

Abstract
Bacillus thuringiensis subsp. israelensis is the most widely used microbial control agent against mosquitoes and blackflies. Its insecticidal success is based on an arsenal of toxins, such as Cry4A, Cry4B, Cry11A, and Cyt1A, harbored in the parasporal crystal of the bacterium. A fifth toxin, Cry10Aa, is synthesized at very low levels; previous attempts to clone and express Cry10Aa were limited, and no parasporal body was formed. By using a new strategy, the whole Cry10A operon was cloned in the pSTAB vector, where both open reading frames ORF1 and ORF2 (and the gap between the two) were located, under the control of the cyt1A operon and the STAB-SD stabilizer sequence characteristic of this vector. Once the acrystalliferous mutant 4Q7 of B. thuringiensis subsp. israelensis was transformed with this construct, parasporal bodies were observed by phase-contrast microscopy and transmission electron microscopy. Discrete, ca. 0.9-μm amorphous parasporal bodies were observed in the mature sporangia, which were readily purified by gradient centrifugation once autolysis had occurred. Pure parasporal bodies showed two major bands of ca. 68 and 56 kDa on sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis. These bands were further characterized by N-terminal sequencing of tryptic fragments using matrix-assisted laser desorption ionization-time of flight mass spectrometry analysis, which identified both bands as the products of ORF1 and ORF2, respectively. Bioassays against fourth-instar larvae of Aedes aegypti of spore-crystal complex and pure crystals of Cry10Aa gave estimated 50% lethal concentrations of 2,061 ng/ml and 239 ng/ml, respectively. Additionally, synergism was clearly detected between Cry10A and Cyt1A, as the synergistic levels (potentiation rates) were estimated at 13.3 for the mixture of Cyt1A crystals and Cry10Aa spore-crystal complex and 12.6 for the combination of Cyt1A and Cry10Aa pure crystals.
The subspecies Bacillus thuringiensis subsp. israelensis (serotype H-14) was discovered by Goldberg and Margalit in 1977 (11). To date, its insecticidal potential has not been overcome by any other bacterium (or any biological control agent) as an effective control measure against mosquito and blackfly larvae (8). Recently, its toxicity spectrum has been expanded to a coleopteran pest, the coffee berry borer (Hypothenemus hampei) (23), indicating that this strain may have potential versatility. Also, the so-called pBtoxis megaplasmid harbored in this strain, containing all the endotoxin-encoding genes found in its parasporal crystal, including cry4A, cry4B, cry10A, cry11A, and cyt1A, was recently sequenced (1). Among many other interesting aspects of this serotype, the occurrence of this mosquitocidal arsenal in one strain and their synergistic interaction make this bacterium scientifically and technologically attractive.
The parasporal crystal of B. thuringiensis subsp. israelensis contains large amounts of Cry4A, Cry4B, Cry11A, and Cyt1A toxins (14), and consequently, most of the knowledge about the toxicity of this strain has been focused on these proteins, acting either as a complex (31) or tested separately (6). Although the cry10Aa gene was originally cloned in 1986 (known then as cryIVC) (30), to date, little is known about cry10Aa and the protein it encodes, mostly due to its very low levels of expression (10) in B. thuringiensis subsp. israelensis. Interestingly, cry10Aa is an operon as it includes two open reading frames (ORFs), previously reported as pBt047 and pBt048 (hereafter referred to only as ORF1 and ORF2, respectively), separated by a 48-bp untranslated gap (1). ORF1 contains the complete δ-endotoxin sequence (active toxin), with a coding capacity for a 78-kDa protein. Interestingly, ORF2 shows high identity with the coding sequence of the C-terminal half of Cry4-type proteins, with a coding capacity for a 56-kDa protein. Therefore, it is believed that a putative ancestral cry10Aa gene is similar in size to the cry4-type genes (ca. 4 kbp), but either a small sequence had been inserted in the middle of the coding sequence or site mutations produced end codons (two end codons flank the gap) in this region (1).
Previous attempts to clone and express the cry10Aa gene included ORF1 and only part of ORF2 (7, 10, 30). This was a reasonable strategy, as most of the so-called “complete” protoxins are partially digested to become active toxins (δ-endotoxins) (28), and ORF1 included the complete sequence to code the Cry10Aa δ-endotoxin. However, in all these cases, the expression levels were very low, and no parasporal body was formed. Similar results were obtained when the promoter was changed and a stabilizing sequence was added to the construction (13). The low expression levels achieved in these cases led to conclusions that assumed low toxic levels of Cry10Aa when tested against mosquito larvae (30). In spite of the low toxicity of Cry10Aa found against mosquito larvae, a synergistic effect was reported between Cry10Aa and Cry4Ba toxins in Culex (7). Obtaining high levels of expression and crystallization of Cry10Aa are required to properly characterize and understand the toxic spectrum of this protein.
In this report, we show the formation of parasporal bodies of Cry10Aa, achieved by cloning the whole Cry10Aa operon under the control of the cyt1A promoter and the STAB-SD sequence. We also show that Cry10Aa is as toxic as most of the other B. thuringiensis subsp. israelensis toxins acting separately, and in synergism with the Cyt1A toxin.
MATERIALS AND METHODS
Cloning of the Cry10Aa operon.
In order to clone the complete sequence of the cry10Aa operon, which includes ORF1, ORF2, and the gap between the two (all in GenBank accession number AL731825), two primers were designed from the reported sequence (1). Sites for SalI and SphI were added to allow the subcloning of the amplicon into the pSTAB vector. The sequences of the designed primers were as follows: cry10D (direct), 5′-AATGTCGACTTGCAACAGAAAAGAGTTGTGTC-3′; and cry10R (reverse), 5′-CGAGCATGCACATTTCCCCACAATTTTCA-3′ (the SalI and SphI sites are underlined, respectively).
