

Summary
Membrane-binding peripheral proteins play important roles in many biological processes, including cell signaling and membrane trafficking. Unlike integral membrane proteins, these proteins bind the membrane mostly in a reversible manner. Since peripheral proteins do not have canonical transmembrane segments, it is difficult to identify them from their amino acid sequences. As a first step toward genome-scale identification of membrane-binding peripheral proteins, we built a kernel-based machine learning protocol. Key features of known membrane-binding proteins, including electrostatic properties and amino acid composition, were calculated from their amino acid sequences and tertiary structures, which were then incorporated into the support vector machine to perform the classification. A data set of 40 membrane-binding proteins and 230 non-membrane-binding ones was used to construct and validate the protocol. Cross-validation and holdout evaluation of the protocol showed that the accuracy of the prediction reached up to 93.7% and 91.6%, respectively. The protocol was applied to the prediction of membrane binding properties of four C2 domains from novel protein kinases C. Although these C2 domains have 50% sequence identity, only one of them was predicted to bind the membrane, which was verified experimentally with surface plasmon resonance analysis. These results suggest that our protocol can be used for predicting membrane-binding properties of a wide variety of modular domains and may be further extended to genome-scale identification of membrane-binding peripheral proteins.
Keywords: protein-membrane interactions, function annotation, support vector machines, peripheral proteins, protein function prediction
Introduction
A plethora of cellular processes including cell signaling and membrane trafficking involve complex arrays of protein-protein and lipid-protein interactions. A large number of cytosolic proteins, collectively known as peripheral proteins, are recruited to different cellular membranes to form these macromolecular interactions 1; 2; 3. The past decade has witnessed a tremendous growth in our understanding of and interests in these peripheral proteins. Unlike integral membrane proteins, peripheral proteins bind the membrane mostly in a reversible manner using different strategies. A large number of peripheral proteins contain one or more modular domains specialized in lipid binding 1; 4. These lipid-binding structural modules, also known as membrane-targeting domains, include C1 5; 6, C2 5; 7; 8, PH 9; 10, FYVE 11, PX 12, ENTH 13, ANTH 13, BAR 14, FERM 15, and tubby domains 16. Also, there are peripheral proteins that do not have separate membrane-targeting domains but utilize a part of their molecular surface or an amphipathic secondary structure to interact with the membrane. In addition, some peripheral proteins have covalently attached lipid anchors that embed in the lipid bilayer 17; 18 and others are intrinsically unstructured proteins whose membrane binding surfaces are induced upon membrane interaction 19.
With the availability of whole genome sequence information for many different organisms, it is expected that an increasing number of membrane targeting domains and peripheral proteins will be identified in the near future. In vitro membrane binding studies and cellular membrane translocation studies have played a main role in identifying new peripheral proteins. Also, structural biology has deciphered the structural basis of specific lipid binding and membrane interactions of membrane targeting domains and peripheral proteins. However, it would be prohibitively time-consuming and expensive to search and identify new peripheral proteins on a genomic-scale by these experimental approaches. Therefore, a fast and accurate bioinformatics-based annotation scheme for peripheral proteins would greatly supplement the effort to identify membrane-binding peripheral proteins on a genomic scale.
Traditional sequence-based computational approaches, such as database searching for homologous proteins or detection of conserved motifs, do not always render credible results, since the sequence homology does not assure similarity in functional behavior. For example, it was shown that in spite of a very high sequence identity, the lipid-binding affinities of FYVE domains differ drastically 20. A more reliable prediction can be achieved from tertiary structure-based methods, such as fold or domain recognition, since it is estimated that two-thirds of proteins with similar structural topologies carry out similar functions 21. However, there are many examples of proteins with similar folds showing different properties. For instance, PH domains share similar structural folds but show a wide range of membrane affinities; a large portion of PH domains do not bind the membrane at all 10; 22. In light of all these observations, it is of great needs to develop a computational method that is not solely based on the sequence homology or the tertiary structural similarity.
In this work, an automated prediction protocol for identifying membrane-binding peripheral proteins was built using a machine-learning algorithm, the support vector machine (SVM) 23. The SVM algorithm has been applied to pattern recognition problems in bioinformatics, including gene expression analysis 24, protein fold recognition 25; 26, protein-protein interactions 27; 28, and protein-DNA interactions 29. When trained with an ensemble of characteristics of lipid-binding proteins, the SVM learned to distinguish membrane-binding proteins from the non-binding ones with high accuracy. The SVM was also able to single out a membrane-binding C2 domain among highly homologous C2 domains, which was experimentally verified by membrane-binding measurements by surface plasmon resonance (SPR) analysis. This protocol can be used to predict the membrane binding properties of a large number of modular domains with unknown properties and can be developed into a more general method for genome-wide prediction of membrane-binding peripheral proteins.
Results
Features of Membrane-Binding Proteins
Our strategy for the SVM-based prediction of membrane-binding peripheral proteins is schematically shown in Fig. 1. A key step in our classification protocol is the generation of features that distinguish lipid-binding proteins from non-binding ones. On the basis of previous studies on membrane binding proteins, we selected three classes of features: net charge, the distribution of surface cationic patches, and amino acid composition.
Fig 1.
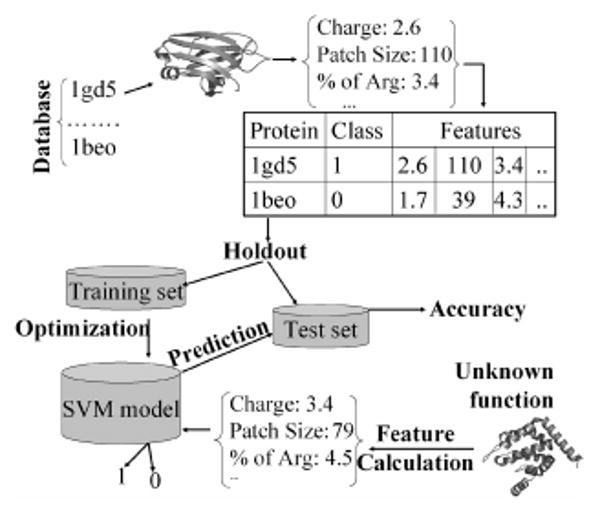
Overall strategy for constructing the protocol for identification of membrane-binding proteins. For each protein in the database (composed of negative and positive cases), features are generated. The dataset is then divided into two parts for holdout evaluation (the complementary cross-validation evaluation is not shown here). The SVM is optimized for best separation on the training set, which is then tested using the testing set. For an unclassified protein, similar features are generated and fed into the SVM for classification (1 or 0).
