

Abstract
Probably the most unusual class of proteins in nature is the intrinsically unstructured proteins (IUPs), because they are not structured yet play essential roles in protein-protein signaling. Many IUPs can bind different proteins, and in many cases, adopt different bound conformations. The p21 protein is a small IUP (164 residues) that is ubiquitous in cellular signaling, for example, cell cycle control, apoptosis, transcription, differentiation, and so forth; it binds to approximately 25 targets. How does this small, unstructured protein recognize each of these targets with high affinity? Here, we characterize residual structural elements of the C-terminal segment of p21 encompassing residues 145–164 using a combination of NMR measurements and molecular dynamics simulations. The N-terminal half of the peptide has a significant helical propensity which is recognized by calmodulin while the C-terminal half of the peptide prefers extended conformations that facilitate binding to the proliferating cell nuclear antigen (PCNA). Our results suggest that the final bound conformations of p21 (145–164) pre-exist in the free peptide even without its binding partners. While the conformational flexibility of the p21 peptide is essential for adapting to diverse binding environments, the intrinsic structural preferences of the free peptide enable promiscuous yet high affinity binding to a diverse array of molecular targets.
Keywords: p21, residual structure, intrinsically unfolded protein, NMR, molecular dynamics
Introduction
Many proteins adopt a well-defined tertiary structure under physiological conditions, and this structure largely determines protein function. However, there is a class of proteins known as intrinsically unstructured proteins (IUPs) that also play specific roles in protein-protein recognition. Some well-known examples include the phosphorylated kinase-inducible domain (pKID) of the cAMP responsive element binding protein (CREB),1 the transcriptional activation domain (TAD) of p53,2 and the GTPase-binding domain (GBD) of the Wiskott-Aldrich syndrome protein (WASP).3 Spectroscopic studies suggest that these IUPs are not completely random, but can exhibit residual secondary structural preferences. For example, NMR studies demonstrated that the linker helix of p27Kip1 has a nascent secondary structure in its free state4 although it is largely unstructured in solution.5 It is increasingly apparent that residual structure of IUPs plays crucial roles in molecular recognition.6–8 In this regard, it has been suggested that the classic protein structure-function paradigm for IUPs be re-assessed and that protein function for these systems can be understood using a formalism that models the IUP structure as an ensemble of distinct conformations.6,9,10
p21Waf1/Cip1/Sdi1 (hereafter referred to as p21) is an IUP involved in the regulation of the cell cycle.11 p21 was first identified as a cyclin-dependent kinase (Cdk) inhibitor12 that mediates the G1/S arrest13 and later was found to function in apoptosis,14 differentiation,15 transcription,16 DNA synthesis control17 and stem cell self-renewal.18 The C-terminal region of p21, which is unique among the Cip/Kip family of Cdk inhibitors, interacts with a large array of proteins, including the proliferating cell nuclear antigen (PCNA),19,20 calmodulin (CaM),21 SET,22 c-Myc,23 and the E7 oncoprotein of human papilloma virus 16 (HPV-16).17
How does p21, a small protein of 164 residues, physically recognize so many structurally dissimilar proteins without sacrificing binding affinity? The binding diversity of the C-terminal segment of p21 has been attributed to its ability to acquire different conformations upon binding to distinct targets.24 Although the far UV CD spectra suggest that residues 145–164 (p21(145–164)) is unstructured, this peptide adopts a well defined structure having a helical N-terminal region and an extended strand C-terminal region when bound to PCNA.25 Based on previous examples of CaM-substrate binding26 and CD measurement of mutant p21(145–164),24 p21(145–164) is likely to acquire a helical conformation when bound to CaM.
In this study, we combine NMR measurements of free p21(145–164) with molecular dynamics (MD) simulations to obtain models for the unfolded ensemble of the free peptide at a physiologically relevant temperature and pH. We found that the N-terminal half of the peptide has a significant amount of residual helical structure and the C-terminal half has a preference for extended conformations in the unbound state of p21(145–164). NMR dipolar coupling measurements of the CaM – p21(145–164) complex indicate that the peptide is helical when bound to CaM, which in turn suggest that the region of the peptide with helical preference is likely to interact with CaM. On the other hand, the C-terminal loop-like region of the peptide adopts an extended conformation when bound to the PCNA.25 Our results show that the structure adopted by p21(145–164) upon binding to CaM or PCNA already exist in the free peptide in significant population and suggest that the pre-formed structural elements of p21(145–164) contribute to its binding specificity.
Results
Residual secondary structure in p21(145–164) detected by NMR spectroscopy
The 1H-15N HSQC spectrum of 15N-labeled p21(145-164) (Fig. 1) showed poor dispersion (< 1 ppm) of amide proton chemical shifts, consistent with a largely unstructured peptide as observed by far-UV CD spectroscopy.24 To investigate whether there is any residual structure in p21(145–164), we measured 13Cα and 1Hα chemical shifts, as well as the 3-bond 3JHN-Hα coupling constants. The 13Cα and 1Hα chemical shift values are very sensitive to local conformation and thus their deviations from the values of random coil, known as secondary chemical shifts, are indicative of secondary structure.27 Secondary shift analysis is the most widely-used method for detecting residual structural elements in largely unstructured polypeptide chains.28–30 The 13Cα secondary chemical shift of p21(145–164) [Fig. 2(A)] shows that the N-terminal half of the peptide, encompassing residues Met147-Lys154 is partially helical on average, while its C-terminal half shows a preference for extended conformations. In agreement with the 13Cα shifts, the 1Hα secondary shifts [Fig. 2(B)] are consistent with an increased propensity for helical conformation for the N-terminal segment Thr148-Arg155. However, the 1Hα shifts for the C-terminal half of the peptide are not characteristic enough to draw any conclusions on the secondary structure preferences. Independent from the chemical shifts, the deviations of the 3JHN-Hα coupling constants from random coil values [Fig. 2(C)] also suggest helical tendency of the N-terminal half of the peptide, Thr145-Lys154.
Figure 1.