The B. thuringiensis subsp. israelensis strain ONR-60A, kindly provided by the International Entomopathogenic Bacillus Center at the Pasteur Institute, Paris, France, was subjected to DNA extraction following the protocol detailed by Jensen et al. (18). The PCR mixture comprised 500 ng template DNA, 12.5 μl double-distilled H2O (ddH2O), 2.5 μl of 10× reaction buffer, 2.5 μl of 225 mM MgCl2, 0.5 μl deoxynucleoside triphosphate mixture, 20 μM of each primer, and 2.5 U Taq polymerase. A Perkin-Elmer GeneAmp PCR system 2400 thermocycler was set at the following conditions: an initial denaturation step of 5 min at 95°C, followed by 30 cycles, with 1 cycle consisting of denaturation at 95°C for 1 min, annealing at 50°C for 1.5 min, and polymerization at 70°C for 3 min. Amplification was completed with an extension step at 70°C for 10 min.
The expected 3,815-bp amplicon was cloned in the pCR2.1TOPO vector (Invitrogen) where, once ligated, it was used to transform competent Escherichia coli TOP-10 cells (Invitrogen) by thermoshock following the manufacturer's protocol. Transformed colonies were selected in plates with LB medium supplemented with 100 μg/ml ampicillin, 20 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal), and 100 μg/ml isopropyl-β-d-thiogalactopyranoside (IPTG). A set of 10 colonies was selected and subjected to plasmid extraction as described earlier (27), and the products were digested with EcoRV. The digestion products were visualized in 1% (wt/vol) agarose gels and compared to those obtained from the in silico analysis (BioEdit) (12) of the expected construct. This construct was called pCR2.1TOPO-cry10.
Both the pCR2.1TOPO-cry10 construct and the pSTAB vector (kindly donated by B. Federici, University of California, Riverside, CA) (25) were digested with SalI and SphI, one enzyme at a time, and their products were visualized on and purified from agarose gels using the QIAquick gel extraction kit (Qiagen). Ligation of the 3,815-bp insert into the pSTAB vector at the specific SalI and SphI sites was carried out as previously described (27), and the new constructs were used to transform competent E. coli TOP-10 cells. Once transformed, colonies were selected and subjected to plasmid extraction. Single EcoRI and double EcoRI-SalI digestions were used to corroborate appropriate insertion of the cry10Aa operon into pSTAB. The new construct was called pSTAB-cry10.
This new construct was used to transform the acrystalliferous B. thuringiensis subsp. israelensis 4Q7 strain (kindly provided by the Bacillus Stock Center, Ohio State University, Columbus, OH) by electroporation following the protocol described by Lereclus et al. (20). Colonies selected in medium supplemented with 50 μg/ml erythromycin were subjected to plasmid purification (20), and the product was used as DNA template for the specific amplification of the cry10Aa gene. For this purpose, the following primers, described earlier (26), were used: 10A5 (direct) (5′-ATATGAAATATTCAATGCTC-3′) and 10A3 (reverse) (5′-ATAAATTCAAGTGCCAAGTA-3′). DNA from nontransformed 4Q7 strain was used as a negative control. Both the reaction mixture preparation and amplification conditions were as described above, except for the polymerization time, which was reduced to 1 min. Once the expected amplicon was obtained and purified, it was sequenced in an ABI PRISM 377 system (Applied Biosystems). The obtained sequence was analyzed by BlastN from the GenBank database.
Parasporal body formation.
Once selected colonies of the B. thuringiensis subsp. israelensis 4Q7 strain transformed with the pSTAB-cry10 construct were obtained (Bt-pSTAB-cry10), they were inoculated in milk broth treated with peptone as previously described (15) to optimize the expression of the Cry10Aa protein. Cultures were monitored by phase-contrast microscopy until they reached the sporulation stage. Photographs were taken at a magnification of ×1,000. A more detailed visualization of the Bt-pSTAB-cry10 spore-crystal complex was obtained by transmission electron microscopy. For this purpose, mature sporangia were collected, washed, and fixed in 3% (vol/vol) glutaraldehyde in phosphate buffer (pH 7.4). Sporangia were sedimented in melted 2% (wt/vol) low-melting-point agarose to form an embedded pellet for further processing. The pellets were then dehydrated in an ethanol series and embedded in Spurr's low-viscosity resin mixture (Polysciences Inc.). Ultrathin sections were obtained with a NOVA LKB ultramicrotome and stained with lead citrate and uranyl acetate. Sections were examined and photographed in a Phillips Morgani electron microscope operated at an accelerating voltage of 80 kV.
Purification and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis of parasporal bodies.
Pure parasporal bodies from Bt-pSTAB-cry10 were obtained as previously described (14) by differential centrifugation in continuous NaBr gradients from an autolyzed culture obtained in milk broth treated with peptone. The band corresponding to the parasporal bodies was extracted and washed by centrifugation three times in ddH2O. Parasporal bodies obtained from a 200-ml culture were suspended in 100 μl ddH2O. A 20-μl aliquot was solubilized in a buffer containing 50 mM NaOH, 25 mM dithiothreitol, and 50 mM 3-(cyclohexylamino)-1-propanesulfonic acid (CAPS) (pH 11.5) and incubated for 1 h at 37°C. Then 10 μl of 4× Laemmli buffer [2% (wt/vol) SDS, 40% (vol/vol) glycerol, 5% (vol/vol) mercaptoethanol, 0.001% (wt/vol) bromophenol blue, 0.0625 M Tris-HCl (pH 8)] were added and incubated for 3 min in boiling water. Finally, 15 μl was loaded on a 10% (wt/vol) polyacrylamide gel and electrophoresed at 80 V for 4 h, basically by the method of Sambrook and Russell (27). The Bio-Rad 161-0304 molecular marker was used as a reference. Gels were stained in 0.25% (wt/vol) Coomassie brilliant blue, 40% (vol/vol) methanol, and 7% (vol/vol) acetic acid and destained in 40% (vol/vol) methanol and 7% (vol/vol) acetic acid.
