

Abstract
Heptameric pores formed by the protective antigen (PA) moiety of anthrax toxin translocate the intracellular effector moieties of the toxin across the endosomal membrane to the cytosol of mammalian cells. We devised a protocol to characterize the effects of individual mutations in a single subunit of heptameric PA prepores (pore precursors) or pores. We prepared monomeric PA containing a test mutation plus an innocuous Cys-replacement mutation at a second residue (Lys563, located on the external surface of the prepore). The introduced Cys was biotinylated, and the protein was allowed to cooligomerize with a 20-fold excess of wild-type PA. Finally, biotinylated prepores were freed from wild-type prepores by avidin affinity chromatography. For the proof of principle, we examined single-subunit mutations of Asp425 and Phe427, two residues where Ala replacements have been shown to cause strong inhibitory effects. The single-subunit D425A mutation inhibited pore formation by >104 and abrogated activity of PA almost completely in our standard cytotoxicity assay. The single-subunit F427A mutation caused ∼100-fold inhibition in the cytotoxicity assay, and this effect was shown to result from a combination of strong inhibition of translocation and smaller effects on pore formation and ligand affinity. Our results show definitively that replacing a single residue in one subunit of the heptameric PA prepore can inhibit the transport activity of the oligomer almost completely—and by different mechanisms, depending on the specific residue mutated.
Keywords: anthrax toxin, translocation, planar lipid bilayer, cytotoxicity, phenylalanine clamp, pore formation
Abbreviations
- ANTXR2
anthrax toxin receptor 2 (referred to as capillary morphogenesis protein 2, CMG2)
- DN
dominant negative
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- DphPC
1,2-diphytanoyl-sn-glycerol-3-phosphocholine
- DTA
the catalytic domain of diphtheria toxin
- EF
edema factor
- HABA
4-hydroxyazobenzene-2-carboxylic acid
- LUV
large unilamellar vesicles; LF, lethal factor
- LFN
residues 1-263 of LF
- PA
protective antigen
- PBS
phosphate buffered saline
- TCEP
tris(2-carboxyethyl)phosphine hydrochloride
- VWA
von Willebrand factor A
- WT
wild type
Introduction
Anthrax toxin is an ensemble of three large, monomeric proteins secreted by Bacillus anthracis, the causative agent of anthrax. Two of these proteins, lethal factor (LF) and edema factor (EF), enzymatically modify molecular targets within the cytosol of mammalian cells, contributing to the symptoms of anthrax infections.1 The third, protective antigen (PA), is a pore-forming protein, which transports EF and LF across the endosomal membrane to the cytosol. The question of how the PA pore functions in translocation is relevant both to the pathogenesis of B. anthracis and to understanding how large globular proteins can unfold and thread through a narrow proteinaceous pore.
After PA binds to a cellular receptor, it is activated by a member of the furin family of proteases to produce an active, N-terminally truncated 63-kDa form (PA63).2,3 PA can also be cleaved and activated by serum proteases.4 Once activated, PA63 self-assembles into a ring-shaped heptamer, termed the prepore, which can bind up to three molecules of EF and/or LF.5 The resulting hetero-oligomeric complexes are then endocytosed and delivered to the endosome. The acidic environment of the endosome induces the prepore to undergo a conformational change to generate a mushroom-shaped, cation-selective pore in the membrane.6 A major facet of this conformational change is the relocation of the 2β2-2β3 loops and flanking β strands of the seven subunits to the base of the prepore, where they combine to form a 14-strand, 100-Å-long β-barrel,7,8 representing the transmembrane stem of the pore. Bound effector proteins (LF and/or EF) are then translocated across the membrane and delivered to the cytosol. The 12–15 Å diameter of the lumen within the stem9,10 necessitates unfolding of translocatable proteins at least to the level of α helix, for the passage through the pore.
Translocation of proteins through the PA pore has recently been replicated in planar phospholipid bilayers.11,12 Upon addition of prepore to a compartment (defined as the cis compartment) of a planar phospholipid bilayer under symmetric acidic conditions (pH 5–6) and a low cis positive potential (e.g., Δψ = +20 mV), cation-selective pores (channels) are formed.11,13 If LFN, the N-terminal PA-binding domain of LF, is then added to the cis compartment, it binds to the PA channel, and the disordered cationic leader sequence of LFN enters the channel, blocking ion conductance.11 Raising the pH of the trans compartment (that opposite the cis) to neutrality generates a transmembrane pH gradient analogous to that across the endosomal membrane and prompts the bound LFN to translocate through the pore in an N-terminal to C-terminal direction,14 as evidenced by release of the ion-conductance block. We have proposed a charge-state-dependent Brownian ratchet mechanism to explain the mechanism of pH-driven translocation.10,14–16 It is also possible to induce translocation of LFN by raising Δψ12,14,15; however, current evidence favors the pH gradient across the endosomal membrane as the primary driving force for this process in vivo.14
How the PA63 pore is formed and how it functions has been investigated in recent years by analyzing mutated forms of the protein. Of particular interest are mutations of certain residues within loops 2β7-2β8 and 2β10-2β11, lying in the lumen of the prepore. For example, Sellman et al.17 reported that mutating D425 to Ala, Asn, or Lys inhibited conversion of the SDS-dissociable prepore to the SDS-resistant pore, and mutating F427 to Ala allowed SDS-resistant pore to form, but inhibited translocation. Subsequent studies of F427 mutants have demonstrated a major role of this residue in promoting passage of translocatable polypeptides through the PA63 pore.15,16,18
To date, the effects of mutations on PA functions have been characterized with homogeneous mutant proteins or with mixtures of mutant and wild-type proteins in various ratios. Studies of mixtures revealed that certain mutations, at D425, F427 and other sites, were dominantly negative, as shown by the strong inhibitory effect of adding small amounts of the mutant protein to the wild-type.15,17–20 Indeed, the degree of inhibition by some mutants suggested that a single mutant subunit within the PA heptamer could block the oligomer's overall function. To test this hypothesis directly, and to provide a way to analyze the effects of individual mutations more precisely, we devised a protocol to generate preparations of heteroheptameric prepore devoid of wild-type homoheptamers and comprised predominantly of heptamers containing only one mutant subunit. Here we describe this protocol and its application to study Ala replacement mutations in D425 and F427.
