

Abstract
A synthesis of orthogonally protected L-threo-β-hydroxyasparagine from L-aspartic acid is reported. Iodocyclization of 3-benzoylaminoaspartic acid provided an intermediate oxazoline dicarboxylate that was efficiently hydrolyzed to L-threo-β-hydroxyaspartic acid. The synthetic route for conversion of the free β-hydroxy-α-amino acid into the target compound is highly efficient and amenable to preparation various orthogonally protected asparagine derivatives, on a multiple gram scale.
Keywords: asymmetric synthesis, amino acids, amides, peptides, protecting groups
Katanosin B is a cyclic depsipeptide antibiotic that was isolated by the Shionogi Research Laboratories from a producing organism related to the genus Cytophaga. 1,2 In a recent evaluation, katanosin B displayed very good antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA), as well as vancomycin-resistant entercocci, with minimum inhibitory concentrations (MIC’s) ranging from 0.39-0.78 mg/mL.3 Given this promising antibacterial activity, katanosin B has emerged as an attractive target for chemical synthesis.
During the course of our ongoing investigation into the synthesis of katanosin B (1), and a structurally related depsipeptide antibiotic, lysobactin,4 we required a dependable and efficient synthetic route to the non-proteinogenic amino acid L-threo-β-OH-Asn (Figure 1). The isomers of β-OH-Asn were originally isolated from human urine or synthesized as a racemate that was separated via resolution.5,6 Several synthetic procedures have been published for the enantioselective synthesis of the isomers of β-hydroxyaspartic acid 7-12 and erythro-β-hydroxyasparagine.13 In contrast, there are few descriptions of an asymmetric synthesis of threo-β-hydroxyasparagine.14,15 Boger reported a procedure that relies upon the Sharpless asymmetric aminohydroxylation reaction of (E)-4-methoxycinnamate, where the methoxy-substituted aromatic ring serves as masking group for the side chain carboxyl group.14 Lectka also reported a procedure involving ring-opening of appropriately derivatized blactams deriving from a cinchona alkaloid catalyzed asymmetric [2+2] cycloaddition reaction of ketenes and imines.15 We sought to develop a synthetic route to threo-hydroxyasparagine derivatives that would take advantage of readily available members of the chiral pool. Herein, we describe an efficient alternative asymmetric synthesis of orthogonally-protected L-threo-FmocNH-β-OH-Asn(Trt)-OH 2 for use in our synthesis of katanosin B.
Figure 1.
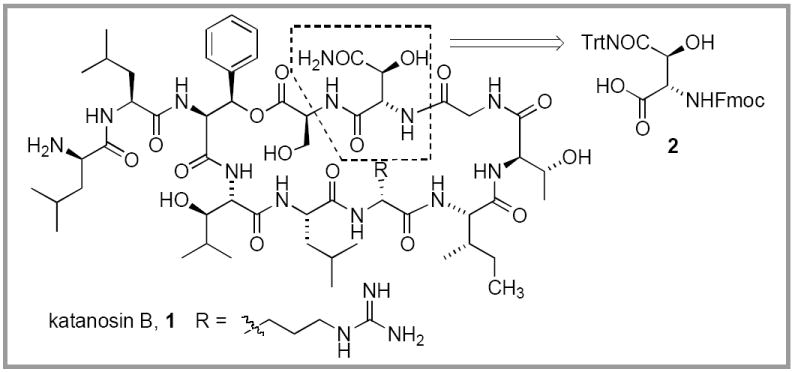
Structure of katanosin B
Our synthetic route (Scheme 1) relied upon the highly stereoselective conversion of L-aspartic acid to L-threo-β-hydroxyaspartic acid 3, as reported by Cardillo.7 A two-step sequence was used to convert 3 into Boc-protected monoester 4. Exposure to acidic methanol at reflux, to provide the side chain monoester,16,17 followed by Boc-protection of the free amino group provided 4 in 62% overall yield. Direct aminolysis of methyl ester 4 (NH3, MeOH, rt, 48 h, 90%) provided carboxamide 5. The free carboxyl group was efficiently protected as its benzyl ester (BnBr, NaHCO3, DMF) and provided 6 in 86% yield. At this juncture, the protecting group scheme we envisaged for our total synthesis of katanosin B required exchange of the amine protective groups. Thus, removal of the Boc group (4N HCl, dioxane) and reprotection of the free amino group (Fmoc-OSu, NaHCO3, dioxane/H2O) as its Fmoc carbamate provided the Fmoc-protected benzyl ester 7 in 87% overall yield.
