

Abstract
Free oxygen radicals are involved in the pathogenesis of necrotizing enterocolitis (NEC) in premature infants. The stress-activated p38 mitogen-activated protein kinase (MAPK) has been implicated in gut injury. Here, we found that phosphorylated p38 was detected primarily in the villus tip of normal intestine, whereas it was expressed in the entire mucosa in NEC. H2O2 treatment resulted in a rapid phosphorylation of p38 MAPK and subsequent apoptosis of rat intestinal epithelial (RIE)-1 cells; this induction was attenuated by treatment with SB203580, a selective p38 MAPK inhibitor, or transfection with p38α siRNA. Moreover, SB203580 also blocked H2O2-induced PKC activation. In contrast, the PKC inhibitor (GF109203x) did not affect p38 activation, indicating that p38 MAPK activation occurs upstream of PKC activation in H2O2-induced apoptosis. H2O2 treatment also decreased mitochondrial membrane potential; pretreatment with SB203580 attenuated this response. Our study demonstrates that the p38 MAPK/PKC pathway plays an important role as a pro-apoptotic cellular signaling during oxidative stress-induced intestinal epithelial cell injury.
Keywords: Oxidative stress, intestinal epithelial cell injury, NEC, p38 MAPK, Apoptosis
Introduction
Necrotizing enterocolitis (NEC), characterized by diffuse inflammation, ischemia and necrosis, is a devastating gastrointestinal (GI) condition in premature infants; the exact etiology remains unknown [1]. Recent advances in the care of premature infants born with respiratory insufficiency have resulted in increased survival for extremely small premature infants and, as a result, the incidence of NEC has steadily risen. In particular, a potential link between reactive oxygen species (ROS) produced by ischemia-reperfusion injury to the premature gut and the development of NEC has been suggested by several studies [1-3].
The cellular responses to oxidative stress can vary from growth arrest to cell death depending upon the stressful stimuli, duration of exposure, cell type, and surrounding cell environment [4]. Consequently, ROS-mediated cell damage is implicated in the pathogenesis of a variety of diseases, including in the colonic inflammation associated with ulcerative colitis [5]. Interestingly, the exact role of ROS during NEC has not been clearly defined. The p38 mitogen-activated protein kinase (MAPK), activated by inflammatory cytokines [tumor necrosis factor-α and interleukin (IL)-1β] and a diverse array of cellular stresses (e.g., ultraviolet light, heat and osmotic shock) contributes to the induction of apoptosis. Oxidative stress is also known to induce apoptosis in a variety of cell types by activating intracellular cell death signaling cascades including p38 MAPK [6]. Recently, endotoxin has been shown to cause a p38-dependent release of the pro-inflammatory molecule cyclooxygenase-2 by enterocytes, to potentiate the systemic inflammatory response during NEC [7]. Prolonged activation of p38 MAPK was also noted in the immature gut and shown to mediate the excessive activation of IL-8 [8]. However, the exact role of p38 MAPK in the regulation of intestinal epithelial cell apoptosis during NEC is not clearly defined.
Since elevated p38 MAPK activity has been implicated in the pathogenesis of NEC as well as in ROS-mediated apoptosis in variety of cell types, we sought to determine the exact role of p38 MAPK in the H2O2-induced rat intestinal epithelial cell (RIE-1) apoptosis. We show that H2O2 induces apoptosis in RIE-1 cells with activation of p38 MAPK. Inhibition of p38 MAPK significantly attenuated H2O2-mediated apoptosis. Moreover, we found that inhibition of p38 MAPK blocked H2O2-induced protein kinase C (PKC) phosphorylation while inhibition of PKC did not have an effect on H2O2-induced p38 MAPK phosphorylation, further suggesting that a p38 MAPK/PKC pathway plays an important role in H2O2-mediated apoptosis in intestinal cells.
Materials and Methods
Reagents
H2O2 were purchased from Sigma (St. Louis, MO). Bis-indolylmaleimide (GF109203x) and SB203580 were from Calbiochem (San Diego, CA). Rabbit polyclonal anti-caspase-3 and rabbit anti-p38α were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-actin antibody was purchased from Sigma. Rabbit polyclonal anti-p38 and rabbit polyclonal anti-phospho-p38 were purchased from Cell Signaling (Beverly, MA). Tissue culture media and reagents were obtained from Life Technologies, Inc. (Grand Island, NY).