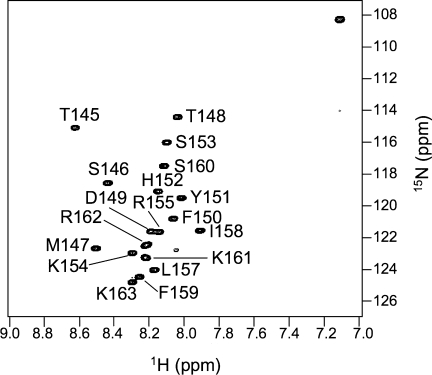
The 1H-15N heteronuclear single quantum correlation (HSQC) spectrum of the 15N-labeled p21(145-164) recorded at 1H frequency of 500 MHz, pH 6.5, and 30°C. All of the backbone 1HN and 15N are assigned except for Arg156.
Figure 2.

NMR measurements of residual secondary structures of p21(145-164). (A) Deviation of 13Cα chemical shifts of p21(145-164) from the random coil values.31 Δ13Cα = 13Cα (p21)-13Cα (random coil). (B) Same as in (A) for the 1Hα chemical shifts. (C) Deviation of the 3JHN-Hα coupling constants of p21 (145-164) from random coil values.32 Δ3JHN-Hα = 3JHN-Hα (p21)-3JHN-Hα (random coil). Data are not available for Arg156 (indicated by *). (D) Secondary structure prediction based on the consensus among the prediction programs AGADIR, APSSP2, HNN, JUFO, nnPredict, Prof, and PSIpred (Supp. Info. Table S1). Shaded and dotted boxes represent the helical and extended conformations, respectively.
The experimental chemical shifts and scalar coupling constants together indicate that the N-terminal half of the peptide is helical, on average, under physiological conditions. While the 13Cα chemical shifts of residues Leu157-Lys163 suggests that this region is extended on average [Fig. 2(A), Supp. Info. Table S1], the 1Hα secondary shifts [Fig. 2(B)] and the 3JHN-Hα deviations [Fig. 2(C)] are less conclusive. Our result is consistent with the secondary structure prediction of p21(145–164) based on the consensus among 7 different prediction programs [Fig. 2(D)], including AGADIR,33 APSSP2,34 HNN (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page = npsa_nn.html), JUFO,35 nnPredict.36,37 and PSIpred.38
Modeling the unfolded state of p21(145–164) with MD simulations
NMR measurements for unfolded proteins correspond to ensemble averages over a number of structurally dissimilar states and therefore do not provide information about the underlying distribution of conformers in the ensemble. To further our understanding of the conformers that make up the unfolded ensemble, we used molecular dynamics (MD) simulations to generate structural ensembles which agree with our experimental data.
Our method, energy-minima mapping and weighting (EMW), associates a statistical weight with each structure that corresponds to the probability that the given protein samples that conformation. The application of EMW presented here is similar to that previously described39 and relies on obtaining NMR chemical shift data for a free peptide in solution, and optimizing structural ensembles containing energetically favorable conformations of that peptide to minimize the error between the calculated chemical shifts and the experimentally measured values. The resulting ensembles agree with the experimental values, while also fulfilling physical constraints imposed by the potential energy function.
As the construction of an unfolded ensemble is an underdetermined problem, there may be several ensembles that agree with any given set of experimental constraints. Therefore EMW does not strive to construct one ensemble that models the unfolded state of p21(145–164). Instead, we generate multiple ensembles that are all consistent with a given set of experimental data, and focus our analysis on local structural motifs that are present across different ensembles.
EMW was used to construct 250 ensembles using absolute 13Cα chemical shifts. The 250 ensembles were then ranked according to their ability to reproduce experimentally determined amide nitrogen chemical shifts, which were not used in the optimization procedure. The 10 ensembles best able to reproduce amide nitrogen chemical shifts were chosen for further analysis.
Calculated 13Cα chemical shifts for these 10 ensembles were in excellent agreement with experiment [Fig. 3(A)]. Moreover, secondary Cα chemical shifts from each of the 10 ensembles were also in excellent agreement with the experimental secondary shift values [Fig. 3(B)]. Experimentally determined nitrogen chemical shifts were all within 1.5 ppm of the experimental values; that is, the error associated with backbone carbon and nitrogen chemical shift predictions40 (Supp. Info. Table S2). To further explore whether these models adequately represent the unfolded state of p21(145–164), we also computed 1Hα, Cβ and 15N absolute chemical shifts for each model and compared these data to the corresponding experimental result. Calculated 1Hα absolute chemical shifts were all within 0.2 ppm of the experimental result, well below the error associated with SHIFTX predictions of proton shifts40 (Supp. Info. Table S2). Similarly, calculated Cβ chemical shifts were all within 1.5 ppm of the experimental values; that is, the error associated with backbone heavy atom chemic shift predictions40 (Supp. Info. Table S2). We note that while the 15N chemical shift errors are the largest, all are less than 1.5 ppm and the SHIFTX error associated with 15N predictions is ∼2.4 ppm.40 Lastly, we calculated scalar 3-bond J-coupling constants from the each of the 10 ensembles and compared these data to the corresponding experimental values.41 Calculated J-coupling constants for p21146–164 had rms errors within 1 Hz from the experimental values. When the C-terminal residue (residue 164) is excluded, the rms error for the non-terminal residues (p21146–163) have an rms error of 0.77 Hz relative to the experimental values (Supp. Info. Table S3). For comparison, prior predictions of scalar J-coupling constants yield an average rms error of 0.73 Hz when the crystallographic structure of the target protein is known, suggesting that even if one knew the precise structure of the protein of interest, an error near 0.73 Hz would not be unexpected.41 In summary, although only Cα chemical shifts were used to create the ensembles, the ensembles themselves have calculated values that are in reasonable agreement with 1Hα, Cβ, and 15N chemical shifts, and scalar J-coupling constants. The final set of ensembles consists of heterogeneous sets of structures (Supp. Info. Fig. S1).
Figure 3.