Scheme 1.
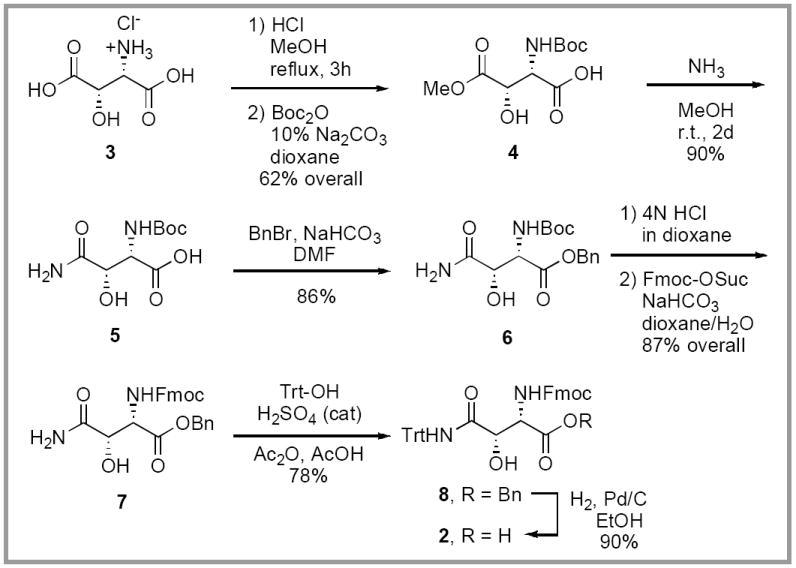
Synthesis of orthogonally protected β-hydroxyasparagine
Unprotected carboxamide groups of asparagine residues are susceptible to various side reactions during peptide coupling, i.e., dehydration to nitriles and intramolecular cyclization onto activated carboxyl groups to provide succinimide by-products. In an effort to avoid these unwanted side reactions during the course of our synthetic effort toward katanosin B, a trityl protecting group was introduced onto the side chain carboxamide. The trityl group was readily installed (Trt-OH, cat. H2SO4, Ac2O, AcOH, 78%) using the Boger modification14 of the protocol initially reported by Sieber and co-workers.18 Finally, careful hydrogenolysis of benzyl ester 8 (H2, Pd/C, EtOH, 90%) provided the target compound L-threo-Fmoc-β-OH-Asn(Trt)-OH.
In summary, we have accomplished an asymmetric synthesis of an orthogonally protected version of L-threo-β-hydroxyasparagine. The synthetic route utilizes a precursor that is readily available from the chiral pool. The synthetic sequence is sufficiently flexible that such that other protecting group schemes could be utilized. The reaction sequence is also quite efficient and provides the possibility for synthesis of these valuable building blocks on a multiple gram scale. The protecting group scheme employed for the synthesis of 2 was selected in order to maximize its utility in our synthetic efforts directed toward katanosin B and lysobactin. Efforts toward the synthesis of each of these depsipeptide antibiotics are in progress and will be presented in due course.
L-threo-BocNH-β-OH-Asp(OMe)-OH (4)
L-threo-β-hydroxyaspartic acid (1.00 g, 5.41mmol) was dissolved in a solution of concentrated HCl (0.89 mL, 10.8 mmol) in methanol (18.0 mL) at 0 °C. The reaction was heated at reflux for 3 h, then cooled to room temperature, and concentrated in vacuo. The crude L-threo-β-OH-Asp(OMe)-OH was triturated with ether, collected by filtration, and used without further purification in the following reaction. An aqueous solution of 10% Na2CO3 (18.0 mL) was added to L-threo-β-OH-Asp(OMe)-OH and the resulting mixture was cooled in an iced bath. A solution of Boc2O (3.65 g, 16.2 mmol) in dioxane (18.0 mL) was added dropwise to the reaction mixture and the resulting solution was stirred overnight at room temperature. After concentration by rotary evaporation, the residue was dissolved in ethyl acetate (150 mL) and washed with 1N HCl solution (100mL × 3). The organic layer was separated, dried over Na2SO4, filtered, and concentrated in vacuo. Flash chromatography (SiO2, 35% EtOAc-hexanes followed by 10-25% MeOH-CHCl3) provided 4 (0.88 g, 62%) as a foamy white solid. mp 109-110°C; [α]23 D+22 (c 1.00, CH3OH); 1H NMR (CD3OD, 500 MHz) δ 4.79 (s, 1H), 4.50 (s, 1H), 3.77 (s, 3H), 1.46 (s, 9H); 13C NMR (CD3OD, 125 MHz) δ 174.2, 170.2, 155.1, 80.4, 73.0, 59.0, 52.8, 28.7, IR(film) νmax 3388, 2981, 2934, 2858, 1739, 1699, 1608, 1512, 1393, 1367, 1251, 1167, 1107, 1061cm-1; HR-EI-MS m/z 263.1001 (M+, C10H17O7N requires 263.1000).
