

Abstract
Familial amyloidosis–Finnish type (FAF) results from a single mutation at residue 187 (D187N or D187Y) within domain 2 of the actin-regulating protein gelsolin. The mutation somehow allows a masked cleavage site to be exposed, leading to the first step in the formation of an amyloidogenic fragment. We have performed NMR experiments investigating structural and dynamic changes between wild-type (WT) and D187N gelsolin domain 2 (D2). On mutation, no significant structural or dynamic changes occur at or near the cleavage site. Areas in conformational exchange are observed between β-strand 4 and α-helix 1 and within the loop region following β-strand 5. Chemical shift differences are noted along the face of α-helix 1 that packs onto the β-sheet, suggesting an altered conformation. Conformational changes within these areas can have an effect on actin binding and may explain why D187N gelsolin is inactive. {1H-15N} nuclear Overhauser effect and chemical shift data suggest that the C-terminal tail of D187N gelsolin D2 is less structured than WT by up to six residues. In the crystal structure of equine gelsolin, the C-terminal tail of D2 lies across a large cleft between domains 1 and 2 where the masked cleavage site sits. We propose that the D187N mutation destabilizes the C-terminal tail of D2 resulting in a more exposed cleavage site leading to the first proteolysis step in the formation of the amyloidogenic fragment.
Amyloid diseases such as Alzheimer's disease are characterized by the deposition of protein fibrils (1). Amyloid formation results from a conformational change of a normal protein because of mutation, aging, a change in the environment, or proteolysis (2–4). The conformational change leads to an amyloidogenic precursor that then may interact with other precursors forming the amyloid fibrils with the characteristic cross-β structure (5). In Alzheimer's disease, the amyloid β precursor protein is cleaved, resulting in a peptide capable of forming amyloid. X-ray diffraction studies of amyloid indicate that the protein forms β-strands perpendicular to the long axis of the fibril, leading to a β-sheet growing along the long axis (5). Amyloid fibrils are insoluble and may deposit themselves in the extracellular space. Recent experiments suggest that amyloid formation is not limited to a small number of proteins and that many proteins may form amyloid under the appropriate conditions (6).
Familial amyloidosis–Finnish type (FAF) is a disease where amyloid deposits can be observed in the cornea, the cranial or peripheral nerves, and a number of other tissues (7). The symptoms of FAF are corneal lattice dystrophy, cranial neuropathy, skin elasticity problems, and renal complications (7). The onset of the disease usually occurs in the third to fifth decade (7). FAF is a hereditary disease that is distributed world wide, although the majority of reported cases occur in Finland (7). In FAF patients, there is a single base mutation of A to G or T at nucleotide 654 of the gelsolin gene leading to the mutation of Asp-187 to Asn or Tyr (8). The amyloid deposits from FAF patients contain either a 71- (residues 173–243) or 55-residue (residues 173–225) fragment of the mutant gelsolin (9, 10).
Gelsolin is a six-domain protein; each domain contains a central β-sheet sandwiched between two α-helices (Fig. 1) (11). In general, gelsolin is involved in actin filament severing, capping, and nucleating (12). There are two forms of gelsolin, cellular and plasma (13). Cellular gelsolin is involved in maintaining the architecture and motility of cells (12). Plasma gelsolin is a component in the actin-scavenging system that removes actin filaments from the bloodstream, reducing the viscosity of the blood (14). The fragments of gelsolin in the amyloid plaques are from plasma gelsolin (15). Plasma gelsolin differs from cellular gelsolin only by a 25-residue amino terminus extension that is believed to target the protein for secretion (13). The mutation at residue 187 in domain 2 (D2, residues 151–266) from Asp to Asn or Tyr results in the protein being susceptible to proteolysis between Arg-172 and Ala-173 during secretion (Fig. 1A) (7). In the plasma, the protein is cleaved again at residue 243 resulting in the 71-residue amyloidogenic fragment (7). This fragment has no residual structure as a monomer in solution and requires a change in pH or another change in environment to lead to amyloid formation (16).
Figure 1.