Immunohistochemical analysis
Paraffin-embedded blocks of intestinal sections from 16 infants who underwent resection for NEC as well as sections from 4 infants who required intestinal resection for non-inflammatory condition of the GI tract (i.e., intestinal atresia) were analyzed as ‘normal’ controls. Sections (4 μm) representing the areas adjacent to the intestinal injury from NEC (necrosis) and ‘normal’ intestine were prepared for hematoxylin and eosin (H&E) staining and immunohistochemical studies using specific antibodies as described previously [9]. A DAKO EnVision™ + System, Peroxidase (DAB) kit was used according to the manufacture's instruction. Briefly, endogenous peroxidase was blocked by placing slides in 0.03% H2O2/sodium azide block solution for 5 min, washed with deionized water, and placed in tris buffered saline (pH 7.4) for 5 min. Slides were incubated at room temperature with primary mouse monoclonal anti-phospho-p38 antibody (1:150, Cell signaling, cat. 9216) for 60 min. Peroxidase-labeled polymer complex amplification and detection system was used. Negative controls (isotype matched rabbit IgG) were used in each assessment.
Cell culture
RIE-1 cells obtained from Dr. Kenneth D. Brown (Cambridge Research Station, Babraham, Cambridge, U.K.) were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum and cultured at 37°C under an atmosphere containing 5% CO2. All experiments were performed on cells within 6 weeks of culture from liquid nitrogen stocks and free of Mycoplasma contamination. In order to eliminate the potential ROS scavenger action of dimethyl sulfoxide (DMSO) [10] in vehicle, we used the same concentrations of DMSO (0.1%) for both inhibitors and vehicle.
DNA fragmentation assay
Cells were plated in 96-well plates 24 h before treatment. DNA fragmentation was evaluated by examination of cytoplasmic histone-associated DNA fragments using a Cell Death Detection ELISAPlus kit (Roche Molecular Biochemicals, Indianapolis, IN) according to the manufacturer's instructions.
Protein extraction and Western blot analysis
Cells were lysed with TNN buffer as described previously [11]. Protein concentrations were determined using the Bradford method [12]. Total protein (100 μg) was resolved on a 10% polyacrylamide gel and transferred onto polyvinylidene difluoride membranes (Millipore Corp, Bedford, MA). Filters were incubated overnight at 4°C in a blocking solution (Tris-buffered saline containing 5% nonfat dried milk and 0.1% Tween 20), followed by a 1 h incubation with primary antibodies at 4°C overnight. Filters were washed three times in a blocking solution and incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. After three washes, the immune complexes were visualized by enhanced chemiluminescence system (Amersham Biosciences, Piscataway, NJ).
p38 MAPK siRNA
Rat p38α siRNA and control siRNA duplexes (Dharmacon, Inc., Lafayette, CO) were introduced into cells by electroporation (400 V, 500 microfarads) using GenePulser XCell (Bio-Rad). After 24 h, cells were trypsinized and seeded into 96-well plates for cell death assay. p38α expression was analyzed by Western blotting.
JC-1 mitochondrial membrane potential detection
The mitochondrial membrane potential was analyzed using MitoProbe™ JC-1 Assay kit (Molecular Probes, Eugene, OR). The collapse in the electrochemical gradient across the mitochondrial membrane was measured using a fluorescent cationic dye 5,5′,6,6,-tetrachloro-1,1′,3,3′-tetraethyl-benzamidazolo-carbocyanin iodide, known as JC-1. This dye exhibits potential dependent accumulation in mitochondrial matrix [13]. Cells (1×106) were incubated with 2 μM JC-1 for 15 min at room temperature in darkness. Cells were washed twice with PBS at 4°C, resuspended in 0.5 ml PBS, and analyzed on a FACSCalibur flowcytometer.
Statistical analysis
Results are expressed as the mean ± SEM. The data in the figures were analyzed using the Kruskal-Wallis test and assessed at the 0.05 level of significance.
Results
Activation of p38 MAPK is associated with NEC in vivo and oxidative stress in vitro
Phosphorylated p38 MAPK in the normal bowel and in the small bowel of infants with NEC was assessed using a specific antibody. Representative sections shown in Fig. 1A demonstrate that phosphorylated (i.e., active) p38 MAPK was mostly localized in the villi in sections from noninflammatory conditions of the GI tract (Fig. 1A), while the intensity of staining was significantly decreased in the crypt. Consistent with our data, active p38 MAPK has been shown expressed primarily in the villi of the human fetal intestine [14]. In contrast, in intestinal sections adjacent to NEC, phosphorylated p38 was present throughout the entire epithelium from the crypt to the villus suggesting an association of p38 activation with NEC.