All intracellular membranes contain varying degrees of anionic lipids with the inner plasma membrane being the most anionic 1; 30. Also, phosphoinositides that function as membrane-recruiting signals for many peripheral proteins carry a high degree of negative change and may form local anionic clusters 31. Thus, electrostatic complementarity between cationic proteins and anionic membranes should be an important factor in membrane binding of peripheral proteins. To assess the importance of electrostatic properties of proteins in membrane binding, we used both the net charge and the size of surface cationic patches of proteins as feature sub-vectors. The net charge was assigned to all the atoms of proteins using the CHARMM force field parameters 32. Histidine residues were assigned a neutral charge. The protocols for the assignment of surface residues to surface cationic patches and their size calculation are described under Methods. The size of the largest patch, the cumulative size of the largest two, three, four patches, and the cumulative size of all patches were separately used as the features. Finally, two types of amino acid composition were evaluated as the third class of features: the ‘overall’ composition where all residues were used for the calculation and the ‘surface’ composition where only the surface residues were included.
We analyzed these features for the proteins in positive and negative data sets. To assess the contribution of each feature, Fisher's score (FS) is calculated for each feature j as
where μi and si are the means and variances of the feature in class i, respectively. The FS value of zero means no discriminative power for a given feature, while higher FS values indicate a higher discriminatory power. Roughly speaking, the FS values of 0.1 and 1 correspond to 69% and 92% accuracy, respectively, using linear separation.
Fig. 2 illustrates the distribution of the features in the two data sets and the FS for each feature. The net charge shows significantly different distributions in the two data sets, and the FS value for this single feature is 0.589. Clearly, membrane-binding proteins have higher net positive charges than non-binding ones. Also, non-binding ones have a significant negative skew, as evident from the fact that their mean (small middle square) is lower than the median (the middle bar) and that the lower whisker is longer than the upper whisker. When the size of the largest cationic patch is compared, membrane-binding proteins have larger patches than non-binding proteins, and the FS value for this feature is 0.169. Similarly, when the cumulative sizes of the largest two, three, and four patches are compared, membrane-binding proteins consistently exhibit larger cationic patch sizes than non-binding proteins, although the FS value decreases slightly with the increase of the cumulative patch size. Also, membrane-binding proteins have highly positively skewed distributions. Interestingly, however, the total cationic patch size has much lower correlation with the membrane binding behavior of protein with FS = 0.066. Therefore, the net charge and the size of the largest, the largest two, the largest three, and the largest four patches were used as features to train SVM for prediction.
Fig 2.
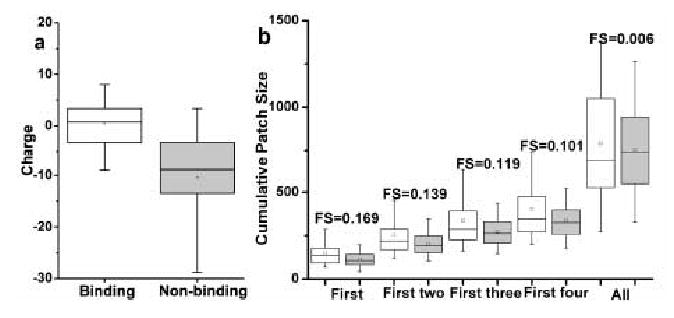
Box and whisker plots showing the distribution of net charge (a) and cumulative positive patch sizes (b) for lipid-binding (clear) and non-binding proteins (shaded). Fisher's score for individual features are also shown.
To validate the use of cationic patches in our prediction, we also investigated the location of the cationic patches relative to the membrane binding surface. Among the five proteins shown in Figure 3, membrane binding surfaces and membrane binding orientations have been experimentally determined for two of them 33; 34 and proposed for the rest 35; 36. Also shown in Figure 3 are the four largest cationic patches (colored in blue, cyan, white, and pink) that are described above. In four out of the five cases, at least two of the cationic patches directly interact with the membrane, while in one case only the largest patch is in contact with the membrane. This good correlation between cationic patches and membrane binding surfaces validates our use of cationic patches in SVM training and prediction. However, it should be noted that the cationic patches by themselves are not a conclusive indicator for membrane binding surface prediction, although their sizes are good indicator for predicting whether a protein can bind to membrane or not. Undoubtedly, more filtering criteria are needed for the prediction of actual membrane binding interface.
Fig 3.
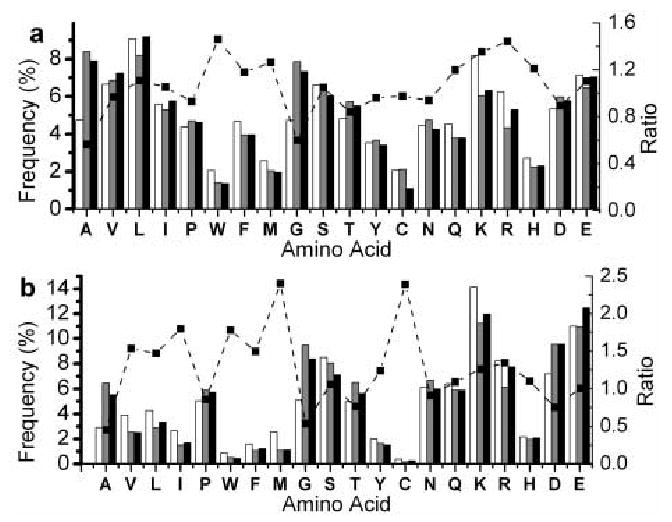
Relative orientation of cationic patches with respect to membrane binding surfaces. For cytosolic phospholipase A2 C2 domain33 (A) and PKCα C2 domain34 (B), membrane binding surfaces were experimentally determined. For β-spectrin PH domain36 (C), CALM-ANTH domain35 (D), and epsin 1-ENTH domain35 (E), membrane binding surfaces have been proposed based on biophysical and mutation studies. Proteins are shown in cartoon representation and cationic patches are displayed in ‘surface’ representation. RGB scale is used to color the patches with blue, cyan, white, and pink representing the largest, the second largest, third largest, and the fourth largest patches, respectively. A solid line indicates the location of the lipid headgroup region in the lipid bilayer. Figure are made with VMD 50.
