.
Median-joining networks for the inference of haplotype genealogy were constructed with NETWORK 4.5.44 Estimate of the time to the most recent common ancestor (TMRCA) was obtained via a phylogeny-based approach implemented in NETWORK, with the use of a mutation rate based on the number of fixed differences between human and chimpanzee and the assumption of a separation time from humans of 6 MY ago.45 A second TMRCA estimate was derived from application of a maximum-likelihood coalescent method implemented in GENETREE.46,47 Again, the mutation rate μ was obtained on the basis of the divergence between human and chimpanzee and under the assumption of a generation time of 25 yrs. Using this μ and θ maximum likelihood (θML), we estimated the effective population-size parameter (Ne). With these assumptions, the coalescence time, scaled in 2Ne units, was converted into years. For the coalescence process, 106 simulations were performed.
All calculations were performed in the R environment.48
Results
Nucleotide Diversity and Neutrality Tests for ADRB Genes
The three ADRB genes have been included in the Seattle SNPs variation-discovery resource (SeattleSNPs, NHLBI Program for Genomic Applications) and have been fully resequenced in samples of two populations with African (either YRI or AA) and European origin (EU).
We exploited the availability of these data to calculate nucleotide-diversity parameters for the three genes in these population samples; in particular, all the analyses reported below have been performed on gene regions extending from the most 5′ resequenced nucleotide to the 3′ UTR end.
Nucleotide diversity was assessed through two indexes: θW,36 an estimate of the expected per-site heterozigosity, and π,37 the average number of pairwise sequence nucleotide differences. Under neutral evolution, values of θW and π are roughly equal. The Tajima's D statistic (DT,34) is commonly used to evaluate their difference and, therefore, departure from neutrality. Positive values of DT indicate an excess of intermediate-frequency variants and are evidence of balancing selection, and negative DT values indicate either purifying selection or a high representation of rare variants as a result of a selective sweep. Fu and Li's F and D (as well as F∗ and D∗35) statistics are also based on SNP frequency spectra and differ from Tajima's D in that they also take into account whether mutations occur in external or internal branches of a genealogy. Because population history, in addition to selective processes, is known to affect frequency spectra and, therefore, all related statistics, we performed coalescent simulations by using a population genetics model that incorporates demographic scenarios.41 Moreover, to the same aim, we exploited the conundrum whereby selection acts on a single locus while demography affects the whole genome, calculating diversity parameters and test statistics for a set of 238 genes resequenced by the NIEHS program; in particular, a 5 kb window was randomly selected for each gene (see Material and Methods).
Data for ADRB genes are reported in Table 1 and indicate that no departure from neutrality is observed for ADRB1. Conversely, significantly positive values of DT and Fu and Li's D and F were obtained for ADRB2 in Europeans. In Africans, the presence of nine singletons in ADRB2 yielded low values for Fu and Li's statistics but DT ranked highly when compared to the distribution of 238 resequenced genes in African populations (Table 2). Moreover, both θW and π results were higher for ADRB2 than for most NIEHS genes (Table 1).
Table 1.
Summary Statistics for ADRB Genes
|
θWd |
πd |
Tajima's D |
Fu & Li's D |
Fu & Li's F |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | La | Pop. | Nb | Sc | Value | Ranke | Value | Ranke | Value | pf | Value | pf | Value | pf |
| ADRB1 | 9.4 | AA | 48 | 35 | 8.73 | 0.50 | 5.75 | 0.31 | −1.16 | 0.17 | 0.72 | 0.037 | 0.02 | 0.16 |
| EU | 46 | 22 | 5.54 | 0.53 | 7.39 | 0.69 | 1.09 | 0.099 | 1.12 | 0.059 | 1.33 | 0.048 | ||
| ADRB2 | 6 | YRI | 48 | 35 | 13.65 | 0.89 | 17.86 | 0.98 | 1.05 | 0.0066 | −0.29 | 0.34 | 0.26 | 0.12 |
| EU | 46 | 27 | 10.63 | 0.93 | 19.17 | 0.98 | 2.68 | 0.0003 | 1.32 | 0.031 | 2.20 | 0.0012 | ||
| ADRB3 | 7.4 | YRI | 48 | 23 | 7.96 | 0.42 | 3.47 | 0.11 | −1.84 | 0.048 | −1.75 | 0.12 | −2.15 | 0.060 |
| EU | 46 | 8 | 2.80 | 0.14 | 1.59 | 0.09 | −1.20 | 0.049 | −1.62 | 0.090 | −1.76 | 0.064 | ||
Length of the analyzed region (from the most 5′ resequenced nucleotide to the end of the 3′ UTR).
Sample size (chromosomes).
Number of segregating sites.
Nucleotide diversity indexes (× 10−4).
Percentile rank relative to the distribution of 238 5 kb windows.
p values obtained from coalescent simulations.
Table 2.
Summary Statistics for ADRB2 in Five Human Populations
|
Tajima's D |
Fu & Li's D∗ |
Fu & Li's F∗ |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pop. | Na | Sb | Value | pc | Rankd | Value | pc | Rankd | Value | pc | Rankd |
| YRI | 48 | 35 | 1.05 | 0.0066 | 0.99 | −0.22 | 0.34 | 0.59 | 0.27 | 0.11 | 0.82 |
| EU | 46 | 27 | 2.68 | 0.0003 | 0.99 | 0.93 | 0.083 | 0.91 | 1.83 | 0.0043 | 0.99 |
| EAS | 50 | 30 | 1.59 | 0.038 | 0.94 | 1.56 | 0.0052 | 0.99 | 1.87 | 0.0053 | 0.99 |
| NSA | 48 | 22 | 1.33 | 0.047 | n.a.e | 1.38 | 0.035 | n.a.e | 1.61 | 0.020 | n.a.e |
| AUA | 24 | 26 | 0.69 | 0.18 | n.a.e | 0.75 | 0.18 | n.a.e | 0.85 | 0.17 | n.a.e |
Sample size (chromosomes).
Number of segregating sites.
p values obtained from coalescent simulations.
Percentile rank relative to the distribution of 5 kb windows.
Not available.
Finally, analysis of ADRB3 revealed negative statistics (with marginally significant p values), as well as low levels of nucleotide variation, in YRI and EU (Table 1).
These findings prompted us to further investigate the evolutionary history of ADRB2 and ADRB3.
The Signature of Selection at ADRB2
As a first step, we wished to verify whether rejection of the neutral model was also verified for other human population samples. We therefore resequenced the 6 kb encompassing ADRB2 coding and promoter regions in three additional populations: EAS, NSA, and AUA. When model-fitting parameters were available (i.e., for EAS), we applied a population genetics model41 for coalescent simulations; in the case of AUA and NSA, the standard neutral model was used. Nucleotide diversity parameters and neutrality tests are shown in Table 2 and indicate that, in analogy to EU, one or more statistics rejected neutrality for all population samples, excluding AUA. In agreement with these data, both DT and Fu and Li's F∗ and D∗ display a high percentile rank in the distribution of NIEHS genes for EU and EAS (Table 2).