Results
Preparation of heteroheptameric prepores
To prepare heteroheptameric prepore comprised predominantly of six WT subunits and one mutated subunit, we took advantage of the fact that native PA contains no Cys residues. Previously, it was shown that replacing a solvent-exposed residue in domain 3, K563, with Cys and then derivatizing the Cys with a thiol-reactive dye did not affect the biological activity of the protein.18 In the current study, we showed that derivatizing the K563C mutant with the thiol-selective biotinylation reagent, Nα-(3-maleimidylpropionyl)biocytin, caused no change in the biological activity of the protein. Next we prepared a double mutant of PA containing the K563C mutation and an Ala mutation at either of the two sites, D425 or F427. We then modified the Cys with the biotinylation reagent and mixed the biotinylated mutant protein in a 1:20 ratio with wild-type PA. After activation of the mixture by limited digestion with trypsin, the PA63 (prepore) fraction was isolated by anion-exchange chromatography. Finally, the heteroheptameric prepore was freed from WT homoheptamer ([WT]7) by two sequential affinity chromatography steps on monomeric avidin columns.
Assuming oligomerization of the WT and mutant forms of PA63 to be completely random, one can calculate from the binomial function that the population of heptamers derived from a 20:1 ratio of WT:mutant monomers would consist of the following: ∼71% with purely WT subunits ([WT]7), ∼25% with a single mutant subunit ([WT]6[mutant]1), ∼3.7% with two mutant subunits ([WT]5[mutant]2), and <1% with three or more mutant subunits. From these numbers, one can calculate that, among the prepores having any mutant subunits, ∼86% would have only a single mutant subunit, and most of the rest would contain only two.
Because the mutations tested were known to cause strong inhibitory effects in the homoheptameric state, the specific activities of [WT]5[mutant]2 and traces of other heteroheptamers present would be expected to be lower (or certainly no higher) than that of [WT]6[mutant]1. Thus our activity measurements would yield good approximations of the specific activities of the predominant species, [WT]6[mutant]1. Neglecting contributions of the minor heteroheptameric species to the measured activity, the specific activity of [WT]6[mutant]1 would be underestimated by ∼14%, which is generally within the error limits of the assays used.
The D425A heteroheptamer
The ability of the [WT]6[D425A]1 preparation to transport an effector protein to the cytosol was determined with LFN-DTA, a chimeric protein derived by fusing the C terminus of LFN to the N terminus of DTA, the enzymatic domain of diphtheria toxin.21,22 DTA inhibits protein synthesis by covalently modifying elongation factor-2, and we measured the incorporation of 3H-leucine into protein in CHO-K1 cells after incubating them for 16 h with varying concentrations of prepore in the presence of 100 nM LFN-DTA. As shown in Figure 1(A), the half maximal effective concentration (EC50) of the heteroheptamer preparation in this assay was over four orders of magnitude lower than that of the wild-type homoheptamer, [WT]7, supporting a strong dominant-negative effect of the single D425A subunit. The D425A homoheptamer, [D425A]7, was at least an order of magnitude less active than [WT]6[D425A]1.
Figure 1.
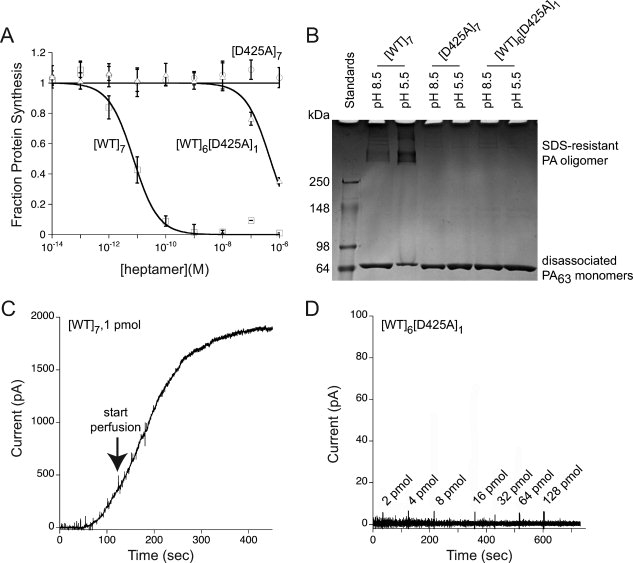
Effect of a single-subunit D425 mutation in the PA prepore. (A) Comparison of [WT]6[D425A]1 and [D425A]7 with [WT]7 in mediating entry of LFN-DTA into CHO-K1 cells. Cells were incubated with [WT]7, [WT]6[D425A]1, and [D425A]7 at indicated concentrations in the presence of 100 nM LFN-DTA. After 16 h, we measured the incorporation of 3H-leucine into protein over a 1-h period. The data plotted are averages of three experiments, with standard error shown. (B) Comparison of [WT]6[D425A]1 and [D425A]7 with [WT]7 in forming a SDS-resistant oligomer under acidic conditions. Lane 1, molecular weight markers; Lane 2, [WT]7, pH 8.5; Lane 3, [WT]7, pH 5.5; Lane 4, [D425A]7, pH 8.5; Lane 5, [D425A]7, pH 5.5; Lane 6, [WT]6[D425A]6, pH 8.5; Lane 7, [WT]6[D425A]1, pH 5.5. Each lane contains ∼3 μg prepore PA, and the gel was stained with Coomassie Brilliant Blue. (C) Monitoring of pore insertion by macroscopic current over time of 1 pmol [WT]7 upon addition to planar lipid bilayers (voltage +20 mV, pH 5.5). The arrow indicates initiation of perfusion of the cis compartment to remove excess PA that had not inserted into the membrane. (D) Monitoring of pore insertion by macroscopic current over time of [WT]6[D425A]1 upon addition to planar lipid bilayers (voltage +20 mV and pH 5.5). The values shown at the arrows represent incremental amounts of [WT]6[D425A]1 added at those time points, giving a total of >200 pmol.
Although [WT]7 maintained at pH > 8 dissociates into its monomeric state in the presence of SDS, exposure to pH < 7 causes the prepore to convert to an SDS-resistant, high molecular weight oligomer.6,23,24 The SDS-resistant state has been suggested to be a property of the pore or of an intermediate in pore formation.19 In contrast to [WT]7, both [WT]6[D425A]1 and [D425A]7 fully dissociated to monomeric PA63 at pH 5.5 when treated with SDS [Fig. 1(B)]. Furthermore, the single D425A subunit in [WT]6[D425A]1 completely blocked the ability of the prepore to form ion-conductive channels in planar lipid bilayers under acidic conditions [Fig. 1(C,D)]. There was no increase in current (limit of detection ∼2 pA) after the cumulative addition of >200 pmol [WT]6[D425A]1 to the cis compartment at pH 5.5, whereas addition of 1 pmol [WT]7 gave a value of ∼2000 pA, even when the cis chamber was perfused to keep the current on scale [Fig. 1(C)(C)]. Thus, a single D425A subunit in the prepore was sufficient to block its conversion to the pore, as assessed by both SDS resistance and permeabilization of bilayers.