Partial protein sequencing.
Specific bands from polyacrylamide gels were sliced, taken out, and subjected to matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF-MS) analysis as described previously (21) in order to obtain the partial amino acid sequences of several tryptic fragments from each band. The amino acid sequences obtained were subjected to Mascot Search analysis, and matches were analyzed individually with the BioEdit sequence analysis editor software (12).
In silico analysis of putative mRNAs from the Cry10Aa operon.
Due to the limited or null expression of Cry10Aa in previous attempts (7, 13), an in silico analysis of the stability of the putative secondary structures of three possible mRNAs obtained from the operon was carried out. Previous attempts to express the Cry10Aa toxin used only ORF1 (and sometimes a fragment of ORF2). In this study, the stability of the secondary structure of the putative mRNA from the pSTAB-cry10 construct (ORF1-gap-ORF2) was compared with the putative mRNAs from each separate ORF using the RNAdraw v1.1 software (22).
Toxicological analysis. (i) Individual toxicity of Cry10Aa.
Because the Cry10Aa toxin was expressed as a discrete inclusion body, an accurate toxicological analysis of its individual insecticidal activity could be performed. Bioassays on early fourth-instar larvae of Aedes aegypti were carried out essentially as previously described (17). Six to 10 concentrations of freeze-dried Bt-pSTAB-cry10 spore-crystal complex or Cry10Aa gradient-purified parasporal bodies were tested on each replicate. Twenty larvae, added to 100 ml dechlorinated water, were assayed per concentration along with a water-only negative control. A minimum of three replicates were carried out for each sample. Larvae were incubated at 28°C and examined after 24 h. The mean 50% lethal concentration (LC50) was estimated for each preparation by probit analysis using the previously established statistical parameters (16).
(ii) Joint action of Cry10Aa and Cyt1Aa.
Because a synergistic effect between the rest of the B. thuringiensis subsp. israelensis toxins has been well established (6, 29), a series of bioassays were performed in order to detect any synergistic effect between Cry10Aa and Cyt1Aa, which is the major synergistic component of the native parasporal body. The bioassay procedure was performed as described above. By using a freeze-dried powder of gradient-purified Cyt1Aa crystals, an LC50 was estimated for this toxin acting alone. Then, by using 1:1 mixtures of Cyt1Aa crystals either with Bt-pSTAB-cry10 spore-crystal complex or with Cry10Aa gradient-purified crystals, an LC50 for each mixture was estimated. These estimates were then compared with the “expected” LC50 calculated using the formula reported by Tabashnik (29), which presumes an additive (nonsynergistic) joint action response between the tested factors. Synergism is inferred if the expected LC50 is greater than the highest fiducial limit (P = 0.95) of the experimentally estimated LC50.
RESULTS
Cloning of the cry10Aa operon.
A 3.8-kb amplification product was detected in agarose gels (see Fig. S1A in the supplemental material) after PCR amplification of ORF1 and ORF2 from the cry10 operon using the cry10D and cry10R primers described in Materials and Methods. The corresponding 3,815-bp amplicon was cloned in the pCR2.1TOPO vector to constitute the new pCR2.1TOPO-cry10 construct, which was subjected to EcoRV digestion. As expected, 2.6-kb and 5.1-kb bands were detected by agarose electrophoresis (see Fig. S1B in the supplemental material), corresponding to a larger part of the insert and the vector with a fragment of the insert, respectively.
Once pCR2.1TOPO-cry10 was subjected to a double SalI-SphI digestion at the new sites added by the designed primers, the 3,815-bp insert was subcloned into the pSTAB vector by ligation at the corresponding SalI and SphI sites. The new construct, pSTAB-cry10, was subjected to single EcoRI and double EcoRI-SalI digestions, showing the expected 4.3-kb and 6.5-kb bands and 0.6-kb, 3.8-kb, and 6.5-kb bands, respectively, on electrophoretic analysis (see Fig. S1C in the supplemental material).
Next, pSTAB-cry10 was used to transform the acrystalliferous B. thuringiensis subsp. israelensis 4Q7 strain, and once selected, transformant colonies were subjected to DNA plasmid purification. When purified plasmids were used as templates to specifically amplify the cry10Aa gene with the previously reported 10A5 and 10A3 primers (26), a distinct ca. 550-bp band was obtained as a PCR product (see Fig. S1D in the supplemental material). The amplicon was sequenced, and its sequence was subjected to BlastN analysis, showing 100% identity with sequences corresponding to the cry10Aa gene previously reported (1, 30) (GenBank accession numbers AL731825.1 and M12662.1). No amplification was detected when DNA extracted from the nontransformed 4Q7 strain was used as a template.
Parasporal body formation.
Once the cry10 operon (ORF1-gap-ORF2) was cloned and transferred to the acrystalliferous 4Q7 strain, one transformant colony, called Bt-pSTAB-cry10, was selected for further analysis. First, this strain was observed by phase-contrast microscopy at the sporangium stage. Observations clearly indicated the formation of obvious parasporal bodies alongside the endospore (Fig. 1a). Parasporal bodies showed an amorphous semispherical shape, and it was common to observe two distinct inclusion bodies within each sporangium, one smaller than the other. These parasporal bodies were released during autolysis. As expected, nontransformed 4Q7 strain sporangia showed only endospores. Inclusion body formation was corroborated by transmission electron microscopy, where discrete parasporal bodies were clearly observed (Fig. 1b and c). Although crystallization of the recombinant protein was assumed in order to explain the formation of a discrete particle, lattice arrangement of molecules was not clear. Only a few limited areas of the inclusion bodies showed unclear paracrystalline arrangements of molecules. Furthermore, no defined shape of the inclusion body was observed, as it displayed diffused borders and scattered fragments, unlike the typical parasporal body of B. thuringiensis subsp. israelensis, where an envelope around the inclusion body is clearly observed. Still, the size of the recombinant parasporal body was somewhat similar to the native crystal, ranging from 0.57 to 0.92 μm, with an average of 0.75 μm at the longest axes.