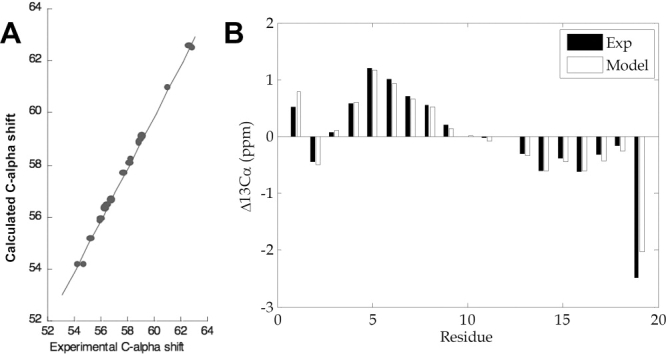
Comparison of experimental Cα chemical shifts and Cα chemical shifts calculated using the “worst” model; that is, the model with the highest rms difference between the calculated and experimental result. (A) Relationship between experimental and calculated absolute Cα chemical shifts. (B) Comparison of experimental and calculated Cα secondary chemical shifts.
To identify local conformations preserved across ensembles, all structures were clustered based on the rms backbone deviation of contiguous six-residue subsequences in p21(145–164). A characteristic length of six residues was chosen as the local region size for this analysis since a crystal structure of a bound state of p21(145–164) contained local structured regions approximately six residues in length.42 Clustering all contiguous six residue segments resulted in 225 distinct clusters. Each cluster is representative of a local conformation within p21(145–164). The total weight associated with a given cluster in an ensemble is given by the sum of the weights of all structures in the cluster. A given cluster is said to be preserved across all 10 ensembles if it has a non-zero weight in each ensemble. Using this definition, only 5.8% of the clusters were preserved across all ensembles.
A preserved structural motif that is present in all independent ensembles is likely required to reproduce the experimental data. Consequently, we consider such locally preserved structures to represent local conformational preferences. Structures of conserved local conformations offer a more detailed view of ensemble characteristics than ensemble averaged experimental secondary chemical shifts.
Conformational preferences for p21(145–164) are shown in Figure 4. Several points are clear from Figure 4. First, every residue in p21(145–164) is found in a six-residue segment that has an extended conformation. Hence, the simulations predict that each residue can adopt an extended state in solution, including the N-terminal residues that have positive secondary Ca chemical shifts [Fig. 2(A)]. What distinguishes the N-terminus is the fact that these residues can also adopt helical conformations (see Fig. 4). Three conserved N-terminal helical clusters were found, corresponding to preformed helical states in the unfolded ensemble, in the six-residue regions corresponding to residues 147–152, 148–153, and 149–154. Residues 153–158 can also adopt a loop/turn conformation in solution. Lastly, the simulations predict that residues 159–163 have a distinct preference for only extended states.
Figure 4.
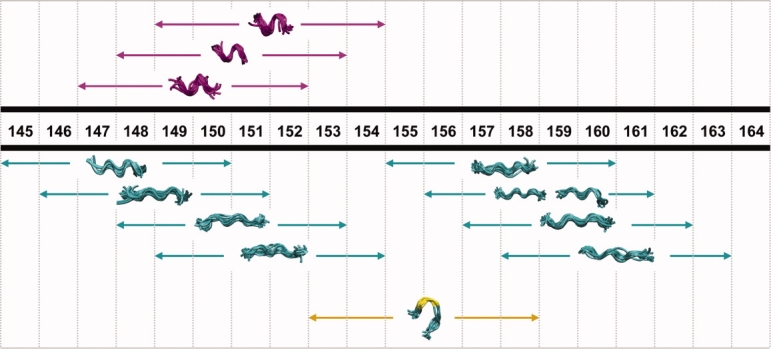
Conserved six-residue structural motifs. Helical conformations are colored purple and extended regions are shown in cyan. The region corresponding to a turn is colored yellow.
It was previously reported that residues (146–151) in PCNA-bound form of p21(139–160) forms a 310 helix, while the C-terminal region spanning residues 152–160 adopts an extended strand which hydrogen bonds to a neighboring β strand in PCNA [Fig. 5(A,B)].42 We sought to determine whether comparable local conformational preferences are predicted for the unfolded ensemble representing the unbound state. We find that the six residue subsequence ranging from residues 147 to 152 can adopt a helical conformation in solution and that the subsequence consisting of residues 155–160 adopts an extended state, suggesting that there local structural preferences in the unfolded ensemble similar to those adopted by the bound form of p21(139–160) [Fig. 5(C)].
Figure 5.

(A) X-ray crystallographic structure of a p21 model peptide (residues 139–160) bound to PCNA [PDB ID 1AXC.43 The helical region (p21 residues 146–151) is highlighted in purple, while the extended C-terminus (p21 residues 152–160) is depicted in cyan. Only residues of p21 included in our model peptide (145–160) are shown. (B) Structure of the p21 model peptide alone. (C) Comparison of corresponding local conformational preferences in models of the unbound form of p21(145–164). Regions for which models of the unbound form include local conformations which match the bound form crystal structure are indicated.
Helical mode of p21(145–164) binding to Ca2+-calmodulin from NMR dipolar couplings
Based on CD measurements of mutant p21(145–164)24 and the previous knowledge of CaM-binding peptides,26 we expected p21(145–164) to be helical upon binding to CaM. Previously, at least 180 CaM recruitment signaling (CRS) motifs were identified and classified based on the spacing of the hydrophobic residues of CRS that make major hydrophobic interaction with CaM.26 However, p21 peptide contains no sequence that conforms to any of these known CRS motifs.
In order to confirm the helical mode of p21(145–164) binding to Ca2+-CaM, two types of residual dipolar couplings (RDCs), 1DNH and 1DCαHα, were measured for Ca2+-CaM in complex with p21(145–164) in a liquid crystalline medium containing 18 mg/mL filamentous phage Pf1. RDCs have been successfully used to determine the binding mode of CaM-interacting peptide to Ca2+-CaM.44,45 The experimentally measured RDCs of CaM in complex with p21(145–164) were fitted to the free Ca2+-CaM structure [Fig. 6(A)] and CaM/peptide complexes using the singular value decomposition (SVD) method.46 The measured RDCs show very poor agreement with the dumbbell-shape crystal structure of the free Ca2+-CaM [Fig. 6(A)] because the conformation of Ca2+-CaM bound to p21(145–164) is very different from that of free Ca2+-CaM. Among the 13 CaM/peptide complexes tested (see Supp. Info. Table S4), including the complexes that show different types of binding mode, the best correlation was obtained for CaM/CaMKII complex that belongs to the 1–10 class47 [Fig. 6(B)]. This suggests that p21(145–164) adopts a known CaM-binding mode (possibly the 1–10 class) in which the CaM-interacting region is helical.