The F427A heteroheptamer
As shown in 2(A), the EC50 of the [WT]6[F427A]1 preparation was approximately two orders of magnitude lower than that of [WT]7 in the LFN-DTA cytotoxicity assay, whereas [F427A]7 was over five orders of magnitude lower. The measured activity of the purified [WT]6[F427A]1 preparation was not due to a trace of contaminating [WT]7, as shown by a control in which identical amounts of [WT]7 and purified heteroheptamer were subjected to an additional chromatography step on monomeric avidin resin [Fig. 2(B)]. Titrations in the LFN-DTA assay demonstrated that exposure to monomeric avidin resin removed all LFN-DTA transport activity associated with the heteroheptamer preparation, but none of that associated with [WT]7 [ Fig. 2(B)]. Furthermore, the specific activity of heteroheptamer eluted from the resin with d-biotin was unchanged. The two successive avidin chromatography steps in our purification protocol had therefore reduced [WT]7 concentration to a level undetectable by our cytotoxicity assay. We conclude that our measurements with the [WT]6[F427A]1 preparation closely approximate the activity solely of the heterooligomeric, biotinylated F427A-containing species.
Figure 2.
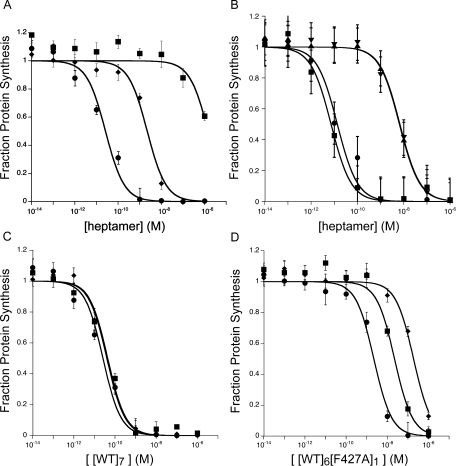
Activity of [WT]6[F427A]1 in mediating entry of LFN-DTA into CHO-K1 cells. (A) Cells were incubated with [WT]7 (circles), [F427A]7 (diamonds), or [WT]6[F427A]1 (squares) at indicated concentrations in the presence of 100 nM LFN-DTA. After 16 h, we measured the incorporation of 3H-leucine into protein over a 1-h period. (B) To demonstrate that the activity of the [WT]6[F427A]1 preparation was due to heteroheptamer, and not to traces of [WT]7, we incubated each preparation with 5 mL monomeric avidin resin, washed the resin with 15 mL buffer, and then eluted with 15 mL 2 mM d-biotin. With the [WT]6[F427A]1 preparation, all of the protein bound to the resin and was eluted with biotin (triangles), and its specific activity was unchanged by the additional purification step. With [WT]7, none of the protein bound to the resin, and the specific activity after resin-treatment (squares and circles) was unchanged. (C, D) Titration of [WT]7 (C) and [WT]6[F427A]1 (D) in CHO-K1 cells in the presence of various concentrations of LFN-DTA (circles = 100 nM, squares = 10 nM, and diamonds = 1 nM) for ability to inhibit protein synthesis in CHO-K1 cells. C: [WT]7 gave EC50 values of 26 ± 7 pM, 44 ± 9 pM, and 40 ± 5 pM for LFN-DTA concentrations of 100 nM, 10 nM, and 1 nM, respectively. D: [WT]6[F427A]1 gave EC50 values of 2.2 ± 0.3 nM, 22 ± 6 nM, and 190 ± 30 nM, respectively, for the same set of LFN-DTA concentrations. The data plotted are averages of three experiments, with standard error shown.
In Figure 2, panels C and D, we show data from a cytotoxicity assay in which [WT]6[F427A]1 and[WT]7 were titrated in the presence of various concentrations of LFN-DTA. The concentration of [WT]7 required for half-maximal inhibition (EC50) remained constant at ∼40 pM, as the concentration of LFN-DTA was varied from 1 nM to 100 nM [Fig. 2(C)]. In contrast, with [WT]6[F427A]1, the EC50 varied from 2 nM to 200 nM over the same range of LFN-DTA concentrations [Fig. 2(D)]. With [F427A]7, inhibition was <50% even at 100 nM LFN-DTA and 1 nM prepore (data not shown). These findings suggest that incorporation of even one F427A subunit into the prepore alters the affinity of LFN-DTA for the prepore and/or the pore.
To determine the effect of the F427A subunit on the rate of formation of ion-conductive pores in membranes, we measured the release of K+ from KCl-loaded large unilamellar vesicles (LUV) at pH 5.0.25 [WT]6[F427A]1, [WT]7 and [F427A]7 prepore were complexed with isolated von Willebrand factor A (VWA) domain from anthrax toxin receptor 2 (ANTXR2), and each complex was mixed with DOPC LUVs prepared in buffer containing 100 mM KCl. The mixture was then diluted into isotonic NaCl buffered to pH 5.0 with acetate/acetic acid, and the release of K+ was monitored with a K+-selective electrode. Complexing the prepores with ANTXR2 VWA, besides creating conditions more closely replicating those of cell-bound prepore, partially stabilized the prepore in solution, allowing the kinetics of pore formation in LUVs to be measured accurately. The release of K+ with the heteroheptamer was slower by a factor of ∼2 than with WT prepore, and the F427A homoheptamer was ∼4-fold slower (see Fig. 3). The result with the F427A homoheptamer is consistent with findings reported elsewhere, in which the [F427A]7 prepore was shown to undergo prepore-to-pore conversion at a significantly lower rate than the WT prepore.26 The rates of K+ release for the heteroheptamer and F427A homoheptamer shown in Figure 3 may under-represent the effects of the mutation, given that the mutation increases single-channel conductance, as described below.
Figure 3.
