

Abstract
The naturally occurring nitroalkenes, nitrolinoleic (NO2-LA) and nitrooleic (NO2-OA) acids, are among the most potent endogenous ligand activators of PPARγ-dependent transcription. In order to understand mechanisms that regulate cellular response to these nitroalkenes, we previously demonstrated that glutathione conjugation of NO2-LA and MRP1-mediated efflux of the conjugates were associated with significant attenuation of PPARγ activation by this nitroalkene (Biochemistry 45: 7889-7896, 2006). Here we show that NO2-OA activation of PPARγ is similarly affected by non-enzymatic conjugation and MRP1-mediated efflux. Moreover, the roles of glutathione S-transferases (GSTs) in the glutathione conjugation and bioactivities of NO2-LA and NO2-OA were investigated. While none of the GST isozymes tested (GSTA1-1, A4-4, M1a-1a, and P1a-1a) enhanced the rate of glutathione conjugation, expression of GSTA1-1, M1a-1a or P1a-1a in MCF7 cells significantly reduced the magnitude of PPARγ-dependent reporter gene transcription in response to NO2-LA and NO2-OA treatment—with GSTP1a-1a expression mediating the most potent inhibition of PPARγ. Although these GSTs failed to catalyze nitroalkene conjugation with glutathione, the nitroalkenes were found to associate avidly with all four GST isozymes as indicated by their ability to inhibit GST activity with Ki's in the nanomolar range. Treatment of purified GSTP1a-1a with excess NO2-LA and NO2-OA resulted in the formation of covalent adducts between GSTP1a monomers and nitroalkenes; although, separate experiments indicated that such covalent bond formation was not necessary for avid GST-nitroalkene interactions. These results suggest that GSTs can inhibit the activation of transcription by nitroalkenes via non-catalytic sequestration of these ligands, and their glutathione conjugates, away from their nuclear target, PPARγ.
The naturally occurring nitroalkenes, including nitrolinoleic (NO2-LA) and nitrooleic (NO2-OA)1 acids, are implicated in the modulation of multiple signaling pathways resulting in several important bioactivities including vasodilation, inhibition of inflammation, and inhibition of platelet activation and function (1-4). In addition, these nitrated unsaturated fatty acids are among the most potent endogenous ligand activators of PPARγ—a nuclear receptor involved in the transcription of genes associated with carbohydrate and lipid homeostasis, cellular differentiation, and inflammation (5-8). The physiological importance of these nitroalkenes is indicated by their relative abundance in vivo with aggregate levels reported to be around 1-2 μM in normal human plasma and erythrocytes (9)—levels in excess of those necessary to significantly activate PPARγ-dependent transcription in vitro (10, 11).
In order to understand how nitroalkene activities are regulated at the cellular level, we previously demonstrated that NO2-LA forms conjugates with glutathione, rapidly and non-enzymatically, under physiological conditions (12). These conjugates are efficiently effluxed by MRP1, a ubiquitous ATP-dependent glutathione conjugate transporter, and combined conjugation plus MRP1-mediated efflux profoundly attenuates PPARγ-dependent transcription activation in response to NO2-LA (12). Here we demonstrate that activation of PPARγ by NO2-OA is similarly affected by non-enzymatic conjugation and efflux. Moreover, we investigated whether expression of representative isozymes of the soluble forms of glutathione S-transferase (GST) would influence NO2-LA and NO2-OA bioactivities either by catalyzing their conjugation with glutathione or by an alternative mechanism. Indeed, we show that while expression of GSTA1-1, GSTM1a-1a, or GSTP1a-1a significantly attenuates the activation of PPARγ-dependent transcription by NO2-LA and NO2-OA, none of these isozymes, or GSTA4-4, is able to catalyze glutathione conjugation of the nitroalkenes. Rather, these GSTs appear to influence transcription via avid covalent and non-covalent interactions with the nitroalkenes and their glutathione conjugates thereby sequestering them from their nuclear target, PPARγ.
Experimental Procedures
Materials
NO2-LA was synthesized as a mixture of four regioisomers from linoleic acid as described previously (12, 13). The 9-NO2 and 10-NO2 regioisomers of OA-NO2 ((E)-9-nitrooctadec-9-enoic acid and (E)-10-nitrooctadec-9-enoic acid, respectively) were synthesized according to Gorczynski and King (14) and combined as a 1:1 mixture for the studies described herein. All solvents were of HPLC grade or better from Fisher Scientific (Pittsburgh, PA). Glutathione, glutathione-agarose, isopropyl-β-D-thiogalactopyranoside, ATP, 5'-(β,γ-methylene)diphosphonate (AMP-PCP), and 1-chloro-2,4-dinitrobenzene were obtained from Sigma-Aldrich (St. Louis, MO). The human PPARγ expression plasmid, pcDNA3-PPARγ, was kindly provided by Dr. V. K. K. Chatterjee (15). Superfect reagent was obtained from Qiagen (Valencia, CA) and the Dual Luciferase assay kit and sequence grade trypsin were from Promega (Madison, WI). Trifluoroacetic acid was obtained from ThermoScientific (Rockford, IL), [glycine-2-3H]-glutathone from PerkinElmer (Waltham, MA), and rosiglitazone from Cayman Chemicals (Ann Arbor, MI).
Cell Lines and Culture
All cell lines were derived from parental MCF7 cells (MCF7/WT). Transgenic cell lines expressing MRP1 (MCF7/MRP1-10) or the soluble GST isozymes GSTA1-1 (MCF7/α-8), GSTM1a-1a (MCF7/μ-5) and GSTP1a-1a (MCF7/π-11) have been described previously (16, 17). Cells were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10 % fetal bovine serum at 37°C, 5 % CO2. Transgenic cells were maintained in selecting drugs, either 0.5 mg/ml G418 (MCF7/MRP1-10) or 0.2 mg/ml hygromycin (MCF7/α-8, /μ-5, and/π-11), until just prior to experiments.
Recombinant GSTs
A bacterial expression vector encoding human GSTA4 was constructed by PCR amplification from a human liver cDNA library. The 5' oligonucleotide used for amplification, 5'- CAAGGGATCCACTTTAAGAAGGAGATATACCATGGCGGCGAGGCCAAAGC TTCACTATCCGAATGGAAGGGGTCGCATGGAGTCCGTGAGA-3', incorporated a bacterial ribosomal binding site and, in order to optimize codon usage for expression in Escherichia coli, ten silent mutations as described by Hubatsch et al. (18). The 3' oligonucleotide was 5'-CAAGCTCGAGGTTTTATGGCCTAAAGAT-3'. The 705 bp GSTA4 fragment amplified was inserted into the BamHI and XhoI multiple cloning sites of pET21(+) (Novagen, Gibbstown, NJ). This vector and the vectors encoding human GSTA1, GSTM1a, and GSTP1a described previously (16, 19) were used to transform competent BL21 star bacteria (Invitrogen, Carlsbad, CA). Recombinant GSTs were prepared as described (19). Purified GSTs were free of contaminating protein (Coomassie staining of SDS-polyacrylamide electrophoresis gels) and their specific activities determined using 1-chloro-2,4-dinitrobenzene as substrate (20) were: GSTA4-4, 5.3 μmol/(min•mg); GSTA1-1, 80 μmol/(min•mg); GSTM1a-1a, 166 μmol/(min•mg); and GSTP1a-1a, 134 μmol/(min•mg).