The analysis of amino acid composition has been used to perform fold recognition 26 and to identify DNA-binding behavior 37. Since a large majority of membrane-binding residues are surface-exposed, the surface amino acid composition should be as important a factor as overall amino acid composition. We therefore analyzed the differences in both overall and surface amino acid compositions between membrane-binding and non-binding proteins. The comparison of surface amino acid composition reveals that noticeably higher proportions of aliphatic (i.e., Val, Leu, Ile, and Met) and aromatic (Trp in particular) residues are present on the surface of membrane-binding proteins (Fig. 4). Importantly, when the overall amino acid composition is compared, many of these residues are found in membrane-binding and non-binding proteins to comparable degrees. Abundance of aliphatic residues and Trp on the surface of membrane-binding proteins is consistent with the notion that these residues are involved in partial penetration of membrane, which plays a key role in kinetics and thermodynamics of membrane binding of peripheral proteins 1. An interesting observation is higher occurrence of surface Cys in membrane-binding proteins. Surface-exposed cysteins are often involved in disulfide bond formation or metal coordination 38, both of which increase the protein stability. In this regard, it is interesting to find that Gly, which is known to confer conformational flexibility and instability 38, is found much less on the surface of membrane-binding proteins than on the surface of non-binding proteins. This suggests that the surface regions of membrane-binding proteins need more conformational stability than their non-binding counterparts because they shuttle between aqueous and membrane environments. Also, more cationic residues (Lys and Arg) are found on the surface of membrane-binding proteins than on the surface of non-binding proteins. Notice that for cationic residues that are mostly surface-exposed, the differences between the two classes of proteins in overall and surface compositions are similar.
Fig 4.
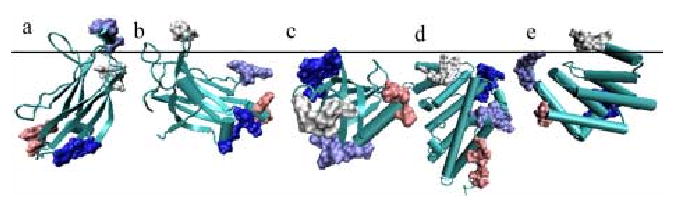
Histograms showing the overall (a) and surface (b) amino acid composition of binding (clear) and non-binding (shaded) proteins. Corresponding compositions for all proteins in the Astral-40 database are also indicated (black bars). The solid line plots the ratio of frequency in binding proteins to that in non-binding proteins. The ratio above 1.0 (see the right Y axis) indicates a higher propensity to be present in membrane-binding proteins.
To ascertain that the non-binding dataset used in this study was an unbiased one, we calculated the overall and surface compositions of the 6600 proteins in the Astral-40 database 39 (http://astral.berkeley.edu/). We found that our non-binding dataset was essentially identical to astral database in terms of amino acid composition (Fig. 4). This affirms that our observations regarding amino acid composition are not due to any inclination in the non-binding dataset but reflect the connatural propensities.
Evaluation of Prediction Protocol
We formulated the above attributes (net charge, cumulative patch sizes and two kinds of amino acid composition) into feature vectors and fed them to the SVM. The SVM was then tuned to maximize either accuracy or NP in the jackknife cross-validation test. Accuracy is defined as % of correct predictions, i.e., 100 × (TP + TN)/(TP + FP + TN + FN), where T, F, P, and, N indicate True, False, Positive and Negative, respectively. Sensitivity and specificity are defined as 100 × TP/(TP + FN) and 100 × TN/(TN+FP), respectively. The performance of the protocol was also appraised using net prediction (NP), which is defined as the average of sensitivity and specificity and can serve as a better measure when the data size of the two states are not balanced. The mathematical formalism of SVM and the validation methods including the jackknife and holdout tests are described in the Methods section.
When the SVM was optimized against accuracy, 93.7% accuracy was achieved (Table 1). Specificity and sensitivity under this condition were 95% and 84.8%, respectively. A relatively low sensitivity value can be attributed to asymmetry in the sizes of the positive and negative dataset. Since accuracy is the ratio of ‘number’ of correct predictions to total predictions made, the separating plane giving the highest accuracy tends to give more weight to the larger negative subset (hence the specificity) at the cost of a higher misclassification in the smaller positive set (thus decreasing the sensitivity). When higher sensitivity is favored, NP, which gives the same weights to sensitivity and specificity, may serve as a better index of performance. When the SVM was optimized against NP, a value of 93.1% NP was achieved. Under these circumstances, the specificity was 86.2% and the model was perfectly sensitive (100% sensitivity; Table 1), meaning that it correctly classified all positive cases. In general, tuning against accuracy will allow more accurate prediction, making it useful for predicting membrane-binding properties of structurally homologous proteins. On the other hand, tuning against NP will yield a larger group of likely candidates, thus making it better suited for screening potential membrane-binding proteins from structurally diverse proteins. The current SVM protocol performed effectively under both conditions.
Table 1.
Evaluation of the prediction protocol by different methods.
| Evaluation methods | Tuned against | Accuracy | Sensitivity | Specificity | Net Prediction |
|---|---|---|---|---|---|
| Jackknife | Accuracy | 93.7% | 84.8% | 95% | 89.9% |
| Jackknife | Net Prediction | 86.4% | 100% | 86.2% | 93.1% |
| 1-pair holdout | Accuracy | 91.6% | 80.9% | 91.3% | 85.7% |
To test if our protocol was over-trained, we evaluated it with a leave-one-pair holdout method in which a random pair of a binding and a non-binding protein was left out as the test set. When a binding protein is selected as test case, we removed the proteins that shared more than 20% sequence identity with it from the training set to preclude that prediction resulted from the sequence homology. With this evaluation, an accuracy of 91.6% could be achieved, which was comparable to the accuracy for the jackknife test (see Table 1). We further assessed the features for their discriminatory power using FS as the criteria. The net charge had the highest FS value, which was followed by the size of the largest cationic patch, the overall and surface composition of Ala and Gly, the overall composition of Trp, Arg and Lys, and the surface composition of Met. Thus, these features represent the most pronounced differences between membrane binding and non-binding proteins.