Potassium release assay for pore formation in liposomes by F427A-containing prepores. Pore formation was assayed by potassium release from DOPC LUVs containing 100 mM KCl into medium containing 100 mM NaCl. [WT]7, [F427A]7, or [WT]6[F427A]1 prepores were preincubated with the PA-binding domain of anthrax receptor 2 and then added to liposomes in buffer at pH 5.0. K+ release was measured with a K+-selective probe. Representative traces are shown. Rates were biphasic, with similar rate constants for one phase (k2 = 0.006 s−1 for all three heptamers), representing nonspecific release, and different for the other phase (k1 = 0.33 s−1 for [WT]7, 0.19 s−1 for [WT]6[F427A]1 and 0.078 s−1 for [F427A]7), representing the pore-mediated release. Representative data are shown from n ≥ 3 trials.
Single-channel measurements (Δψ = +20 mV; symmetric pH of 5.5) in planar phospholipid bilayers yielded conductance values of 55 pS and 110 pS, respectively, for [WT]7 and [F427A]7, consistent with earlier results.15 [WT]6[F427A]1 exhibited a single channel conductance of ∼70 pS. As previously reported,15 addition of LFN (7 nM) to the cis chamber inhibited single-channel conductance almost completely in the case of [WT]7, but inhibition of channels formed with [F427A]7 was lower and characterized by flickering among open, partially closed, and fully closed states [Fig. 4(A,B)]. Addition of LFN to [WT]6[F427A]1 induced a similar block to that of [F427A]7, but residence within the open and intermediate flickering states was increased [Fig. (C)4]. The flickering may reflect rapid transitions among various interaction states of the disordered and highly charged N-terminal region of bound LFN with residues at position Phe427 and perhaps other sites in the pore lumen.
Figure 4.
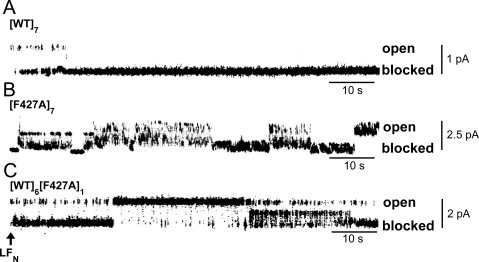
Effect of LFN on single-channel conductance of F427A-containing pores. Single-channel current records after adding LFN (7 nM, final concentration) to the cis chamber (time 0). The membrane potential was +20 mV and the pH was symmetric, at pH 5.5. The traces shown in panels A and B were reported earlier15 and are included for comparison purposes.
Macroscopic conductance measurements suggested that the single F427A subunit of [WT]6[F427A]1 affects the dissociation of LFN from channels (see Fig. 5). After channels were formed with [WT]6[F427A]1, [WT]7 or [F427A]7 in planar bilayers under symmetric, pH 5.5 conditions, a nontranslocatable complex of C-terminally biotinylated LFN with streptavidin was added and allowed to block the channels. The cis compartment was then perfused to remove free blocking agent. With [WT]7 channels, the conductance block declined by <10% over 300 s, whereas with [WT]6[F427A]1 channels the block was alleviated significantly faster. The release of block under these conditions reflects dissociation of LFN into the cis compartment, as the bound streptavidin prevented translocation through the pore. The inability of the LFN:streptavidin complex to translocate was confirmed by raising the pH of the trans compartment to neutrality immediately after terminating perfusion of the cis compartment (data not shown). Similar dissociation occurred in studies with LFN and LFN-DTA (data not shown).
Figure 5.
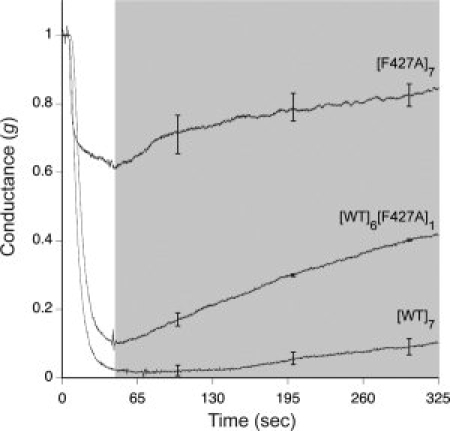
Normalized macroscopic channel current traces monitoring dissociation of LFN-streptavidin from F427A-containing mutants during perfusion of the cis compartment. Streptavidin was bound to LFN through a C-terminal biotin tag on the latter. LFN-streptavidin (10 nM) was added at time zero to channels formed by [WT]7, [F427A]7, or [WT]6[F427A]1. After channel blockage approached its maximum value, we perfused the cis compartment and monitored current while holding the membrane potential at Δψ = +5 mV. The shaded area shows the period during which perfusion (∼ 7 mL/min) was conducted. LFN-streptavidin dissociation is manifested by the rise in current during perfusion. The data plotted are averages of three experiments, with standard error shown for every 1000 data points.
If one raises the pH of the trans compartment to neutrality (pH 7.2) with uncomplexed LFN bound to the [WT]7 channel, this ligand rapidly translocates to the trans compartment, and the conductance rises as LFN exits the channel. Figure 6 shows representative time courses of LFN translocation through [WT]7, [F427A]7, and [WT]6[F427A]1 channels. Translocation through [WT]7 channels was complete within about a minute, whereas no translocation through [F427A]7 channels was observed. With [WT]6[F427A]1 channels, translocation of LFN was very much slower than with [WT]7 channels. Thus, the presence of one F427A subunit in the pore caused a strong inhibition of translocation. LFN-DTA behaved identically to LFN with respect to blockage of channels and translocation (data not shown).
Figure 6.
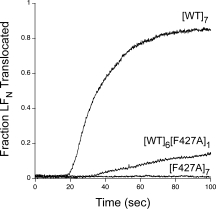
pH-driven translocation of LFN across planar bilayers at Δψ = +5 mV. Macroscopic current values were obtained for [WT]7, [F427A]7, and [WT]6[F427A]1 at symmetrical pH of 5.5 and Δψ = +5 mV. The channels were blocked with 10 nM LFN and the cis compartment was then perfused to remove excess LFN (90 s). At time 0, translocation was initiated by adding 2M KOH to the trans compartment to raise the pH to 7.2; there was an approximately 20-s mixing delay in this system. Both compartments were stirred continuously. Representative data are shown from n ≥ 3 trials.