Fig. 1. Expression of p38 MAPK in NEC.
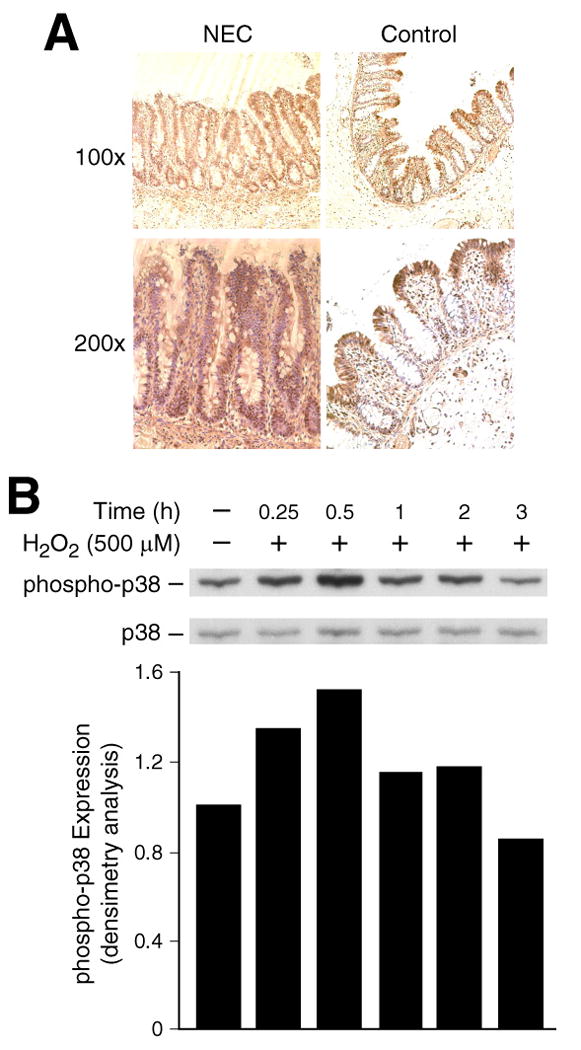
(A) Representative sections show strong crypt and villus phospho-p38 MAPK expression (brown staining) in intestine adjacent to NEC, whereas no immunoreactivity is noted in ‘normal’ sections (Control). (B) RIE-1 cells were treated with H2O2 (500 μM) over a time course for Western blotting using anti-phospho-p38 (p-p38) antibody. The membranes were stripped, reprobed with anti-p38 antibody. Densitometric analysis of signals is expressed as relative phospho-p38 expression (corrected for total p38). A representative blot from three separate experiments is shown.
We have previously found that RIE-1 cells undergo apoptosis when exposed to H2O2 in a dose- and time-dependent manner [11]. Oxidative stress is known to activate multiple signal transduction pathways [4, 6]. To further identify the signaling mechanisms activated by H2O2 in RIE-1 cells, we assessed levels of phosphorylated p38. Treatment with H2O2 resulted in rapid increases in phosphorylated p38, without affecting the total protein expression levels (Fig. 1B). The activation of p38 occurred at 15 min after the addition of H2O2 (500 μM), peaked at 30 min, and returned to basal level by 3 h. Phosphorylated p38 was also assessed by densitometric analysis and corrected using total p38 expression. This rapid induction of p38 MAPK, a protein kinase associated with increased apoptosis, suggested a role for p38 in oxidative stress-induced intestinal epithelial cell death.
Inhibition of p38 attenuates H2O2-induced apoptosis and mitochondrial depolarization
To delineate the role of p38 in H2O2-induced apoptosis, RIE-1 cells were pretreated for 30 min with a selective p38 inhibitor, SB203580 (10 μM), prior to treatment with H2O2 (500 μM) for 3 h and then DNA fragmentation was determined. H2O2 treatment induced significant RIE-1 cell death which was reversed by pretreatment with SB203580 (Fig. 2A). Consistent with the cell death ELISA results, treatment with H2O2 for 3 h resulted in increased cleavage (i.e., activation) of caspase-3, and PARP (Mr 85,000; cleavage product); this increase was also attenuated by SB203580 (Fig. 2B). These results suggest a pro-apoptotic role for the p38 MAPK pathway in H2O2-induced RIE-1 cell death.
Fig. 2. Effect of SB203580, a specific p38 inhibitor, on H2O2-induced RIE-1 cell death.