Prediction and Validation of Membrane Binding Properties of C2 Domains
The C2 domain is one of the most common domains and has been found in more than 200 proteins 5; 7; 8. Although the C2 domain was first discovered as the Ca2+-dependent membrane binding site in conventional PKCs, there are many C2 domain that have been shown to have little to no affinity for Ca2+ and membranes. This functional diversity of C2 domains makes them an attractive model system to test the reliability of our prediction protocol. In this study, we selected C2 domains from Ca2+-independent (or novel) PKCs, the biological functions of which are not fully understood. Among four mammalian novel PKC C2 domains, membrane binding affinities of PKCδ-C2 and PKCε-C2 remain controversial 40; 41, whereas membrane affinities of the C2 domains of PKCη and PKCθ have not been tested. Among four mammalian novel PKC C2 domains, crystal structures of PKCδ-C2 42 and PKCε-C2 43 have been determined. Since each of PKCδ-C2/PKCθ-C2 and PKCε-C2/PKCη-C2 pairs has strong sequence homology, we were able to build with high confidence the model structures of PKCθ-C2 and PKCη-C2 by homology modeling using the crystal structures of PKCδ-C2 and PKCε-C2, respectively, as template. We then computed all the aforementioned features for the four C2 domains. Since all these C2 domains are from Ca2+-independent PKCs, Ca2+-induced changes in electrostatic potentials of the domains were not taken into account.
The analysis of individual features for these C2 domains yielded conflicting results. In terms of net charge, PKCθ-C2 is most likely to be a membrane-binding protein because it is positively charged (+2) whereas PKCδ-C2, PKCε-C2, PKCη-C2 are negatively charged (-1, -8, and -1.6, respectively; Fig 5a). As far as the cumulative cation patch size is concerned, however, PKCδ-C2 is closer to membrane-binding proteins, while the other three are closer to non-binding ones (Fig 5b). The four C2 domains also showed conflicting patterns in terms of amino acid composition (Fig 5c & d). For example, PKCδ-C2 is similar to membrane-binding proteins with respect to the surface and overall frequency of Gly and Lys, whereas PKCθ-C2 is likely to be a membrane-binding protein in terms of the overall frequency of Ala, Val and Arg, and the surface frequency of Ala, Cys, Leu, and Ile. Because of these contradictory results, we could not predict with confidence by simple graphic evaluation of propensities. We therefore formulated the above characteristics of the four C2 domains into feature vectors and classified the domains using our SVM-based classification protocol (see Fig 1). Using the parameters that were optimized for either maximal accuracy or NP, SVM classified PKCδ-C2, PKCε-C2, and PKCη-C2 as non-binding and PKCθ-C2 as a membrane-binding peripheral protein.
Fig 5.
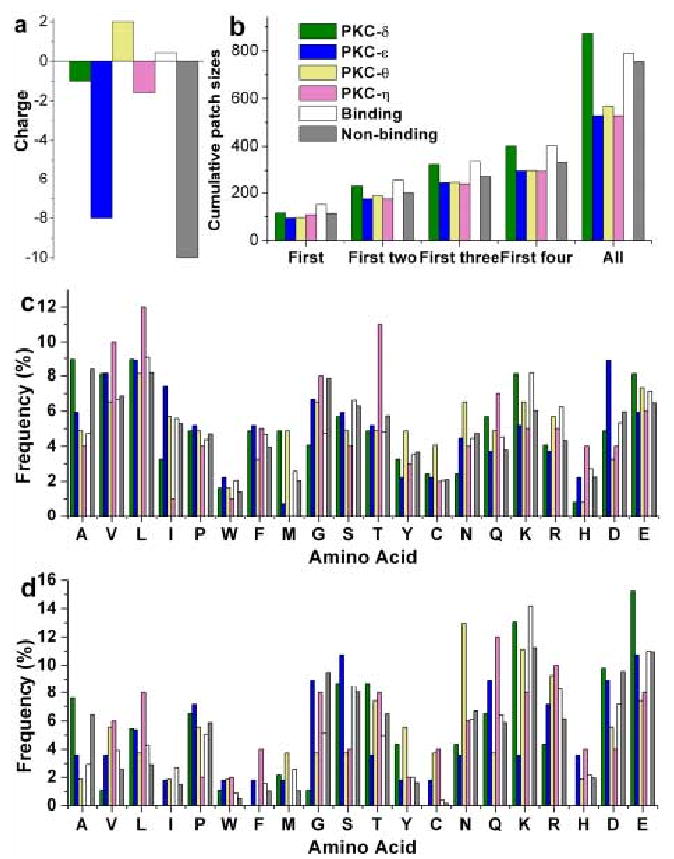
Distribution of various features for the C2 domains of PKCδ, ε, η and θ. The average value of corresponding features from the known binding and non-binding groups are shown for a direct comparison of the former with the any of the latter two cases.
To verify the prediction, we measured the binding of the four C2 domains to phospholipid vesicles with various compositions by SPR analysis. PKCδ-C2, PKCε-C2, and PKCη-C2 showed little to no binding to 1-palimitoyl-2-oleoyl-sn-3-phosphochline (POPC) vesicles containing 5 to 30 mol% of various anionic phospholipids, including 1-palimitoyl-2-oleoyl-sn-3-phosphoserine (POPS) and phosphatidylinositol-4,5-bisphosphate (PtdIns(4,5)P2) (see Fig. 6). However, PKCθ-C2 exhibited strong binding to POPC/POPS/PtdIns(4,5)P2 (65:30:5) vesicles with a Kd ∼ 80 nM (Fig. 7), which is comparable to that of other PtdIns(4,5)P2-binding domains 35. These results support the validity of our prediction method.
Fig 6.