Under neutral evolution, the amount of within-species diversity is predicted to correlate with levels of between-species divergence, because both depend on the neutral mutation rate.49 We performed a multilocus HKA test by using the HKA software, which allows estimation of statistical significance through coalescent simulations (see Material and Methods). As above, the latter were performed with the use of a previously described demographic model;41 due to the lack of a demographic model, the test was not performed for AUA and NSA. Significant results were obtained for EU, YRI, and EAS (Table 3), and in all cases, the test of maximum cell value50 indicated ADRB2 as an outlier, its removal from the gene set yielding nonsignificant tests (Table 3).
Table 3.
Multilocus HKA Results for ADRB2
|
Multilocus HKA |
Multilocus HKA Excluding ADRB2 |
|||
|---|---|---|---|---|
| Pop. | Likelihooda | Max Cell Valueb | Likelihooda | Max Cell Valueb |
| YRI | 16.94 (0.008) | 5 (<0.001) | 10.66 (0.19) | 1.84 (0.16) |
| EU | 28.54 (0.008) | 6.52 (0.010) | 20.53 (0.07) | 2.55 (0.28) |
| EAS | 30.07 (0.019) | 10.38 (<0.001) | 19.05 (0.17) | 3.71 (0.18) |
Sum of deviations, p values in parentheses.
Maximum cell value test, p values in parentheses.
Next, we wished to analyze the structure of ADRB2 inferred haplotypes. Construction of a median-joining network44 (Figure 1) revealed the presence of three major clades (haplogroups A, B, and C, hereafter referred to as HA, HB, and HC) separated by long branch lengths, each containing common inferred haplotypes. HC is further split into two subclades (referred to as HC 1 and HC2). In order to estimate the TMRCA of the three inferred haplotype clades, we applied a phylogeny-based method44 based on the measure ρ, the average pairwise difference between the two haplotype clusters. The result of ρ was equal to 10.58, so that, with the use of a mutation rate based on 77 fixed differences between chimpanzee and human and a separation time of 6 MY,45 we estimated a TMRCA of 1.65 MY (SD: 352 KY). In order to obtain a second TMRCA estimate, we used GENETREE, which is based on a maximum-likelihood coalescent analysis.46,47 The method assumes an infinite-site model without recombination, and, therefore, haplotypes and sites that violate these assumptions need to be removed. In this case, 16 single segregating sites had to be removed. The resulting gene tree, rooted with the use of the chimpanzee sequence, is partitioned into three major branches (Figure S1). A maximum-likelihood θ estimate (θML) of 4 was obtained, resulting in an estimated effective population size (Ne) of 10667, a value comparable to most figures reported in the literature.51 With this method, the TMRCA amounted to 1.05 MY (SD: 293 KY). A third TMRCA estimate of 1.56 MY was obtained by applying a previously described method52 that calculates the average sequence divergence separating the MRCA and each of the chromosomes. All TMRCAs are not unusually deep; estimates for neutrally evolving autosomal human loci range between 0.8 and 1.5 MY.51
Figure 1.
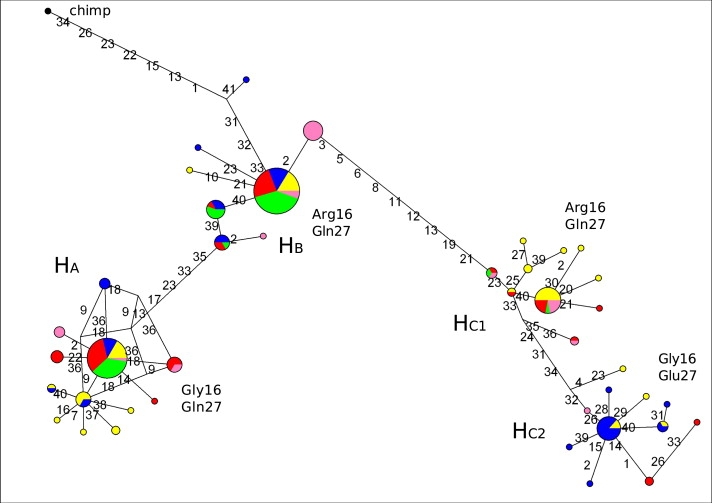
Genealogy of ADRB2 Inferred Haplotypes Reconstructed through a Median-Joining Network
Each node represents a different inferred haplotype, the size of the circle proportional to frequency. Nucleotide differences between haplotypes are indicated on the branches of the network. Circles are color coded according to population (yellow: YRI, green: NSA, blue: EU, red: EAS, pink: AUA). The chimpanzee sequence is also shown (black circle). Note that the relative position of mutations along a branch is arbitrary. The amino acid status at positions 16 and 27 is shown for the four major inferred haplotypes.
The structure of the inferred haplotype genealogy deserves some comments. The allelic status at amino acid positions 16 and 27 is shown above each haplogroup and indicates that chromosomes carrying Arg16/Gln27 alleles are split among HB and HC1 in all population samples except for EU (there are no EU chromosomes in haplogroup C1); conversely, both Gly16/Gln27 and Gly16/Glu27 chromosomes cluster together to HA and HC2, respectively. Therefore, we analyzed the location and possible functional significance of variants along the branches separating HA-HB, HB-HC1, and HC1-HC2.
HA and HB are separated by five mutations (Figure 1). In addition to codon 16, one of them (rs12654778) falls within a noncoding region, upstream of the 5′ UTR, that is highly conserved among mammals. With respect to variants separating HB and HC1, all of them are located upstream of the transcription start site, suggesting that the two clades have been maintained by transcription-regulatory variants. Analysis of sequence conservation along this genomic region indicated that variant 13 (rs17778257) is located within an element highly conserved among placental mammals. Further supporting their role as regulatory variants, both of these SNPs located within highly conserved sequences have previously been identified as cis-acting expression QTLs in a genome-wide analysis.53
Drysdale and colleagues54 had previously analyzed ADRB2 haplotype structure: their haplotypes 2 and 4, corresponding to HB and HC1, showed differential promoter activity, in line with the vision whereby variants along branch HB/HC1 affect gene expression levels. Finally, HC1 and HC2 differ with respect to codons 16 and 27.
There are different possible reasons underlying selection signatures at human loci. Therefore, we calculated the ratio of observed heterozygosity to expected gene diversity. In order to obtain an estimate of this parameter in the human genome, ratios were also calculated for 5 kb windows deriving from NIEHS genes. Surprisingly, the observed heterozygosity to expected gene diversity for ADRB2 in EU and EAS falls below the fifth percentile of values obtained from NIEHS genes (Table S3). We therefore wished to verify whether any deviation from Hardy-Weinberg equilibrium (HWE) could be observed. In EU, who lack inferred haplotypes in clade HC1, typing of rs1042714 (Arg16Gly) and rs1042713 (Gln27Glu) allows unambiguous reconstruction of the six possible genotypes deriving from the segregation of the three ADRB2 inferred haplotypes. Analysis of the 60 unrelated CEPH individuals in HapMap indicated a marginally significant deviation from HWE and an excess of homozygotes (Table 4). Therefore, we searched the literature, looking for ADRB2 genotypes at both polymorphisms in large European cohorts. In the two available studies,55,56 we observed a marginally significant deviation in women in one study only, which we replicated in our own genotyped Italian sample (Table 4). Because deviations from HWE are not observed in all cohorts and display marginally significant p values (with the exception of the French female sample), we cannot reach definitive conclusions, although the possibility that the results are due to genotyping artifacts or other unrecognized biases (e.g., a deletion segregating in the populations) is unlikely, given that (1) HWE deviation was observed in women but not in men, (2) different studies used different genotyping techniques, and (3) by analyzing 73 Italian trios, we did not detected inconsistent segregation of ADRB2 inferred haplotypes (not shown).