Nitroalkene Conjugation with Glutathione: Synthesis, Characterization and Kinetics
Formation of glutathione conjugates of NO2-OA, NO2-OA-SG, was accomplished as described for NO2-LA with some modifications (12). Briefly, NO2-OA (40 μM) was reacted with 2 mM glutathione in 0.1 M sodium phosphate buffer (pH 7.5) for 5 min at 25°C. Reactants and products were separated by HPLC using a C18 column (Beckman, Ultrasphere ODS, 5 μm, 4.6 mm × 250 mm). The mobile phase consisted of solvent A (10 % acetonitrile, 0.05 % trifluoroacetic acid) and solvent B (100 % acetonitrile, 0.05 % trifluoroacetic acid). Chromatography was at 1 ml/min with 45 % solvent A for 10 min followed by a linear gradient from 45 % solvent A to 90 % solvent B over 1 min and then held at 90 % solvent B. The incubation of NO2-OA with glutathione resulted in quantitative conversion of NO2-OA (eluting at ~20 min) to a peak eluting at 4 min (chromatography monitored at 274 nm). The peak eluting at 4 min was identified as NO2-OA-SG by electrospray ionization mass spectrometry (ESI/MS) (see below).
Kinetic analysis of non-enzymatic NO2-OA-SG formation was accomplished spectrophotometrically as previously described for NO2-LA-SG formation (12). Progression of reactions containing 50 μM NO2-OA, 2 mM glutathione, and 0.1 M sodium phosphate (pH 7.5) at 25°C was monitored as increasing absorbance at 245 nm. Data was fitted to the first-order rate equation to obtain a pseudo-first-order rate constant, k1, from which the second-order rate constant, k2, was calculated (12).
Kinetic analysis of nitroalkene glutathione conjugate formation in the presence of GST was done as follows. Reactions contained 1 mM glutathione, 0.1 M sodium phosphate (pH 6.5), GST (recombinant purified GSTA1-1, 0.5 μg/ml; GSTA4-4, 2.6 μg/ml; GSTM1a-1a, 0.2 μg/ml; or GSTP1a-1a, 0.2 μg/ml) or vehicle control and were initiated at 25°C by the addition of NO2-LA (40 μM) or NO2-OA (80 μM). Reaction progress was monitored spectrophotometrically at 245 nm as described above.
Radiolabeled NO2-OA-SG was purified by HPLC (above) from reactions (1 min, 25°C) in 10 mM sodium phosphate (pH 8.5) containing 200 μM NO2-OA and 50 μM total glutathione (including [glycine-2-3H]-glutathione, final specific activity = 4 μCi/nmol).
ESI/MS Analysis of NO2-OA-SG
The HPLC purified NO2-OA-SG fraction eluting at 4 min (above) was dried under N2, dissolved in methanol, and injected directly into a Micromass Quattro II mass spectrometer equipped with a z spray source and triple quadrapole analyzer. The instrument was operated in the positive ion mode with capillary voltage = 3.3 kV, sampling cone = 50 V, and the desolvation temperature = 200°C. For daughter ion analysis, the collision energy was 20 eV.
For the analysis of NO2-OA-SG formed in cells, acidified methanol extracts derived from NO2-OA treated cells (see below) were analyzed by HPLC/ESI/MS/MS as follows. Acidified methanol soluble extract components were separated by reverse phase HPLC using a Phenomenex (Torrance, CA) C18-BD 3 μ 150 mm × 2 mm column. The mobile phase consisted of solvent A, 99.9 % acetonitrile and 0.1 % formic acid, and solvent B, 9.9 % acetonitrile and 0.1 % formic acid. Separation was begun at 200 μl/min with a linear gradient from 90 % solvent A to 20 % solvent A over 5 min and then held at 20 % solvent A. Approximately 20 % of the HPLC eluate was diverted to the electrospray source for multiple reaction monitoring with the instrument settings noted above. The precursor ion m/z 635 (NO2-OA-SG) was selected and scanned for the daughter ion m/z 506 as discussed in the text.
MRP1-Mediated Transport of the Glutathione Conjugate of NO2-OA (NO2OA-SG)
ATP-dependent uptake of 3H-NO2-OA-SG into inside-out membrane vesicles isolated from MCF7/WT (MRP1-poor) and MCF7/MRP1-10 (transduced, MRP1 expressing) cells was accomplished as described previously (19, 21). Results reported as ATP-dependent uptake were calculated by subtracting transport in the presence of non-hydrolyzable ATP analog, AMP-PCP, from transport observed in the presence of ATP.
To examine NO2-OA-SG formation and MRP1-mediated efflux in cells, MCF7/WT and MCF7/MRP1-10 cells were seeded at densities of 2 × 106 cells per 100 mm tissue culture dish. The following day, cells were treated with 2 μM OA-NO2 for 30 min in Hank's buffered saline solution. At the end of these incubations, cells were washed with ice cold Hank's solution and lysed in a solution of methanol and 0.1 % acetic acid. Insoluble debris was pelleted by centrifugation and the soluble cellular extracts were dried under N2. Dried extracts were dissolved in 90 % acetonitrile and NO2-OA-SG was detected by HPLC/ESI/MS/MS as described above.
PPARγ Transcription Activation
Analysis of activation of PPARγ-dependent transcription by nitroalkenes and rosiglitazone was accomplished using the following reporter gene assay. Parental (MCF7WT) or transduced (MCF7/MRP1-10, MCF7/α-8, MCF7/μ-5, or MCF7/π-11) MCF7 cells were plated at a density of 1.0-1.5 × 105 cells per well in six-well dishes. Twenty-four hours later cells were co-transfected with 1 μg of the PPRE-containing firefly luciferase reporter gene, PPRE-3x-TK-Luc (22), 50 pg control CMV-Renilla luciferase reporter gene, pGL4.75 (Promega, Madison, WI), and either 0.2 μg of the PPARγ expression vector, pcDNA3-PPARγ (15), or pUC8 control vector. Transfections were accomplished using Superfect reagent (Qiagen, Valencia, CA) according to the manufacturer's recommendations. The PPRE-3x-TK-Luc plasmid contains three PPAR responsive elements (PPRE) upstream of an inducible thymidine kinase promotor controlling transcription of the firefly luciferase gene. The internal control pGL4.75 plasmid contains the constitutively active cytomegalovirus promoter driving transcription of the Renilla luciferase gene. Twenty-four hr after the start of transfections, medium was replaced with 3 ml Optimem (Invitrogen, Carlsbad, CA) containing vehicle (control) or varying concentrations of inducing agent (NO2-LA or NO2-OA) for 1 hr at which time medium was replaced with DMEM supplemented with 10% fetal bovine serum minus inducing agent. In some experiments, cells were treated with 0.5 μM rosiglitazone for 3 hours in DMEM supplemented with 10% fetal bovine serum after which the medium was replaced with medium minus drug. Cells were harvested 24 hours after the start of exposure to vehicle or inducing agent. Luciferase assays were accomplished using the Dual Luciferase Assay System (Promega, Madison, WI) and values were corrected for variations in transfection efficiencies and non-specific induction as described previously (23).