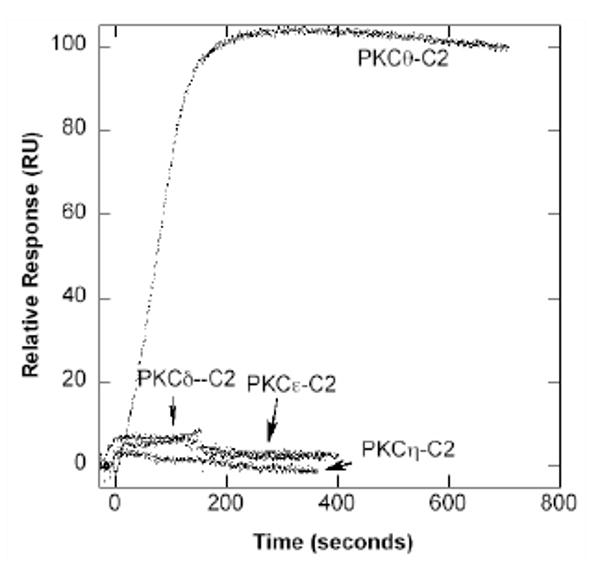
SPR Sensorgrams four PKC C2 domains. SPR sensorgrams were obtained by monitoring resonance unit (RU) changes after injecting each C2 domain (25 nM for PKCθ-C2 and 5 μM for other C2 domains) to the sensor chip coated with POPC/POPS/PtdIns(4,5)P2 (65:30:5) vesicles at 30 μl/min. The control surface was coated with 100% POPC vesicles. 10 mM HEPES buffer, pH 7.4, with 0.16 M NaCl was used for these measurements.
Discussion
The protocol described herein represents the first attempt to identify and predict membrane-binding proteins using the SVM, a machine-learning classification algorithm. Compilation of the dataset consisting of both positive and negative cases is the important first step in the classification. Our positive database is composed of sequentially and structurally diverse domains, which ensures that their properties properly represent general behaviors of membrane binding peripheral proteins. Our analysis of three groups of feature, net charge, the size of cationic patches, and amino acid composition, for the proteins in the positive and negative dataset reveals clear differences between membrane-binding and non-binding proteins. Membrane-binding proteins have a net positive charge, contain large cationic patches, and have more aliphatic and aromatic residues on the molecular surface. Also, they have more ‘stabilizing’ residues and less ‘flexible’ residues on the surface than non-binding ones. These salient features of membrane-binding proteins allow the SVM to readily distinguish membrane-binding proteins from non-binding ones after being trained with the features.
Several independent tests demonstrate the accuracy and reliability of our prediction protocol. First, both cross-validation and holdout evaluations using our dataset show that our prediction is highly reliable. The cross-validation test results in 93.7% accuracy and 93.1% NP while the holdout test gives 91.6% accuracy. In spite of the difference in the size of positive and negative data sets, comparable values of sensitivity and specificity are achieved for the former test. Furthermore, high FS values are obtained for several features that are used to distinguish membrane-binding and non-binding proteins. Lastly, the ability of our protocol to correctly classify the four C2 domains with a sequence identity higher than 50% demonstrates its robustness. Most important, these results suggest that our protocol can be used to predict the membrane-binding properties of modular domains, including other membrane targeting domains, with high accuracy.
There are a large number of membrane targeting domains whose structure and function of have not been characterized 1. There are also a myriad of other modular domains that may interact with membranes. For a majority of these modular domains, the tertiary structure is solved for at least one prototype domain 1. Successful prediction of membrane binding properties of the C2 domains using the structures constructed by homology modeling shows that the membrane binding properties of other domains can be predicted by the same protocol. Since many cellular proteins contain one or more modular domains, our protocol should be able to predict the membrane-binding properties of these proteins by predicting the membrane-binding properties of their modular domains. For those proteins without modular domains and homology to known proteins, our protocol can still serve as initial screening for potential candidates based primarily on the net charge and the overall amino acid composition. Undoubtedly, the progress in ab initio protein tertiary structure prediction will greatly help our protocol develop into a more general method for genomic scale annotation of membrane-binding proteins.
There are at least two factors the current protocol hasn't taken into account. The first factor is the membrane binding induced by ligand binding and protein phosphorylation. It has been shown that calcium binding and phosphorylation (or dephosphorylation) greatly enhances the membrane affinity of peripheral proteins1. Although this factor is not explicitly considered in our protocol, due to a relatively small size of database, it will be included in the future building of machine learning protocols. The second factor is the presence of intrinsically unstructured proteins whose membrane binding surfaces may be induced upon membrane interaction 19. Our current prediction protocol, which is optimized for predicting peripheral proteins with modular structural domains, may not apply to these proteins. At present, the database for these proteins is too small to allow for developing a robust prediction protocol. A priori, a sequence-based machine learning classification algorithm would be better suited to these proteins. As we learn more about intrinsically unstructured proteins and collect a sizable database, we will be able to develop a more reliable protocol for predicting their membrane binding properties.
Methods
Dataset
A list of membrane-binding proteins was built as follows: PDB database was searched with the names of all modular membrane-targeting domains as the keywords, which resulted in a total of 146 proteins. To reduce redundancy, only proteins with less than 40% sequence identity within each domain were selected. This resulted in a total of 40 proteins in the positive dataset (Table 2), which includes all the major lipid-binding domains. Non-binding protein dataset was adopted from a 250-strong control dataset used in a previous study 44. From this list, proteins containing the following keywords in their PDB header or description were removed: lipid, membrane, signaling, trafficking, and the names of all the domains used in the positive dataset. This led to a negative dataset with 230 proteins. The list is shown here: 1a53, 1a8e, 1a8p, 1a8y, 1aac, 1abe, 1ac5, 1aew, 1ah7, 1aho, 1air, 1ajj, 1al3, 1aly, 1amf, 1amk, 1amm, 1amp, 1amx, 1aoa, 1aol, 1aqb, 1arb, 1aru, 1ash, 1ass, 1at0, 1atg, 1auk, 1av4, 1axn, 1ayl, 1az9, 1b0b, 1b51, 1b6a, 1ba3, 1bb9, 1bd8, 1bdb, 1bdo, 1bea, 1beo, 1bfd, 1bg2, 1bg6, 1bgc, 1bhe, 1bhp, 1bj7, 1bk0, 1bob, 1bpi, 1bqk, 1br9, 1bs9, 1bv1, 1bx7, 1byb, 1c52, 1ca1, 1cec, 1cem, 1cfb, 1chd, 1ciy, 1clc, 1cnv, 1cot, 1cpo, 1cpq, 1cpt, 1csh, 1csn, 1ctj, 1ctt, 1cv8, 1cvl, 1cyo, 1czj, 1ddt, 1dfx, 1dhn, 1dhr, 1din, 1doi, 1dpe, 1drw, 1dun, 1dxy, 1eaf, 1ecy, 1edg, 1esc, 1ezm, 1fce, 1fds, 1fit, 1fkj, 1fmk, 1fnc, 1frb, 1fua, 1fus, 1g3p, 1gai, 1gca, 1gen, 1gky, 1gnd, 1gof, 1gpr, 1gsa, 1hcz, 1hfc, 1hka, 1hoe, 1hpm, 1htp, 1hxn, 1hyp, 1iae, 1ido, 1ifc, 1inp, 1iov, 1jdw, 1jer, 1lam, 1lfo, 1lki, 1mrp, 1ndh, 1nsj, 1opr, 1oyc, 1pbv, 1pda, 1php, 1ppn, 1rcb, 1sur, 1sym, 1klo, 1koe, 1kpf, 1kte, 1kuh, 1lbu, 1lcl, 1led, 1lit, 1lki, 1lst, 1ltm, 1mat, 1maz, 1mba, 1mla, 1moq, 1mpp, 1mrp, 1msk, 1mup, 1nar, 1neu, 1nfp, 1ng1, 1nif, 1nkr, 1nls, 1nnc, 1nox, 1npk, 1obr, 1ops, 1opy, 1osa, 1pbe, 1pbv, 1pdo, 1pea, 1pgs, 1phd, 1phk, 1phr, 1pht, 1plc, 1pmi, 1pne, 1poa, 1poc, 1pot, 1prn, 1ptf, 1puc, 1ra9, 1rb9, 1rcb, 1rec, 1rfs, 1rh4, 1rhs, 1rie, 1rkd, 1rmg, 1rzl, 1sbp, 1sek, 1sfp, 1skf, 1smd, 1sra, 1svb, 1tca, 1tde, 1ten, 1tfe, 1thv, 1tml, 1tmy, 1tn3, 1ton, 1try, 1tul, 1uch, 1uok, 1ush, 1utg, 1vls.