FIG. 1.

Phase-contrast microscopy (a) and transmission electron microscopy (b) of mature sporangia (a and b) and sporangia close to autolysis (c) from the Bt-pSTAB-cry10 recombinant strain. sp, spore; pb, parasporal body. Bars, 2 μm (a) and 1 μm (b and c).
Purification and SDS-PAGE analysis of parasporal bodies.
Autolyzed cultures of the Bt-pSTAB-cry10 strain were subjected to NaBr continuous gradient centrifugation. A distinctive band was formed at about two-thirds from the top of the tube (Fig. 2a), made up of >99% pure parasporal bodies as corroborated by phase-contrast microscopy. The pellet at the bottom of the gradient showed a mixture of spores and parasporal bodies. The protein composition of the purified parasporal crystals was analyzed by SDS-PAGE. For this purpose, pure crystals were washed, freeze-dried, dissolved in solubilization buffer (see Materials and Methods), and subjected to SDS-PAGE analysis. Pure crystals showed two major bands of ca. 68 kDa and 56 kDa and a minor band of ca. 78 kDa (Fig. 2b).
FIG. 2.
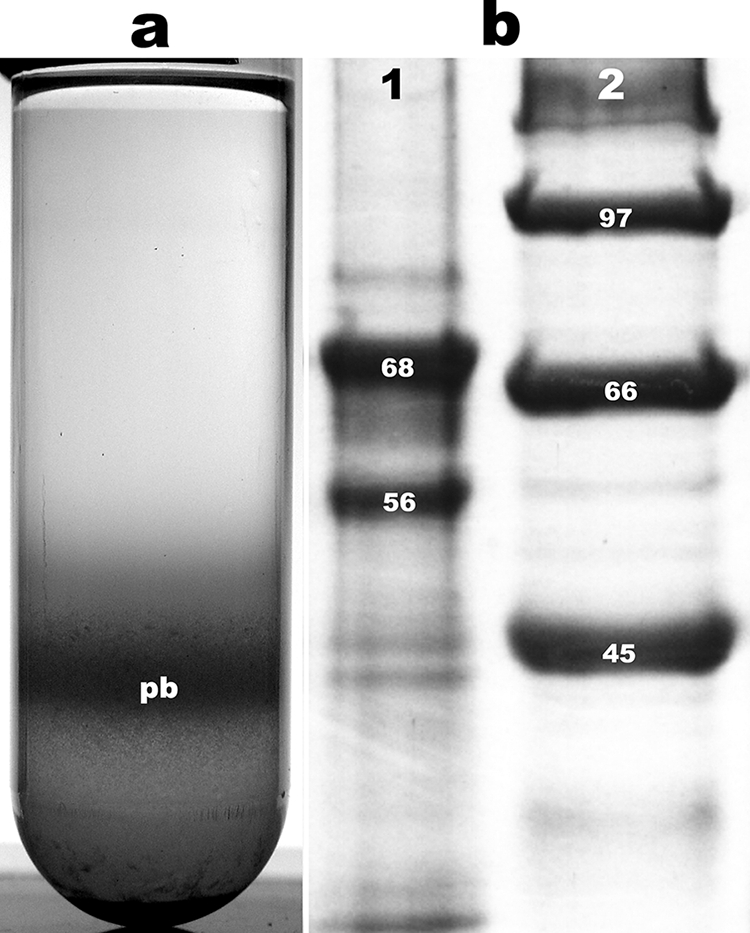
(a) NaBr continuous gradient centrifugation of a Bt-pSTAB-cry10 recombinant strain autolyzed culture. pb, band containing the recombinant parasporal bodies. (b) SDS-PAGE analysis of gradient-purified Bt-pSTAB-cry10 recombinant strain parasporal bodies (lane 1) and molecular mass markers (lane 2). The numbers on the bands are the estimated molecular masses (in kilodaltons).
Partial protein sequencing.
Precise identification of the major bands observed in the SDS-polyacrylamide gel was achieved by partial sequencing by MALDI-TOF-MS analysis. Each of the two major bands was excised from the gel and subjected to trypsin digestion. MALDI-TOF-MS sequence analysis of six and seven tryptic fragments from the ca. 68-kDa and 56-kDa bands, respectively, showed a perfect match with the amino acid sequences of the expected translations from Cry10Aa ORF1 and ORF2, respectively (GenBank accession numbers M12662.1 and CAD30099) (Table 1).
TABLE 1.
MALDI-TOF-MS sequences from tryptic fragments of the ca. 68-kDa and 56-kDa bands obtained from a polyacrylamide gela
| Band | Amino acid sequence of tryptic fragment | GenBank accession no. (gene) |
|---|---|---|
| 68-kDa | SNNYSRYPLANKPNQPLK | M12662.1 (ORF1 of cry10Aa) |
| ELIREVYTNVNSDTFRTITELENGLTR | ||
| FPFYRNKPIDKVEIVR | ||
| TDNYIFSVVR | ||
| FLKNVSR | ||
| IRYATNAPKTTVFLTGIDTISVELPSTTSRONPNATDLTYADFGYVTFPR | ||
| 56-kDa | LMLWDQVK | CAD30099 (ORF2 of cry10Aa) |
| GNYLNISGAR | ||
| GFVGSSKDVELVVSR | ||
| DVELVVSR | ||
| QIVCQDSHQFK | ||
| RSETQQAYVAK | ||
| YLYDTR |
Bands of ca. 68 kDa and 56 kDa were obtained from a polyacrylamide gel, when pure parasporal crystals from strain Bt-pSTAB-cry10 were electrophoresed.