L-threo-BocNH-β-OH-Asn (5)
A sample of 4 (1.30 g, 4.94 mmol) was dissolved in MeOH (16.5 mL) and treated with NH3 gas until the solution became saturated. The resulting mixture was stirred at room temperature for 3 days. The reaction was concentrated by rotary evaporation and the crude material was purified by flash chromatography (SiO2, 25 --> 50% MeOH-CHCl3), which provided 5 (1.10 g, 90%) as a crystalline solid. mp 165-166 °C; [α]23 D-11 (c 1.0 , CH3OH); 1H NMR (CD3OD, 400 MHz) δ 4.63 (s, 1H), 4.48 (s, 1H), 1.40 (s, 9H); 13C NMR (CD3OD, 125 MHz) δ 177.8, 177.7,157.6, 80.4, 73.6, 59.4, 28.8 IR(KBr) νmax 3369, 2980, 2933, 1694, 1602, 1510, 1395, 1251, 1168, 1109, 1102, 1059, 1029 cm-1; HR-EI-MS m/z 249.1002 (M+, C9H16O6N2 requires 249.1003).
L-threo-BocNH-β-OH-Asn-OBn (6)
A solution of 5 (0.910 g, 3.67 mmol) in DMF (18.3 mL) at 0°C was treated with NaHCO3 (0.616 g, 7.33 mmol) and benzyl bromide (1.74 mL, 14.3 mmol). The resulting solution was stirred at 0 °C for 2 h and then for 24h at room temperature. The solution was cooled to 0 °C and water (100 mL) was slowly added. The resulting mixture was extracted with EtOAc (50 mL × 3). The combined organic extracts were combined and washed with H2O (100 mL). The The organic layer was dried over Na2SO4 and concentrated in vacuo. The crude product was purified by flash chromatography (SiO2, 0 --> 5% MeOH-CHCl3), which provided 6 (1.06 g, 86%) as a crystalline solid. mp 135-136°C; [α]23 D-17 (c 1.0, CHCl3); 1H NMR (CD3OD, 400 MHz) δ 7.38-7.30 (m, 5H), 5.19 (s, 2H), 4.70 (d, 1H, 2Hz), 4.59 (d, 1H, J = 2.5 Hz), 1.40 (s, 9H); 13C NMR (CD3OD, 125 MHz) δ 176.2, 171.7, 157.8, 136.9, 129.3, 129.2, 129.0, 128.9, 80.8, 72.7, 68.2, 58.1, 28.7 IR(film) νmax 3358, 2979, 1685, 1653, 1559 cm-1 HR-EI-MS m/z 338.1471 (M+, C16H22O6N2 requires 338.1472)
L-threo-FmocNH-β-OH-Asn-OBn (7)
Compound 6 (0.740 g, 2.19 mmol) was dissoled in 4M HCl-dioxane (16.4 mL) and stirred at room temperature for 2 h. The solution was concentrated by rotary evaporation and dried under high vacuum. The residue obtained was dissolved in 50% dioxane-H2O (32.4 mL, 1:1) and the resulting solution was cooled to 0 °C. Fmoc-OSuc (1.14 g, 3.28 mmol) and NaHCO3 (0.368 g, 4.37 mmol) were added to the solution and the resulting mixture was stirred at room temperature for 12 h. The reaction solution was diluted with EtOAc (100 mL) and saturated aqueous NaHCO3 (150 mL). The aqueous layer was separated and extracted with EtOAc (75 mL × 2). The combined organic extracts were dried over Na2SO4 and concentrated by rotary evaporation. The crude product was dissolved in the minimal amount of 20% MeOH-CHCl3 solution. Precipitation with hexanes and collection