(A) Structure of gelsolin D2 excised from the crystal structure of whole equine gelsolin (11). The secondary elements are a five-stranded β-sheet (strand 1, residues 161–166; strand 2, residues 171–176; strand 3, residues 187–193; strand 4, residues 196–201; strand 5, residues 230–235) and 2 α-helices (helix 1, residues 206–223; helix 2, residues 241–247). The mutation site, Asp-187 (red), is at the N-terminal end of β-strand 3, and the masked cleavage site, Arg-172–Ala-173 (magenta), is in β-strand 2. In full-length gelsolin, β-strand 2 forms additional sheet structure with 2 strands from domain 1 and is not exposed. (B) HSQC chemical shift differences are mapped onto the crystal structure of gelsolin D2. The differences are calculated as: Diff = |(δN-WT - δN-D187N)|/7 + |(δH-WT − δH-D187N)|. If the residue is a glycine, the difference between the 15N chemical shifts is divided by 5. Differences are 0–0.1 (cyan), 0.1–0.2 (yellow), and >0.2 (red). Prolines and exchange-broadened residues are gray. (C) The Cα chemical shift differences are displayed. The differences are 0–0.2 (cyan), 0.2–0.4 (yellow), and >0.4 (red). Cαs that could not be assigned are gray. The results from the relaxation experiments are displayed for WT (D) and D187N (E). Residues that had high or low Jeff(0) values indicative of exchange or increased mobility, respectively, are colored magenta. Also, those residues that are exchanged broadened in the HSQC spectra are colored green (in WT and D187N) or orange (in D187N only). Figs. 1, 3, and 4 were produced by using the program molmol (41).
D2 can be removed from the other five domains of gelsolin and can fold to a reasonably stable structure (ΔGD−N = 3.67 kcal/mol, 25°C) (17). Equilibrium and kinetic studies on the wild-type (WT) and the two amyloidogenic mutants (D187N and D187Y) of gelsolin D2 show that these fold through a two-state mechanism, and no intermediates are observed (17). The D187N and D187Y mutants are destabilized compared with WT by 1.22 kcal/mol and 2.16 kcal/mol, respectively (17). These results suggest that mutant gelsolin is more susceptible to the protease at Arg-172–Ala-173 because of a greater population of denatured protein at equilibrium, which is susceptible to proteolysis, and not from the buildup of partially folded intermediates (17). However, there is a possibility that the mutation at 187 can modify the native state such that the native state is a digest target for proteolysis at 172–173. One argument in favor of modification of the native state is that the amyloidogenic mutations also result in a loss of the actin-binding, -severing, and -nucleating functions (18).
NMR is an ideal tool for investigating the structural and dynamic differences between the native and mutant proteins. In this study, we have used NMR spectroscopy to study structural and dynamic aspects of both the WT and D187N gelsolin D2. NMR triple resonance experiments were used to obtain backbone assignments for WT and D187N NMR spectra. Additionally, 15N NMR dynamics experiments [R1, R2, and heteronuclear nuclear Overhauser effect (NOE)] have been performed to look at the differences in the dynamics between the two proteins. Our results reflect that D187N is a more mobile protein that is undergoing local areas of conformational exchange. This conformational exchange may affect the actin binding and may help make the D187N protein more susceptible to cleaving at residues 172–173.
Materials and Methods
Expression and Purification of WT and D187N Gelsolin D2.
Expression of WT and D187N gelsolin D2 was described previously (17). Briefly, for producing the 15N and 13C/15N samples, the Escherichia coli cells with the appropriate vectors were grown at 37°C in minimal media with ampicillin (150 μg/ml) and with 15N ammonium chloride and 13C glucose as the sole source of N or C where required. The cells were induced with isopropyl-d-thiogalactoside at A600 = 0.4. Cells were harvested by centrifugation. For the WT protein, the cell pellets were resuspended in Pi buffer (20 mM phosphate, pH 7.2, with NaCl to 0.2 M calculated ionic strength, 100 ml per liter of culture). The cells were lysed by sonication, and the cell debris was pelleted by centrifugation. The supernatant was purified by using Ni affinity chromatography, and thrombin was used to cleave the histidine tag. A final gel filtration step removed residual impurities and the soluble aggregated gelsolin fraction that eluted within the void volume of the column. For the D187N protein, the same steps were followed with the addition of 8 M urea. The D187N protein was refolded on the Ni affinity column before elution with an imidazole solution.
NMR Spectroscopy.
Spectra were recorded at 298 K on Bruker (Karlsruhe, Germany) AMX 500 and DRX 600 NMR spectrometers. 1H-15N correlated spectroscopy (HSQC) experiments with WATERGATE (19) water suppression were obtained for 15N-labeled gelsolin D2 samples. Sequence-specific resonance assignments for 13C/15N-labeled gelsolin D2 samples were made based on the HNCACB (20) and CBCA(CO)NH (21) triple-resonance experiments. 15N-edited total correlation spectroscopy and NOE spectroscopy (22) experiments were acquired to confirm and extend the sequential assignments.
Data were processed and analyzed with nmrpipe (23) and nmrview (Merck). Typically, shifted sinesquare window functions were applied before Fourier transformation. In three-dimensional experiments, linear prediction was performed in the 15N dimension.