Table 4.
Genotype Counts, Inferred Haplotype Frequencies, and HWE p Values in Four Populations
|
Genotype Counts |
Haplotype Frequencies |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pop. | Count | HAHA | HBHB | HCHC | HAHB | HAHC | HBHC | Sample Size | HA | HB | HC | HWE p Value |
| CEUd | ||||||||||||
| All | OBSa | 5 | 7 | 19 | 11 | 5 | 13 | 60 | 0.22 | 0.32 | 0.46 | 0.0356 |
| EXPb | 2.82 | 6.02 | 13.07 | 8.23 | 12.13 | 17.73 | ||||||
| Dutche | ||||||||||||
| All | OBSa | 315 | 1376 | 1776 | 1257 | 1481 | 2986 | 9191 | 0.18 | 0.38 | 0.44 | 0.256 |
| EXPb | 308.5 | 1330.9 | 1749.1 | 1281.6 | 1469.3 | 3051.5 | ||||||
| RFc | 0.99 | 1 | 0.99 | 0.96 | 0.99 | 0.96 | ||||||
| Females | OBSa | 175 | 759 | 996 | 700 | 845 | 1603 | 5078 | 0.19 | 0.38 | 0.44 | 0.094 |
| EXPb | 177.6 | 717.9 | 969.7 | 714.1 | 829.9 | 1668.8 | ||||||
| RFc | 0.93 | 1 | 0.97 | 0.93 | 0.96 | 0.91 | ||||||
| Males | OBSa | 140 | 608 | 780 | 557 | 637 | 1383 | 4105 | 0.18 | 0.38 | 0.44 | 0.874 |
| EXPb | 133.0 | 605.3 | 780.3 | 567.5 | 644.3 | 1374.6 | ||||||
| RFc | 1 | 0.95 | 0.95 | 0.93 | 0.94 | 0.96 | ||||||
| Frenchf | ||||||||||||
| All | OBSa | 58 | 157 | 181 | 158 | 190 | 385 | 1129 | 0.205 | 0.380 | 0.415 | 0.053 |
| EXPb | 47.67 | 162.63 | 194.41 | 176.11 | 192.55 | 355.63 | ||||||
| RFc | 1 | 0.79 | 0.77 | 0.74 | 0.81 | 0.89 | ||||||
| Females | OBSa | 30 | 85 | 89 | 63 | 98 | 197 | 562 | 0.20 | 0.38 | 0.42 | 0.009 |
| EXPb | 21.8 | 82.4 | 99.6 | 84.8 | 93.2 | 181.2 | ||||||
| RFc | 1 | 0.75 | 0.65 | 0.54 | 0.76 | 0.79 | ||||||
| Males | OBSa | 28 | 72 | 92 | 95 | 92 | 188 | 567 | 0.21 | 0.38 | 0.41 | 0.421 |
| EXPb | 26.0 | 80.6 | 94.8 | 91.5 | 99.3 | 174.9 | ||||||
| RFc | 1 | 0.83 | 0.90 | 0.96 | 0.86 | 1 | ||||||
| Italiang | ||||||||||||
| All | OBSa | 35 | 109 | 78 | 132 | 135 | 165 | 654 | 0.26 | 0.39 | 0.35 | 0.110 |
| EXPb | 43.4 | 101.4 | 79.5 | 132.7 | 117.5 | 179.5 | ||||||
| RFc | 0.70 | 0.94 | 0.85 | 0.87 | 1 | 0.83 | ||||||
| Females | OBSa | 13 | 53 | 40 | 63 | 69 | 72 | 310 | 0.25 | 0.39 | 0.36 | 0.0368 |
| EXPb | 20.1 | 46.8 | 39.4 | 61.4 | 56.3 | 85.9 | ||||||
| RFc | 0.53 | 0.92 | 0.83 | 0.84 | 1 | 0.68 | ||||||
| Males | OBSa | 22 | 56 | 38 | 69 | 66 | 93 | 344 | 0.26 | 0.40 | 0.34 | 0.876 |
| EXPb | 23.3 | 54.6 | 40.1 | 71.3 | 61.1 | 93.6 | ||||||
| RFc | 0.88 | 0.95 | 0.88 | 0.90 | 1 | 0.92 | ||||||
A Selective Sweep at ADRB3
As reported above, negative values of frequency-spectrum-based statistics were obtained for ADRB3. This result can be interpreted as evidence of either purifying selection or selective sweep, given that both processes result in an excess of low-frequency variants. Fay and Wu's H is usually applied to distinguish between the two possibilities.38 Negative H values indicate an excess of high-frequency derived alleles, a finding consistent with the action of directional but not purifying selection. Given that selective sweeps are expected to affect genomic regions surrounding the selected variant(s), we calculated H (and performed all successive analyses) over the whole resequenced region (which extends ∼3.8 kb downstream of the 3′ UTR). A significantly negative H value was obtained for YRI and EU (H = −6.72, p = 0.014 and H = −7.33, p = 0.045, respectively).
In order to obtain a further confirmation, we applied a composite-likelihood ratio test (CLR57), which evaluates the local reduction of variation and skew of the frequency spectrum. At first, we evaluated statistical significance of likelihood ratio (LR) values in two ways: distinguishing ancestral from derived alleles and then not distinguishing allele states (Test 1 and Test 2, respectively). Results are summarized in Table 5 and indicate that, after coalescent simulations, data for Africans fit significantly better a hitchhiking event for both conditions; conversely, for Europeans, only Test 1 gave significant results. Given that CLR is not robust to demographic history, we applied a goodness of fit (GOF) test;58 this method specifically tests how well a selective sweep model fits the data, as opposed to a generalized alternative model, by simulating genealogies under directional selection. Thus, nonsignificant p values represent a good fit of the sweep model to the data, whereas low p values fall within the range of effects that can also be generated by demographic events such as population bottlenecks or undetected population structure. For YRI, the GOF p values suggest that rejection of neutrality by the CLR test is more likely due to a selective sweep than to demographic history (Table 5). In contrast, the GOF test fails to discard a relevant demographic effect for Europeans.
Table 5.
Composite Likelihood Ratio Test for ADRB3
|
Test 1a |
Test 2b |
Test 3c |
|||
|---|---|---|---|---|---|
| Pop. | LRd | GOFe | LRd | GOFe | LRd |
| YRI | 12.74 (0.002) | 453 (0.38) | 12.45 (<0.0001) | 3438 (0.060) | 7.26 (0.60) |
| EU | 9.34 (0.002) | 550 (0.040) | 3.25 (0.07) | 1615 (0.044) | n.a.f |
Distinguishing ancestral and derived allele.
Nondistinguishing ancestral and derived allele.
Distinguishing ancestral and derived allele only for inferred haplotypes carrying the putative selected allele (W64).
Likelihood ratio, p values in parentheses.
Goodness-of-fit test, p values in parentheses.
Not analyzed.