Figure 6.
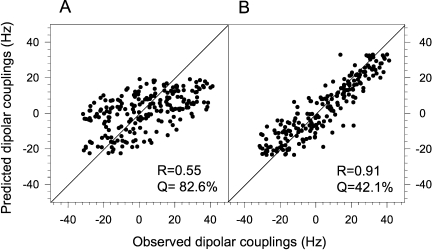
Determination of the CaM/p21 binding mode using RDCs. Best-fitting of observed backbone RDCs of the Ca2+-CaM/p21(145-164) complex to (A) the free Ca2+-CaM crystal structure (PDB code: 1EXR) and (B) the crystal structure of Ca2+-CaM/CaMKII (PDB code: 1CDM). All RDCs are normalized to 1DNH.
Discussion
The NMR data and molecular dynamics simulations are used to explain the binding promiscuity and specificity of p21(145–164) mediated protein-protein signaling. The binding promiscuity can be attributed to the structural plasticity, or unstructured nature of the peptide, which allows it to adapt to the distinct structural environments of many different target proteins. A fundamental question remains-if the peptide is largely unstructured, how does it bind multiple proteins with high affinity?
We first examined the residual structure of p21(145–164) in solution and compared locally preferred conformations to regions of structure in bound forms of p21(145–164). The crystal structure of p21(139–160) bound to PCNA shows that residues Ser146 – Tyr151 of p21 forms a 310-helix which is involved in hydrophobic interaction with PCNA, whereas residues His152 – Ser160 of p21 adopt an extended β-strand conformation which interacts with PCNA.25 We have shown the helical preference of the N-terminal region of the p21(145–154) which corresponds to the helical region of the PCNA-bound structure. The chemical shifts and scalar couplings for residues Arg155-Lys163 show no sign of residual helical structure and the MD simulations suggest that the helical and extended portions of p21 that bind PCNA exist in the free state prior to binding (see Fig. 5). The recognition for PCNA in this case comes from the fact that residues His152-Ser160 of p21 have a preference for conformational arrangements which readily expose the positively charged residues for specific interactions with PCNA. In the extended, uncoiled conformation of the p21 peptide, residues Arg155 and Arg156 readily interact with the negatively charged Asp122 and Asp29 of PCNA, respectively.
In addition, the NMR structure of Cdk4-bound p21(141–160) also includes a helical region (residues 149–156).48 Similarly, model ensembles of the unbound state generated by EMW suggest that the region corresponding to residues 149–154 can adopt helical conformations (see Fig. 4).
We then examined the mechanism of recognition between p21 and Ca2+-CaM. Although the NMR resonances of the p21(145–164) peptide bound to CaM are extremely broad due to chemical exchange, the resonances of CaM are much less affected by the peptide and thus allow accurate structure measurement. An extensive set of RDCs measured for CaM bound to the p21(145–164) peptide indicates a CaM-substrate interaction mode in which the substrate adopts an α-helical conformation. For the free p21(145–164) peptide, the NMR chemical shifts and scalar couplings together show that residues 147–154 have significant helical propensity, and MD simulations suggest that the N-terminal region has a strong preference for helical conformations while helical structure is absent in the C-terminal region. We believe the residual helical segments observed in the free p21(145–164) peptide is responsible for its initial recognition with Ca2+-CaM. The helical structure is then stabilized by the binding. Overall, the structural propensities of the free p21(145–164) peptide correlate well with the different structures adopted by p21 upon binding to different targets. This observation suggests that pre-existing residual conformations of p21 provide the initial recognition for the target proteins. Binding then further stabilizes these residual conformations. The p21 peptide provides an interesting example of how residual structural elements of an IUP are involved in specific and diverse protein-protein signaling.
Our data suggest that p21's binding promiscuity is explained by the fact that its target selects the appropriate preformed p21conformation. This “conformational selection” mechanism has been described for a number of proteins and therefore may represent a general method that enables proteins to bind structurally dissimilar targets.49–52 This is to be contrasted with an induced fit mechanism, where the association of two proteins leads to concomitant binding and folding.53–55 For proteins that associate via induced fit, binding and folding are coupled.55 One difference between conformational selection and induced fit is that conformational selection presupposes that the protein conformation, which is complementary to a given binding partner, exists in solution even when the binding partner is absent. By contrast, this need not be the case with induced fit; that is, complementary protein conformations are formed when the protein of interest interacts with its binding partner. Hence information as to what mechanism is at play for any given protein may be obtained from an analysis of the conformational thermodynamics of the protein of interest in the absence of its binding partner. Consequently, methods that combine both experiment and simulation to provide insight into the conformational thermodynamics of IUPs like p21, may provide insight into the binding mechanism of a number of disordered systems.
Methods
Cloning, protein expression and purification
The peptide p21(145–164) was expressed as a C-terminal in-frame fusion to the trpLE protein containing the N-terminal 9-His tag. A pair of Asp-Pro residues was engineered between trpLE and p21(145–164) for acid catalyzed cleavage to release the p21 peptide from the fusion protein. The expression vector was constructed by inserting the DNA fragment of p21(145–164) into the C264 vector, a gift from Dr. M.E. Call, Harvard Medical School, Boston.
Escherichia coli strain BL21 (DE3) that express the trpLE-fused p21(145–164) were cultured in M9 minimal media for isotope labeling. The cell cultures were grown at 37°C to OD600 of 0.6–0.8 before overnight induction at 25°C with 0.4 mM IPTG. Inclusion bodies were dissolved with a buffer containing 50 mM Tris, pH 8.0, 0.2 M NaCl, 6 M guanidine HCl, 10 mM imidazole. The cleared solution was bound to Ni2+ affinity column (Sigma) and eluted in 50 mM Tris, pH 8.0, 0.2 M NaCl, 6 M guanidine HCl, 400 mM imidazole. The eluted fusion protein was dialyzed against water to remove guanidine HCl. The precipitant was pelleted by centrifugation at 3000 rpm for 30 min. Incubation of the pellet in 0.1 N HCl at 37°C for 3 days released the p21(145–164) peptide from the trpLE fusion partner. The released peptide was dialyzed against water, lyophilized, and purified by reverse-phase HPLC on a C18 column (Grace-Vydac) with a gradient of water containing 0.1% trifluoroacetic acid (TFA) to acetonitrile containing 0.1% TFA. The resulting peptide was lyophilized and dissolved in 100 mM KCl, 10 mM CaCl2, pH 6.5.