15N R1, R2, and {1H-15N} NOE experiments (24, 25) were recorded at a 15N frequency of 50.13 MHz on a Bruker AMX500 spectrometer. R1 relaxation delays were set to 16, 64, 160, 208, 256, 304, 400, 448, 496, 544, 592, and 640 ms. R2 experiments featured a CPMG pulse train with a spin echo delay, 2τ, of 1.25 ms. R2 relaxation delays were set to 10, 20, 40, 60, 80, 100, 120, 140, 160, 180, 200, 220, and 240 ms. Peak intensities were fitted to a monoexponential decay function by using kaleidagraph 3.0 (Synergy Software, Reading, PA). The standard errors from the Levenberg–Marquardt fitting routine were taken as the uncertainties in the obtained R1 and R2 values. {1H-15N} NOEs were determined from spectra obtained with and without presaturation of amide proton resonances. Presaturation was achieved by applying a comb of 120° 1H pulses for 2 s. Reference spectra were obtained by replacing the presaturation sequence with a simple decay. For both experiments, the recycle delay was set to 5 s. The {1H-15N} NOE values were calculated by NOE = (Isat − Iref)/Iref, where Isat and Iref are peak intensities in the spectrum with and without presaturation, respectively (26).
Reduced Spectral Density Mapping.
The distribution of frequencies contained in the orientational fluctuation of the NH vectors relative to the static external magnetic field is described by the spectral density function J(ω) (27, 28). By using the high-frequency approximation J(ωH) ≈ J(ωH + ωN) ≈ J(ωH − ωN), the classically used 15N transverse (R2), longitudinal (R1) relaxation rates, and heteronuclear {1H-15N} NOEs yield the following expressions for J(ω) at frequencies 0, ωN, and ωH + ωN:
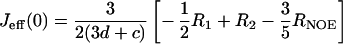 |
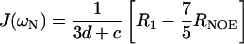 |
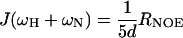 |
Where
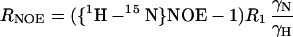 |
where d = γ2Hγ2N(h/2π)2/4r6HN, c = Δ2ω2N/3; Δ is the chemical shift anisotropy of the amide nitrogen; γH and γN are magnetogyric ratios for nuclei 1H and 15N, respectively; h is Planck's constant; and rHN is the NH bond length. For rHN = 1.02 Å and Δ = −160 ppm, the constant d is approximately 1.3 × 109 (rad/s)2, and the constant c is approximately 0.87 × 109 (29).
Jeff(0) is used instead of J(0) to denote that contributions from other pseudo first order processes, such as chemical exchange with its associated rate Rex (30) that increase the R2 rate, are not considered explicitly in the present calculation.
Results
Assignments.
Triple resonance experiments were used to assign the backbone of the WT and D187N HSQC spectrum. Backbone resonances were assigned by using correlations in the HNCACB and CBCA(CO)NH spectra between i and i − 1 residues. These assignments were then further compared with 15N-edited total correlation spectroscopy and NOE spectroscopy spectra for confirmation. Assignments are listed in the BioMagResBank [BMRB-4794 (WT) and BMRB-4795 (D187N)].
The most observable difference between the WT and D187N spectra is the absence of peaks in the D187N HSQC because of exchange broadening. The WT spectrum was completely assignable except for Arg-169, Val-170, Asn-195, and Asp-259. Like WT, Arg-169, Val-170, and Asn-195 were not seen in the D187N spectrum; however, Val-152, Val-153, Trp-180, Asn-185, Gly-186, Asn-187, Phe-189, Ser-203, Glu-235, Glu-236, Gly-237, Thr-238, and Lys-250 were also not observed (Fig. 1 D and E).
Structural Differences.
From the Cα chemical shift deviations from random coil, the secondary structure of the WT and D187N gelsolin D2 appears strikingly similar to that of the equine crystal structure (supplemental Fig. 5; see www.pnas.org) (31). A sequence comparison of D2 of both human and equine gelsolin has only a 14-residue difference (88% sequence identity). The program talos (32), which predicts dihedral angles from a database of chemical shifts and protein sequences, suggests that the secondary structure between the equine crystal structure and the human proteins is similar (supplemental Fig. 6). All of these data suggest that the equine crystal structure is a good structural model for human gelsolin D2.
Chemical shift differences between WT and D187N were measured for the residues visible in the HSQC (Figs. 1B and 2A). Large differences were observed for residues 164–167 (β-strand 1), 202–207 (loop between β-strand 4 and α-helix 1), and 255–258 (C-terminal tail). Residues 165 and 167 had higher differences than residues 164 and 166. In the crystal structure, residues 165 and 167 had NH vectors that pointed at the mutation site (Asp-187). Moderate chemical shift differences were seen in α-helix 1 for residues Glu-209, Lys-212, Ala-213, Val-216, Ile-220, and Glu-224.