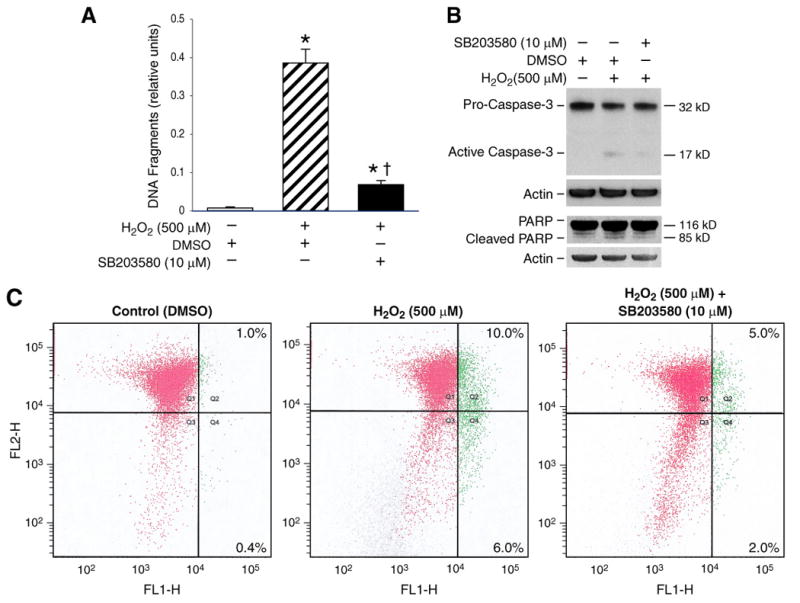
RIE-1 cells were pretreated with the p38 inhibitor, SB203580 (10 μM), for 30 min prior to H2O2 treatment (500 μM) for 3 h. (A) Apoptosis was estimated by an ELISA assay (Data represent triplicate determinations; mean ± SEM; * = p< 0.05 vs. control; † = p< 0.05 vs. H2O2 alone). (B) Cleaved protein products for caspase-3 and PARP were determined by Western blotting. (C) Mitochondrial depolarization by JC-1 assay after treatment with vehicle (control; left panel), H2O2 alone (500 μM; middle panel) and H2O2 + SB203580 (10 μM; right panel). Percentage in the right two quadrants (Q2, Q4) indicate proportion of cells with depolarized mitochondria. Representative data from three separate experiments are shown.
Mitochondrial membrane depolarization, which increases permeability and releases pro-apoptotic factors into the cytosol, was shown to be an early sign of apoptosis in human intestinal epithelial cell lines; inhibition of PKC attenuates this decrease [15]. To determine if the mitochondria-initiated pathway is related to p38 MAPK in H2O2-induced apoptosis, we measured the effect of p38 MAPK inhibition on H2O2-induced alterations in mitochondrial membrane potential. In healthy cells, the lipophilic cation JC-1 exists as a monomer in the cytosol (FL-1 positive; green) and also accumulates as aggregates in the mitochondria (FL-2 positive; red). In apoptotic and necrotic cells, JC-1 exists exclusively in a monomer form and produces a green cytosolic signal. As shown in Figure 2C, treatment with H2O2 decreased mitochondrial membrane potential when compared to control cells (Fig. 2C; left and center panels), implicating altered mitochondrial membrane permeability as an important event for H2O2-induced apoptosis in RIE-1 cells. Pretreatment with the p38 MAPK inhibitor SB203580 attenuated H2O2-induced mitochondrial depolarization (Fig. 2C; right panel). These results suggest that regulation of H2O2-induced apoptosis by p38 MAPK may be upstream of mitochondria.
p38 siRNA attenuates H2O2-induced apoptosis
To further confirm the role of p38 MAPK, RIE-1 cells were transfected with nontargeting (control) siRNA or siRNA targeting p38α siRNA. Transfection with the p38 MAPK siRNA attenuated H2O2-induced apoptosis (Fig. 3A). p38 MAPK protein knockdown with p38α siRNA treatment was confirmed by Western blot (Fig. 3B). In addition, knockdown of p38 MAPK attenuated H2O2-induced cleavage of caspase-3 and PARP (Fig. 3C). Taken together, these results, using complementary approaches (i.e., chemical inhibition of p38 and transfection with p38 MAPK siRNA), demonstrate a contributory role for p38 MAPK in H2O2-induced apoptosis in RIE-1 cells.