In silico analysis of putative mRNAs from the cry10Aa operon.
According to the results obtained from the RNAdraw v1.1 software, a single mRNA that includes both ORF1 and ORF2 is 48% and 59% more stable at 37°C than separate mRNAs from ORF1 and ORF2, respectively. This may explain the success of cloning both ORFs in tandem in the expression of the Cry10Aa toxin.
Toxicity. (i) Individual toxicity of Cry10Aa.
The mosquitocidal activity of the Cry10Aa toxin, expressed in strain Bt-pSTAB-cry10 acting alone, was estimated by bioassays against fourth-instar larvae of Aedes aegypti. For this purpose, both spore-crystal complexes and gradient-purified crystals were tested as freeze-dried powders. Once replicates fulfilled the minimal statistical requirements previously established (16), LC50s of 2,061 ng/ml and 239 ng/ml were estimated when spore-crystal complexes and purified crystals, respectively, were tested (Table 2).
TABLE 2.
Probit analysis data from the bioassays of the recombinant strain Bt-pSTAB-cry10 spore-crystal complex and pure crystals and their synergism with Cyt1Aa pure crystals against fourth-instar larvae of Aedes aegypti
| Samplea | Exptl LC50 (FL) (ng/ml)b | Slope (±SE) | Expected LC50 (ng/ml)c |
|---|---|---|---|
| Cry10Aa s-c | 2,061 (1,451-2,927) | 1.05 (±0.15) | |
| Cry10Aa p-c | 239 (182-312) | 1.28 (±0.11) | |
| Cyt1Aa p-c | 739 (630-866) | 2.41 (±0.21) | |
| Cry10Aa s-c/Cyt1Aa p-c | 81.8 (73.7-91.2) | 3.49 (±0.31) | 1,088 |
| Cry10Aa p-c/Cyt1Aa p-c | 28.6 (26.1-31.3) | 4.46 (±0.46) | 362 |
s-c, spore-crystal complex; p-c, pure crystals; Cry10Aa s-c/Cyt1Aa p-c, combination of Cry10Aa s-c and Cyt1Aa p-c.
LC50 (FL), lethal concentration causing 50% mortality and its fiducial limits at P = 0.95.
Expected LC50 calculated by the method of Tabashnik (29).
(ii) Joint action of Cry10Aa and Cyt1Aa.
In order to test a possible synergism between the Cry10Aa toxin expressed in strain Bt-pSTAB-cry10 and the Cyt1Aa toxin, as previously found with other B. thuringiensis subsp. israelensis toxins (6, 29), a series of bioassays were conducted to test mixtures of pure Cyt1Aa crystals with either Bt-pSTAB-cry10 spore-crystal complexes or Cry10Aa pure parasporal bodies. Because synergism is established as a result of tests conducted first with the toxic factors acting separately and then acting together, an LC50 of 739 ng/ml was estimated for pure Cyt1Aa crystals against fourth-instar larvae of A. aegypti (Table 2).
When a 1:1 mixture of the pure Cyt1Aa crystals and the Bt-pSTAB-cry10 spore-crystal complex was tested, an LC50 of 81.8 ng/ml was estimated. Likewise, when the same proportion was used for the pure Cyt1Aa and Cry10Aa parasporal bodies, an LC50 of 28.6 ng/ml was estimated. By using these data in the formula reported by Tabashnik (29), the expected LC50 for the joint action of the Cyt1Aa crystals and the spore-crystal complex was calculated to be 1,088 ng/ml if an additive (nonsynergistic) relationship between toxins was expected (Table 2). Likewise, the expected LC50 for the joint action of the Cyt1Aa and Cry10Aa purified parasporal bodies was calculated to be 362 ng/ml for an additive joint action response. When the expected and experimentally estimated LC50s were compared, a clear synergistic effect was observed in both cases, with a synergistic level (potentiation rate) of 13.3 for the first combination and a potentiation rate of 12.6 for the second combination.
DISCUSSION
Previous attempts to clone and express the cry10Aa gene from B. thuringiensis subsp. israelensis (7, 10, 13, 30) failed to achieve sufficient levels of expression to allow the formation of a parasporal inclusion body as usually occurs with most B. thuringiensis endotoxins. With the parasporal inclusion body formation achieved in this study, a precise toxicity level was estimated against mosquito larvae. Also, it was established that Cry10Aa acted synergistically with Cyt1Aa toxin, as occurs with the other endotoxins of B. thuringiensis subsp. israelensis (29).
The parasporal body formation of the Cry10Aa protein may be related to two factors. The first factor is the use of the pSTAB vector, which is under control of the cyt1Aa gene promoters and the STAB-SD stabilizer sequence from the cry3A gene just upstream of the coding sequence, in addition to a potential role of a modestly high vector copy number (ca. four), compared to the megaplasmid where the gene is originally harbored. This vector has been shown to increase up to 10 times the expression of other cry genes cloned under the control of these sequences (25). However, an earlier attempt to express the cry10Aa gene using this vector failed to form parasporal crystals (13), which might have been related to the cloning of only ORF1.