Figure 2.

Results from NMR experiments. The differences in the HSQC (A) and Cα (B) chemical shifts between WT and D187N are displayed. The {1H-15N} NOEs are displayed as (Isat − Iref)/Iref for WT (C) and D187N (D). The more negative values are indicative of motion on the nanosecond time scale. The C-terminal tail of D187N begins falling off at Ala-252 compared with Glu-258 in WT, suggesting that six more residues are unstructured in the mutant.
Cα chemical shift differences between WT and D187N showed four areas of deviation: residues 163–168 (β-strand 1), 180–190 (loop between β-strands 2 and 3 and β-strand 3), 238–240 (loop between β-strand 5 and α-helix 2), and 254–258 (C-terminal tail) (Figs. 1C and 2B). Two of these areas coincide with differences in the HSQC (residues 163–168 and 254–258). The differences in the C-terminal tail are more evident in the Cα chemical shifts. Further, a large Cα chemical shift difference is seen for Pro-251 that cannot be observed in the HSQC as the residue lacks a NH vector. In the equine crystal structure, Pro-251 packs against Trp-180 that is exchange broadened in D187N.
Dynamic Characterization.
To investigate further the effects of the D187N mutation on gelsolin D2, a set of 15N dynamics experiments (R1, R2, and heteronuclear NOE) were performed on both species. Changes in the dynamics of a protein because of mutation can make it more susceptible to proteases. From an overview of the results, the WT type protein shows less dynamic fluctuations than the D187N mutant, but the increased dynamic variations in D187N are isolated to regions of the protein.
In the reduced spectral density map (supplemental Fig. 7A), the WT gelsolin D2 had six regions with interesting relaxation properties. These six regions included: region 1 (Gly-186 and Asp-187), region 2 (Gly-194), region 3 (Arg-225, Gly-227, and Ala-229), region 4 (Ser-234 and Glu-235), region 5 (Glu-241, Met-243), and region 6 (Gly-248 and Leu-253). Region 1 contains the mutation site at the end of β-strand 3. Regions 2 and 3 consist of turn-and-loop regions between β-strands 3 and 4 and α-helix 1 and β-strand 5, respectively. Region 4 is at the end of β-strand 5. Region 5 is contained in the first turn of α-helix 2, and region 6 is within the long C-terminal tail. Asp-187, Arg-225, Gly-227, Glu-235, and Gly-248 all have elevated Jeff(0) values, indicative of chemical exchange behavior. The high values for these residues can be observed in the R2 values. Gly-186, Met-243, and Leu-253 have depressed Jeff(0) values, reflecting internal motion. These values are mirrored in the R2 values as well. Gly-194 and Ala-229 have depressed J(50) and J(450), indicative of fast motion. The values result from depressed R1 values. Ala-229 also has a slightly depressed Jeff(0) from a low R2. Ser-234 has a slightly elevated J(50) and J(450) from an elevated R1 value. Glu-241 has an elevated J(450) from a depressed heteronuclear NOE. Graphs for the R1 and R2 values may be found in supplemental Fig. 7.
The reduced spectral density map (supplemental Fig. 7B) of D187N gelsolin D2 has some similarities to that of WT although it has more areas that appear to be in exchange. The D187N spectral density map can be broken into seven areas: region 1 (Val-165), region 2 (Asn-193), region 3 (Cys-201, Gly-202, and Ser-205), region 4 (Glu-224, Arg-225, and Ala-229), region 5 (Val-233 and Ser-234), region 6 (Gln-245 and Leu-247), and region 7 (Ala-252 and Gly-256). Region 1 is in the middle of β-strand 1. Regions 2 and 4 make up turn or loop regions between secondary structure regions. Region 3 in the D187N mutant consists of the end of β-strand 4 and part of the loop region. Region 5 is the same as region 4 in WT and is at the end of β-strand 5. Region 6 is within the C-terminal end of α-helix 2, and region 7 is part of the long C-terminal tail. Val-165, Cys-201, Gly-202, Ser-205, Glu-224, Arg-225, Val-233, Gln-245, Leu-247, Ala-252, and Gly-256 have elevated Jeff(0) values resulting from high R2 values. Asn-193 had a low Jeff(0), suggesting it is flexible. This result is also observed in the R2 values. Ala-229 has depressed values for J(50) and J(450) as in WT. These values result from a largely decreased R1 value. Ser-234 has an elevated J(50) value that appears because of its high R1 value.