CaM was expressed and purified as previously described.56 Isotropic NMR samples were prepared in 100 mM KCl, 10 mM CaCl2, pH 6.5 in 93% H2O/7% D2O. The aligned sample contained 18 mg/mL filamentous phage Pf1 (Asla Labs, Riga, Latvia), 100 mM KCl, 10 mM CaCl2, and 1 mM sodium azide, pH 6.5 in 93% H2O/7% D2O.
NMR spectroscopy
All NMR spectra were collected at 30°C on Bruker and Varian spectrometers operating at 1H frequencies of 500 MHz or 600 MHz and equipped with cryogenic probes. The sequence-specific backbone assignments were accomplished using pairs of HNCACB/CBCA(CO)NH and HNCA/HNCACB on the 15N, 13C-labeled CaM in complex with unlabeled p21(145–164) and 15N, 13C-labeled p21(145–164), respectively. Two types of backbone RDCs, 1DNH and 1DCαHα, were measured on the 15N, 13C-labeled CaM in complex with unlabeled p21(145–164). The 1H-15N RDCs were obtained from 1JNH/2 and (1JNH+1DNH)/2, which were measured at 600 MHz (1H frequency) by interleaving a regular gradient enhanced HSQC and a gradient-selected TROSY, both acquired with 80 ms of 15N evolution. The 1H-13Cα RDCs were measured at 600 MHz (1H frequency) using the 3D CT-(H)CA(CO)NH without 1H-decoupling.57 Measurement of 3JHN-Hα coupling constant for determining backbone ϕ angle was carried out on the 15N, 13C-labeled p21(145–164) using the 3D HNHA experiment.41 The 1H chemical shifts were referenced directly to external 2,2-dimethyl-2-silapentane-5-sulfonic acid (DSS) in D2O and 13C chemical shifts are indirectly referenced to 0 ppm proton using the method in ref.58.
Data processing and spectra analyses were done in NMRPipe,59 CARA,60 and Sparky (http://www.cgl.ucsf.edu/home/sparky). RDCs were extracted by subtracting isotropic couplings from the aligned couplings. Fitting of RDCs to structures was done by singular value decomposition,46 using the program PALES61 The goodness of fit was assessed by both Pearson correlation coefficient (R) and the quality factor (Q).62 The 1H-13Cα RDCs were normalized to the 1H-15N RDCs by a scaling factor of 0.5.
Molecular dynamics simulations
Energy-minima mapping and weighting (EMW)
To construct an ensemble that represents the unfolded state of p21(145–164), we employ EMW method. Details of EMW are described in detail below.
Conformational sampling
The goal of conformational sampling was to generate a library of energy-minimized structures with representatives from all regions of conformational space accessible to the peptide. This was done using quenched molecular dynamics (QMD), as diagrammed in Figure S2 (Supp. Info.). To ensure that both compact and extended structures were adequately sampled, QMD was carried out at 50 different end-to-end distance constraints, spanning a range from 4 to 53 Å. At each distance constraint, a polar hydrogen model of an extended peptide having the sequence TSMTDFYHSKRRLIFSKRKP (p21(145–164)) was constructed using CHARMM, and a harmonic penalty was introduced to enforce the desired distance between 13Cα of T145 and 13Cα of P164. The structure was then minimized using 500 steps of steepest descent minimization followed by 10,000 steps of minimization using the Adopted Basis Newton Rhapson algorithm. Next, the structure was heated to 1000 K for 10 ps and allowed to equilibrate for 10 ps, before high temperature MD was run for 3 ns. Throughout the simulation, a Berendsen heat bath was used to maintain the temperature,63 and the EEF1 energy function (a Gaussian solvent exclusion model for the solvation free energy) was used to assign energies.64 The SHAKE algorithm was employed to hold bonds to hydrogen atoms fixed near their equilibrium values, allowing for a 2 fs time step during high temperature MD simulations.65 The peptide's coordinates were saved after every 5000 steps (10 ps) of high temperature MD simulation, yielding 300 structures per end-to-end distance constraint. Thus, 15000 structures were created using high-temperature molecular dynamics.
With end-to-end distance constraints still in place, each of these structures was coupled to a Berendsen heat bath at 298 K and cooled for 40 ps, at which point the end-to-end constraint was removed and the system was minimized using 10000 steps of Adopted Basis Newton Rhapson minimization.63 Cooling and equilibrating each structure before minimization gave the system a chance to escape shallow local energy minima, thereby making more stable structures accessible. The 15,000 structures obtained in this manner comprised our structure library.
Model optimization
A goal of this procedure is to find ensembles that represent the solubilized p21 peptide, where each ensemble consists of 15 structures and their associated weights. Accordingly, the optimization stage of EMW involved generating such ensembles by choosing structures from the conformational library generated in the first step and assigning weights to these structures. Experimentally determined 13Cα NMR chemical shifts for the peptide were used to determine what constituted an optimal ensemble; structures and weights were assigned to minimize the root mean square error between 13Cα chemical shifts computed from the model using SHIFTX and 13Cα chemical shifts that were experimentally measured.40 We focused on the Cα chemical shifts for the model construction and used the remaining experimental data (13CO, 15N, Hα, scalar J-couplings) to screen the models to ensure that they could reproduce data that was not used to construct the model. Sequestering part of the experimental data (and not using it in model construction) helps to ensure that our models are not overly fit to a given set of experimental results.