Table 2. Positive dataset of membrane binding proteins used in this study.
| Domains | PDB codes |
|---|---|
| C1 | 1kbf, 1r79 |
| C2 | 1bci, 1cfg, 1czs, 1dqv, 1dsy, 1rsy, 1v27, 1iqd_A, 1iqd_B, 1ab8, 1dji, 1d5r |
| PX | 1gd5, 1h6h, 1kmd, 1ocs, 1rlw |
| PH | 1btn, 1bwn, 1eaz, 1fao, 1fgy, 1mai, 1v5p, 1w1g, and 2dyn, 1djx, 1mi1, 1pls |
| FYVE | 1joc, 1vfy, 1dvp |
| ENTH | 1edu |
| ANTH | 1hfa |
| FERM | 1h4r, 1mix, 1ni2 |
| TUBBY | 1i7e |
Calculation of cationic patches
The distribution of surface cationic patches was determined by calculating the electrostatic potentials at the site of every atom of the protein using the Delphi package 45. Protein-bound lipid molecules, wherever present in the complex, were not included in the calculations. The CHARMM force-field was used for assignment of partial charges to all the atoms of the protein and Debye-Huckel boundary conditions were employed. A probe radius of 1.4 Å and a stern (ion-exclusion) layer of 2Å were specified. Salt concentration and temperature were fixed at 145 mM and 298 K, respectively. The dielectric constants used were 2.0 and 80.0 for protein interior and the solvent. A fine-resolution grid structure with a scale (grids/Å) of 2 was employed. The percentage fill specified was 50%, meaning that protein fills half of the total volume of the grid cubic. The center of the grid architecture was translated to the geometric center of the protein. An iterant expanding algorithm was then employed to locate the cationic patches on the ‘surface’ residues, which were defined as the ones having more than 40% of their area exposed to solvent. Solvent accessible area was calculated using DSSP 46. All atoms belonging to surface residue was designated as surface atoms. Every atom satisfying the following two criteria was chosen as the ‘start seed’ of the patch: 1) the atom has an electrostatic potential larger than 100 kT/e and 2) the average potential of surrounding surface atoms within 3.5Å is non-negative. Each of these atoms was then used as the ‘expansion seed’ for growing the patch further. The patch was grown until the atoms satisfying the second criterion could no longer be found. The procedure was iterated with all isolated start seeds. The size of the patch was defined as the number of atoms within the patch.
SVM and evaluation methods
The features were formulated into feature vectors with a class label attached to every vector (1 for binding and 0 for non-binding) and were fed into SVM, which is a classification engine based on statistical learning theory. During training, input vectors were mapped into a higher dimensional space called the ‘feature space’ and a hyperplane separating the input vectors of the two classes was then sought as the one that maximized the margin between the two classes and minimized the error. During testing, when the class of the input vectors was not known, the vectors were mapped on to the feature space and were classified according to the side of the decision plane on which they fell.
For evaluation of the classification performance, the leave-one-out (jackknife) cross-validation test was used. In this test, all but one of the proteins was used for training and the left-out protein was tested. This was done until every protein is tested exactly once. In addition, holdout evaluation was used, where the dataset was randomly divided into two parts. SVM was trained on one part (training set) to search for the best hyperplane with no regard to the other set (test set), which was subsequently tested. In contrast to the jackknife evaluation in which the best plane was sought with regard to both training and testing sets, the optimizing of parameters was done purely on the training set in the holdout evaluation.
A server is provided for online submission of proteins for prediction of their membrane-binding properties (http://proteomics.bioengr.uic.edu/pro-mem).
Model Building and Membrane Binding Measurements of C2 Domains
The 3D structures of the C2 domains of protein kinases C (PKC) η and θ were constructed by homology modeling using the crystal structures of PKCε 43 and PKCδ 42 C2 domains as templates, respectively. Both pairs exhibit more than 50% sequence identity as calculated from the sequence alignment obtained from ClustalW 47. 3D structure was built using MODELLER 48. Since the modeled proteins have high sequence similarities with respective templates, resulting model structure were expected to be reliable. After the model building, features for all these C2 domains were calculated for prediction (see Fig. 1).
The four PKC C2 domains were expressed in Escherichia coli and purified as previously described 40; 41. Binding of these C2 domains to different lipid vesicles was measured by SPR analysis 49 using a Biacore X biosensor system. Lipid vesicles were prepared and coated on the L1 biosensor chip (Biacore AB) as previously described 49. Data was analyzed using BIAevaluation software (Biacore) to determine the rate constants of association (ka) and dissociation (kd) as described previously 49. The equilibrium dissociation constant (Kd) was calculated as kd/ka assuming 1:1 protein-membrane surface binding.