Fig. 3. Effects of p38 MAPK inhibition on H2O2-induced RIE-1 cell death.
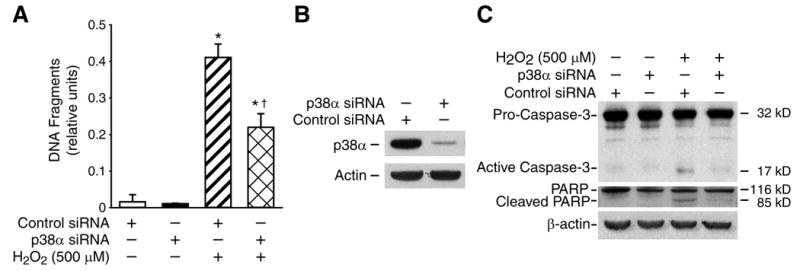
RIE-1 cells were transiently transfected with duplex siRNA targeted against p38α or with control siRNA. Cells were treated with H2O2 (500 uM) for 3 h. (A) Apoptosis measured by an ELISA assay (Data represent triplicate determinations; mean ± SEM; * =p< 0.05 vs. control siRNA; †=p< 0.05 vs. control siRNA + H2O2). (B, C) Expression of p38 and cleavage of caspase-3, PARP were determined by Western blotting. Representative data from three separate experiments are shown.
p38 activation is upstream of PKC activation during H2O2-induced apoptosis
Recently, we showed that PKC plays an important pro-apoptotic role during H2O2-induced RIE-1 cell death and that the inhibition of PKC significantly inhibited H2O2-induced RIE-1 cell death [11]. Intracellular signaling mechanisms regulating cell death are complex and may involve p38 MAPK and PKC [16]. To clarify the causative relationship between p38 MAPK and PKC activation during H2O2-induced apoptosis, we first determined the activation of PKC by H2O2 treatment by demonstrating increased phosphorylation of total PKC (phospho-pan-PKC) (Fig. 4A). Next, we analyzed the regulation of PKC phosphorylation by p38 MAPK. Interestingly, pre-treatment with SB203580 blocked H2O2-induced PKC phosphorylation (Fig. 4B). These results suggested that significant attenuation of H2O2-mediated apoptosis with p38 inhibition may act through the inhibition of PKC activation. PKC-dependent p38 activation occurs in H2O2-induced cell death in some cells [17]; therefore, we determined whether p38 activation by H2O2 requires PKC activation. RIE-1 cells were pretreated with the PKC inhibitor GF109203x (5 μM) for 30 min prior to treatment with H2O2 for an additional 30 min. As shown in Fig. 4C, H2O2 induced p38 phosphorylation; treatment with GF109203x did not affect the induction of p38 phosphorylation by H2O2. These results suggest that PKC is a downstream target of p38 in H2O2-induced cell death. Taken together, our results suggest that H2O2-induced intestinal cell apoptosis requires p38-dependent PKC activation.
Fig. 4. p38 MAPK activation is upstream of PKC activation in H2O2-induced RIE-1 cell death.
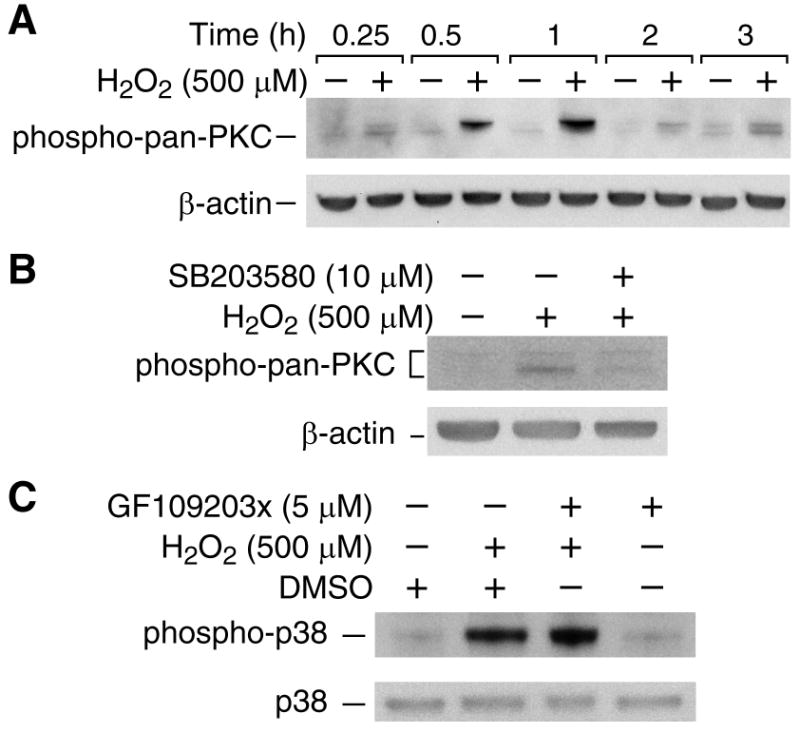
(A) RIE-1 cells were treated with H2O2 to determine expression of phospho-PKC (pan) and β-actin by immunoblotting. (B) RIE-1 cells were pretreated with the p38 inhibitor SB203580 (10 μM) for 30 min prior to H2O2 (500 μM) and phospho-PKC (pan) and β-actin were determined as above. (C) RIE-1 cells were pretreated with the PKC inhibitor GF109203x (5 μM) for 30 min prior to treatment with H2O2 (500 μM) for 3 h. Western blotting performed using anti-phospho-p38 and anti-β-actin. Representative blots of at least three repeats at all data points are shown.