Nitroalkene-GST Interactions: Inhibition of GST Activity by Nitroalkenes
Inhibition of GST catalysis by NO2-LA, NO2-OA, and their glutathione conjugates was assessed using 1-chloro-2,4-dinitrobenzene as the variable substrate as described (19). For analysis of inhibition by NO2-LA and NO2-OA, mixtures containing varying concentrations of 1-chloro-2,4-dinitrobenzene (0-2 mM) and nitroalkenes or vehicle control in 0.1 M sodium phosphate (pH 6.5) were prepared and incubated at 25°C. Reactions were initiated by the addition of mixtures containing glutathione (final concentration = 1 mM) and GSTA1-1, A4-4, M1a-1a, P1a-1a, or vehicle control. Inhibition of GST by the glutathione conjugates, NO2-OA-SG and NO2-LA-SG, was assessed similarly except that the nitroalkenes were preincubated with 1 mM glutathione for 5 min at room temperature prior to the addition of GST. This strategy enabled quantitative conversion of the nitroalkenes to their glutathione conjugates prior to initiation of the reaction. Due to solubility and absorbance limitations on achievable substrate (1-chloro-2,4- dinitrobenzene) concentrations and high Km values for some GST isozymes, especially GSTA4-4, kinetic data were fitted to the equation, , which allowed accurate estimation of Kcat/Km even when these parameters could not be accurately determined separately (discussed by Kolm et al. (24)). Inhibitory constants, Ki, were determined from secondary plots of Kmapp/kcatapp versus inibitor (nitroalkene) concentrations (25).
Nitroalkene-GST Interactions: Formation and Analysis of Nitroalkene Adducts of GSTP1a-1a
Recombinant GSTP1a-1a (200 pmol) was incubated with a 20-fold molar excess of NO2-OA or NO2-LA, or vehicle control, in 10 mM Tris (pH 7.5) for one hour at 25°C. Reactions were precipitated for several hours at -20°C with four volumes acetone plus formic acid added to a final concentration of 1 %. Protein was pelleted by microfuge centrifugation, rinsed with 90 % acetone/1 % formic acid, decanted, and allowed to air dry.
For MALDI-TOF MS, acetone precipitated samples were resuspended in 0.1% trifluoroacetic acid to a final concentration of 2 μg/mL. Samples were mixed with an equal volume of saturated sinapinic acid matrix in acetonitrile:water (1:2) and 0.1% trifluoroacetic acid and were spotted on the target and air-dried. External calibrations were performed using a modified protein calibration standard I (Bruker Daltonics) and were analyzed in linear mode. Positive ion mode was used with 20 Hz laser frequency and 400 ns pulsed ion extraction.
For ESI/Q-TOF mass spectrometry analysis of GSTP1a-nitroalkene adducts, acetone precipitates (200 pmol protein treated as described above) were dissolved in methanol:water:formic acid (50:50:0.1) for direct injection into the Micromass Q-TOF API-US mass spectrometer equipped with an Advion TriVersa NanoMate sprayer. Data was obtained in the positive ion mode with the cone set at 55 V, the electrospray source at 1.7 kV, and the source temperature at 95°C. Protein masses were determined from raw ion profiles using maximum entropy calculations (Masslynx software, Waters, Milford, MA).
Results
Non-enzymatic formation of NO2-OA-SG
The ability of NO2-OA to form conjugates with glutathione at physiological pH and glutathione concentrations was examined. Incubation of 50 μM NO2-OA with 2 mM glutathione at pH 7.5, 25°C, resulted in rapid and quantitative conversion of the nitroalkene to NO2-OA-SG as indicated by absorbance change at 245 nm (Figure 1) and ESI/MS analysis of HPLC fractionated reaction products (Figure 2). Using the kinetic data shown in Figure 1, a pseudo-first order rate constant of 0.16 s-1 was determined from which the second-order rate constant of 80 s-1M-1 was calculated. Similar second-order constants were calculated from experiments using 1 mM and 5 mM glutathione (not shown). This second-order rate constant is comparable to the value of 190 s-1M-1 we previously determined for NO2-LA-SG formation (12) and the values later reported by Baker et al. for NO2-LA-SG and NO2-OA-SG formation under slightly different conditions (26). ESI/MS identified the parent ion of NO2-OA-SG [M+H] with the expected m/z of 635 (Figure 2A). Analysis of daughter ions from collision-induced dissociation (Figure 2B) revealed several predicted fragments including a prominent ion at m/z = 506, corresponding to M-glutamate, which was used in subsequent ESI/MS/MS analysis (below).
Figure 1. Non-enzymatic kinetics of NO2-OA-SG formation.

NO2-OA (50 μM) was reacted with 2 mM glutathione at 25°C, pH 7.5. Reaction progress was monitored by change in absorbance at 245 nm and analyzed as described in Experimental Procedures.
Figure 2. Mass spectrometry analysis of the glutathione conjugate of NO2-OA, NO2-OA-SG.

Shown are spectra of the parent ion M+H at m/z = 635 (panel A) and daughter ions generated by collision-induced dissociation (panel B). Evident in panel B are peaks corresponding to the parent ion and the daughter ions: m/z 506 [M-glutamate], m/z 560 [M-glycine], and m/z 459 [M-glutamate-NO2].
MRP1-mediated transport of NO2-OA-SG
The ability of the efflux transporter, MRP1, to support ATP-dependent transport of NO2-OA-SG was examined using inside-out membrane vesicles derived from MRP1-poor MCF7/WT and MRP1-rich MCF7/MRP1-10 cells. Data shown in Figure 3A demonstrate that MRP1-containing vesicles, but not MRP1-poor vesicles, mediate efficient transport of NO2-OA-SG as we have previously shown for NO2-LA-SG (12). To determine the effect of MRP1 expression on the levels of NO2-OA-SG accumulated in intact cells, MCF7/WT and MCF7/MRP1-10 cells were treated for 30 min with 2 μM NO2-OA and the relative levels of intracellular NO2-OA-SG retained in the two cell lines at the end of these incubations were assessed by ESI/MS/MS analysis of cell lysates. As shown in Figure 3B, NO2-OA-SG was readily detected in MRP1-poor MCF7/WT cells (upper panel) while none was observed in MRP1-rich MCF7/MRP1-10 cells (lower panel). These data indicate that MRP1 interferes with intracellular NO2-OA-SG accumulation by active efflux of NO2-OA-SG. In addition, we cannot rule out the possibility that MRP1 may also transport the parent lipid, NO2-OA.
Figure 3. MRP1-mediated transport of NO2-OA-SG.