Discussion
In the search for residues crucial to functioning of PA, the 2β7-2β8 and 2β10-2β11 loops in the lumen of the prepore have emerged as major points of interest. Replacing any of several residues in these loops causes drastic inhibition—several orders of magnitude in some cases—of pore formation and/or pore functioning. As a route to describing the mechanisms of inhibition and dominance in greater detail, we devised a protocol to characterize the activity of heteroheptameric PA prepores with an inhibitory mutation in only one subunit ([WT]6[mutant]1).
To validate the protocol, we chose Ala substitutions for D425 and F427, in the 2β10-2β11 loop. Both D425A and F427A were known to cause strong, dominant-negative inhibition of PA activity, but by different mechanisms. D425A strongly blocked the conformational transition of the prepore to the pore.17 F427A partially impeded pore formation,26 and the pores formed were devoid of ability to translocate protein substrates. Also, binding of substrates was perturbed by this mutation.15
Recent studies of a PA homolog, a component of Clostridium botulinum C2 toxin termed C2II, confirm the importance of the corresponding residues, F428 and D426, as well as other luminal residues.27,28 Mutations of C2II F428 to Ala and other residues altered the single-channel conductance properties of pores and the binding of chloroquine and its analogs to the pore.28 Likewise, mutating C2II D426 to Ala blocked pore formation.27
In the current study, we showed that, remarkably, [WT]6[D425A]1 was about five orders of magnitude less active than [WT]7 in the standard cytotoxicity assay. The single D425A subunit in the prepore blocked conversion of the prepore to the SDS-resistant oligomer in solution and ablated pore formation in planar lipid bilayers under low pH conditions (see Fig. 1). These findings, which are consistent with earlier results obtained with D425A homoheptamer, illustrate in a dramatic fashion the strength and dominance of a single D425A mutation within the prepore.
Understanding the structural role played by D425 will require a higher resolution structure than that currently available. Consistent with its pivotal role in prepore-to-pore transition, D425 is highly conserved in PA homologs. Glu is found in PA homologues from Xanthomonas campestris, X. oryzae, and X. axonopodis, suggesting the need for a negative charge at this site. There is evidence from covariation and mutatational analyses that a neighboring residue, D426, forms a salt bridge with K397, in 2β10-2β11,16 an interaction that would place constraints on the D425 side chain. The cognate interactions of the D425 side chain have not been identified.
In the case of F427, Ala in a single subunit caused a less dramatic overall effect on cytotoxicity: ∼100-fold inhibition. Homoheptameric F427A channels formed in planar phospholipid bilayers were shown earlier to be unable to translocate cargo under the influence of ΔpH.14,15 In the current study, we induced translocation by raising the trans pH from pH 5.5 to neutrality at Δψ = +5 mV and observed a strong reduction in translocation rate by [WT]6[F427A]1 (see Fig. 6). This effect on translocation presumably accounts for much of the overall effect on cytotoxicity, but effects were also found on pore formation and affinity of LFN for the pore (Figs. 3–5). Thus the dominance of the F427A mutation was multifactorial and weaker than that of D425A. Assigning precise values to the relative inhibition of pore formation, ligand affinity and translocation is not possible, given that the value of Δψ across the endosomal membrane is unknown. The effect on ligand binding could be substantial, but is more complex to evaluate, because it depends on both the ligand concentration and Δψ.
How do these findings relate to current concepts of how F427 functions in protein transport? When LFN interacts with WT pores, a seal is formed that blocks the passage of current-carrying ions. The pore is cation-selective, and in our planar bilayers, where KCl is the predominant electrolyte, blockage of current implies a seal against K+. However, it is reasonable to assume that the passage of protons is also blocked. If this is the case, the blockage could preserve the pH gradient within the individual pore and perhaps the microenvironment in the local vicinity of the Phe clamp. The major effect on translocation of replacing a single F427 residue in the pore with Ala may therefore result from disruption of the seal, and thus dissipation of ΔpH as the driving force for translocation.
In considering how F427 contributes to binding of LFN, we note first that in the neutral pH range LFN binds with high affinity (Kd ∼ 1 nM) to sites on domain 1′ of the prepore formed at the intersection of adjacent subunits.16 A good understanding of this recognition has come from mutation-based mapping, energy minimization, and hydrogen–deuterium exchange measurements.29–31 Under acidic conditions, free LFN transitions to a molten globule state,10 which may have a weaker affinity for the binding sites on the surface of domain 1′. However, evidence from planar bilayer experiments indicates that the disordered ∼30-residue N-terminal segment of LFN interacts with the Phe clamp under acidic conditions, at which this segment is positively charged.11,14,15 The suggestion of weaker affinities for the single Ala at the Phe clamp (see Fig. 5) is supported by the strong dependence of cytotoxicity on LFN-DTA concentration found when we used [WT]6[F427A]1 [Fig. 2(D)]. We suggest that the weaker affinity of the N-terminal segment of LFN for the F427A-containing Phe clamp causes LFN-DTA to bind less strongly as the complex transitions to an acidic environment in the endosome.
The postulated effect of a single F427A subunit on prepore-to-pore conversion are consistent with results with F427A homoheptamers,26 and we invoke the model put forth in that report to explain those findings. Evidence from electron paramagnetic measurements of F427C prepore and pore indicates that the F427 side chains, which are ∼20 Å apart in the prepore, come into close proximity, and perhaps even direct contact, in the pore or an intermediate state in prepore-to-pore conversion.15,16 In this case, hydrophobic interactions between the F427 side chains may facilitate the conformational change of prepore to pore, and the substitution of Ala for one or more of the seven F427 residues may hinder the conversion.
In the final analysis, it is clear that F427 and D425 play pivotal roles in a highly evolved bacterial toxin translocation system. The importance of F427 and D425 in the functioning of PA is consistent with the conservation of these residues in all known homologs of PA.10,15 Our protocol to isolate oligomers containing predominantly single-subunit mutations will be useful in analyzing other residues in PA and should be applicable to other systems, allowing more detailed investigation of inhibitory mutations.