The second factor that can explain the high levels of expression obtained in this work is the cloning of the whole operon, including ORF1, ORF2, and the sequence between the two. A sequence analysis of ORF2 showed a high identity with the C-terminal halves of Cry4 toxins (3), indicating that cry10Aa may be the result of a natural mutation of a cry4-type gene which was truncated at approximately the middle of the gene (1, 9). Additionally, it has been proposed that, although the N-terminal halves of the toxins contain the δ-endotoxins (activated toxins), the C-terminal halves may have some effect on the crystallization of the protoxin (2, 28). If this is true, cloning both ORFs in tandem might explain the crystal formation in strain Bt-pSTAB-cry10. Other evidence that may support this hypothesis is the analysis of the secondary structure stability of the putative mRNAs from separate ORF1 and ORF2 and a single mRNA from the whole operon, the latter being significantly more stable. However, taking into account the facts that the entire Cry10A operon is present in the wild-type strain and its expression levels are still very low (10), expression competence among the other endotoxin genes in the wild-type strain should also be considered. Therefore, the expression levels observed in this work may be the effect of a combination of factors: the vector (more precisely, the cyt1A promoter and the STAB-SD sequence), the cloning of the whole operon, and the lack of expression competence in the recombinant strain.
It is important to note that the occurrence of this type of arrangement among the cry genes (ORF1-gap-ORF2), where ORF2 shows high identity with the C-terminal halves of so-called “complete” endotoxins, is found in some other instances, such as cry30A, cry30B, cry30C, cry39Aa, and cry40Aa (9; Bacillus thuringiensis toxin nomenclature website http://www.lifesci.sussex.ac.uk/Home/Neil_Crickmore/Bt/). More interesting is the high identity shown by the gaps between the two ORFs in all these cases. Excluding cry30A, a Kimura analysis (19) of these gaps showed an average genetic distance of 0.2, indicating a possible common origin. The results obtained in this research may be the basis to explore how the expression of these other cry genes may be affected if both ORFs are cloned in tandem using a powerful regulating sequence.
Parasporal bodies observed in the recombinant strain Bt-pSTAB-cry10 showed an amorphous, sometimes semispherical, inclusion body and an additional smaller fragment within the same sporangium. Both inclusion bodies may be the result of a separate crystallization process of each protein expressed from each ORF, as they were also detected separately in the SDS-PAGE analysis. However, more detailed studies are required to prove this, as both large and small inclusion bodies may be made up of a mixture of both proteins. In fact, this might be the reason why these crystals were so difficult to solubilize, as a special buffer was necessary to achieve solubilization (see Materials and Methods). It is known that the typical B. thuringiensis subsp. israelensis parasporal inclusion body is made up of three protoxins, Cry4A, Cry4B, and Cyt1A, whereas Cry11A is formed slightly separated in a bar-shaped inclusion body (14). Also, the typical bipyramidal crystals of lepidopteran-active protoxins of B. thuringiensis are frequently made up of a homogeneous blend of Cry1-type proteins (4, 32). In spite of its seemingly low density, the size of the Bt-pSTAB-cry10 parasporal body is somewhat similar to the sizes of most B. thuringiensis crystals (28), which indicates a normal expression level of the protein. However, the native cry10Aa gene is normally expressed in such low levels in B. thuringiensis subsp. israelensis that its detection as part of the crystal or in polyacrylamide gels is very difficult (10).
It is worth noting that actual crystallization (lattice formation) of the recombinant proteins were not clearly observed by transmission electron microscopy. The parasporal bodies showed a more diffuse and loose structure, rather than a compact particle, but could still be purified by differential centrifugation. Nevertheless, it is worth mentioning that observation of a lattice by transmission electron microscopy of B. thuringiensis crystals is very unusual, and in the case of B. thuringiensis subsp. israelensis, a lattice has been observed only in the attached bar-shaped inclusion body formed by Cry11A proteins (14). Another feature typical of the native B. thuringiensis subsp. israelensis crystal, which is missing in the recombinant Cry10Aa parasporal body, is the surrounding envelope that keeps the different observed inclusion bodies together. However, a loose envelope was usually observed around the recombinant parasporal bodies, which might be either part of the mother cell wall or a projection of the exosporium (Fig. 1b and c). Scanning electron microscopy analysis of the recombinant parasporal bodies might have helped to resolve their particulate nature. Unfortunately, several attempts to do this resulted in ambiguous and inconsistent observations that made it difficult to draw sound conclusions.
As mentioned above, SDS-PAGE analysis of the pure Cry10Aa parasporal bodies revealed that ORF1 and ORF2 were being translated effectively and separately, as two major bands (ca. 68 kDa and 56 kDa) appeared on the gel. The expected molecular mass for the translated ORF2 was precisely 56.3 kDa, which matches the bottom band in the gels and with the partial amino acid sequences from its tryptic fragments. However, the expected molecular mass of the translated ORF1 is 77.8 kDa, almost a 10-kDa difference from the observed top band in the gels in spite of its perfect match with the partial amino acid sequences from its tryptic fragments. Perhaps the original expression product is being processed either postranscriptionally or postranslationally, or it is simply degraded to a smaller protein. According to MALDI-TOF-MS analysis, there must be a loss of the 22 N-terminal residues and the 63 C-terminal residues to leave a 67.9-kDa protein which matches the observed band. Degradation of the original protein may be the most feasible reason, as there are reports indicating that this phenomenon occurs in other Cry proteins (5, 24), although its insecticidal activity remains in this fragment. Removal of N-terminal residues during activation of the protoxin into the δ-endotoxin is common in most Cry proteins. Also it is worth noting the following: first, that polyacrylamide gels showed a minor band of ca. 78 kDa (Fig. 2b), which may correspond to the undegraded product from ORF1; and second, that both of these two expression products are very different from the 58-kDa protein reported earlier as Cry10Aa (10). Additionally, the fact that ORF2 is translated separately from ORF1 may indicate that a putative ancestral Cry4-type gene was subdivided, but some kind of adaptive benefit selected for reactivation of the nontoxic part of the gene.