A. Shown are time courses of ATP-dependent transport of radiolabeled NO2-OA-SG into inside-out plasma membrane vesicles (pmol transported/mg membrane protein) derived from MRP1-expressing (+ MRP1, MCF7/MRP1-10) and MRP1-poor (- MRP1, MCF7/WT) cells. B. MRP1-poor (WT, MCF7/WT, upper panel) or MRP1-expressing (MRP1-10, MCF7/MRP1-10, lower panel) cells were treated with 2 μM NO2-SG for 30 min. Intracellular NO2-OA-SG accumulated at the end of these 30 min incubations were determined by ESI/MS/MS as described in Experimental Procedures. The NO2-OA-SG precursor ion, m/z 635, was scanned for the daughter ion, m/z 506, corresponding to M-glutamate.
MRP1 expression attenuates activation of PPARγ-dependent transcription in response to NO2-OA
To evaluate the biological consequences of MRP1-mediated efflux of NO2-OA-SG, we examined the effect of MRP1 expression on the transcription activation of a PPARγ-dependent reporter gene (Figure 4). Treatment of MCF7/WT cells (MRP-) with 2 μM NO2-OA resulted in significant, 3-fold, induction of reporter gene activity mediated by endogenous PPARγ (PPARγ-). The level of induction was augmented to ~10-fold when MCF7/WT cells were co-transfected with a PPARγ expression vector (PPARγ+). However, similar treatment of MCF7/MRP1-10 cells (MRP1+) resulted in minimal induction of reporter gene whether or not additional PPARγ was expressed by co-transfection.
Figure 4. Expression of MRP1 attenuates NO2-OA induced activation of PPARγ-dependent reporter gene transcription.
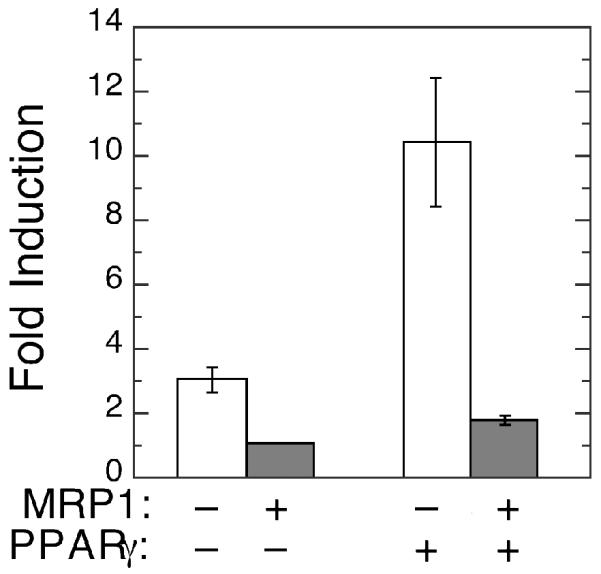
Shown are inductions of PPARγ-dependent reporter gene (PPRE3x-TK-Luc) expression in MCF7 cells by treatment with 2 μM NO2-OA. Reporter gene firefly luciferase activity was corrected for variations in transfection efficiencies and for non-specific induction using the co-transfected CMV-Renilla luciferase reporter gene (pGL4.75) as described in Experimental Procedures. Fold induction is defined as corrected firefly luciferase activities following NO2-OA treatment divided by corrected luciferase activities of control cells treated with vehicle alone. The bars on the left represent fold inductions with cells expressing endogenous PPARγ only (PPARγ-) and bars on the right represent inductions with cells expressing the co-transfected PPARγ vector, pcDNA3-PPARγ (PPARγ+). Open bars denote transfections of MRP1-poor (MRP1-) MCF7/WT cells and the shaded bars transfections of MRP1-expressing (MRP1+) MCF7/MRP1-10 cells. Fold inductions are expressed as the mean values calculated from triplicate independent transfections ± 1 sd.
In summary, these data indicate that, as shown previously for NO2-LA (12), glutathione conjugation and MRP1-mediated efflux can profoundly attenuate the ability of NO2-OA to activate PPARγ-dependent transcription. In the following studies, we examined whether GSTs—either by enhancing the rate of glutathione conjugation or by other mechanisms—can also influence PPARγ activation by nitroalkenes.
GST-Nitroalkene Interactions: Catalysis?
To determine whether the various soluble isozymes of GST could catalyze glutathione conjugation with nitroalkenes, NO2-OA and NO2-SG reaction mixtures were supplemented with GSTA1-1, GSTA4-4, GSTM1a-1a, and GSTP1a-1a. We were particularly interested in the catalytic activity of GSTA4-4 because this enzyme, of the human isozymes of GST, has among the best activities towards conjugation of α,β-unsaturated electrophiles such as 4-hydroxynonenal and some cyclopentenone prostaglandin derivatives (18, 27). However, addition of any of the isozymes of GST failed to enhance the rates of NO2-OA or NO2-LA conjugation (Figure 5). In fact, at even higher enzyme concentrations (up to 10 μ/ml), GSTA4-4 inhibited NO2-LA-SG formation (not shown) suggesting that GSTs may interact with nitroalkenes, but non-catalytically.
Figure 5. Effect of isozymes of GST on the rates of glutathione conjugation with nitroalkene fatty acids.

The rates of NO2-OA-SG (panel A) and NO2-LA-SG (panel B) formation were determined in the absence of GST (open circles) and in the presence of GSTA1-1 (open squares), GSTA4-4 (open diamond), GSTM1a-1a (closed circles), or GSTP1a-1a (closed triangle) as described in Experimental Procedures. Reaction progression was monitored by increasing absorbance at 245 nm.
GST-Nitroalkene Interactions: Inhibition
To investigate these potential interactions further, the effect of the nitroalkenes, NO2-OA and NO2-LA, and their glutathione conjugates, on GST activity was examined in the kinetic experiments summarized in Figure 6 and Table 1. Using 1-chloro-2,4-dinitrobenzene as the variable substrate, these experiments revealed that both nitroalkenes are potent inhibitors of all GSTs tested with Ki values in the nanomolar range. Moreover, glutathione conjugates of the nitroalkenes were similarly effective inhibitors of GST activity that, with the exception of GSTA1-1, had Ki values nearly equal to those of the parent nitroalkenes (Table 1). These data show that nitroalkenes and their glutathione conjugates interact avidly, and non-catalytically, with the four isozymes of GST. Both nitroalkenes and their conjugates had the lowest Ki values in GSTP1a-1a and GSTM1a-1a mediated reactions.
Figure 6. Inhibition of GST activity by nitroalkene fatty acids.

Shown are the kinetics of inhibition of the indicated isozymes of GST by NO2-OA (panels A, C, E, and G) and by NO2-LA (panels B, D, F, and H). GST activities are expressed as the initial velocities of glutathione conjugation with 1-chloro-2,4-dintrobenzene (CDNB) (mol•min-1•mg enzyme-1) as a function of CDNB concentration. Open circles represent no added nitroalkene while open squares, open diamonds, closed circles, closed triangles, open triangles, and crosses represent increasing concentrations of nitroalkene fatty acids (panels A and B: 0.1, 0.5, 1.0, 2.5 and 5.0 μM; panels C and D: 0.1, 0.25, 0.5, 1.0, 2.5 μM; and panels E-H: 0.005, 0.01, 0.05, 0.1, 0.25, and 0.5 μM NO2-OA or NO2-LA).