Discussion
Transient mesenteric ischemia-reperfusion injury resulting in the production of ROS has been implicated as a contributing factor for NEC [1-3]. Extensive apoptosis also occurs in enterocytes in the apical villi of infants with NEC [7]. In our current study, we show, for the first time, the differential localization of active p38 MAPK between ‘normal’ neonatal intestinal sections, in which the localization of p38 MAPK activity was found mostly in the villi, and the intestinal sections adjacent to NEC, where active p38 MAPK was noted in both crypt and villi. Furthermore, we found that H2O2 treatment resulted in rapid induction of p38 MAPK phosphorylation in rat intestinal cells; inhibition of p38 by the selective inhibitor SB203580 or transfection with p38 siRNA attenuated H2O2-induced intestinal cell death. Moreover, the attenuation of H2O2-induced PKC activation by p38 inhibition suggested that a p38 MAPK/PKC pathway may play an important role in NEC.
We showed that H2O2 increases p38 MAPK phosphorylation in intestinal epithelial cells. The role of MAPK cascades as an apoptotic signal transduction pathway has recently attracted considerable attention. Oxidative stress can trigger the activation of multiple signaling pathways that influence the cytotoxicity observed in affected cells, including the activation of the three major MAPK cascades (i.e., ERK1/2, JNK, p38 MAPK). ERK1/2 is activated mainly by growth factors and is critically involved in the regulation of mitogenesis [18], while JNK and p38 MAPKs are activated mainly by cytotoxic insult and are often associated with apoptosis [19]. Recently, we reported that H2O2 treatment results in increased JNK and ERK1/2 phosphorylation in intestinal epithelial cells; however, inhibition of either ERK or JNK did not completely block H2O2-induced cell death [11]. In our current study, we show that H2O2 activates p38 MAPK. Conversely, the inhibition of this process protects intestinal epithelial cells from oxidative stress-induced apoptosis. These results are highly suggestive of a role for p38 MAPK in the apoptotic pathway. In addition, the blockage of H2O2 -induced caspase-3 cleavage and PARP cleavage by p38 inhibition suggested that p38 MAPK activation may act as an upstream molecule for caspase-3 activation. In agreement with our findings, p38 MAPK-dependent caspase-3 activation has been found in the zinc-induced apoptosis in human leukemia HL-60 cells [20].
Although we have shown that inhibition of PKC, which resulted in the activation of Akt, attenuated H2O2-induced RIE-1 cell death [11]; the signaling pathways related to PKC activation in this process are unknown. PKC was found to activate p38 MAPK in the regulation of cell death (eg, PKC promotes apoptosis in LNCaP prostate cancer cells through activation of p38 MAPK) [21]. p38 MAPK mediated nitric oxide-induced apoptosis of chondrocytes through the inhibition of PKC-ζ [22]. However, p38 MAPK activation was found to be upstream of PKC activation during apoptosis induced by 8-chloro-cyclic AMP [23]. Our results showed that the p38 inhibitor, SB203580, blocked H2O2-induced PKC activation, while the PKC inhibitor, GF109203x, could not attenuate p38 activation, indicating that p38 MAPK activation is upstream of PKC activation in H2O2-induced intestinal cell apoptosis.