As reported in the Introduction, the ancestral R64 allele has been associated with obesity and insulin resistance in some populations, and its frequency is relatively low in Europeans, Asians, and Africans.29 As a consequence, the W64 derived allele, the only high-frequency derived allele in the ADRB3 coding sequence, might be a good candidate to represent the selected variant. Therefore, we performed an additional test for YRI under the assumption that an incomplete sweep has been ongoing, with the W64 allele being the selected site. To this aim, only inferred haplotypes carrying the W64 allele were selected and LR value was compared with those obtained by simulating data under a complete sweep model59 (Test 3). Nonsignificant p values (Table 5) make this evolutionary hypothesis very appealing.
Haplotype analysis indicated that a total of 27 ADRB3 inferred haplotypes can be identified (Figure 2), but one inferred haplotype carrying the W64 allele occurred 55 times and accounted for 45.8% and 71% of African and European chromosomes, respectively. The Hudson haplotype test60 for the W64 variant indicated that significantly fewer (p = 0.0046) inferred haplotypes than expected under neutrality are observed in YRI. We then constructed a median-joining network for ADRB3 inferred haplotypes (Figure 3). Given the presence of a few recombination events immediately downstream of the transcription end site, the gene region extending from the most 5′ resequenced nucleotide to the 3′ UTR end was used for this analysis. As shown in Figure 3, all haplotypes carrying the W64 allele (mutation 11 in the network) cluster together or are few steps away from the major haplotype, except for a divergent branch of African haplotypes.
Figure 2.
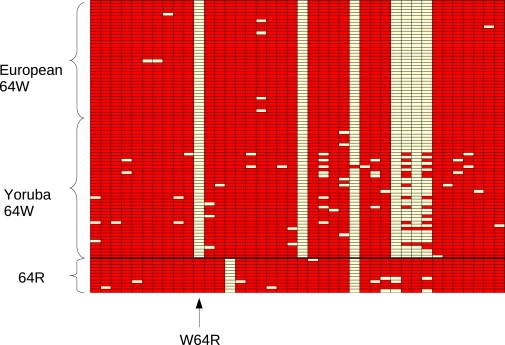
Analysis of ADRB3 Inferred Haplotypes
Polymorphic positions are color coded according to their allelic state (white: derived, red: ancestral). The position of the W64R variant is shown, and inferred haplotypes are ordered on the basis of the allele at codon 64.
Figure 3.
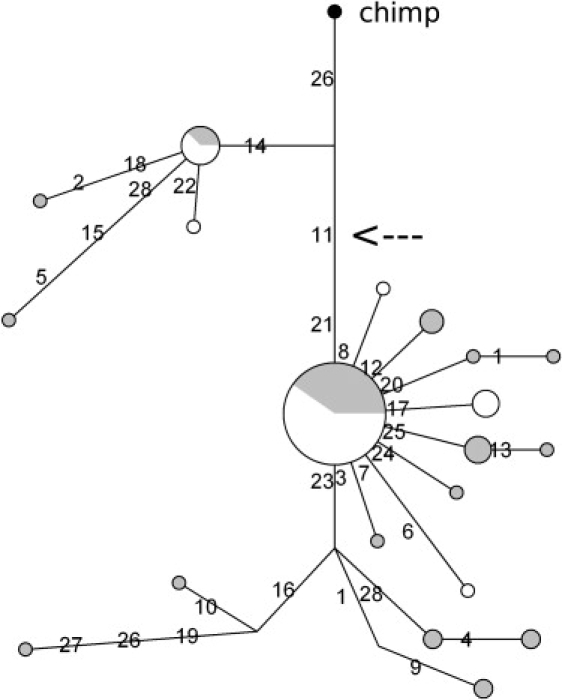
Genealogy of ADRB3 Inferred Haplotypes Reconstructed through a Median-Joining Network
Each node represents a different inferred haplotype, the size of the circle proportional to frequency. Nucleotide differences between haplotypes are indicated on the branches of the network. Circles are color-coded according to population (gray: YRI, white: EU). The chimpanzee sequence is also shown (black circle), and the arrow indicates the W64 polymorphism.
These data are consistent with directional selection at the ADRB3 locus in YRI. With respect to the European sample, most tests failed to reject neutrality; yet, it should be noted that only eight SNPs are segregating in this population, possibly resulting in insufficient power to detect deviations from the neutral expectations.
Discussion
In this work, we wished to analyze the evolutionary history of ADRB genes in humans, with particular concern to selective patterns. Indeed, it has been proposed61,62 that population genetics approaches can be regarded as a counterpart to classic association studies, in that by focusing on the effect of susceptibility alleles on evolutionary fitness, they allow inference of functional relevance. Consistently, it has been shown63 that genes targeted by selection acting on segregating variants are more likely to be associated with human diseases.
Data reported herein indicated that the genomic region covering the promoter and coding sequences of ADRB2 displays high nucleotide diversity, an excess of intermediate-frequency alleles, and a higher level of within-species diversity in comparison to interspecific divergence. All of these features strongly suggest the action of balancing selection. In particular, analysis of inferred haplotypes indicated that the three clades are likely to be maintained by both coding and regulatory balanced SNPs, being consistent with a model of multiallelic balancing selection. As shown by the median-joining network (Figure 1), three lineages are observed (as opposed to two expected clades in the case of a biallelic model). This is in line with values of the test statistics (Tajima's D and Fu and Li's F∗ and D∗), which are not strikingly positive; the skew toward intermediate frequency variants tends to be less marked in a multiallelic selection model than in the case of biallelic selection. Estimation of TMRCAs for ADRB2 inferred haplotype clades indicated that they are not unusually deep. Thus, an alternative possibility is that variants in the gene are going through a selective sweep; indeed, both balancing and positive selection are initiated by the spread in a population of a newly selected allele(s) until either selection opposes (balanced situation) or promotes (complete sweep) its fixation. Population genetics analyses such as the one reported herein provide a snapshot of a dynamic evolutionary process, making it difficult to precisely determine the underlying selective regime.
It is worth mentioning that the ADRB2 inferred haplotype structure might harbor implications for association studies. Indeed, whereas in Europeans, all chromosomes carrying alleles Arg16 and Gln27 display the same promoter structure (clade HB), in all other population samples, inferred haplotypes harboring these coding variants are split into two groups (HB and HC1) with different alleles in the promoter region and, possibly, different transcriptional activity.53,54 This observation might partially explain the low consistency among association studies in different population samples.