of the solid by filtration provided 7 as a white solid. mp 173-174°C; [α]23 D -18 (c 0.80, CHCl3); 1H NMR (CD3OD, 400 MHz) δ 7.78 (d, 2H, J = 7.2 Hz), 7.64 (d, 2H, J = 6 Hz), 7.38-7.26 (m, 9H), 5.21 (d, 2H, J = 3.2 Hz), 4.64 (d, 1H, 2.4 Hz), 4.35 (dd, 1H, J = 6 Hz, 3.6 Hz), 4.22 (m, 2H); 13C NMR (CD3OD, 125 MHz) δ 176.4, 171.7, 158.8, 145.2, 145.1, 142.5, 142.4, 137.0, 129.5, 129.2, 129.1, 128.8, 128.7, 128.2, 128.1, 126.3, 126.2, 120.1, 72.8, 68.4, 68.3, 58.6, 48.2, IR(film) νmax 3328, 1724, 1695, 1655, 1539, 1451, 1299, 1255, 1109 cm-1 HR-EI-MS m/z 460.1638 (M + H+, C26H24O6N2 requires 460.1629)
L-threo-FmocNH-β-OH-Asn(Trt)-OBn (8)
A solution of 7 (0.863 g, 1.87 mmol) and Trt-OH (5.05g, 18.7mmol) in HOAc (6.52mL) was heated to 50 °C and treated with concentrated H2SO4 (65 μL, 1.12mmol) and acetic anhydride (0.443 mL, 4.69 mmol). The resulting mixture was stirred at 50 °C for 2.5 h. After cooling to room temperature, the reaction solution was diluted with EtOAc (100mL) and saturated aqueous NaHCO3 (150mL). The organic layer was separated and aqueous layer was extracted with EtOAc (50 mL × 2). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. Purification by flash chromatography chromatography (SiO2, 15 --> 30% EtOAc-hexanes) provided 8 (1.03g, 1.46 mmol, 78%) as a white solid. mp 74-75°C; [α]23 D-14 (c 0.93, CHCl3); 1H NMR (CD3OD, 400 MHz) δ 7.79 (d, 1H, J = 3.2 Hz), 7.77 (d, 1H, J = 3.2 Hz), 7.67 (d, 1H, J = 7.6 Hz), 7.62 (d, 1H, J = 7.2 Hz), 7.38-7.17 (m, 24H), 5.20 (s, 2H), 4.81 (d, 1H, 2 Hz), 4.65 (d, 1H, 2Hz), 4.51 (dd, 1H, J = 9.6Hz, 4Hz), 4.17 (m, 2H); 13C NMR (CHCl3, 125 MHz) δ 172.2, 171.8, 158.9, 145.7, 145.3, 145.0, 142.5, 142.4, 137.1, 129.9, 129.7, 129.5, 129.2, 129.0, 128.9, 128.8, 128.7, 128.2, 128.1, 126.5, 126.2, 120.9, 73.4, 71.5, 68.6, 68.3, 58.6, 48.2, IR(film) νmax 3347, 3061, 3022, 2915, 2522, 1723, 1709, 1670, 1493, 1451, 1333, 1218cm-1 HR-EI-MS m/z 702.2721 (M+, C45H38O6N2 requires 702.2724)
L-threo-FmocNH-β-OH-Asn(Trt)-OH (2)
Pd/C (0.086 g) was cautiously added to a solution of 8 (0.860 g 1.22 mmol) in ethanol (12.2 mL). The solution was placed under an atmosphere of hydrogen gas for 1 h. The catalyst was removed by filtration through Celite and the filter cake was washed with EtOH. The combined filtrates were concentrated in vacuo and purified by flash chromatography (SiO2, 25%-->50% MeOH-CHCl3), which provided 2 (0.674g, 90%) as a white solid. mp 189-190°C decomposition; [α]23 D -17 (c 1.00, MeOH/CHCl3,1:1); 1H NMR (DMSO-d6, 400 MHz) δ 8.29 (s, 1H), 7.88 (d, 2H, J = 7.5 Hz), 7.76 (d, 1H, J = 6.5 Hz), 7.65 (d, 1H, J = 7 Hz), 7.38 (q, 2H, J = 7.0, 5.0 Hz), 7.27-7.08 (m, 18H), 4.52 (d, 1H, 12 Hz), 4.35 (m, 3H), 4.17 (t, 1H, 6.5Hz); 13C NMR (DMSO, 125 MHz) δ 173.8, 171.0, 156.1, 144.7, 143.8, 143.7, 140.7, 128.4, 127.7, 127.1, 126.7, 125.3, 120.1, 72.8, 69.1, 65.8, 57.7, 46.7, IR(KBr) νmax 3387, 3062, 2926, 2857, 1954, 1695, 1601, 1510, 1448, 1404, 1328, 1248, 1106, 1058 cm-1 HR-EI-MS m/z 612.2256 (M+, C38H32O6N2 requires 612.2255)