Each p38 MAPK isoform (α, β, γ, δ) phosphorylates a diverse array of intracellular proteins including stress-responsive transcription factors [24]. Although transfection of p38α siRNA almost completely blocked p38α expression, the inhibition of H2O2-induced cell death was less effective when compared with SB203580 treatment. These results suggest that other isoforms of p38 MAPK may also participate in the H2O2-induced intestinal cell death. Indeed, p38β and p38δ are required specifically for anoikis (detachment-associated cell death) in human intestinal epithelial cells [25]. Further studies are needed to delineate the expression and involvement of these p38 isoforms in oxidative stress-induced intestinal cell death. Based on our observations, we propose the signaling cascade in H2O2-induced intestinal cell death is as follows: H2O2 activates p38 MAPK and ultimately leads to PKC activation. PKC, in turn, inactivates Akt. In this manner, p38/PKC/Akt pathway participates in the regulation of cell death. Activation of p38 MAPK/PKC results in mitochondrion-dependent apoptosis. Ultimately, breakdown of mitochondrial integrity leads to the activation of caspases, PARP cleavage, and cell death. p38 MAPK may act to alter mitochondrial function via a direct pathway. This is at least possible, since p38α causes apoptosis in a lymphoblastoid B-cell line by translocating directly to the mitochondria [26].
In conclusion, our study demonstrates that p38 MAPK is involved in NEC. A better understanding of signaling pathways during oxidative stress-induced intestinal cell injury may allow us to elucidate molecular mechanisms contributing to NEC and potentially provide a novel treatment for this devastating disease which predominantly occurs in premature infants.
Acknowledgments
The authors thank Karen Martin for manuscript preparation and Tatsuo Uchida for assistance with statistical analysis. This work was supported by grants RO1 DK61470, RO1 DK48498 and PO1 DK35608 from the National Institutes of Health and 8580 from the Shriners Burns Hospital.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Clark DA, Fornabaio DM, McNeill H, Mullane KM, Caravella SJ, Miller MJ. Contribution of oxygen-derived free radicals to experimental necrotizing enterocolitis. Am J Pathol. 1988;130:537–542. [PMC free article] [PubMed] [Google Scholar]
- 2.Kelly N, Friend K, Boyle P, Zhang XR, Wong C, Hackam DJ, Zamora R, Ford HR, Upperman JS. The role of the glutathione antioxidant system in gut barrier failure in a rodent model of experimental necrotizing enterocolitis. Surgery. 2004;136:557–566. doi: 10.1016/j.surg.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 3.Okur H, Kucukaydin M, Kose K, Kontas O, Dogam P, Kazez A. Hypoxia-induced necrotizing enterocolitis in the immature rat: the role of lipid peroxidation and management by vitamin E. J Pediatr Surg. 1995;30:1416–1419. doi: 10.1016/0022-3468(95)90395-x. [DOI] [PubMed] [Google Scholar]
- 4.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 5.Sedghi S, Fields JZ, Klamut M, Urban G, Durkin M, Winship D, Fretland D, Olyaee M, Keshavarzian A. Increased production of luminol enhanced chemiluminescence by the inflamed colonic mucosa in patients with ulcerative colitis. Gut. 1993;34:1191–1197. doi: 10.1136/gut.34.9.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 7.Hackam DJ, Upperman JS, Grishin A, Ford HR. Disordered enterocyte signaling and intestinal barrier dysfunction in the pathogenesis of necrotizing enterocolitis. Semin Pediatr Surg. 2005;14:49–57. doi: 10.1053/j.sempedsurg.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 8.Nanthakumar NN, Young C, Ko JS, Meng D, Chen J, Buie T, Walker WA. Glucocorticoid responsiveness in developing human intestine: possible role in prevention of necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G85–92. doi: 10.1152/ajpgi.00169.2004. [DOI] [PubMed] [Google Scholar]