In analogy to the underlying selective regimes, the nature of selective pressures acting on human genes is often difficult to identify. Our analyses of different European cohorts suggests that ADRB2 inferred haplotypes deviate from HWE in females but not in males. Deviations from HWE in a sex-dependent manner have previously been reported for variants in the BRCA2 gene (MIM 600185) in humans.64 Still, in the case of ADRB2, deviations from HWE often display marginally significant p values, and genotypes with maximum and minimum relative fitness are different across the three European populations (Table 4), an observation difficult to reconcile with simple selection models. Previous theoretical modeling has demonstrated that different situations, including maternal (parental) selection65,66 and epistasis,67 can result in complex genotype distributions and oscillations in genotype frequencies. In theory, both effects might apply to ADRB2, although maternal selection is supported by previous findings indicating that both fetal18,22 and maternal19–21 genotype at single ADRB2 SNPs predispose to preterm delivery and cerebral palsy. Unfortunately, none of these studies addressed the role of sex in pregnancy outcome. A known example of genetic variation influencing intrauterine viability concerns the methylenetetrahydrofolate reductase gene (MTHFR [MIM 607093]),68 and, in analogy to what we observed for ADRB2, deviations from HWE in different human populations have been observed, as an excess of either homozygotes or heterozygotes in Italy and Finland, respectively.69 Another instance involves the phosphoglucomutase 1 (PGM1 [MIM 171900]) gene. The joint maternal-neonatal distribution of PGM1 genotypes has been shown to deviate from that expected under HWE,70 and the same authors reported that a significant association is observed between birth weight and mother-newborn PGM1 genotype in infant females but not males.71
Additional studies will be required for verifying whether selection for intrauterine viability (or viability in adult life) does occur on ADRB2 haplotypes and determining whether it is responsible for the selection signature that we observed. Indeed, the central role played by ADRB2 in many physiologic pathways suggests that it might have been a target of diverse selective pressures.
Polymorphic variations in the gene have been associated with three extremely frequent conditions: asthma, hypertension, and obesity (or insulin resistance). Some recent theories29,30 proposed these common diseases to be widespread in human populations because susceptibility alleles have been selected for during human evolution and have more recently become unfavorable as a consequence of changed environmental condition and lifestyle. In particular, it has been suggested29 that alleles responsible for type 2 diabetes might have evolved as “thrifty” variants in ancient populations; similarly, variants predisposing to asthma and atopy possibly conferred increased resistance to parasites in endemic areas. Finally, susceptibility alleles for hypertension were proposed to have conferred selective advantage to populations adapted to hot and humid climates in order to maximize sodium retention or vascular reactivity.29,30 These evolutionary-framework hypotheses might well fit ADRB2 coding variants. Indeed, the ancestral Glu27 allele has been associated with obesity in a number of studies and the three β-adrenergic receptors are known to play pivotal roles in regulating metabolic rate and thermogenesis.72 Also, the ancestral Gly16 variant has been associated with both asthma and resistance to A. lumbricoides infection,23 and a latitudinal cline has been identified for Gly16 and Glu27, the two ancestral variants being possibly adapted to hot climates.73 The ancestral amino acid state at codons 16 and 27 might therefore have conferred some selective advantage with respect to energy storage, ascariasis, and vascular reactivity. Yet, some recent evidence suggests that additional selective pressures might have been acting on ADBR2 variants. Indeed, signaling via ADRB2 and heterotrimeric guanine nucleotide-binding proteins has been shown to regulate the entry74,75 and intracellular maturation75 of Plasmodium falciparum in erythrocytes. Malaria (MIM 611162) is thought to have exerted a strong selective pressure on humans, resulting in the selection of hundreds of genetic variants conferring some disease protection; it is therefore conceivable that Plasmodium infection has exerted some selective pressure on ADRB2 polymorphisms, as well.
In analogy to ADRB2 the ancestral susceptibility model might apply to ADRB3; other authors29 suggested that the ADRB3 Arg64 allele might represent a thrifty variant. Although the contribution of ADRB3 genotypes to the etiology of obesity and type 2 diabetes is still controversial, a meta-analysis76 indicated that the Arg64 variant plays a modest but significant role in the susceptibility to non-insulin-dependent diabetes mellitus. Moreover, a consistent association with younger age at onset of diabetes has been reported in several analyses.76–79 Similarly, a study80 based on paired sibling analysis indicated a significant association between the ADRB3 Arg64 allele and obesity. In line with these findings, homozygotes for the Arg64 allele display lower metabolic resting rates as compared to those of subjects carrying at least one Trp64 variant,81 and the Arg64 allele might associate with mild gestational diabetes and increased weight gain in pregnancy.82 Despite its appeal, the thrifty-genotype hypothesis has received limited experimental support, and a limited number of studies83–85 have attempted to apply population genetics approaches to detect selection signatures at thrifty variants. Here, we have shown that the the derived Trp64 variant, which possibly represents a nonthrifty allele, has raised in frequency in human populations as a result of directional selection.
The specific selective pressure underlying this event remains to be identified, although changes in diet, which are thought to have occurred during recent human history, might have resulted in widespread selection of genes involved in energy metabolism.86
Supplemental Data
Supplemental Data include one figure and three tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
MaxDip, http://genapps.uchicago.edu
NIEHS Environmental Genome Project, http://egp.gs.washington.edu
Online Mendelian Inheritance in Man, www.ncbi.nlm.nih.gov/Omim/
R Project, www.r-project.org
Seattle SNPs, http://pga.mbt.washington.edu
Acknowledgments
We are grateful to Alessandra Frigerio, Cristina Barlassina, Roberto Giorda, and Massimo Molteni for collecting DNA samples and for helpful discussion on the manuscript, and we also wish to thank Roberto Del Bo and Dario Ronchi for providing and purifying DNA samples. This work was supported by the Italian Ministry of Health (grant RF-IEM-2007-633627). M.S. is part of the Doctorate School in Molecular Medicine, University of Milan.