Acknowledgments
This work was supported by the NIH grants P01 AI69015 (H.L.), GM68849 (W.C.), and GM 52987 (W.C), and UIC Bioengineering Startup Funds (H.L). N.B. acknowledges the support from the FMC Fellowship. R.E.L. was supported by the NIH predoctoral training grant T32HL07692 (Cellular Signaling in the Cardiovascular System).
Abbreviations
- FS
Fisher's score
- NP
net prediction
- PKC
protein kinase C
- POPC
1-palimitoyl-2-oleoyl-sn-3-phosphochline
- POPS
1-palimitoyl-2-oleoyl-sn-3-phosphoserine
- PtdIns(4,5)P2
phosphatidylinositol-4,5-bisphosphate
- SPR
surface plasmon resonance
- SVM
support vector machine
References
- 1.Cho W, Stahelin RV. Membrane-protein interactions in cell signaling and membrane trafficking. Annu Rev Biophys Biomol Struct. 2005;34:119–51. doi: 10.1146/annurev.biophys.33.110502.133337. [DOI] [PubMed] [Google Scholar]
- 2.Teruel MN, Meyer T. Translocation and reversible localization of signaling proteins: a dynamic future for signal transduction. Cell. 2000;103:181–4. doi: 10.1016/s0092-8674(00)00109-4. [DOI] [PubMed] [Google Scholar]
- 3.Hurley JH, Meyer T. Subcellular targeting by membrane lipids. Curr Opin Cell Biol. 2001;13:146–52. doi: 10.1016/s0955-0674(00)00191-5. [DOI] [PubMed] [Google Scholar]
- 4.DiNitto JP, Cronin TC, Lambright DG. Membrane recognition and targeting by lipid-binding domains. Sci STKE. 2003;2003:re16. doi: 10.1126/stke.2132003re16. [DOI] [PubMed] [Google Scholar]
- 5.Cho W. Membrane targeting by C1 and C2 domains. J Biol Chem. 2001;276:32407–10. doi: 10.1074/jbc.R100007200. [DOI] [PubMed] [Google Scholar]
- 6.Yang C, Kazanietz MG. Divergence and complexities in DAG signaling: looking beyond PKC. Trends Pharmacol Sci. 2003;24:602–8. doi: 10.1016/j.tips.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 7.Nalefski EA, Falke JJ. The C2 domain calcium-binding motif: structural and functional diversity. Protein Sci. 1996;5:2375–90. doi: 10.1002/pro.5560051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rizo J, Sudhof TC. C2-domains, structure and function of a universal Ca2+-binding domain. J Biol Chem. 1998;273:15879–82. doi: 10.1074/jbc.273.26.15879. [DOI] [PubMed] [Google Scholar]
- 9.Ferguson KM, Kavran JM, Sankaran VG, Fournier E, Isakoff SJ, Skolnik EY, Lemmon MA. Structural basis for discrimination of 3-phosphoinositides by pleckstrin homology domains. Mol Cell. 2000;6:373–84. doi: 10.1016/s1097-2765(00)00037-x. [DOI] [PubMed] [Google Scholar]
- 10.Lemmon MA, Ferguson KM. Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem J. 2000;350(Pt 1):1–18. [PMC free article] [PubMed] [Google Scholar]
- 11.Stenmark H, Aasland R, Driscoll PC. The phosphatidylinositol 3-phosphate-binding FYVE finger. FEBS Lett. 2002;513:77–84. doi: 10.1016/s0014-5793(01)03308-7. [DOI] [PubMed] [Google Scholar]
- 12.Xu Y, Seet LF, Hanson B, Hong W. The Phox homology (PX) domain, a new player in phosphoinositide signalling. Biochem J. 2001;360:513–30. doi: 10.1042/0264-6021:3600513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Camilli P, Chen H, Hyman J, Panepucci E, Bateman A, Brunger AT. The ENTH domain. FEBS Lett. 2002;513:11–8. doi: 10.1016/s0014-5793(01)03306-3. [DOI] [PubMed] [Google Scholar]
- 14.Habermann B. The BAR-domain family of proteins: a case of bending and binding? EMBO Rep. 2004;5:250–5. doi: 10.1038/sj.embor.7400105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bretscher A, Edwards K, Fehon RG. ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol. 2002;3:586–99. doi: 10.1038/nrm882. [DOI] [PubMed] [Google Scholar]
- 16.Carroll K, Gomez C, Shapiro L. Tubby proteins: the plot thickens. Nat Rev Mol Cell Biol. 2004;5:55–63. doi: 10.1038/nrm1278. [DOI] [PubMed] [Google Scholar]
- 17.McLaughlin S, Aderem A. The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem Sci. 1995;20:272–6. doi: 10.1016/s0968-0004(00)89042-8. [DOI] [PubMed] [Google Scholar]
- 18.Magee T, Seabra MC. Fatty acylation and prenylation of proteins: what's hot in fat. Curr Opin Cell Biol. 2005;17:190–6. doi: 10.1016/j.ceb.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 19.McLaughlin S, Murray D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature. 2005;438:605–11. doi: 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- 20.Blatner NR, Stahelin RV, Diraviyam K, Hawkins PT, Hong W, Murray D, Cho W. The molecular basis of the differential subcellular localization of FYVE domains. J Biol Chem. 2004;279:53818–27. doi: 10.1074/jbc.M408408200. [DOI] [PubMed] [Google Scholar]
- 21.Koppensteiner WA, Lackner P, Wiederstein M, Sippl MJ. Characterization of novel proteins based on known protein structures. J Mol Biol. 2000;296:1139–52. doi: 10.1006/jmbi.1999.3501. [DOI] [PubMed] [Google Scholar]
- 22.Singh SM, Murray D. Molecular modeling of the membrane targeting of phospholipase C pleckstrin homology domains. Protein Sci. 2003;12:1934–53. doi: 10.1110/ps.0358803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vapnik V, Cortes C. Support vector networks. Machine Learning. 1995;20:273–93. [Google Scholar]
- 24.Brown MP, Grundy WN, Lin D, Cristianini N, Sugnet CW, Furey TS, Ares M, Jr, Haussler D. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc Natl Acad Sci U S A. 2000;97:262–7. doi: 10.1073/pnas.97.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding CH, Dubchak I. Multi-class protein fold recognition using support vector machines and neural networks. Bioinformatics. 2001;17:349–58. doi: 10.1093/bioinformatics/17.4.349. [DOI] [PubMed] [Google Scholar]