Table 1.
Inhibition of Glutathione S-Transferases by Nitroalkene Fatty Acids.
| GST | Ki (μM)1 | |||
|---|---|---|---|---|
| NO2-LA | NO2-LA-SG | NO2-OA | NO2-OA-SG | |
| GSTA4-4 | 0.46 ± 0.01 | 0.32 ± 0.03 | 0.53 ± 0.10 | 0.57 ± 0.05 |
| GSTA1-1 | 0.15 ± 0.02 | 0.78 ± 0.004 | 0.11 ± 0.02 | 0.67 ± 0.04 |
| GSTM1a-1a | 0.007 ± 0.002 | 0.010 ± 0.04 | 0.028 ± 0.007 | 0.026 ± 0.005 |
| GSTP1a-1a | 0.069 ± 0.014 | 0.079 ± 0.001 | 0.033 ± 0.005 | 0.048 ± 0.001 |
Ki were determined from nitroalkene (Figure 6) and nitroalkene-glutathione conjugate (not shown) inhibition data as described in Experimental Procedures. Shown are mean values ± 1 sd from 3 independent determinations.
GST-Nitroalkene Interactions: Biological Consequences
To examine the effect of GST expression on nitroalkene activation of PPARγ, reporter gene studies were conducted in control MCF7/WT cells (- GST) and in MCF7 cells stably expressing GSTA1-1 (MCF7/ α-10), GSTM1a-1a (MCF7/μ-5) and GSTP1a-1a (MCF7/π-11). The concentrations of GST expressed in these three transgenic cell lines were approximately 18 μM, 10 μM, and 5 μM, respectively (calculated as dimer concentrations; see legend, Figure 7). Expression of any of the three GSTs resulted in significant inhibition of NO2-OA-mediated (Figure 7A) and NO2-LA-mediated (Figure 7B) activation of the PPARγ-dependent reporter gene. Expression of GSTP1a-1a—even though present at the lowest concentration of the three isozymes—resulted in the greatest inhibition (80-90 %) of nitroalkene-mediated activation.
Figure 7. Expression of isozymes of GST attenuate nitroalkene-induction of PPARγgg-dependent transcription.

MCF7 cells were transiently co-transfected with the PPARγ-dependent reporter gene (PPRE-3x-TK-Luc), the control reporter gene (pGL4.75), and the PPARγ expression vector (pcDNA3-PPARγ). Cells were treated with 3 μM NO2-OA (panel A), 3 μM NO2-LA (panel B), or vehicle control. PPARγ dependent reporter gene activities were corrected for variations in transfection efficiencies and fold induction was calculated as described in Figure 4 and Experimental Procedures. Nitroalkene-mediated induction of PPARγ-dependent reporter gene activities in cells expressing GSTA1-1 (MCF7/α-8, GSTA1), GSTM1a-1a (MCF7/μ-5, GSTM1) and GSTP1a-1a (MCF7/π-11, GSTP1) are represented as a percentage of induction observed in cells lacking GST expression (MCF7/WT, -GST). Bars show the mean values of 6 (A) or 9 (B) independent transfections ± 1 sem. Inductions in cells expressing isozymes of GST were significantly less that in cells lacking GST (*, P<0.05; **, P<0.01; ***, P<0.001; one way ANOVA). GST concentrations in the stably transduced cell lines were estimated from the cellular GST activities (activity/mg total cellular protein), molecular weights of the GST homodimers, and the specific activities of the purified of GSTs (Experimental Procedures) assuming the concentrations of total cellular proteins were 150 mg/ml. Accordingly, intracellular concentrations of the GST isozymes were calculated as: GSTA1-1 (MCF7/α-8, GSTA1), 18 μM; GSTM1a-1a (MCF7/μ-5, GSTM1), 10 μM; and GSTP1a-1a (MCF7/π-11, GSTP1), 5 μM.
The specificity, towards nitroalkene-induced activation of PPARγ, of GST attenuation was examined by additional experiments using the pharmaceutical ligand, rosiglitazone. There are several reports indicating that GSTs, especially of the pi class (e.g. GSTP1-1), can interfere with mitogen-activated protein kinase signaling (28-30). Perturbation of such signaling could, then—indirectly and independently of GST-ligand interactions—affect ligand-activated PPARγ-dependent transcription through altered phosphorylation of PPARγ or other ancillary transcription factors. Rosiglitazone was chosen to test this idea because it is a potent ligand activator of PPARγ which: does not inhibit GST activity (not shown) and, hence, is unlikely to interact avidly with GST, and, secondly, does not readily form conjugates with glutathione. We examined the effect of GSTP1a-1a, the isozyme with the greatest inhibition of nitroalkene-mediated PPARγ transcription activation, on rosiglitazone-mediated transcription induction. As shown in Figure 8, expression of GSTP1a-1a does not inhibit rosiglitazone activation of PPARγ suggesting that the effect of GST expression is selective for PPARγ ligands, such as NO2-OA and NO2-LA, that interact directly with GST.
Figure 8. Effect of GSTP1a-1a expression on rosiglitazone-induced PPARγ-dependent transcription.

Parental GST-poor MCF7/WT (- GSTP1-1) and transgenic cells expressing GSTP1a-1a, MCF/π-11 (+ GSTP1-1) were transiently co-transfected with the PPARγ-responsive PPRE-3x-TK-Luc reporter, the control pGL4.75 reporter, and the pcDNA3-PPARγ expression plasmids. Transfected cells were treated with 0.5 μM rosiglitazone or vehicle control as described in Experimental Procedures. Fold induction was calculated from corrected firefly luciferase activies as outlined in Figure 4. Bars represent the mean values ± 1 sd from 9 independent transfections.
GST-Nitroalkene Interactions: Covalent?
To determine whether the avid interactions between nitroalkenes and the isozymes of GST involved covalent adduct formation, the GSTs were incubated with a 20-fold molar excess of NO2-OA or NO2-LA for 1 hour at 25°C, pH 7.5. These reactions were processed for analysis by MALDI-TOF and ESI/Q-TOF mass spectrometry. Repeated attempts failed to detect nitroalkene adducts with either GSTA1-1 or GSTM1a-1a (not shown). MALDI-TOF/MS analysis of GSTP1a-1a without the addition of nitroalkene revealed two peaks with approximate masses consistent with GSTP1a monomers plus (23,354 Da) and minus (23,223 Da) the N-terminal methionine (Figure 9A and B, thin black lines). The identities of these peaks as GSTP1a monomers ± N-terminal methionine was verified by ion-trap mass spectrometry of GSTP1a tryptic digestions (see Supplemental Material). Incubation of GSTP1a-1a with NO2-OA (Figure 9A, thick black line) or NO2-LA (Figure 9B, thick black line) resulted in a shift of the two (± methionine) peaks ~325 Da higher, a shift consistent with the formation of stable covalent adducts with the nitroalkenes. For better resolution and more accurate mass determinations, GSTP1a-1a samples prepared in the absence (Figure 10A) or presence (Figure 10B) of NO2-LA were analyzed by ESI/Q-TOF mass spectrometry. These data show unequivocally that incubation of GSTP1a-1a with NO2-LA results in the formation of GSTP1a monomers (± methionine) stably adducted with one (23,548 and 23,679 Da peaks) and two (23,873 and 24,004 Da peaks) molecules of NO2-LA.