References
- 1.Guimaraes S., Moura D. Vascular adrenoceptors: an update. Pharmacol. Rev. 2001;2:319–356. [PubMed] [Google Scholar]
- 2.Litonjua A.A. The significance of beta2-adrenergic receptor polymorphisms in asthma. Curr. Opin. Pulm. Med. 2006;1:12–17. doi: 10.1097/01.mcp.0000198068.50457.95. [DOI] [PubMed] [Google Scholar]
- 3.Sandilands A.J., O'Shaughnessy K.M. The functional significance of genetic variation within the beta-adrenoceptor. Br. J. Clin. Pharmacol. 2005;3:235–243. doi: 10.1111/j.1365-2125.2005.02438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liggett S.B. Polymorphisms of beta-adrenergic receptors in heart failure. Am. J. Med. 2004;7:525–527. doi: 10.1016/j.amjmed.2004.07.039. [DOI] [PubMed] [Google Scholar]
- 5.Brodde O.E. Beta-1 and beta-2 adrenoceptor polymorphisms: functional importance, impact on cardiovascular diseases and drug responses. Pharmacol. Ther. 2008;1:1–29. doi: 10.1016/j.pharmthera.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Arner P., Hoffstedt J. Adrenoceptor genes in human obesity. J. Intern. Med. 1999;6:667–672. doi: 10.1046/j.1365-2796.1999.00495.x. [DOI] [PubMed] [Google Scholar]
- 7.Mason D.A., Moore J.D., Green S.A., Liggett S.B. A gain-of-function polymorphism in a G-protein coupling domain of the human beta1-adrenergic receptor. J. Biol. Chem. 1999;18:12670–12674. doi: 10.1074/jbc.274.18.12670. [DOI] [PubMed] [Google Scholar]
- 8.Dionne I.J., Garant M.J., Nolan A.A., Pollin T.I., Lewis D.G., Shuldiner A.R., Poehlman E.T. Association between obesity and a polymorphism in the beta(1)-adrenoceptor gene (Gly389Arg ADRB1) in Caucasian women. Int. J. Obes. Relat. Metab. Disord. 2002;5:633–639. doi: 10.1038/sj.ijo.0801971. [DOI] [PubMed] [Google Scholar]
- 9.Ryden M., Hoffstedt J., Eriksson P., Bringman S., Arner P. The Arg 389 Gly beta1-adrenergic receptor gene polymorphism and human fat cell lipolysis. Int. J. Obes. Relat. Metab. Disord. 2001;11:1599–1603. doi: 10.1038/sj.ijo.0801815. [DOI] [PubMed] [Google Scholar]
- 10.Mialet Perez J., Rathz D.A., Petrashevskaya N.N., Hahn H.S., Wagoner L.E., Schwartz A., Dorn G.W., Liggett S.B. Beta 1-adrenergic receptor polymorphisms confer differential function and predisposition to heart failure. Nat. Med. 2003;10:1300–1305. doi: 10.1038/nm930. [DOI] [PubMed] [Google Scholar]
- 11.Green S.A., Turki J., Innis M., Liggett S.B. Amino-terminal polymorphisms of the human beta 2-adrenergic receptor impart distinct agonist-promoted regulatory properties. Biochemistry. 1994;32:9414–9419. doi: 10.1021/bi00198a006. [DOI] [PubMed] [Google Scholar]
- 12.Green S.A., Cole G., Jacinto M., Innis M., Liggett S.B. A polymorphism of the human beta 2-adrenergic receptor within the fourth transmembrane domain alters ligand binding and functional properties of the receptor. J. Biol. Chem. 1993;31:23116–23121. [PubMed] [Google Scholar]
- 13.Johnatty S.E., Abdellatif M., Shimmin L., Clark R.B., Boerwinkle E. Beta 2 adrenergic receptor 5′ haplotypes influence promoter activity. Br. J. Pharmacol. 2002;8:1213–1216. doi: 10.1038/sj.bjp.0704935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brodde O.E., Leineweber K. Beta2-adrenoceptor gene polymorphisms. Pharmacogenet. Genomics. 2005;5:267–275. doi: 10.1097/01213011-200505000-00001. [DOI] [PubMed] [Google Scholar]
- 15.Taylor M.R. Pharmacogenetics of the human beta-adrenergic receptors. Pharmacogenomics J. 2007;1:29–37. doi: 10.1038/sj.tpj.6500393. [DOI] [PubMed] [Google Scholar]
- 16.Connors S.L., Crowell D.E., Eberhart C.G., Copeland J., Newschaffer C.J., Spence S.J., Zimmerman A.W. Beta2-Adrenergic Receptor Activation and Genetic Polymorphisms in Autism: Data from Dizygotic Twins. J. Child Neurol. 2005;11:876–884. doi: 10.1177/08830738050200110401. [DOI] [PubMed] [Google Scholar]
- 17.Cheslack-Postava K., Fallin M.D., Avramopoulos D., Connors S.L., Zimmerman A.W., Eberhart C.G., Newschaffer C.J. beta2-Adrenergic receptor gene variants and risk for autism in the AGRE cohort. Mol. Psychiatry. 2007;3:283–291. doi: 10.1038/sj.mp.4001940. [DOI] [PubMed] [Google Scholar]
- 18.Gibson C.S., MacLennan A.H., Dekker G.A., Goldwater P.N., Dambrosia J.M., Munroe D.J., Tsang S., Stewart C., Nelson K.B. Genetic polymorphisms and spontaneous preterm birth. Obstet. Gynecol. 2007;2:384–391. doi: 10.1097/01.AOG.0000252712.62241.1a. [DOI] [PubMed] [Google Scholar]
- 19.Doh K., Sziller I., Vardhana S., Kovacs E., Papp Z., Witkin S.S. Beta2-adrenergic receptor gene polymorphisms and pregnancy outcome. J. Perinat. Med. 2004;5:413–417. doi: 10.1515/JPM.2004.138. [DOI] [PubMed] [Google Scholar]
- 20.Landau R., Xie H.G., Dishy V., Stein C.M., Wood A.J., Emala C.W., Smiley R.M. beta2-Adrenergic receptor genotype and preterm delivery. Am. J. Obstet. Gynecol. 2002;5:1294–1298. doi: 10.1067/mob.2002.128524. [DOI] [PubMed] [Google Scholar]
- 21.Ozkur M., Dogulu F., Ozkur A., Gokmen B., Inaloz S.S., Aynacioglu A.S. Association of the Gln27Glu polymorphism of the beta-2-adrenergic receptor with preterm labor. Int. J. Gynaecol. Obstet. 2002;3:209–215. doi: 10.1016/s0020-7292(02)00035-8. [DOI] [PubMed] [Google Scholar]
- 22.Gibson C.S., Maclennan A.H., Dekker G.A., Goldwater P.N., Sullivan T.R., Munroe D.J., Tsang S., Stewart C., Nelson K.B. Candidate genes and cerebral palsy: a population-based study. Pediatrics. 2008;5:1079–1085. doi: 10.1542/peds.2007-3758. [DOI] [PubMed] [Google Scholar]
- 23.Ramsay C.E., Hayden C.M., Tiller K.J., Burton P.R., Hagel I., Palenque M., Lynch N.R., Goldblatt J., LeSouef P.N. Association of polymorphisms in the beta2-adrenoreceptor gene with higher levels of parasitic infection. Hum. Genet. 1999;3:269–274. doi: 10.1007/s004390050947. [DOI] [PubMed] [Google Scholar]
- 24.Xu B.Y., Arlehag L., Rantapaa-Dahlquist S.B., Lefvert A.K. beta2 Adrenoceptor gene single nucleotide polymorphisms are associated with rheumatoid arthritis in northern Sweden. Ann. Rheum. Dis. 2005;5:773–776. doi: 10.1136/ard.2004.027532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malysheva O., Pierer M., Wagner U., Wahle M., Wagner U., Baerwald C.G. Association between beta2 adrenergic receptor polymorphisms and rheumatoid arthritis in conjunction with human leukocyte antigen (HLA)-DRB1 shared epitope. Ann. Rheum. Dis. 2008;12:1759–1764. doi: 10.1136/ard.2007.083782. [DOI] [PubMed] [Google Scholar]
- 26.Diatchenko L., Anderson A.D., Slade G.D., Fillingim R.B., Shabalina S.A., Higgins T.J., Sama S., Belfer I., Goldman D., Max M.B. Three major haplotypes of the beta2 adrenergic receptor define psychological profile, blood pressure, and the risk for development of a common musculoskeletal pain disorder. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2006;5:449–462. doi: 10.1002/ajmg.b.30324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia-Rubi E., Calles-Escandon J. Insulin resistance and type 2 diabetes mellitus: its relationship with the beta 3-adrenergic receptor. Arch. Med. Res. 1999;6:459–464. doi: 10.1016/s0188-4409(99)00077-6. [DOI] [PubMed] [Google Scholar]