- 9.Chung DH, Ethridge RT, Kim S, Owens-Stovall S, Hernandez A, Kelly DR, Evers BM. Molecular mechanisms contributing to necrotizing enterocolitis. Ann Surg. 2001;233:835–842. doi: 10.1097/00000658-200106000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zegura B, Lah TT, Filipic M. The role of reactive oxygen species in microcystin-LR-induced DNA damage. Toxicology. 2004;200:59–68. doi: 10.1016/j.tox.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 11.Zhou Y, Wang Q, Evers BE, Chung DH. Oxidative Stress-Induced Intestinal Epithelial Cell Apoptosis is Mediated by P38 Mitogen-Activated Protein Kinase Activation. Pediatric Research. 2005 in press. [Google Scholar]
- 12.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 13.Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, Steele GD, Jr, Chen LB. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci U S A. 1991;88:3671–3675. doi: 10.1073/pnas.88.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Houde M, Laprise P, Jean D, Blais M, Asselin C, Rivard N. Intestinal epithelial cell differentiation involves activation of p38 mitogen-activated protein kinase that regulates the homeobox transcription factor CDX2. J Biol Chem. 2001;276:21885–21894. doi: 10.1074/jbc.M100236200. [DOI] [PubMed] [Google Scholar]
- 15.Li JM, Zhou H, Cai Q, Xiao GX. Role of mitochondrial dysfunction in hydrogen peroxide-induced apoptosis of intestinal epithelial cells. World J Gastroenterol. 2003;9:562–567. doi: 10.3748/wjg.v9.i3.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vrana JA, Grant S. Synergistic induction of apoptosis in human leukemia cells (U937) exposed to bryostatin 1 and the proteasome inhibitor lactacystin involves dysregulation of the PKC/MAPK cascade. Blood. 2001;97:2105–2114. doi: 10.1182/blood.v97.7.2105. [DOI] [PubMed] [Google Scholar]
- 17.Ryer EJ, Sakakibara K, Wang C, Sarkar D, Fisher PB, Faries PL, Kent KC, Liu B. Protein kinase C delta induces apoptosis of vascular smooth muscle cells through induction of the tumor suppressor p53 by both p38-dependent and p38-independent mechanisms. J Biol Chem. 2005;280:35310–35317. doi: 10.1074/jbc.M507187200. [DOI] [PubMed] [Google Scholar]
- 18.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 19.Callsen D, Brune B. Role of mitogen-activated protein kinases in S-nitrosoglutathione-induced macrophage apoptosis. Biochemistry. 1999;38:2279–2286. doi: 10.1021/bi982292a. [DOI] [PubMed] [Google Scholar]
- 20.Requirement of caspase and p38MAPK activation in zinc-induced apoptosis in human leukemia HL-60 cells. Eur J Biochem. 2002;269:6204–6211. doi: 10.1046/j.1432-1033.2002.03339.x. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka Y, Gavrielides MV, Mitsuuchi Y, Fujii T, Kazanietz MG. Protein kinase C promotes apoptosis in LNCaP prostate cancer cells through activation of p38 MAPK and inhibition of the Akt survival pathway. J Biol Chem. 2003;278:33753–33762. doi: 10.1074/jbc.M303313200. [DOI] [PubMed] [Google Scholar]
- 22.Kim JS, Park ZY, Yoo YJ, Yu SS, Chun JS. p38 kinase mediates nitric oxide-induced apoptosis of chondrocytes through the inhibition of protein kinase C zeta by blocking autophosphorylation. Cell Death Differ. 2005;12:201–212. doi: 10.1038/sj.cdd.4401511. [DOI] [PubMed] [Google Scholar]
- 23.Ahn YH, Jung JM, Hong SH. 8-Chloro-cyclic AMP-induced growth inhibition and apoptosis is mediated by p38 mitogen-activated protein kinase activation in HL60 cells. Cancer Res. 2005;65:4896–4901. doi: 10.1158/0008-5472.CAN-04-3122. [DOI] [PubMed] [Google Scholar]
- 24.Paul A, Wilson S, Belham CM, Robinson CJ, Scott PH, Gould GW, Plevin R. Stress-activated protein kinases: activation, regulation and function. Cell Signal. 1997;9:403–410. doi: 10.1016/s0898-6568(97)00042-9. [DOI] [PubMed] [Google Scholar]
- 25.Vachon PH, Harnois C, Grenier A, Dufour G, Bouchard V, Han J, Landry J, Beaulieu JF, Vezina A, Dydensborg AB, Gauthier R, Cote A, Drolet JF, Lareau F. Differentiation state-selective roles of p38 isoforms in human intestinal epithelial cell anoikis. Gastroenterology. 2002;123:1980–1991. doi: 10.1053/gast.2002.37072. [DOI] [PubMed] [Google Scholar]
- 26.Rosini P, De Chiara G, Lucibello M, Garaci E, Cozzolino F, Torcia M. NGF withdrawal induces apoptosis in CESS B cell line through p38 MAPK activation and Bcl-2 phosphorylation. Biochem Biophys Res Commun. 2000;278:753–759. doi: 10.1006/bbrc.2000.3871. [DOI] [PubMed] [Google Scholar]