Model generation was performed by minimizing an appropriate error function, f, given by:
| 1.1 |
where N is the number of structures in the ensemble (N = 15), Xi is the ith structure, ωi is the weight of the ith structure, r is the number of residues in the peptide for which experimental chemical shift data is available (r = 18), 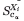 (j) is the calculated Cα chemical shift of residue j in structure Xi, and
(j) is the calculated Cα chemical shift of residue j in structure Xi, and 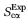 (j) is the experimentally determined 13Cα chemical shift of residue j.
(j) is the experimentally determined 13Cα chemical shift of residue j.
A simulated annealing protocol using a cooling schedule based upon that described by Nulton et al. was implemented.66,67 Each ensemble was generated from an initial ensemble consisting of 15 Boltzmann-weighted structures chosen at random from the conformational library.68 This initial ensemble was subjected to an iterative simulated annealing protocol that minimized the rmse between measured and predicted 13Cα chemical shifts. Each step of the annealing protocol consisted of carrying out Monte Carlo steps at a given value of the control parameter T, which is analogous to the temperature in physical systems, until the system had equilibrated.
A Monte Carlo step consists of perturbing the ensemble by replacing one conformer in that ensemble with a conformer from our structure library. Weights for all structures are then reassigned to minimize the overall error, f. The number of Monte Carlo steps for a given value of the control parameter, as well as the schedule used to decrease the overall temperature, is as described in a previous work.67 Overall, 250 ensembles were generated using this simulated annealing protocol.
Model validation
The rmse between predicted nitrogen chemical shifts and measured nitrogen chemical shifts for each ensemble was calculated for each of the 250 models generated, and those ensembles in which this error was less than 1.5 ppm were taken to be valid models based upon their ability to predict experimentally measured amide nitrogen chemical shifts. Ten valid ensembles were found that reproduced the NMR chemical shift data well, due to the underdetermined nature of the problem, but we accounted for this by using all 10 of these independently generated, validated structural ensembles in our analysis and focusing on those structural motifs that were preserved across all of them. Calculated 3-bond scalar J-couplings were also computed using the Karplus relations as previously described.41,69
Procedure for identifying conserved preformed structures
Two different methods were employed to find structural motifs that were present across all validated model ensembles of the unfolded state. Since studies have shown that bound states of p21(145–164) adopt helical conformations, we looked for evidence of preformed helical motifs in the unfolded ensemble. We then sought to identify other structural motifs suggested by the model ensembles.
Identification of conserved local structure by clustering
We sought to identify other types of local structural motifs in the peptide. To this end, all conformers were clustered based on local conformational preferences. Since the helical region in the crystal structure of PCNA-bound p21 is six residues in length, we defined the characteristic size of a local structural motif to be six residues. To account for all local motifs, every six residue subsequence of p21(145–164) was analyzed to find preserved conformations. This was accomplished by clustering based upon backbone atom RMS deviations within each six-residue window of interest. Clustering was carried out in MATLAB (© Mathworks) such that the maximum rmsd between any two structures was 2.5 Å. Clustering based on other window sizes (five, seven, and eight residues in length) was performed to ensure that analysis was relatively insensitive to the choice of window size. Clusters that were represented in all 10 ensembles were identified as preserved local structural motifs.
Supplemental material
References
- 1.Radhakrishnan I, Perez-Alvarado GC, Dyson HJ, Wright PE. Conformational preferences in the Ser133-phosphorylated and non-phosphorylated forms of the kinase inducible transactivation domain of CREB. FEBS Lett. 1998;430:317–322. doi: 10.1016/s0014-5793(98)00680-2. [DOI] [PubMed] [Google Scholar]
- 2.Dawson R, Muller L, Dehner A, Klein C, Kessler H, Buchner J. The N-terminal domain of p53 is natively unfolded. J Mol Biol. 2003;332:1131–1141. doi: 10.1016/j.jmb.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 3.Kim AS, Kakalis LT, Abdul-Manan N, Liu GA, Rosen MK. Autoinhibition and activation mechanisms of the Wiskott-Aldrich syndrome protein. Nature. 2000;404:151–158. doi: 10.1038/35004513. [DOI] [PubMed] [Google Scholar]
- 4.Lacy ER, Filippov I, Lewis WS, Otieno S, Xiao L, Weiss S, Hengst L, Kriwacki RW. p27 binds cyclin-CDK complexes through a sequential mechanism involving binding-induced protein folding. Nat Struct Mol Biol. 2004;11:358–364. doi: 10.1038/nsmb746. [DOI] [PubMed] [Google Scholar]
- 5.Bienkiewicz EA, Adkins JN, Lumb KJ. Functional consequences of preorganized helical structure in the intrinsically disordered cell-cycle inhibitor p27(Kip1) Biochemistry. 2002;41:752–759. doi: 10.1021/bi015763t. [DOI] [PubMed] [Google Scholar]
- 6.Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuxreiter M, Simon I, Friedrich P, Tompa P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J Mol Biol. 2004;338:1015–1026. doi: 10.1016/j.jmb.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 8.Tompa P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005;579:3346–3354. doi: 10.1016/j.febslet.2005.03.072. [DOI] [PubMed] [Google Scholar]