- 28.Ioannidis J.P., Ntzani E.E., Trikalinos T.A. 'Racial' differences in genetic effects for complex diseases. Nat. Genet. 2004;12:1312–1318. doi: 10.1038/ng1474. [DOI] [PubMed] [Google Scholar]
- 29.Di Rienzo A., Hudson R.R. An evolutionary framework for common diseases: the ancestral-susceptibility model. Trends Genet. 2005;11:596–601. doi: 10.1016/j.tig.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 30.Barnes K.C., Grant A.V., Gao P. A review of the genetic epidemiology of resistance to parasitic disease and atopic asthma: common variants for common phenotypes? Curr. Opin. Allergy Clin. Immunol. 2005;5:379–385. doi: 10.1097/01.all.0000182543.37724.7b. [DOI] [PubMed] [Google Scholar]
- 31.Chio A., Schymick J.C., Restagno G., Scholz S.W., Lombardo F., Lai S.L., Mora G., Fung H.C., Britton A., Arepalli S. A two-stage genome-wide association study of sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 2009;18:1524–1532. doi: 10.1093/hmg/ddp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stephens M., Smith N.J., Donnelly P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001;4:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stephens M., Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am. J. Hum. Genet. 2005;3:449–462. doi: 10.1086/428594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;3:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu Y.X., Li W.H. Statistical tests of neutrality of mutations. Genetics. 1993;3:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watterson G.A. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 1975;2:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- 37.Nei M., Li W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA. 1979;10:5269–5273. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fay J.C., Wu C.I. Hitchhiking under positive Darwinian selection. Genetics. 2000;3:1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thornton K. Libsequence: a C++ class library for evolutionary genetic analysis. Bioinformatics. 2003;17:2325–2327. doi: 10.1093/bioinformatics/btg316. [DOI] [PubMed] [Google Scholar]
- 40.Hudson R.R. Two-locus sampling distributions and their application. Genetics. 2001;4:1805–1817. doi: 10.1093/genetics/159.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schaffner S.F., Foo C., Gabriel S., Reich D., Daly M.J., Altshuler D. Calibrating a coalescent simulation of human genome sequence variation. Genome Res. 2005;11:1576–1583. doi: 10.1101/gr.3709305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wright S. Genetical structure of populations. Nature. 1950;4215:247–249. doi: 10.1038/166247a0. [DOI] [PubMed] [Google Scholar]
- 43.Hudson R.R., Slatkin M., Maddison W.P. Estimation of levels of gene flow from DNA sequence data. Genetics. 1992;2:583–589. doi: 10.1093/genetics/132.2.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bandelt H.J., Forster P., Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999;1:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 45.Glazko G.V., Nei M. Estimation of divergence times for major lineages of primate species. Mol. Biol. Evol. 2003;3:424–434. doi: 10.1093/molbev/msg050. [DOI] [PubMed] [Google Scholar]
- 46.Griffiths R.C., Tavare S. Sampling theory for neutral alleles in a varying environment. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1994;1310:403–410. doi: 10.1098/rstb.1994.0079. [DOI] [PubMed] [Google Scholar]
- 47.Griffiths R.C., Tavare S. Unrooted genealogical tree probabilities in the infinitely-many-sites model. Math. Biosci. 1995;1:77–98. doi: 10.1016/0025-5564(94)00044-z. [DOI] [PubMed] [Google Scholar]
- 48.R Development Core Team . R Foundation for Statistical Computing; Vienna, Austria: 2008. R: A language and environment for statistical computing.http://www.R-project.org [Google Scholar]
- 49.Kimura M. Cambridge University Press; Cambridge: 1983. The Neutral Theory of Molecular Evolution. [Google Scholar]
- 50.Wang R.L., Hey J. The speciation history of Drosophila pseudoobscura and close relatives: inferences from DNA sequence variation at the period locus. Genetics. 1996;3:1113–1126. doi: 10.1093/genetics/144.3.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tishkoff S.A., Verrelli B.C. Patterns of human genetic diversity: implications for human evolutionary history and disease. Annu. Rev. Genomics Hum. Genet. 2003;4:293–340. doi: 10.1146/annurev.genom.4.070802.110226. [DOI] [PubMed] [Google Scholar]
- 52.Evans P.D., Gilbert S.L., Mekel-Bobrov N., Vallender E.J., Anderson J.R., Vaez-Azizi L.M., Tishkoff S.A., Hudson R.R., Lahn B.T. Microcephalin, a gene regulating brain size, continues to evolve adaptively in humans. Science. 2005;5741:1717–1720. doi: 10.1126/science.1113722. [DOI] [PubMed] [Google Scholar]
- 53.Stranger B.E., Nica A.C., Forrest M.S., Dimas A., Bird C.P., Beazley C., Ingle C.E., Dunning M., Flicek P., Koller D. Population genomics of human gene expression. Nat. Genet. 2007;10:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drysdale C.M., McGraw D.W., Stack C.B., Stephens J.C., Judson R.S., Nandabalan K., Arnold K., Ruano G., Liggett S.B. Complex promoter and coding region beta 2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. Proc. Natl. Acad. Sci. USA. 2000;19:10483–10488. doi: 10.1073/pnas.97.19.10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sethi A.A., Tybjaerg-Hansen A., Jensen G.B., Nordestgaard B.G. 164Ile allele in the beta2-Adrenergic receptor gene is associated with risk of elevated blood pressure in women. The Copenhagen City Heart Study. Pharmacogenet. Genomics. 2005;9:633–645. doi: 10.1097/01.fpc.0000172243.27299.78. [DOI] [PubMed] [Google Scholar]
- 56.Dallongeville J., Helbecque N., Cottel D., Amouyel P., Meirhaeghe A. The Gly16→Arg16 and Gln27→Glu27 polymorphisms of beta2-adrenergic receptor are associated with metabolic syndrome in men. J. Clin. Endocrinol. Metab. 2003;10:4862–4866. doi: 10.1210/jc.2003-030173. [DOI] [PubMed] [Google Scholar]
- 57.Kim Y., Stephan W. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics. 2002;2:765–777. doi: 10.1093/genetics/160.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jensen J.D., Kim Y., DuMont V.B., Aquadro C.F., Bustamante C.D. Distinguishing between selective sweeps and demography using DNA polymorphism data. Genetics. 2005;3:1401–1410. doi: 10.1534/genetics.104.038224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meiklejohn C.D., Kim Y., Hartl D.L., Parsch J. Identification of a locus under complex positive selection in Drosophila simulans by haplotype mapping and composite-likelihood estimation. Genetics. 2004;1:265–279. doi: 10.1534/genetics.103.025494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hudson R.R., Bailey K., Skarecky D., Kwiatowski J., Ayala F.J. Evidence for positive selection in the superoxide dismutase (Sod) region of Drosophila melanogaster. Genetics. 1994;4:1329–1340. doi: 10.1093/genetics/136.4.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Di Rienzo A. Population genetics models of common diseases. Cur. Opin. Genet. Dev. 2006;6:630–636. doi: 10.1016/j.gde.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 62.Nielsen R., Hellmann I., Hubisz M., Bustamante C., Clark A.G. Recent and ongoing selection in the human genome. Nat. Rev. Genet. 2007;11:857–868. doi: 10.1038/nrg2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bustamante C.D., Fledel-Alon A., Williamson S., Nielsen R., Hubisz M.T., Glanowski S., Tanenbaum D.M., White T.J., Sninsky J.J., Hernandez R.D. Natural selection on protein-coding genes in the human genome. Nature. 2005;7062:1153–1157. doi: 10.1038/nature04240. [DOI] [PubMed] [Google Scholar]