The structured regions of the protein differ between WT and D187N, on the basis of the {1H-15N} NOE results (Fig. 2 C–D). In the WT gelsolin D2, the {1H-15N} NOE retains high values until after Glu-258. The D187N {1H-15N} NOE suggests a less structured C-terminal tail, with the structure ending after Ala-252. This would suggest that at least six more residues are unstructured in the mutant protein.
Discussion
The D187N mutation makes plasma gelsolin susceptible to proteolysis between Arg-172 and Ala-173 (7). This fragment is then cleaved at residue 243 in the plasma resulting in a 71-residue amyloidogenic peptide that leads to the disease, FAF. Although there is no high-resolution structure of the human form of gelsolin D2, NMR chemical shift deviations from random coil and the program talos (32) suggest that the crystal structure of equine gelsolin D2 (11) is very similar (supplemental Figs. 5 and 6; see www.pnas.org). Asp-187 forms a cross-strand salt bridge with Arg-166 in β-strand 1 within the crystal structure and is part of a hydrogen-bonding network with Asn-184 and Gln-164. These interactions have led to previous speculation that the mutation at Asp-187 destroys this network, leading to a disruption of β-strand 1 and the loop region between β-strands 2 and 3 (11). The loss of structure in these regions was thought to allow a protease access to the cleavage site in β-strand 2. We have performed NMR experiments to test the possible structural and dynamic changes to gelsolin D2 on mutation of Asp-187 to Asn-187.
No Major Conformational Changes Around Cleavage Site in D187N.
On mutating Asp-187 to Asn-187, no significant changes in chemical shifts or relaxation data were observed at the cleavage site (Arg-172–Ala-173), suggesting that β-strand 2 is not affected by the mutation. Further, the hypothesis that the mutation leads to disruption of β-strand 1 and the loop region between β-strands 2 and 3 exposing the cleavage site to proteases is not supported by the data. Although chemical shift differences are seen in β-strand 1, larger differences are observed for those residues with their NH vectors pointed at the mutation site (residues 165 and 167) than those pointed away (residues 164 and 166) in the equine crystal structure suggesting that β-strand 1 is still formed (Fig. 1B). Further, chemical shift deviations from random coil suggest β-strand 1 is formed in both WT and D187N gelsolin D2 (supplemental Fig. 5). Although residues 180–190, which include some of the loop between β-strands 2 and 3, had Cα chemical shift differences between WT and D187N, these differences were not large (0.3–0.4 ppm) (Fig. 1C). No major differences were observed between the WT and D187N chemical shifts or relaxation data for the loop region between β-strands 2 and 3, suggesting that no structural change was occurring. These results suggest that the D187N mutation is exposing the cleavage site through another mechanism.
Proposed Mechanism for Exposing Cleavage Site.
Large chemical shift differences between WT and D187N are observed in the C-terminal region (residues 254–258), suggesting a conformational change. This is further supported by low {1H-15N} NOE values (Fig. 2 C–D) and high Jeff(0) values (Fig. 1 D and E and supplemental Fig. 7 A and B; see www.pnas.org) in this region. In D187N, low {1H-15N} NOE values start after Ala-252, whereas in the WT the values begin to fall only after Glu-258. The drop in {1H-15N} NOE values indicates motion at the nanosecond time scale, indicative of that seen for residues beyond structural domain boundaries. The high Jeff(0) values for Ala-252 and Gly-256 suggest they are participating in chemical exchange. The exchange process could be an interaction with solvent or a conformational transition, such as structured ⇌ unstructured. Further, in WT, Gly-256 has distinct chemical shifts for each Hα (3.807 and 4.031 ppm), whereas in D187N the protons are indistinguishable, with a shift of 3.951 ppm because of conformational averaging. Interestingly, the less structured D187N residues (residues 253–258) follow Pro-251 that has a large Cα chemical shift difference resulting possibly from a change in packing with Trp-180. We propose that in D187N the C-terminal tail becomes less structured compared with WT by up to six residues. In the equine crystal structure, residues 253–258 help protect the cleavage site from exposure. With a loss of structure in this region, the cleavage site would be susceptible to proteases, leading to the initial proteolysis step in the formation of the amyloidogenic fragment (Fig. 3).
Figure 3.

Space-filling model of full-length equine gelsolin. Domain 2 (gray) makes contacts with domains 1, 3, and 6 (black). The cleavage site (magenta) sits in a cleft that is covered by the C-terminal tail of domain 2 (green and orange). In A, the C-terminal tail is present and the portion of the tail with low {1H-15N} NOE values in both WT, and D187N is colored green. The additional residues that are believed to be unstructured or in some form of conformational exchange in D187N only are colored orange. In B, the mobile portion of the C-terminal tail in D187N (residues 253–266) has been removed, and the exposed cleavage site is visible. We propose that the C-terminal tail of domain 2 is more unstructured in the D187N mutant, resulting in better access to the masked cleavage site for proteases. The proteolysis fragment can be hydrolyzed further at residue 243, producing the amyloidogenic fragment.