Methods
Materials
Biochemical reagents were from Sigma unless indicated otherwise. Mutagenic oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). LFN-DTA was prepared as described,18 the VWA domain of CMG2 was prepared as described,32 3H-Leu was obtained from New England Nuclear (Waltham, Massachusetts), Nα-(3-maleimidylpropionyl)-biocytin was obtained from Invitrogen—Molecular Probes (Carlsbad, California), UltraLink Immobilized Monomeric avidin and immunopure streptavidin were from Pierce (Rockford, IL), and HABA reagent used to measure protein-linked biotin groups was from the Pierce EZ Biotin Quantitation Kit. E. coli used for expression of proteins was grown in ECPM1 medium.33
Methods
Expression and purification of proteins
Recombinant WT PA and PA mutants were over-expressed in the periplasm of E. coli BL21 (DE3) and purified by anion-exchange chromatography.6 Recombinant LFN was expressed in the cytoplasm of E. coli BL21 (DE3) as an N-terminally hexa-His tagged protein. It was purified over Ni-NTA resin and treated with bovine α-thombin to cleave off the His-tag, as described.11 As a negative control for translocation in vitro, LFN was biotinylated on its C-terminal residue and bound to streptavidin as described.11
Biotinylation of PA containing K563C
Monomeric PA mutants (∼50 mg) containing K563C and either F427A or D425A in PBS (100 mM phosphate, 150 mM NaCl, pH 7.2) were treated with 5 mM TCEP for 1 h at room temperature before biotinylation with a 10-fold molar excess of Nα-(3-maleimidylpropionyl)biocytin at 4°C overnight. Excess biotinylation reagent was removed by desalting resin (DG50, BioRad) before passing the biotinylated protein over 25 mL of UltraLink monomeric avidin resin (Pierce) and eluting with 2 mM d-biotin. Free biotin was removed by an additional pass by desalting resin.
Isolation of PA prepore
WT PA prepore ([WT]7), PA F427A/K563C prepore ([F427A]7), and PA D425A/K563C prepore ([D425A]7) were formed by limited trypsin digestion and purified by anion-exchange chromatography, as described.32 Heteroheptamers containing predominantly one mutant subunit were prepared as follows. We developed the methods using PA F427A/K563C as a prototype. First, we mixed WT PA with biotinylated PA F427A/K563C in a 20:1 molar ratio, nicked the mixture with trypsin, and purified the resulting heptamers over anion-exchange chromatography (5 mL HiTrap Q HP, GE Healthcare). The resulting preparation was then passed over monomeric avidin (25 mL of UltraLink monomeric avidin resin, Pierce), and bound biotinylated heptamers were eluted with 2 mM d-biotin. The product was desalted to remove free biotin and then passed over a second monomeric avidin column to ensure the removal of [WT]7. A parallel control experiment was done in which purified [WT]7 was passed over two sequential avidin columns and the percentage of [WT]7 retained was quantified. The molar ratio of biotin per heteroheptamer was determined quantitatively by using 4-hydroxyazobenzene-2-carboxylic acid (HABA) reagent, which forms a colored product when bound to avidin (ɛ500 nm = 35,500 M−1cm−1).34,35 Because biotin binds tighter to avidin than does HABA, biotinylated protein displaces HABA, reducing the amount of colored product. A standard curve was constructed using 1–25 nmol d-biotin.
Although affinity chromatography on monomeric avidin resin effectively removed [WT]7, it did not remove heteroheptamers containing more than one mutant subunit. Thus, when we mixed WT and biotinylated F427A PA in various ratios, isolated the prepore population formed from each mixture, and passed it over a monomeric avidin column, the number of biotinyl groups per unit protein in the eluted fraction, as measured by the HABA assay, increased from 1.0 ± 0.1 with a 20:1 ratio of WT:F427A to 4.4 ± 0.2 with a 2:1 ratio of WT:F427A. Prepore with ≥1 biotinyl group was therefore retained on the column, and at least some containing multiple biotinyl groups was eluted under the conditions used. Regardless, the value of 1.0 ± 0.1 biotinyl group per heptamer measured with heteroheptameric product yielded by WT:F427A at a 20:1 ratio is consistent with [WT]6[F427A]1 being the predominant molecular species and shows that prepores with >1 F427A subunit were not over-represented in the final product.
Cytotoxicity assays
Cytotoxicity assays were performed as described.17,19 PA-dependent translocation of LFN-DTA is reported by the DTA domain, the enzymatic domain of diphtheria toxin, which inhibits protein synthesis. Translocation of the DTA domain into CHO-K1 cells was determined by measuring the incorporation of 3H-leucine into protein after a 16 h incubation with varying concentrations of [PA]7 and 100 nM LFN-DTA at 37°C under 5% CO2.
SDS-resistance assay
An SDS-resistant oligomer is formed when WT PA prepore is subjected to acidic pH.6 This condition has been shown to promote pore formation and translocation of cargo.23 Thus, a test for susceptibility to pore formation by any given PA mutant is to examine its resistance to SDS under acidic conditions. PA prepores (5 μg) made of [WT]7, [D425A]7, or [WT]6[D425A]1 were incubated in 50 mM HEPES with 150 mM NaCl at either pH 8.5 or pH 5.5 for 30 min at room temperature before the addition of SDS sample buffer and being electrophoresed on a 4–12% Tris-Glycine gradient gel by the method of SDS PAGE.
Membrane insertion assay
Pore formation in liposomes was assayed using the potassium release (K+ release) assay, as described.25 Liposomes composed of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) were prepared 2–4 h before use, as described.25 Immediately before performing the K+ release assay, the liposomes maintained in a K+ buffer (10 mM HEPES, 100 mM KCl, pH 7.4) were exchanged into a Na+ buffer (10 mM HEPES, 100 mM NaCl, pH 7.4) in order to establish liposomes with K+ inside and Na+ outside. PA prepore (10 μg), at pH 8.5, was incubated with 10 μg von Willebrand factor A (VWA) domain of anthrax toxin receptor 2 (ANTXR2) for 1 h on ice before addition of 150 μL of the freshly prepared liposomes. The liposome and protein mixtures were incubated on ice for 30 min and then diluted into 5 mL 10 mM sodium acetate buffer, pH 5.0, containing 100 mM NaCl. The mixtures were stirred continuously with a magnetic stirrer, and release of K+ from the liposomes was monitored with an electrode selective for K+ (Orion Research). The background, determined with buffer alone, was subtracted, to allow comparison of the different samples. The data were fitted to a biphasic sum of two exponentials equation with KaliedaGraph software.