Figure 9. MALDI-TOF mass spectrometry of nitroalkene fatty acid adducts of GSTP1a.

Recombinant purified GSTP1a-1a (200 pmol) was incubated at pH 7.5 for 1 hour at room temperature with (heavy dark spectra) or without (thin dark spectra) a 20-fold molar excess of NO2-OA (panel A) or NO2-LA (panel B). Shown are the MALDI-TOF MS analyses of these incubations. In the samples not treated with nitroalkenes, the major species corresponding to GSTP1a monomers with (+ met, 23,354 Da) and without (- met, 23,223 Da) the N-terminal methionine are indicated by vertical arrows. The horizontal arrows indicate the ~325 Da increase in size of these major peaks associated with nitroalkene fatty acid treatments.
Figure 10. ESI/Q-TOF mass spectrometry of NO2-LA adducts with GSTP1a.

Recombinant GSTP1a-1a samples incubated without (panel A) and with (panel B) a 20-fold molar excess of NO2-LA were prepared as described in Figure 9 and processed for mass spectrometry according to Experimental Procedures. Raw ion data were transformed by maximum entropy calculations to yield masses corresponding to unmodified GSTP1a monomers (23,223 [-met] and 23,354 [+met]) and to GSTP1a monomers adducted with one NO2-LA (23,548 [-met] and 23,679 [+met]) or two NO2-LA (23,873 [-met] and 24,004 [+met]) molecules.
Discussion
GSTs—a large superfamily of isozymes expressed differentially in alternative cells and tissues—possess several identified activities and functions. They catalyze the conjugation of many electrophiles with glutathione, generally rendering the electrophile less chemically reactive and marked for cellular efflux and excretion by specific membrane-associated transport proteins (31, 32). More recently, several reports have indicated that GSTs, especially those of the pi class, can regulate the activities of several mitogen-activated and other stress kinases (28-30): perhaps the best characterized of these is the inhibition of c-Jun N-terminal kinase (JNK) activity by direct interactions between GSTp monomers and JNK (28). Lastly, one of the earliest properties of GSTs described is their ability to bind, non-covalently, hydrophobic and amphiphilic intracellular ligands—hence, the name “ligandin” first applied to members of the GST family (33-35). From this discussion, it is evident that GSTs have the potential to influence the intracellular activities of amphiphilic electrophiles, such as nitroalkene fatty acids, by several alternative mechanisms.
Nitroalkene fatty acids are abundant, potent ligand activators of PPARγ as well as mediators of other activities including inhibition of NFkB-dependent transcription and activation of transcription via the Nrf2/Keap1 pathway (1, 4). Previous studies (12) and those described herein indicate that non-enzymatic conjugation with glutathione and MRP1-mediated efflux of these conjugates can attenuate NO2-OA- and NO2-LA-induced activation of PPARγ. The main goal of the current studies was to determine if GSTs could, by enhancing the rate of glutathione conjugation or by alternative mechanisms, contribute to the overall modulation of nitroalkene activities. Indeed, GSTs have been reported to catalyze the glutathione conjugation of other electrophilic fatty acids including prostaglandins A1, A2, and J2 (36, 37). Although we previously failed to observe any significant catalysis of glutathione conjugation with the cyclopentenone prostaglandin, 15-deoxy-Δ12,14prostaglandin J2, by GSTA1-1, M1a-1a, or P1a-1a (16), we speculated here that nitroalkenes might be preferred GST substrates. GSTA4-4 was of particular interest because it, among the human isozymes of GST, has been shown to have the most robust activity towards the conjugation of the α,β-unsaturated electrophiles, 4-hydroxynonenal, prostaglandin A2 and 15-A2T-isoprostane (18, 27)—suggesting that GSTA4-4 would be a particularly promising candidate for catalysis of nitroalkene fatty acid conjugation with glutathione. However, as demonstrated in Figure 5, no activity towards either NO2-OA or NO2-LA was apparent with any of the four GSTs tested.
Nevertheless, during the course of these studies, the nitroalkenes were observed to interact avidly with GSTs as evidenced by their abilities to inhibit GST activity at nanomolar concentrations (Figure 6 and Table 1). These observations mirror previous results reported by our laboratory involving the less abundant electrophilic PPARγ ligand, 15-deoxy-Δ12,14prostaglandin J2 (16). Moreover, expression of GSTs, at levels readily observed in many cells and tissues, resulted in significant attenuation of PPARγ-dependent transcription activation by nitroalkene fatty acids (Figure 7). These results suggest that GSTs—that are expressed at various levels in most normal cells and tissues and that often are induced by electrophilic stress (38, 39)—may provide a physiologically relevant layer of regulation for nitroalkene-mediated transcription activation via sequestering the nitroalkenes away from their nuclear target, PPARγ.
While the issue is not completely resolved, our studies shed some light on the potential reversibility of GST-nitroalkene interactions and nitroalkene inhibition of GSTs. It is well documented that nitroalkene fatty acids can form covalent bonds with thiols of proteins such as glyceraldehyde-3-phosphate dehydrogenase and NFkB (1, 40). Although such Michael addition reactions have limited reversibility, the forward reactions are kinetically favored at physiological pH and, hence, the thioether bonds formed between nitroalkenes and proteins are expected to be relatively stable. It is notable that of the GSTs tested, GSTP1a-1a was the only isozyme with which we detected stable nitroalkene adducts and was also the most potent inhibitor of nitroalkene-induced PPARγ activation. It is tempting to speculate that the greater inhibitory potency of GSTP1a-1a is related to its propensity to form covalent adducts or, alternatively, GSTP1a-1a may have more nitroalkene fatty acid binding sites than the other GSTs tested. However, while the present study demonstrates that a 20-fold molar excess of NO2-OA or NO2-LA can result in stable adducts with purified GSTP1a-1a in vitro, this occurs under rather extreme conditions not encountered in intact cells. Although we cannot rule out that covalent adducts may also occur with other GSTs, our failure to observe them with GSTA1-1 and GSTM1a-1a suggests that such adducts do not from readily. In addition, glutathione conjugates of nitroalkenes, NO2-OA-SG and NO2-LA-SG, also interact strongly with GSTs. Indeed, with the exception of GSTA1-1, the avidities of these interactions were equal to those with the parent, unconjugated nitroalkene (Table 1). As the glutathione conjugates of nitroalkenes have lost their reactive electrophilic centers, these results indicate that avid interactions do not require covalent bond formation with GSTs. Thus, it is most likely that GSTs regulate the availability of free nitroalkene fatty acids and their glutathione conjugates via reversible interactions between the GSTs and the lipid moieties of these fatty acid ligands.