Exchange Behavior in the D187N Spectrum.
The major difference between WT and D187N HSQC spectra was the amount of exchange broadening in the mutant spectrum. The broadening in D187N gelsolin D2 appeared mainly in two regions (residues 185–187, 189, and residues 235–238) (Fig. 1E). The first region encompasses the mutation site and appears to be undergoing some conformational transitions. Cys-188, which was the only residue found in the HSQC in this region, does not show the same exchange characteristics because it is held rigid by the disulfide bond with Cys-201. The second region defines part of the loop region between β-strand 5 and α-helix 2. Interestingly, there are no direct interactions between the two regions in exchange, as residues 202–204 separate them. Interestingly, Ser-203 is also exchange broadened, and residues 201–202 and 205 have high Jeff(0) values (supplemental Fig. 7B; www.pnas.org) suggestive of chemical exchange along with chemical shift differences in the HSQC.
In the crystal structure of equine gelsolin, the main chain nitrogen of Gly-202 is within hydrogen-bonding distance with the main chain carbonyl oxygen of Asp-187. Further, the amide nitrogen and the side chain oxygen of Ser-203 are in hydrogen-bonding distance with the side chain of Glu-236, which is the only residue in the region of 235–238 that forms major interactions with the rest of the protein (Fig. 4). We propose the mutation of Asp-187 to Asn causes a conformational exchange process in residues 185–187 and 189, resulting in the loss of the hydrogen bond with Gly-202. The hypothesis is that the loss of the hydrogen bond and conformational changes lead to exchange behavior observed for residues 201–203 and 205, resulting in the loss of the hydrogen bond between Ser-203 and Glu-236, accounting for the exchange broadening seen in that region.
Figure 4.

Hydrogen-bonding network connecting the mutation site to residues 235–238. In the equine crystal structure, Asp-187 forms a cross-strand hydrogen bond to Gly-202, most likely stabilizing the mobile Gly. The neighboring Ser-203 has an amide hydrogen that is hydrogen bonded to the side chain 236. This hydrogen bond appears to be the only interaction between the loop region of residues 235–238 with the rest of the protein. The mutation at 187 could destroy this hydrogen-bond network and lead to a conformational transition for residues 235–238 that causes the exchange broadening in the HSQC of D187N.
Possible D187N Effects on Function.
The D187N gelsolin D2 has no actin-severing or -nucleating activity (18). The putative binding site for F-actin encompasses residues 161–172 and 197–226 (33–36). On Ca2+ binding, a portion of domain 6 is proposed to move away, allowing residues 197–226 access to bind to actin (11, 37). Residues 197–226 contain one of the areas in conformational exchange according to the D187N relaxation data (residues 201–203 and 205; Fig. 1E and supplemental Fig. 7B; see www.pnas.org). These residues also had chemical shift differences in the HSQC, suggesting a possible change in structure from WT. The motion or conformational change of this area may not allow the protein to bind F-actin. Further, residues 197–226 encompass α-helix 1 where one side of the helix had moderate chemical shift differences (Fig. 1C), suggesting a slightly altered packing arrangement for the helix against the β-sheet in D187N. A recent study suggests that basic residues within α-helix 1 are important for actin binding (36). An altered helix packing conformation could decrease the binding affinity. Also, residues 235–238 that appear to be in conformational exchange in D187N and the more mobile C-terminal tail may be adopting conformations that block the actin-binding site.
Comparison with Other Actin-Binding Proteins.
Asn-195 is exchange broadened in the HSQC of both WT and D187N gelsolin D2 (Fig. 1 D and E). Furthermore, Gly-194 has depressed J(50) and J(450) values (supplemental Fig. 7A; www.pnas.org) in the WT relaxation experiments suggesting that it is undergoing some fast motion. In the D187N, Leu-193 has a depressed Jeff(0) value (supplemental Fig. 7B), suggesting increased flexibility. These residues are all part of the turn between β-strands 3 and 4. This turn is analogous to the turn between β-strands 4 and 5 in the structurally homologous domain villin 14T (38, 39). NMR relaxation experiments of villin 14T reveal that the analogous turn is mobile (40). With exchange broadening of Asn-195 and motion of nearby residues, we believe this turn in gelsolin is also mobile and undergoing local conformational exchange.
A recent study investigating the structure of severin domain 2 and the actin binding of gelsolin domain 2 suggested that the basic residues from residues 168–170 (turn between β-strands 1 and 2) are important for binding to the side of actin (36). In both, WT and D187N, Arg-169 and Val-170 were exchange broadened and could not be assigned in the HSQC. These residues appear to be flexible and undergoing conformational exchange that may be important for binding.