- 9.Wright PE. Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 10.Dunker AK, et al. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 11.Kriwacki RW, Hengst L, Tennant L, Reed SI, Wright PE. Structural studies of p21(Waf1/Cip1/Sdi1) in the free and Cdk2-bound state: conformational disorder mediates binding diversity. Proc Natl Acad Sci USA. 1996;93:11504–11509. doi: 10.1073/pnas.93.21.11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 13.Waga S, Hannon GJ, Beach D, Stillman B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature. 1994;369:574–578. doi: 10.1038/369574a0. [DOI] [PubMed] [Google Scholar]
- 14.Levkau B, Koyama H, Raines EW, Clurman BE, Herren B, Orth K, Roberts JM, Ross R. Cleavage of p21Cip1/Waf1 and p27Kip1 mediates apoptosis in endothelial cells through activation of Cdk2: role of a caspase cascade. Mol Cell. 1998;1:553–563. doi: 10.1016/s1097-2765(00)80055-6. [DOI] [PubMed] [Google Scholar]
- 15.Dotto GP. p21(WAF1/Cip1): more than a break to the cell cycle? Biochim Biophys Acta. 2000;1471:M43–M56. doi: 10.1016/s0304-419x(00)00019-6. [DOI] [PubMed] [Google Scholar]
- 16.Delavaine L, La Thangue NB. Control of E2F activity by p21Waf1/Cip1. Oncogene. 1999;18:5381–5392. doi: 10.1038/sj.onc.1202923. [DOI] [PubMed] [Google Scholar]
- 17.Funk JO, Waga S, Harry JB, Espling E, Stillman B, Galloway DA. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev. 1997;11:2090–2100. doi: 10.1101/gad.11.16.2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 19.Li R, Waga S, Hannon GJ, Beach D, Stillman B. Differential effects by the p21 CDK inhibitor on PCNA-dependent DNA replication and repair. Nature. 1994;371:534–537. doi: 10.1038/371534a0. [DOI] [PubMed] [Google Scholar]
- 20.Chen J, Jackson PK, Kirschner MW, Dutta A. Separate domains of p21 involved in the inhibition of Cdk kinase and PCNA. Nature. 1995;374:386–388. doi: 10.1038/374386a0. [DOI] [PubMed] [Google Scholar]
- 21.Taules M, Rodriguez-Vilarrupla A, Rius E, Estanyol JM, Casanovas O, Sacks DB, Perez-Paya E, Bachs O, Agell N. Calmodulin binds to p21(Cip1) and is involved in the regulation of its nuclear localization. J Biol Chem. 1999;274:24445–24448. doi: 10.1074/jbc.274.35.24445. [DOI] [PubMed] [Google Scholar]
- 22.Estanyol JM, Jaumot M, Casanovas O, Rodriguez-Vilarrupla A, Agell N. Bachs O. The protein SET regulates the inhibitory effect of p21(Cip1) on cyclin E-cyclin-dependent kinase 2 activity. J Biol Chem. 1999;274:33161–33165. doi: 10.1074/jbc.274.46.33161. [DOI] [PubMed] [Google Scholar]
- 23.Kitaura H, Shinshi M, Uchikoshi Y, Ono T, Iguchi-Ariga SM, Ariga H. Reciprocal regulation via protein-protein interaction between c-Myc and p21(cip1/waf1/sdi1) in DNA replication and transcription. J Biol Chem. 2000;275:10477–10483. doi: 10.1074/jbc.275.14.10477. [DOI] [PubMed] [Google Scholar]
- 24.Esteve V, Canela N, Rodriguez-Vilarrupla A, Aligue R, Agell N, Mingarro I, Bachs O. Perez-Paya E. The structural plasticity of the C terminus of p21Cip1 is a determinant for target protein recognition. Chembiochem. 2003;4:863–869. doi: 10.1002/cbic.200300649. [DOI] [PubMed] [Google Scholar]
- 25.Gulbis JM, Kelman Z, Hurwitz J, O'Donnell M. Kuriyan J. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell. 1996;87:297–306. doi: 10.1016/s0092-8674(00)81347-1. [DOI] [PubMed] [Google Scholar]
- 26.Yap KL, Kim J, Truong K, Sherman M, Yuan T, Ikura M. Calmodulin target database. J Struct Funct Genomics. 2000;1:8–14. doi: 10.1023/a:1011320027914. [DOI] [PubMed] [Google Scholar]
- 27.Wishart DS, Sykes BD. Chemical shifts as a tool for structure determination. Methods Enzymol. 1994;239:363–392. doi: 10.1016/s0076-6879(94)39014-2. [DOI] [PubMed] [Google Scholar]
- 28.Zhang O, Forman-Kay JD. NMR studies of unfolded states of an SH3 domain in aqueous solution and denaturing conditions. Biochemistry. 1997;36:3959–3970. doi: 10.1021/bi9627626. [DOI] [PubMed] [Google Scholar]
- 29.Yao J, Chung J, Eliezer D, Wright PE, Dyson HJ. NMR structural and dynamic characterization of the acid-unfolded state of apomyoglobin provides insights into the early events in protein folding. Biochemistry. 2001;40:3561–3571. doi: 10.1021/bi002776i. [DOI] [PubMed] [Google Scholar]
- 30.Williamson JA, Miranker AD. Direct detection of transient alpha-helical states in islet amyloid polypeptide. Protein Sci. 2007;16:110–117. doi: 10.1110/ps.062486907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwarzinger S, Kroon GJ, Foss TR, Wright PE, Dyson HJ. Random coil chemical shifts in acidic 8 M urea: implementation of random coil shift data in NMRView. J Biomol NMR. 2000;18:43–48. doi: 10.1023/a:1008386816521. [DOI] [PubMed] [Google Scholar]
- 32.Smith LJ, Bolin KA, Schwalbe H, MacArthur MW, Thornton JM, Dobson CM. Analysis of main chain torsion angles in proteins: prediction of NMR coupling constants for native and random coil conformations. J Mol Biol. 1996;255:494–506. doi: 10.1006/jmbi.1996.0041. [DOI] [PubMed] [Google Scholar]
- 33.Lacroix E, Viguera AR, Serrano L. Elucidating the folding problem of alpha-helices: local motifs, long-range electrostatics, ionic-strength dependence and prediction of NMR parameters. J Mol Biol. 1998;284:173–191. doi: 10.1006/jmbi.1998.2145. [DOI] [PubMed] [Google Scholar]
- 34.Raghava GPS. APSSP2: A combination method for protein secondary structure prediction based on neural network and example based learning. CASP. 2002:5. [Google Scholar]
- 35.Meiler J, Baker D. Coupled prediction of protein secondary and tertiary structure. Proc Natl Acad Sci USA. 2003;100:12105–12110. doi: 10.1073/pnas.1831973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kneller DG, Cohen FE, Langridge R. Improvements in protein secondary structure prediction by an enhanced neural network. J Mol Biol. 1990;214:171–182. doi: 10.1016/0022-2836(90)90154-E. [DOI] [PubMed] [Google Scholar]