Acknowledgments
Financial support provided by the National Institutes of Health (AI 059327), The Hellman Foundation, and the Regents of the University of California, is gratefully acknowledged. MSV also thanks Eli Lilly and Company for support in the form of an Eli Lilly and Company New Faculty Award. AGM thanks the University of California at San Diego for support in the form of GAANN Fellowship.
References
- 1.Shoji J, Hinoo H, Matsumoto K, Hattori T, Yoshida T, Matsuura S, Kondo E. J Antibiot. 1988;41:713–718. doi: 10.7164/antibiotics.41.713. [DOI] [PubMed] [Google Scholar]
- 2.Kato T, Hinoo H, Terui Y, Kikuchi J, Shoji J. J Antibiot. 1988;41:719–725. doi: 10.7164/antibiotics.41.719. [DOI] [PubMed] [Google Scholar]
- 3.Maki H, Miura K, Yamano Y. Antimicrob Agents Chemother. 2001;45:1823–1827. doi: 10.1128/AAC.45.6.1823-1827.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tymiak AA, McCormick TJ, Unger SE. J Org Chem. 1989;54:1149–1157. [Google Scholar]
- 5.Okai H, Izumiya N. Bull Chem Soc Jpn. 1969;42:3550–3555. [Google Scholar]
- 6.Singerman A, Liwschitz Y. Tetrahedron Lett. 1968;46:4733–4734. doi: 10.1016/s0040-4039(00)75943-0. [DOI] [PubMed] [Google Scholar]
- 7.Cardillo G, Gentilucci L, Tolomelli A, Tomasini C. Synlett. 1999:1727–1730. [Google Scholar]
- 8.Cho GY, Ko SY. J Org Chem. 1999;64:8745–8747. [Google Scholar]
- 9.De Angelis M, Campiani G. Tetrahedron Lett. 2004;45:2355–2357. [Google Scholar]
- 10.Deng J, Hamada Y, Shioiri T. J Am Chem Soc. 1995;117:7824–7825. [Google Scholar]
- 11.Dudding T, Hafez AM, Taggi AE, Wagerle TR, Lectka T. Org Lett. 2002;4:387–390. doi: 10.1021/ol017087t. [DOI] [PubMed] [Google Scholar]
- 12.Hanessian S, Vanasse B. Can J Chem. 1993;71:1401–1406. [Google Scholar]
- 13.Tohdo K, Hamada Y, Shioiri T. Synlett. 1994:247–249. [Google Scholar]
- 14.Boger DL, Lee RJ, Bounaud P-Y, Meier P. J Org Chem. 2000;65:6770–6772. doi: 10.1021/jo000628s. [DOI] [PubMed] [Google Scholar]
- 15.Hafez AM, Dudding T, Wagerle TG, Shah MH, Taggi AE, Lectka T. J Org Chem. 2003;68:5819–5825. doi: 10.1021/jo034150e. [DOI] [PubMed] [Google Scholar]
- 16.Liwschitz Y, Singerman A. J Chem Soc C. 1967:1696–1700. [PubMed] [Google Scholar]
- 17.Mattingly PG, Miller MJ, Cooper RDG, Daugherty BW. J Org Chem. 1983;48:3556–3559. [Google Scholar]
- 18.Sieber P, Riniker B. Tetrahedron Lett. 1991;32:739–742. [Google Scholar]