Supplementary Material
Acknowledgement
The authors are grateful for expert guidance from Michael Samuel in the design and execution of mass spectrometry experiments.
This work was supported by NIH grants CA70338 (CSM) and HL62198 (SBK).
Footnotes
Supplemental Information Available. The description of and results from ion-trap mass spectrometry analysis of tryptic digests derived from unmodified, recombinant GSTP1a-1a can be found as supplemental material. This information is available, free of charge, via the Internet at http://pubs.acs.org.
Abbreviations: DMEM, Dulbecco's modified Eagle medium; GST, glutathione S-transferase; JNK, c-Jun N-terminal kinase; Keap1, kelch-like ECH-associated protein 1; MRP1, multidrug resistance (or resistance-associated) protein 1 (ABCC1); NFkB, nuclear factor kappa B; NO2-LA, nitrolinoleic or nitrooctadeca-9,12-dienoic acid; NO2-LA-SG, glutathione conjugate of nitrolinoleic acid; NO2-OA, nitrooleic or nitrooctadec-9-enoic acid; NO2-OA-SG, glutathione conjugate of nitrooleic acid; Nrf2, NF-E2-related factor 2; PPARγ, peroxisomal proliferator-activated receptor γ.
References
- 1.Cui T, Schopfer FJ, Zhang J, Chen K, Ichikawa T, Baker PRS, Batthyany C, Chacko BK, Feng X, Patel RP, Agarwal A, Freeman BA, Chen YE. Nitrated Fatty Acids: Endogenous Anti-inflammatory Signaling Mediators. J. Biol. Chem. 2006;281:35686–35698. doi: 10.1074/jbc.M603357200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freeman BA, Baker PRS, Schopfer FJ, Woodcock SR, Napolitano A, d'Ischia M. Nitro-fatty Acid Formation and Signaling. J. Biol. Chem. 2008;283:15515–15519. doi: 10.1074/jbc.R800004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lima ES, Bonini MG, Augusto O, Barbeiro HV, Souza HP, Abdalla DS. Nitrated lipids decompose to nitric oxide and lipid radicals and cause vasorelaxation. Free Radic. Biol. Med. 2005;39:532–539. doi: 10.1016/j.freeradbiomed.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 4.Villacorta L, Zhang J, Garcia-Barrio MT, Chen X.-l., Freeman BA, Chen YE, Cui T. Nitro-linoleic acid inhibits vascular smooth muscle cell proliferation via the Keap1/Nrf2 signaling pathway. Am. J. Physiol. Heart Circ. Physiol. 2007;293:H770–776. doi: 10.1152/ajpheart.00261.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear Receptors and Lipid Physiology: Opening the X-Files. Science. 2001;294:1866–1870. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- 6.Evans R, Barish G, Wang Y. PPARs and the complex journey to obesity. Nat. Med. 2004;10:355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 7.Kliewer SA, Lehmann J, Willson TM. Orphan Nuclear Receptors: Shifting Endocrinology into Reverse. Science. 1999;284:757–760. doi: 10.1126/science.284.5415.757. [DOI] [PubMed] [Google Scholar]
- 8.Szanto A, Nagy L. The many faces of PPARγ: anti-inflammatory by any means? Immunobiology. 2008;213:789–803. doi: 10.1016/j.imbio.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 9.Baker PRS, Schopfer FJ, Sweeney S, Freeman BA. Red cell membrane and plasma linoleic acid nitration products: Synthesis, clinical identification, and quantitation. Proc. Natl. Acad. Sci. (USA) 2004;101:11577–11582. doi: 10.1073/pnas.0402587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alexander R, Wright M, Gorczynski M, Smitherman P, Akiyama T, Wood H, Berger J, King S, Morrow C. Differential potencies of naturally occurring regioisomers of nitrolinoleic acid in PPARγ activation. Biochemistry. 2009;48:492–498. doi: 10.1021/bi8016747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schopfer FJ, Lin Y, Baker PRS, Cui T, Garcia-Barrio M, Zhang J, Chen K, Chen YE, Freeman BA. Nitrolinoleic acid: An endogenous peroxisome proliferator-activated receptor γ ligand. Proc. Natl. Acad. Sci. (USA) 2005;102:2340–2345. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alexander RL, Bates DJ, Wright MW, King SB, Morrow CS. Modulation of Nitrated Lipid Signaling by Multidrug Resistance Protein 1 (MRP1): Glutathione Conjugation and MRP1-Mediated Efflux Inhibit Nitrolinoleic Acid-Induced, PPARγ-Dependent Transcription Activation. Biochemistry. 2006;45:7889–7896. doi: 10.1021/bi0605639. [DOI] [PubMed] [Google Scholar]
- 13.Baker PRS, Lin Y, Schopfer FJ, Woodcock SR, Groeger AL, Batthyany C, Sweeney S, Long MH, Iles KE, Baker LMS, Branchaud BP, Chen YE, Freeman BA. Fatty Acid Transduction of Nitric Oxide Signaling: Multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator-activated receptor ligands. J. Biol. Chem. 2005;280:42464–42475. doi: 10.1074/jbc.M504212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gorczynski M, Huang J, King S. Regio- and stereospecific syntheses and nitric oxide donor properties of (E)-9- and (E)-10-nitrooctadec-9-enoic acids. Org. Lett. 2006;8:2305–2308. doi: 10.1021/ol060548w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gurnell M, Wentworth JM, Agostini M, Adams M, Collingwood TN, Provenzano C, Browne PO, Rajanayagam O, Burris TP, Schwabe JW, Lazar MA, Chatterjee VKK. A Dominant-negative Peroxisome Proliferator-activated Receptor γ (PPARγ) Mutant Is a Constitutive Repressor and Inhibits PPARgamma -mediated Adipogenesis. J. Biol. Chem. 2000;275:5754–5759. doi: 10.1074/jbc.275.8.5754. [DOI] [PubMed] [Google Scholar]
- 16.Paumi C, Smitherman P, Townsend A, Morrow C. Glutathione S-transferases (GSTs) inhibit transcriptional activation by the peroxisomal proliferator-activated receptor γ (PPARγ) ligand, 15-deoxy-Delta(12,14)prostaglandin J(2) (15-d-PGJ(2)) Biochemistry. 2004;43:2345–2352. doi: 10.1021/bi035936+. [DOI] [PubMed] [Google Scholar]
- 17.Paumi CM, Wright M, Townsend AJ, Morrow CS. Multidrug Resistance Protein (MRP) 1 and MRP3 Attenuate Cytotoxic and Transactivating Effects of the Cyclopentenone Prostaglandin, 15-Deoxy-Delta(12,14)Prostaglandin J(2) in MCF7 Breast Cancer Cells. Biochemistry. 2003;42:5429–5437. doi: 10.1021/bi027347u. [DOI] [PubMed] [Google Scholar]