- 37.Ouali M, King RD. Cascaded multiple classifiers for secondary structure prediction. Protein Sci. 2000;9:1162–1176. doi: 10.1110/ps.9.6.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 39.Huang A, Stultz CM. The Effect of a K280 Mutation on the unfolded state of a microtubule-binding repeat in tau. PLoS Comput Biol. 2008;4:e100155, 1–12. doi: 10.1371/journal.pcbi.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neal S, Nip AM, Zhang HY. Wishart DS. Rapid and accurate calculation of protein H-1, C-13 and N-15 chemical shifts. J Biomol NMR. 2003;26:215–240. doi: 10.1023/a:1023812930288. [DOI] [PubMed] [Google Scholar]
- 41.Vuister GW, Bax A. Quantitative J correlation: a new approach for measuring homonuclear three-bond J(HNH.alpha) coupling constants in 15N-enriched proteins. J Am Chem Soc. 1993;115:7772–7777. [Google Scholar]
- 42.Gulbis JM, Kelman Z, Hurwitz J, Odonnell M, Kuriyan J. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell. 1996;87:297–306. doi: 10.1016/s0092-8674(00)81347-1. [DOI] [PubMed] [Google Scholar]
- 43.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mal TK, Skrynnikov NR, Yap KL, Kay LE, Ikura M. Detecting protein kinase recognition modes of calmodulin by residual dipolar couplings in solution NMR. Biochemistry. 2002;41:12899–12906. doi: 10.1021/bi0264162. [DOI] [PubMed] [Google Scholar]
- 45.Contessa GM, Orsale M, Melino S, Torre V, Paci M, Desideri A, Cicero DO. Structure of calmodulin complexed with an olfactory CNG channel fragment and role of the central linker: residual dipolar couplings to evaluate calmodulin binding modes outside the kinase family. J Biomol NMR. 2005;1:185–199. doi: 10.1007/s10858-005-0165-1. [DOI] [PubMed] [Google Scholar]
- 46.Losonczi JA, Andrec M, Fischer MW, Prestegard JH. Order matrix analysis of residual dipolar couplings using singular value decomposition. J Magn Reson. 1999;138:334–342. doi: 10.1006/jmre.1999.1754. [DOI] [PubMed] [Google Scholar]
- 47.Meador WE, Means AR, Quiocho FA. Modulation of calmodulin plasticity in molecular recognition on the basis of x-ray structures. Science. 1993;262:1718–1721. doi: 10.1126/science.8259515. [DOI] [PubMed] [Google Scholar]
- 48.Sung YH, Shin J, Shin JH, Lee W. Solution structure of p21(Waf1/Cip1/Sdi1) C-terminal domain bound to Cdk4. J Biomol Struct Dyn. 2001;19:419–427. doi: 10.1080/07391102.2001.10506751. [DOI] [PubMed] [Google Scholar]
- 49.Tsai CJ, Ma BY, Sham YY, Kumar S, Nussinov R. Structured disorder and conformational selection. Proteins Struct Funct Genet. 2001;44:418–427. doi: 10.1002/prot.1107. [DOI] [PubMed] [Google Scholar]
- 50.Dobbins SE, Lesk VI, Sternberg MJE. Insights into protein flexibility: The relationship between normal modes and conformational change upon protein-protein docking. Proc Natl Acad Sci USA. 2008;105:10390–10395. doi: 10.1073/pnas.0802496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dunker AK, Oldfield C, Meng J, Romero P, Yang J, Chen J, Vacic V, Obradovic Z, Uversky V. The unfoldomics decade: an update on intrinsically disordered proteins. BMC Genomics. 2008:9. doi: 10.1186/1471-2164-9-S2-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lange OF, Lakomek NA, Fares C, Schroder GF, Walter KFA, Becker S, Meiler J, Grubmuller H, Griesinger C, de Groot BL. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320:1471–1475. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- 53.Pontius BW. Close encounters—Why unstructured, polymeric domains can increase rates of specific macromolecular association. Trends Biochem Sci. 1993;18:181–186. doi: 10.1016/0968-0004(93)90111-y. [DOI] [PubMed] [Google Scholar]
- 54.Shoemaker BA, Portman JJ, Wolynes PG. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc Natl Acad Sci USA. 2000;97:8868–8873. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sugase K, Dyson HJ. Wright PE. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature. 2007;447:U1021–U1011. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- 56.Hayashi N, Matsubara M, Takasaki A, Titani K, Taniguchi H. An expression system of rat calmodulin using T7 phage promoter in Escherichia coli. Protein Expr Purif. 1998;12:25–28. doi: 10.1006/prep.1997.0807. [DOI] [PubMed] [Google Scholar]
- 57.Tjandra N, Bax A. Large variations in 13c chemical shift anisotropy in proteins correlate with secondary structure. J Am Chem Soc. 1997;119:9576–9577. [Google Scholar]
- 58.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 59.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 60.Keller R. The computer aided resonance assignment tutorial. Switzerland: Goldau; 2004. [Google Scholar]
- 61.Zweckstetter M, Bax A. Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR. J Am Chem Soc. 2000;122:3791–3792. [Google Scholar]
- 62.Cornilescu G, Marquardt JL, Ottiger M, Bax A. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J Am Chem Soc. 1998;120:6836–6837. [Google Scholar]
- 63.Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR. Molecular-dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 64.Lazaridis T, Karplus M. Effective energy function for proteins in solution. Proteins: Struct Funct Genet. 1999;35:133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 65.Vangunsteren WF, Berendsen HJC. Algorithms for macromolecular dynamics and constraint dynamics. Mol Phys. 1997;34:1311–1327. [Google Scholar]
- 66.Nulton JD, Salamon P. Statistical-mechanics of combinatorial optimization. Phys Rev A. 1988;37:1351–1356. doi: 10.1103/physreva.37.1351. [DOI] [PubMed] [Google Scholar]
- 67.Stultz CM, Karplus M. Dynamic ligand design and combinatorial optimization: Designing inhibitors to endothiapepsin. Proteins: Struct Funct Genet. 2000;40:258–289. [PubMed] [Google Scholar]
- 68.Huang A, Stultz CM. Conformational sampling with implicit solvent models: application to the PHF6 peptide in tau protein. Biophys J. 2007;92:34–45. doi: 10.1529/biophysj.106.091207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Karplus M, Anderson DH. Valence-bond interpretation of electron-coupled nuclear spin interactions; Application to methane. J Chem Phys. 1959;30:11–15. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.