The complex toxin arsenal of B. thuringiensis subsp. israelensis (6, 30) makes it difficult to assess the contribution of each component by testing them separately. Still, it has been possible to test individual contributions of Cry4A, Cry4B, Cry11A, and Cyt1A (6) and their synergistic interactions. Individual toxicity of Cry10A has been tested under considerable limitations (7, 30), as no parasporal bodies had been obtained before, leading to erroneous conclusions on its low toxicity (7, 13). Inclusion body formation of this toxin made it possible to obtain a precise toxicity level and, even more significantly, to prove its synergistic interaction with the Cyt1A toxin. Interestingly and in contrast to previous reports (10, 30), the toxicity level of Cry10A acting individually is similar to that observed with other Cry proteins from B. thuringiensis subsp. israelensis (6). It should also be pointed out that the toxicity estimated for Cyt1Aa acting alone was comparable to the toxicity in previous reports (6), with an LC50 1.6 times lower than previously estimated.
Based on the difference between the LC50s estimated for the Bt-pSTAB-cry10 spore-crystal complex and the pure crystals, much of the spore-crystal complex consists of spores and cell debris (85 to 88%). Still, in spite of the great difference in toxicity between these two preparations, the synergistic levels (potentiation rates) are about the same (13.3 and 12.6, respectively), which should be expected as the potentiation rate is independent of the concentration of each tested factor. While the toxicity of pure crystals should be higher than the toxicity of spore-crystal complex (as it occurred), potentiation rates should remain similar, as these only indicate the level of synergism in the mixture. Interestingly, when potentiation rates were calculated from data reported previously (6, 29), they were comparable only with those observed between Cry4-type toxins and Cyt1Aa, that is, the highest potentiation rates obtained so far. These results may suggest that if Cry10Aa were expressed at higher levels in the wild-type strain, its toxicity may be significantly increased. Also, in the case of a possible development of resistance to the wild-type strain by mosquito larvae (a situation that has not occurred up till now), the overexpression of Cry10Aa in the wild-type strain may be considered a control alterative. However, it is worth noting that even if the expression level of Cry10Aa observed in strain Bt-pSTAB-cry10 ensures the formation of parasporal bodies, the yield in terms of total biomass is low, as most of the dry weight is made up of spores and cell debris (see above). Also, the low growth rate of the recombinant strain that was observed may be due to the presence of erythromycin in the medium, as its elimination restores the normal growth rate.
In conclusion, parasporal body formation was achieved from a relatively unknown toxin of B. thuringiensis subsp. israelensis. Its toxicity is comparable to the other toxins in this serotype, and like those, it also shows synergism with the Cyt1A toxin. The potential of Cry10Aa is high and must be added to the arsenal of mosquitocidal toxins from B. thuringiensis. The results reported here may form the basis for novel approaches to delaying potential insect resistance, increasing the toxicity of the wild-type strain, studying the complex toxin interaction in this strain, and broadening the toxic spectrum toward other insect species.
Supplementary Material
Acknowledgments
We thank Regina Basurto, Javier Luévano, Aurora Verver, Lourdes Palma, and Alicia Chagolla for their excellent technical support. We also thank the Institut Pasteur and B. Federici for providing key materials.
This project was partially supported by the United Nations University/BIOLAC (Venezuela), CYTED project III.5 (Spain), the Ministry of Foreign Affaires (SRE/PMCID, Mexico), and CONACYT (Mexico).
Footnotes
Published ahead of print on 22 May 2009.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Berry, C., S. O'Neil, E. Ben-Dov, A. F. Jones, L. Murphy, M. A. Quail, M. T. G. Holden, D. Harris, A. Zaritsky, and J. Parkhill. 2002. Complete sequence and organization of pBtoxis, the toxin-coding plasmid of Bacillus thuringiensis subsp. israelensis. Appl. Environ. Microbiol. 68:5082-5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bietlot, H., I. Vishnubhatla, R. Carey, M. Pozsgay, and H. Kaplan. 1990. Characterization of the cysteine residues and disulfide linkages in the protein crystal of Bacillus thuringiensis. Biochem. J. 267:309-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bravo, A. 1997. Phylogenetic relationships of Bacillus thuringiensis δ-endotoxin family proteins and their functional domains. J. Bacteriol. 179:2793-2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bulla, L., K. Kramer, and L. Davidson. 1977. Characterization of the entomocidal parasporal crystal of Bacillus thuringiensis. J. Bacteriol. 130:375-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chilcott, C. N., J. Kalmakoff, and J. S. Pillai. 1983. Characterization of proteolytic activity associated with Bacillus thuringiensis var. israelensis crystals. FEMS Microbiol. Lett. 18:37-41. [Google Scholar]
- 6.Crickmore, N., E. J. Bone, J. A. Williams, and D. Ellar. 1995. Contribution of the individual components of the δ-endotoxin crystal to the mosquitocidal activity of Bacillus thuringiensis subsp. israelensis. FEMS Microbiol. Lett. 131:249-254. [Google Scholar]
- 7.Delécluse, A., C. Bourgouin, A. Klier, and G. Rapoport. 1988. Specificity of action on mosquito larvae of Bacillus thuringiensis israelensis toxins encoded by two different genes. Mol. Gen. Genet. 214:42-47. [DOI] [PubMed] [Google Scholar]
- 8.Delécluse, A., V. Juárez-Pérez, and C. Berry. 2000. Vector-active toxins: structure and diversity, p. 101-125. In J. F. Charles, A. Delecluse, and C. Nielsen-LeRoux (ed.), Entomopathogenic bacteria: from laboratory to field application. Kluwer Academic Publishers, Dordrecht, The Netherlands.