Conclusions
Chemical shift analysis and relaxation experiments suggest that human WT gelsolin D2 folds to a well-ordered structure with some flexible turns and loops between secondary structure elements. Although the overall stability is not large, the structure does not have locally unfolded regions and maintains all the secondary structure that is observed for the same domain in the equine crystal structure. With the D187N mutation, the structure becomes more flexible, and the overall stability is decreased. Although the chemical shifts suggest that the secondary structure is not greatly affected by the mutation, residues near the mutation site in and around β-strands 3, 4, and 5 appear to be in conformational exchange (Fig. 1). Further, α-helix 1, although formed, has chemical shift differences between WT and D187N that suggest it docks against the β-sheet in a slightly different conformation (Fig. 1B). These conformational differences are observed in areas of the protein that are necessary for actin binding and could explain the inactivity of D187N gelsolin. Previous hypotheses that the mutation causes conformational changes in β-strand 1 or the loop region between β-strands 2 and 3 resulting in an exposed cleavage site (11) are not supported by the data in this study. We propose two possibilities: (i) As previously suggested, the D187N mutation increases the population of the denatured state at equilibrium leading to a more exposed cleavage site (17); (ii) the C-terminal tail of D187N gelsolin D2 is more unstructured by up to six residues. This loss of structure can expose the cleavage site to proteases, leading to the amyloidogenic fragment and the resulting disease, FAF (Fig. 3).
Supplementary Material
Acknowledgments
We thank Mr. Mark Proctor for assistance in purifying NMR samples. We thank Dr. Mark Bycroft for helpful discussions and advice and for reviewing the manuscript. We also thank Drs. Stefan M. V. Freund and Alexander Buchberger for help in analyzing the NMR data. This research was funded by the MRC. S.L.K. was funded by a Hitchings–Elion Fellowship from the Burroughs–Wellcome Fund.
Abbreviations
- FAF
familial amyloidosis–Finnish type
- WT
wild type
- D2
domain 2
- NOE nuclear Overhauser effect
HSQC, 1H-15N heteronuclear single quantum correlation
Footnotes
Data deposition: The NMR chemical shifts have been deposited in the BioMagResBank, www.bmrb.wisc.edu [accession nos. BMRB-4794 (WT) and BMRB-4795 (D187N)].
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.180310097.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.180310097
References
- 1.Thomas P J, Qu B H, Pedersen P L. Trends Biochem Sci. 1995;20:456–459. doi: 10.1016/s0968-0004(00)89100-8. [DOI] [PubMed] [Google Scholar]
- 2.Kelly J W. Curr Opin Struct Biol. 1996;6:11–17. doi: 10.1016/s0959-440x(96)80089-3. [DOI] [PubMed] [Google Scholar]
- 3.Kelly J W. Structure (London) 1997;5:595–600. doi: 10.1016/s0969-2126(97)00215-3. [DOI] [PubMed] [Google Scholar]
- 4.Kelly J W. Curr Opin Struct Biol. 1998;8:101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 5.Blake C, Serpell L. Structure (London) 1996;4:989–998. doi: 10.1016/s0969-2126(96)00104-9. [DOI] [PubMed] [Google Scholar]
- 6.Chiti F, Webster P, Taddei N, Clark A, Stefani M, Ramponi G, Dobson C M. Proc Natl Acad Sci USA. 1999;96:3590–3594. doi: 10.1073/pnas.96.7.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kiuru S. Amyloid: Int J Exp Clin Invest. 1998;5:55–66. [Google Scholar]
- 8.de la Chapelle A, Tolvanen R, Boysen G, Santavy J, Bleeker-Wagemakers L, Maury C P, Kere J. Nat Genet. 1992;2:157–160. doi: 10.1038/ng1092-157. [DOI] [PubMed] [Google Scholar]
- 9.Maury C P J, Nurmiaho-Lassila E-L, Rossi H. Lab Invest. 1994;70:558–564. [PubMed] [Google Scholar]
- 10.Maury C P J, Sletten K, Totty N, Kangas H, Liljestrom M. Lab Invest. 1997;77:299–304. [PubMed] [Google Scholar]