Electrophysiology
Planar lipid bilayers were painted36 onto a 200 μm aperture of a Delrin cup sitting in a Lucite chamber, with 3% 1,2-diphytanoyl-sn-glycerol-3-phosphocholine (DPhPC) in n-decane (Avanti Polar Lipids, Alabaster, AL). The volumes of the cup and the chamber were each 1 mL, and both compartments were stirred constantly. Cis (side to which PA prepore and LFN were added) and trans compartments were generally bathed in a solution containing 100 mM KCl, 1 mM EDTA and 10 mM each of potassium oxalate, potassium phosphate, and MES, pH 5.5. Macroscopic current responses to steps in voltage were recorded as described.12 The membrane potential, Δψ, is defined as Δψ = ψcis − ψtrans, where ψtrans ≡ 0 mV. Single-channel measurements were obtained using DphPC/decane painted on a 50 μM aperture of a 1 mL polystyrene cup.37 Single-channel conductance measurements were carried out at Δψ = +20 mV in symmetric 100 mM KCl, 1 mM EDTA, 25 mM potassium succinate, pH 5.5.
PA63 channel formation and LFN conductance block
Once a membrane was formed in the planar lipid bilayer system, PA prepore (25 pM) was added to the cis compartment, which was held at a Δψ of +5 mV with respect to the trans compartment. After appropriate current increase, the cis chamber was perfused with non-PA-containing buffer at a flow rate of ∼3 mL/min. Once the current was constant, ligand (LFN, LFN-DTA or LFN-biotin-streptavidin) was added to the cis compartment, and the progress of binding to PA channels was monitored by the fall in conductance. Finally, the cis chamber was perfused again to eliminate free ligand.
To measure the rate of dissociation in real time (koff), ligand (LFN or LFN-biotin-streptavidin) was added to the cis chamber (Δψ = +5 mV), allowed to bind PA pores, and then perfusion was initiated. The rate of increase in conductance was used to estimate the rate of ligand dissociation.
Translocation under the influence of ΔpH
After ligand was added and conductance stabilized, translocation was initiated by raising the pH of the trans chamber to pH 7.2 with 2M KOH, while maintaining the cis chamber pH at 5.5. The kinetics of translocation was monitored at a membrane potential of +5 mV by the rise in current.
Acknowledgments
We thank Ruth-Anne Pimental for assistance in protein preparation, Zhengyan Wu for help in single-channel measurements, and Adam Barker and Roman Melnyk for helpful discussions. One of us (RJC) holds equity in PharmAthene, Inc., and consults for CombinatoRx, Inc.
References
- 1.Young JA, Collier RJ. Anthrax toxin: receptor binding, internalization, pore formation, and translocation. Annu Rev Biochem. 2007;76:243–265. doi: 10.1146/annurev.biochem.75.103004.142728. [DOI] [PubMed] [Google Scholar]
- 2.Klimpel KR, Molloy SS, Thomas G, Leppla SH. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc Natl Acad Sci USA. 1992;89:10277–10281. doi: 10.1073/pnas.89.21.10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Molloy SS, Bresnahan PA, Leppla SH, Klimpel KR, Thomas G. Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J Biol Chem. 1992;267:16396–16402. [PubMed] [Google Scholar]
- 4.Ezzell JW, Jr, Abshire TG. Serum protease cleavage of Bacillus anthracis protective antigen. J Gen Microbiol. 1992;138:543–549. doi: 10.1099/00221287-138-3-543. [DOI] [PubMed] [Google Scholar]
- 5.Mogridge J, Cunningham K, Collier RJ. Stoichiometry of anthrax toxin complexes. Biochemistry. 2002;41:1079–1082. doi: 10.1021/bi015860m. [DOI] [PubMed] [Google Scholar]
- 6.Miller CJ, Elliott JL, Collier RJ. Anthrax protective antigen: prepore-to-pore conversion. Biochemistry. 1999;38:10432–10441. doi: 10.1021/bi990792d. [DOI] [PubMed] [Google Scholar]
- 7.Nassi S, Collier RJ, Finkelstein A. PA63 channel of anthrax toxan extended beta-barrel. Biochemistry. 2002;41:1445–1450. doi: 10.1021/bi0119518. [DOI] [PubMed] [Google Scholar]
- 8.Katayama H, Janowiak BE, Brzozowski M, Juryck J, Falke S, Gogol EP, Collier RJ, Fisher MT. GroEL as a molecular scaffold for structural analysis of the anthrax toxin pore. Nat Struct Mol Biol. 2008;15:754–760. doi: 10.1038/nsmb.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blaustein RO, Finkelstein A. Diffusion limitation in the block by symmetric tetraalkylammonium ions of anthrax toxin channels in planar phospholipid bilayer membranes. J Gen Physiol. 1990;96:943–957. doi: 10.1085/jgp.96.5.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krantz BA, Trivedi AD, Cunningham K, Christensen KA, Collier RJ. Acid-induced unfolding of the amino-terminal domains of the lethal and edema factors of anthrax toxin. J Mol Biol. 2004;344:739–756. doi: 10.1016/j.jmb.2004.09.067. [DOI] [PubMed] [Google Scholar]
- 11.Zhang S, Finkelstein A, Collier RJ. Evidence that translocation of anthrax toxin's lethal factor is initiated by entry of its N terminus into the protective antigen channel. Proc Natl Acad Sci USA. 2004a;101:16756–16761. doi: 10.1073/pnas.0405754101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang S, Udho E, Wu Z, Collier RJ, Finkelstein A. Protein translocation through anthrax toxin channels formed in planar lipid bilayers. Biophys J. 2004b;87:3842–3849. doi: 10.1529/biophysj.104.050864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blaustein RO, Koehler TM, Collier RJ, Finkelstein A. Anthrax toxchannel-forming activity of protective antigen in planar phospholipid bilayers. Proc Natl Acad Sci USA. 1989;86:2209–2213. doi: 10.1073/pnas.86.7.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krantz BA, Finkelstein A, Collier RJ. Protein translocation through the anthrax toxin transmembrane pore is driven by a proton gradient. J Mol Biol. 2006;355:968–979. doi: 10.1016/j.jmb.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 15.Krantz BA, Melnyk RA, Zhang S, Juris SJ, Lacy DB, Wu Z, Finkelstein A, Collier RJ. A phenylalanine clamp catalyzes protein translocation through the anthrax toxin pore. Science. 2005;309:777–781. doi: 10.1126/science.1113380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Melnyk RA, Collier RJ. A loop network within the anthrax toxin pore positions the phenylalanine clamp in an active conformation. Proc Natl Acad Sci USA. 2006;103:9802–9807. doi: 10.1073/pnas.0604000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sellman BR, Nassi S, Collier RJ. Point mutations in anthrax protective antigen that block translocation. J Biol Chem. 2001;276:8371–8376. doi: 10.1074/jbc.M008309200. [DOI] [PubMed] [Google Scholar]