- 9.de Maagd, R., A. Bravo, C. Berry, N. Crickmore, and E. Schnepf. 2003. Structure, diversity and evolution of protein toxins from spore forming entomopathogenic bacteria. Annu. Rev. Genet. 37:409-433. [DOI] [PubMed] [Google Scholar]
- 10.Garduno, F., L. Thorne, A. Walfield, and T. Pollock. 1988. Structural relatedness between mosquitocidal endotoxins of Bacillus thuringiensis subsp. israelensis. Appl. Environ. Microbiol. 54:277-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldberg, L. J., and J. Margalit. 1977. A bacterial spore demonstrating rapid larvicidal activity against Anopheles sergentii, Uranotaenia unguiculata, Culex univitattus, Aedes aegypti and Culex pipiens. Mosq. News 37:355-358. [Google Scholar]
- 12.Hall, T. A. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41:95-98. [Google Scholar]
- 13.Hughes, P., M. Stevens, H. Park, B. Federici, E. Dennis, and R. Akhurst. 2005. Response of larval Chironomus tepperi (Diptera: Chironomidae) to individual Bacillus thuringiensis var. israelensis toxins and toxin mixtures. J. Invertebr. Pathol. 88:34-39. [DOI] [PubMed] [Google Scholar]
- 14.Ibarra, J. E., and B. A. Federici. 1986. Isolation of a relatively nontoxic 65-kilodalton protein's inclusion from the parasporal body of Bacillus thuringiensis subsp. israelensis. J. Bacteriol. 165:527-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ibarra, J. E., and B. A. Federici. 1986. Parasporal bodies of Bacillus thuringiensis subsp. morrisoni (PG-14) and Bacillus thuringiensis subsp. israelensis are similar in protein composition and toxicity. FEMS Microbiol. Lett. 34:79-84. [Google Scholar]
- 16.Ibarra, J. E., and B. A. Federici. 1987. An alternative bioassay for determining the toxicity of suspended particles to mosquito larvae. J. Am. Mosq. Control Assoc. 3:187-192. [PubMed] [Google Scholar]
- 17.Ibarra, J. E., M. C. del Rincon, S. Orduz, D. Noriega, G. Benintende, R. Monnerat, L. Regis, C. M. F. de Oliveira, H. Lanz, M. H. Rodriguez, J. Sanchez, G. Pena, and A. Bravo. 2003. Diversity of Bacillus thuringiensis strains from Latin America with insecticidal activity against different mosquito species. Appl. Environ. Microbiol. 69:5269-5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jensen, G., A. Wilkins, S. Petersen, J. Damgaard, J. Baum, and L. Andrup. 1995. The genetic basis of the aggregation system in Bacillus thuringiensis subsp. israelensis is located on the large conjugative plasmid pX016. J. Bacteriol. 17:2914-2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kimura, M. 1980. A simple model for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16:111-120. [DOI] [PubMed] [Google Scholar]
- 20.Lereclus, D., O. Arantes, J. Chaufax, and M. Lecadet. 1989. Transformation and expression of a cloned δ-endotoxin gene in Bacillus thuringiensis. FEMS Microbiol. Lett. 60:211-218. [DOI] [PubMed] [Google Scholar]
- 21.López, M. G., N. A. Mancilla-Margalli, and G. Mendoza-Diaz. 2003. Molecular structures of fructans from Agave tequilana Weber var. azul. J. Agric. Food Chem. 51:7835-7840. [DOI] [PubMed] [Google Scholar]
- 22.Matzura, O., and A. Wennborg. 1996. RNAdraw: an integrated program for RNA secondary structure calculation and analysis under 32-bit Microsoft Windows. Bioinformatics 12:247-249. [DOI] [PubMed] [Google Scholar]
- 23.Méndez-López, I., R. Basurto, and J. E. Ibarra. 2003. Bacillus thuringiensis serovar israelensis is highly toxic to the coffee berry borer, Hyphotenemus hampei Ferr. (Coleoptera: Scolytidae). FEMS Microbiol. Lett. 226:73-76. [DOI] [PubMed] [Google Scholar]
- 24.Oppert, B. 1999. Protease interactions with Bacillus thuringiensis insecticidal toxins. Arch. Insect Biochem. Physiol. 42:1-12. [DOI] [PubMed] [Google Scholar]
- 25.Park, H.-W., B. Ge, L. S. Bauer, and B. A. Federici. 1998. Optimization of Cry3A yields in Bacillus thuringiensis by use of sporulation-dependent promoters in combination with the STAB-SD mRNA sequence. Appl. Environ. Microbiol. 64:3932-3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porcar, M., and V. Juárez-Pérez. 2003. PCR-based identification of Bacillus thuringiensis pesticidal crystal genes. FEMS Microbiol. Rev. 26:419-432. [DOI] [PubMed] [Google Scholar]
- 27.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 28.Schnepf, E., N. Crickmore, J. Van Rie, D. Lereclus, J. Baum, J. Feitelson, D. R. Zeigler, and D. H. Dean. 1998. Bacillus thuringiensis and its pesticidal proteins. Microbiol. Mol. Biol. Rev. 68:775-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tabashnik, B. E. 1992. Evaluation of synergism among Bacillus thuringiensis toxins. Appl. Environ. Microbiol. 58:3343-3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thorne, L., F. Garduno, T. Thompson, D. Decker, M. Zounes, M. Wild, A. Walfield, and T. Pollock. 1986. Structural similarity between the lepidoptera- and diptera-specific insecticidal endotoxin genes of Bacillus thuringiensis subsp. “kurstaki” and “israelensis.” J. Bacteriol. 166:801-811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tyrell, D. J., L. I. Davidson, L. A. Bulla, Jr., and W. A. Ramoska. 1979. Toxicity of parasporal crystals of Bacillus thuringiensis subsp. israelensis to mosquitoes. Appl. Environ. Microbiol. 38:656-658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tyrell, D. J., L. Bulla, R. Andrews, K. Kramer, L. Davidson, and P. Nordin. 1981. Comparative biochemistry of entomocidal parasporal crystals of selected Bacillus thuringiensis strains. J. Bacteriol. 145:1052-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.