- 64.Healey C.S., Dunning A.M., Teare M.D., Chase D., Parker L., Burn J., Chang-Claude J., Mannermaa A., Kataja V., Huntsman D.G. A common variant in BRCA2 is associated with both breast cancer risk and prenatal viability. Nat. Genet. 2000;3:362–364. doi: 10.1038/81691. [DOI] [PubMed] [Google Scholar]
- 65.Gavrilets S. One-locus two-allele models with maternal (parental) selection. Genetics. 1998;2:1147–1152. doi: 10.1093/genetics/149.2.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Spencer H.G. Further properties of Gavrilets' one-locus two-allele model of maternal selection. Genetics. 2003;4:1689–1692. doi: 10.1093/genetics/164.4.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hastings A. Stable cycling in discrete-time genetic models. Proc. Natl. Acad. Sci. USA. 1981;11:7224–7225. doi: 10.1073/pnas.78.11.7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Munoz-Moran E., Dieguez-Lucena J.L., Fernandez-Arcas N., Peran-Mesa S., Reyes-Engel A. Genetic selection and folate intake during pregnancy. Lancet. 1998;9134:1120–1121. doi: 10.1016/s0140-6736(05)79761-0. [DOI] [PubMed] [Google Scholar]
- 69.Wilcken B., Bamforth F., Li Z., Zhu H., Ritvanen A., Renlund M., Stoll C., Alembik Y., Dott B., Czeizel A.E. Geographical and ethnic variation of the 677C>T allele of 5,10 methylenetetrahydrofolate reductase (MTHFR): findings from over 7000 newborns from 16 areas world wide. J. Med. Genet. 2003;8:619–625. doi: 10.1136/jmg.40.8.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gloria-Bottini F., Lucarini N., Palmarino R., La Torre M., Nicotra M., Borgiani P., Cosmi E., Bottini E. Phosphoglucomutase genetic polymorphism of newborns. Am. J. Hum. Biol. 2001;1:9–14. doi: 10.1002/1520-6300(200101/02)13:1<9::AID-AJHB1001>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 71.Gloria-Bottini F., Lucarini N., La Torre M., Lucarelli P., Bottini E. Birth weight and parental PGM1 alleles. Am. J. Hum. Biol. 2001;3:417–420. doi: 10.1002/ajhb.1066. [DOI] [PubMed] [Google Scholar]
- 72.Lowell B.B., Bachman E.S. Beta-Adrenergic receptors, diet-induced thermogenesis, and obesity. J. Biol. Chem. 2003;32:29385–29388. doi: 10.1074/jbc.R300011200. [DOI] [PubMed] [Google Scholar]
- 73.Young J.H., Chang Y.P., Kim J.D., Chretien J.P., Klag M.J., Levine M.A., Ruff C.B., Wang N.Y., Chakravarti A. Differential susceptibility to hypertension is due to selection during the out-of-Africa expansion. PLoS Genet. 2005;6:e82. doi: 10.1371/journal.pgen.0010082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harrison T., Samuel B.U., Akompong T., Hamm H., Mohandas N., Lomasney J.W., Haldar K. Erythrocyte G protein-coupled receptor signaling in malarial infection. Science. 2003;5640:1734–1736. doi: 10.1126/science.1089324. [DOI] [PubMed] [Google Scholar]
- 75.Murphy S.C., Harrison T., Hamm H.E., Lomasney J.W., Mohandas N., Haldar K. Erythrocyte G protein as a novel target for malarial chemotherapy. PLoS Med. 2006;12:e528. doi: 10.1371/journal.pmed.0030528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fujisawa T., Ikegami H., Yamato E., Takekawa K., Nakagawa Y., Hamada Y., Oga T., Ueda H., Shintani M., Fukuda M. Association of Trp64Arg mutation of the beta3-adrenergic-receptor with NIDDM and body weight gain. Diabetologia. 1996;3:349–352. doi: 10.1007/BF00418352. [DOI] [PubMed] [Google Scholar]
- 77.Widen E., Lehto M., Kanninen T., Walston J., Shuldiner A.R., Groop L.C. Association of a polymorphism in the beta 3-adrenergic-receptor gene with features of the insulin resistance syndrome in Finns. N. Engl. J. Med. 1995;6:348–351. doi: 10.1056/NEJM199508103330604. [DOI] [PubMed] [Google Scholar]
- 78.Walston J., Silver K., Bogardus C., Knowler W.C., Celi F.S., Austin S., Manning B., Strosberg A.D., Stern M.P., Raben N. Time of onset of non-insulin-dependent diabetes mellitus and genetic variation in the beta 3-adrenergic-receptor gene. N. Engl. J. Med. 1995;6:343–347. doi: 10.1056/NEJM199508103330603. [DOI] [PubMed] [Google Scholar]
- 79.Silver K., Mitchell B.D., Walston J., Sorkin J.D., Stern M.P., Roth J., Shuldiner A.R. TRP64ARG beta 3-adrenergic receptor and obesity in Mexican Americans. Hum. Genet. 1997;3:306–311. doi: 10.1007/s004390050633. [DOI] [PubMed] [Google Scholar]
- 80.Mitchell B.D., Blangero J., Comuzzie A.G., Almasy L.A., Shuldiner A.R., Silver K., Stern M.P., MacCluer J.W., Hixson J.E. A paired sibling analysis of the beta-3 adrenergic receptor and obesity in Mexican Americans. J. Clin. Invest. 1998;3:584–587. doi: 10.1172/JCI512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Walston J., Andersen R.E., Seibert M., Hilfiker H., Beamer B., Blumenthal J., Poehlman E.T. Arg64 beta3-adrenoceptor variant and the components of energy expenditure. Obes. Res. 2003;4:509–511. doi: 10.1038/oby.2003.71. [DOI] [PubMed] [Google Scholar]
- 82.Festa A., Krugluger W., Shnawa N., Hopmeier P., Haffner S.M., Schernthaner G. Trp64Arg polymorphism of the beta3-adrenergic receptor gene in pregnancy: association with mild gestational diabetes mellitus. J. Clin. Endocrinol. Metab. 1999;5:1695–1699. doi: 10.1210/jcem.84.5.5650. [DOI] [PubMed] [Google Scholar]
- 83.Vander Molen J., Frisse L.M., Fullerton S.M., Qian Y., Del Bosque-Plata L., Hudson R.R., Di Rienzo A. Population genetics of CAPN10 and GPR35: implications for the evolution of type 2 diabetes variants. Am. J. Hum. Genet. 2005;4:548–560. doi: 10.1086/428784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ruiz-Narvaez E. Is the Ala12 variant of the PPARG gene an “unthrifty allele”? J. Med. Genet. 2005;7:547–550. doi: 10.1136/jmg.2004.026765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Helgason A., Palsson S., Thorleifsson G., Grant S.F., Emilsson V., Gunnarsdottir S., Adeyemo A., Chen Y., Chen G., Reynisdottir I. Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nat. Genet. 2007;2:218–225. doi: 10.1038/ng1960. [DOI] [PubMed] [Google Scholar]
- 86.Haygood R., Fedrigo O., Hanson B., Yokoyama K.D., Wray G.A. Promoter regions of many neural- and nutrition-related genes have experienced positive selection during human evolution. Nat. Genet. 2007;9:1140–1144. doi: 10.1038/ng2104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.