- 26.Langlois RE, Diec A, Perisic O, Dai Y, Lu H. Improved protein fold assignment using support vector machines. Int J Bioinformatics Research and Applications. 2005;1:319–334. doi: 10.1504/IJBRA.2005.007909. [DOI] [PubMed] [Google Scholar]
- 27.Bordner AJ, Abagyan R. Statistical analysis and prediction of protein-protein interfaces. Proteins. 2005;60:353–66. doi: 10.1002/prot.20433. [DOI] [PubMed] [Google Scholar]
- 28.Miller JP, Lo RS, Ben-Hur A, Desmarais C, Stagljar I, Stafford Noble W, Fields S. Large-scale identification of yeast integral membrane protein interactions. Proc Natl Acad Sci U S A. 2005 doi: 10.1073/pnas.0505482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhardwaj N, Langlois RE, Zhao G, Lu H. Kernel-based machine learning protocol for predicting DNA-binding proteins. Nucleic Acids Res. 2005;33:6486–93. doi: 10.1093/nar/gki949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okeley NM, Gelb MH. A designed probe for acidic phospholipids reveals the unique enriched anionic character of the cytosolic face of the mammalian plasma membrane. J Biol Chem. 2004;279:21833–40. doi: 10.1074/jbc.M313469200. [DOI] [PubMed] [Google Scholar]
- 31.McLaughlin S, Wang J, Gambhir A, Murray D. PIP(2) and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct. 2002;31:151–75. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 32.Brooks B, Bruccoleri R, Olafson B, States D, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 33.Malmberg NJ, Van Buskirk DR, Falke JJ. Membrane-docking loops of the cPLA2 C2 domain: detailed structural analysis of the protein-membrane interface via site-directed spin-labeling. Biochemistry. 2003;42:13227–40. doi: 10.1021/bi035119+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kohout SC, Corbalan-Garcia S, Gomez-Fernandez JC, Falke JJ. C2 domain of protein kinase C alpha: elucidation of the membrane docking surface by site-directed fluorescence and spin labeling. Biochemistry. 2003;42:1254–65. doi: 10.1021/bi026596f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stahelin RV, Long F, Peter BJ, Murray D, De Camilli P, McMahon HT, Cho W. Contrasting membrane interaction mechanisms of AP180 N-terminal homology (ANTH) and epsin N-terminal homology (ENTH) domains. J Biol Chem. 2003;278:28993–9. doi: 10.1074/jbc.M302865200. [DOI] [PubMed] [Google Scholar]
- 36.Lemmon MA, Ferguson KM, Abrams CS. Pleckstrin homology domains and the cytoskeleton. FEBS Lett. 2002;513:71–6. doi: 10.1016/s0014-5793(01)03243-4. [DOI] [PubMed] [Google Scholar]
- 37.Bhardwaj N, Langlois RE, Zhao G, Lu H. Structure based prediction of binding residues on DNA-binding proteins. The Proceedings of 27th Annual International Conference of the Engineering in Medicine and Biology Society; 2005. In Press. [DOI] [PubMed] [Google Scholar]
- 38.Richardson JS. The anatomy and taxonomy of protein structure. Adv Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 39.Chandonia JM, Hon G, Walker NS, Lo Conte L, Koehl P, Levitt M, Brenner SE. The ASTRAL Compendium in 2004. Nucleic Acids Res. 2004;32:D189–92. doi: 10.1093/nar/gkh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stahelin RV, Digman MA, Medkova M, Ananthanarayanan B, Melowic HR, Rafter JD, Cho W. Diacylglycerol-induced membrane targeting and activation of protein kinase Cepsilon: mechanistic differences between protein kinases Cdelta and Cepsilon. J Biol Chem. 2005;280:19784–93. doi: 10.1074/jbc.M411285200. [DOI] [PubMed] [Google Scholar]
- 41.Stahelin RV, Digman MA, Medkova M, Ananthanarayanan B, Rafter JD, Melowic HR, Cho W. Mechanism of diacylglycerol-induced membrane targeting and activation of protein kinase Cdelta. J Biol Chem. 2004;279:29501–12. doi: 10.1074/jbc.M403191200. [DOI] [PubMed] [Google Scholar]
- 42.Pappa H, Murray-Rust J, Dekker LV, Parker PJ, McDonald NQ. Crystal structure of the C2 domain from protein kinase C-delta. Structure. 1998;6:885–94. doi: 10.1016/s0969-2126(98)00090-2. [DOI] [PubMed] [Google Scholar]
- 43.Ochoa WF, Garcia-Garcia J, Fita I, Corbalan-Garcia S, Verdaguer N, Gomez-Fernandez JC. Structure of the C2 domain from novel protein kinase Cepsilon. A membrane binding model for Ca(2+)-independent C2 domains. J Mol Biol. 2001;311:837–49. doi: 10.1006/jmbi.2001.4910. [DOI] [PubMed] [Google Scholar]
- 44.Stawiski EW, Gregoret LM, Mandel-Gutfreund Y. Annotating nucleic acid-binding function based on protein structure. J Mol Biol. 2003;326:1065–79. doi: 10.1016/s0022-2836(03)00031-7. [DOI] [PubMed] [Google Scholar]
- 45.Gilson MK, Honig BH. Energetics of charge-charge interactions in proteins. Proteins. 1988;3:32–52. doi: 10.1002/prot.340030104. [DOI] [PubMed] [Google Scholar]
- 46.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 47.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sali A, Potterton L, Yuan F, van Vlijmen H, Karplus M. Evaluation of comparative protein modeling by MODELLER. Proteins. 1995;23:318–26. doi: 10.1002/prot.340230306. [DOI] [PubMed] [Google Scholar]
- 49.Stahelin RV, Cho W. Differential roles of ionic, aliphatic, and aromatic residues in membrane-protein interactions: a surface plasmon resonance study on phospholipases A2. Biochemistry. 2001;40:4672–8. doi: 10.1021/bi0020325. [DOI] [PubMed] [Google Scholar]
- 50.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–8. 27–8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]