- 18.Hubatsch I, Ridderstrom M, Mannervik B. Human glutathione transferase A4-4: an alpha class enzyme with high catalytic efficiency in the conjugation of 4-hydroxynonenal and other genotoxic products of lipid peroxidation. Biochem. J. 1998;330(Pt 1):175–179. doi: 10.1042/bj3300175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paumi CM, Ledford BG, Smitherman PK, Townsend AJ, Morrow CS. Role of Multidrug Resistance Protein 1 (MRP1) and Glutathione S-Transferase A1-1 in Alkylating Agent Resistance. Kinetics of glutathione conjugate formation and efflux govern differential cellular sensitivity to chlorambucil versus melphalan toxicity. J. Biol. Chem. 2001;276:7952–7956. doi: 10.1074/jbc.M009400200. [DOI] [PubMed] [Google Scholar]
- 20.Habig W, Pabst M, Jakoby W. Glutathione S-transferase: the first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974;249:7130–7139. [PubMed] [Google Scholar]
- 21.Smitherman PK, Townsend AJ, Kute TE, Morrow CS. Role of Multidrug Resistance Protein 2 (MRP2, ABCC2) in Alkylating Agent Detoxification: MRP2 Potentiates Glutathione S-Transferase A1-1-Mediated Resistance to Chlorambucil Cytotoxicity. J. Pharmacol. Exp. Ther. 2004;308:260–267. doi: 10.1124/jpet.103.057729. [DOI] [PubMed] [Google Scholar]
- 22.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 23.Sibhatu MB, Smitherman PK, Townsend AJ, Morrow CS. Expression of MRP1 and GSTP1-1 modulate the acute cellular response to treatment with the chemopreventive isothiocyanate, sulforaphane. Carcinogenesis. 2008;29:807–815. doi: 10.1093/carcin/bgn013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kolm R, Danielson U, Zhang Y, Talalay P, Mannervik B. Isothiocyanates as substrates for human glutathione transferases: structure-activity studies. Biochem. J. 1995;311:453–459. doi: 10.1042/bj3110453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cornish-Bowden A. Fundamentals of Enzyme Kinetics. Portland Press; London: 1995. [Google Scholar]
- 26.Baker LMS, Baker PRS, Golin-Bisello F, Schopfer FJ, Fink M, Woodcock SR, Branchaud BP, Radi R, Freeman BA. Nitro-fatty Acid Reaction with Glutathione and Cysteine: Kinetic Analysis of Thiol Alkylation by a Michael Addition Reaction. J. Biol. Chem. 2007;282:31085–31093. doi: 10.1074/jbc.M704085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hubatsch I, Mannervik B, Gao L, Roberts LJ, Chen Y, Morrow JD. The cyclopentenone product of lipid peroxidation, 15-A2t-isoprostane (8-isoprostaglandin A2), is efficiently conjugated with glutathione by human and rat glutathione transferase A4-4. Chem.Res.Toxicol. 2002;15:1114–1118. doi: 10.1021/tx020027r. [DOI] [PubMed] [Google Scholar]
- 28.Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, Tew KD, Pincus MR, Sardana M, Henderson CJ, Wolf CR, Davis RJ, Ronai Z. Regulation of JNK signaling by GSTp. Embo. J. 1999;18:1321–1334. doi: 10.1093/emboj/18.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elsby R, Kitteringham NR, Goldring CE, Lovatt CA, Chamberlain M, Henderson CJ, Wolf CR, Park BK. Increased Constitutive c-Jun N-terminal Kinase Signaling in Mice Lacking Glutathione S-Transferase Pi. J. Biol. Chem. 2003;278:22243–22249. doi: 10.1074/jbc.M301211200. [DOI] [PubMed] [Google Scholar]
- 30.Yin ZM, Ivanov VN, Habelhah H, Tew K, Ronai Z. Glutathione S-transferase p elicits protection against H2O2-induced cell death via coordinated regulation of stress kinases. Cancer Res. 2000;60:4053–4057. [PubMed] [Google Scholar]
- 31.Hayes JD, Pulford DJ. The glutathione S-transferase supergene family: Regulation of GST and the contribution of the isozymes to cancer chemoprevention and drug resistance. Crit. Rev. Biochem. Mol. Biol. 1995;30:445–600. doi: 10.3109/10409239509083491. [DOI] [PubMed] [Google Scholar]
- 32.Cole SPC, Deeley RG. Transport of glutathione and glutathione conjugates by MRP1. Trends in Pharmacological Sciences. 2006;27:438. doi: 10.1016/j.tips.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 33.Hayes JD, Strange RC, Percy-Robb IW. Identification of two lithocholic acid-binding proteins. Separation of ligandin from glutathione S-transferase B. Biochem. J. 1979;181:699–708. doi: 10.1042/bj1810699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Litwack G, Ketterer B, Arias IM. Ligandin: a hepatic protein which binds steroids, bilirubin, carcinogens and a number of exogenous organic anions. Nature. 1971;234:466–467. doi: 10.1038/234466a0. [DOI] [PubMed] [Google Scholar]
- 35.Mannervik B, Jensson H. Binary combinations of four protein subunits with different catalytic specificities explain the relationship between six basic glutathione S-transferases in rat liver cytosol. J. Biol. Chem. 1982;257:9909–9912. [PubMed] [Google Scholar]
- 36.Bogaards JJ, Venekamp JC, van Bladeren PJ. Stereoselective conjugation of prostaglandin A2 and prostaglandin J2 with glutathione, catalyzed by the human glutathione S-transferases A1-1, A2-2, M1a-1a, and P1-1. Chem. Res. Toxicol. 1997;10:310–317. doi: 10.1021/tx9601770. [DOI] [PubMed] [Google Scholar]
- 37.Cagen LM, Pisano JJ, Ketley JN, Habig WH, Jakoby WB. The conjugation of prostaglandin A1 and glutathione catalyzed by homogeneous glutathione S-transferases from human and rat liver. Biochim. Biophys. Acta. 1975;398:205–208. doi: 10.1016/0005-2760(75)90184-8. [DOI] [PubMed] [Google Scholar]
- 38.Kim SG, Lee SJ. PI3K, RSK, and mTOR Signal Networks for the GST Gene Regulation. Toxicol. Sci. 2007;96:206–213. doi: 10.1093/toxsci/kfl175. [DOI] [PubMed] [Google Scholar]
- 39.Rushmore T, Pickett C. Glutathione S-transferases, structure, regulation, and therapeutic implications. J. Biol. Chem. 1993;268:11475–11478. [PubMed] [Google Scholar]
- 40.Batthyany C, Schopfer FJ, Baker PRS, Duran R, Baker LMS, Huang Y, Cervenansky C, Branchaud BP, Freeman BA. Reversible Posttranslational Modification of Proteins by Nitrated Fatty Acids in Vivo. J. Biol. Chem. 2006;281:20450–20463. doi: 10.1074/jbc.M602814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.