- 18.Mourez M, Yan M, Lacy DB, Dillon L, Bentsen L, Marpoe A, Maurin C, Hotze E, Wigelsworth D, Pimental RA, Ballard JD, Collier RJ, Tweten RK. Mapping dominant-negative mutations of anthrax protective antigen by scanning mutagenesis. Proc Natl Acad Sci USA. 2003;100:13803–13808. doi: 10.1073/pnas.2436299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sellman BR, Mourez M, Collier RJ. Dominant-negative mutants of a toxin subunit: an approach to therapy of anthrax. Science. 2001;292:695–697. doi: 10.1126/science.109563. [DOI] [PubMed] [Google Scholar]
- 20.Singh Y, Khanna H, Chopra AP, Mehra V. A dominant negative mutant of Bacillus anthracis protective antigen inhibits anthrax toxin action in vivo. J Biol Chem. 2001;276:22090–22094. doi: 10.1074/jbc.M010222200. [DOI] [PubMed] [Google Scholar]
- 21.Blanke SR, Milne JC, Benson EL, Collier RJ. Fused polycationic peptide mediates delivery of diphtheria toxin A chain to the cytosol in the presence of anthrax protective antigen. Proc Natl Acad Sci USA. 1996;93:8437–8442. doi: 10.1073/pnas.93.16.8437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milne JC, Blanke SR, Hanna PC, Collier RJ. Protective antigen-binding domain of anthrax lethal factor mediates translocation of a heterologous protein fused to its amino-or carboxy-terminus. Mol Microbiol. 1995;15:661–666. doi: 10.1111/j.1365-2958.1995.tb02375.x. [DOI] [PubMed] [Google Scholar]
- 23.Milne JC, Furlong D, Hanna PC, Wall JS, Collier RJ. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J Biol Chem. 1994;269:20607–20612. [PubMed] [Google Scholar]
- 24.Benson EL, Huynh PD, Finkelstein A, Collier RJ. Identification of residues lining the anthrax protective antigen channel. Biochemistry. 1998;37:3941–3948. doi: 10.1021/bi972657b. [DOI] [PubMed] [Google Scholar]
- 25.Sun J, Vernier G, Wigelsworth DJ, Collier RJ. Insertion of anthrax protective antigen into liposomal membranes: effects of a receptor. J Biol Chem. 2007;282:1059–1065. doi: 10.1074/jbc.M609869200. [DOI] [PubMed] [Google Scholar]
- 26.Sun J, Lang AE, Aktories K, Collier RJ. Phenylalanine-427 of anthrax protective antigen functions in both pore formation and protein translocation. Proc Natl Acad Sci USA. 2008;105:4346–4351. doi: 10.1073/pnas.0800701105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lang AE, Neumeyer T, Sun J, Collier RJ, Benz R, Aktories K. Amino acid residues involved in membrane insertion and pore formation of Clostridium botulinum C2 toxin. Biochemistry. 2008;47:8406–8413. doi: 10.1021/bi800615g. [DOI] [PubMed] [Google Scholar]
- 28.Neumeyer T, Schiffler B, Maier E, Lang AE, Aktories K, Benz R. Clostridium botulinum C2 toxin. Identification of the binding site for chloroquine and related compounds and influence of the binding site on properties of the C2II channel. J Biol Chem. 2008;283:3904–3914. doi: 10.1074/jbc.M709807200. [DOI] [PubMed] [Google Scholar]
- 29.Lacy DB, Lin HC, Melnyk RA, Schueler-Furman O, Reither L, Cunningham K, Baker D, Collier RJ. A model of anthrax toxin lethal factor bound to protective antigen. Proc Natl Acad Sci USA. 2005;102:16409–16414. doi: 10.1073/pnas.0508259102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lacy DB, Mourez M, Fouassier A, Collier RJ. Mapping the anthrax protective antigen binding site on the lethal and edema factors. J Biol Chem. 2002;277:3006–3010. doi: 10.1074/jbc.M109997200. [DOI] [PubMed] [Google Scholar]
- 31.Melnyk RA, Hewitt KM, Lacy DB, Lin HC, Gessner CR, Li S, Woods VL, Jr, Collier RJ. Structural determinants for the binding of anthrax lethal factor to oligomeric protective antigen. J Biol Chem. 2006;281:1630–1635. doi: 10.1074/jbc.M511164200. [DOI] [PubMed] [Google Scholar]
- 32.Wigelsworth DJ, Krantz BA, Christensen KA, Lacy DB, Juris SJ, Collier RJ. Binding stoichiometry and kinetics of the interaction of a human anthrax toxin receptor, CMG2, with protective antigen. J Biol Chem. 2004;279:23349–23356. doi: 10.1074/jbc.M401292200. [DOI] [PubMed] [Google Scholar]
- 33.Bernard A, Payton M. Current protocols in protein science. Hoboken, NJ: Wiley; 1995. Fermentation and Growth of Escherichia coli for Optimal Protein Production. [DOI] [PubMed] [Google Scholar]
- 34.Livnah O, Bayer EA, Wilchek M, Sussman JL. The structure of the complex between avidin and the dye, 2-(4′-hydroxyazobenzene) benzoic acid (HABA) FEBS Lett. 1993;328:165–168. doi: 10.1016/0014-5793(93)80986-5. [DOI] [PubMed] [Google Scholar]
- 35.Wang ZX, Kumar NR, Srivastava DK. A novel spectroscopic titration method for determining the dissociation constant and stoichiometry of protein-ligand complex. Anal Biochem. 1992;206:376–381. doi: 10.1016/0003-2697(92)90381-g. [DOI] [PubMed] [Google Scholar]
- 36.Mueller P, Rudin DO. Resting and action potentials in experimental bimolecular lipid membranes. J Theor Biol. 1963;4:268–280. doi: 10.1016/0022-5193(68)90163-x. [DOI] [PubMed] [Google Scholar]
- 37.Blaustein RO, Lea EJ, Finkelstein A. Voltage-dependent block of anthrax toxin channels in planar phospholipid bilayer membranes by symmetric tetraalkylammonium ions. Single-channel analysis. J Gen Physiol. 1990;96:921–942. doi: 10.1085/jgp.96.5.921. [DOI] [PMC free article] [PubMed] [Google Scholar]