- 11.Burtnick L D, Koepf E K, Grimes J, Jones E Y, Stuart D I, McLaughlin P J, Robinson R C. Cell. 1997;90:661–670. doi: 10.1016/s0092-8674(00)80527-9. [DOI] [PubMed] [Google Scholar]
- 12.Yin H. BioEssays. 1987;7:176–179. doi: 10.1002/bies.950070409. [DOI] [PubMed] [Google Scholar]
- 13.Kwiatkowski D J, Stossel T P, Orkin S H, Mole J E, Colten H R, Yin H L. Nature (London) 1986;323:455–458. doi: 10.1038/323455a0. [DOI] [PubMed] [Google Scholar]
- 14.Lee W M, Galbraith R M. N Engl J Med. 1992;326:1335–1341. doi: 10.1056/NEJM199205143262006. [DOI] [PubMed] [Google Scholar]
- 15.Kangas H, Paunio T, Kalkkinen N, Jalanko A, Peltonen L. Hum Mol Genet. 1996;3:2223–2239. doi: 10.1093/hmg/5.9.1237. [DOI] [PubMed] [Google Scholar]
- 16.Ratnaswamy G, Koepf E, Bekele H, Yin H, Kelly J W. Chem Biol. 1999;6:293–304. doi: 10.1016/S1074-5521(99)80075-1. [DOI] [PubMed] [Google Scholar]
- 17.Isaacson R L, Weeds A G, Fersht A R. Proc Natl Acad Sci USA. 1999;96:11247–11252. doi: 10.1073/pnas.96.20.11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weeds A G, Gooch J, McLaughlin P, Maury C P J. FEBS Lett. 1993;335:119–123. doi: 10.1016/0014-5793(93)80452-z. [DOI] [PubMed] [Google Scholar]
- 19.Piotto M, Saudek V, Sklenar V. J Biomol NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 20.Wittekind M, Mueller L. J Magn Reson B. 1993;101:201–205. [Google Scholar]
- 21.Muhandiram D R, Kay L E. J Magn Reson B. 1994;103:203–216. [Google Scholar]
- 22.Norwood T J, Boyd J, Heritage J E, Soffe N, Campbell I D. J Magn Reson. 1990;87:488–501. [Google Scholar]
- 23.Delaglio F, Grzesiek S, Vuister G W, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1996;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 24.Wagner G. Curr Opin Struct Biol. 1993;3:748–754. [Google Scholar]
- 25.Palmer A G. Curr Opin Struct Biol. 1997;7:732–737. doi: 10.1016/s0959-440x(97)80085-1. [DOI] [PubMed] [Google Scholar]
- 26.Noggle J H, Schirmer R E. The Nuclear Overhauser Effect: Chemical Applications. New York: Academic; 1971. [Google Scholar]
- 27.Abragam A. The Principles of Nuclear Magnetism. Oxford: Clarendon; 1961. [Google Scholar]
- 28.Wittebort R J, Szabo A. J Chem Phys. 1978;69:1722–1736. [Google Scholar]
- 29.Peng J W, Wagner G. Methods Enzymol. 1994;239:563–596. doi: 10.1016/s0076-6879(94)39022-3. [DOI] [PubMed] [Google Scholar]
- 30.Bloom M, Reeves L W, Wells E J. J Chem Phys. 1965;42:1615–1642. [Google Scholar]
- 31.Wishart D S, Sykes B D. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 32.Cornilescu G, Delaglio F, Bax A. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 33.Sun H-Q, Wooten D C, Janmey P A, Yin H L. J Biol Chem. 1994;269:9473–9479. [PubMed] [Google Scholar]
- 34.Feinberg J, Benyamin Y, Roustan C. Biochem Biophys Res Commun. 1995;209:426–432. doi: 10.1006/bbrc.1995.1520. [DOI] [PubMed] [Google Scholar]
- 35.Van Troys M, Dewitte D, Goethals M, Vanderkerckhove J, Ampe C. FEBS Lett. 1996;397:191–196. doi: 10.1016/s0014-5793(96)01086-1. [DOI] [PubMed] [Google Scholar]
- 36.Puius Y A, Fedorov E V, Eichinger L, Schleicher M, Almo S C. Biochemistry. 2000;39:5322–5331. doi: 10.1021/bi992364d. [DOI] [PubMed] [Google Scholar]
- 37.Robinson R C, Mejillano M, Le V P, Burtnick L D, Yin H L, Choe S. Science. 1999;286:1939–1942. doi: 10.1126/science.286.5446.1939. [DOI] [PubMed] [Google Scholar]
- 38.Markus M A, Matsudaira P, Wagner G. Protein Sci. 1997;6:1197–1209. doi: 10.1002/pro.5560060608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Markus M A, Nakayama T, Matsudaira P, Wagner G. Protein Sci. 1994;3:70–81. doi: 10.1002/pro.5560030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Markus M A, Dayie K T, Matsudaira P, Wagner G. Biochemistry. 1996;35:1722–1732. doi: 10.1021/bi951933o. [DOI] [PubMed] [Google Scholar]
- 41.Koradi R, Billeter M, Wüthrich K. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}