

Abstract
The genomically encoded fosfomycin resistance protein from Pseudomonas aeruginosa (FosAPA) utilizes Mn(II) and K+ to catalyze the addition of glutathione (GSH) to C1 of the antibiotic rendering it inactive. Although this protein has been structurally and kinetically characterized with respect to the substrate, fosfomycin, questions remain regarding the how the enzyme binds the thiol substrate, GSH. Computational studies have revealed a potential GSH binding site in FosAPA that involves six electrostatic or hydrogen-bonding interactions with protein side-chains as well as six additional residues that contribute van der Waals interactions. A strategically placed tyrosine residue, Y39, appears to be involved in the ionization of GSH during catalysis. The Y39F mutant exhibits a 13-fold reduction of catalytic activity (kcat = 14 ± 2 s−1), suggesting a role in the ionization of GSH. Mutation of five other residues (W34, Q36, S50, K90 and R93) implicated in ionic of hydrogen bonding interactions resulted in enzymes with reduced catalytic efficiency, affinity for GSH or both. The mutant enzymes were also found to be less effective resistant proteins in the biological context of E. coli. The more conservative W34H mutant has native-like catalytic efficiency suggesting that the imidazole NH group can replace the indole group of W34 that is important for GSH binding. In the absence of co-crystal structural data with the thiol substrate, these results provide important insights into the role of GSH in catalysis.
Keywords: Antibiotic resistance, Glutathione, Fosfomycin, Metalloenzyme
INTRODUCTION
Development of resistance to antibiotics by microorganisms remains a critical problem in treatment of infectious disease (1–3). Understanding the mechanisms by which bacteria process therapeutic agents can lead to development of novel effective treatments. Fosfomycin, a natural product originally isolated from certain strains of Streptomyces (4,5), was identified as an excellent antibiotic in that it was effective against both Gram positive and negative organisms and produced virtually no side effects with treatment (6). The antibiotic covalently inhibits the enzyme MurA, the pyruvyl transferase that catalyzes the first step in cell wall biosynthesis. However, several avenues of microbial resistance appeared shortly after its introduction into the clinic. These include mutations in enzyme MurA (7), mutations in phosphonate uptake pathways (8,9), and the inactivation of fosfomycin by resistance proteins that cleave the carbon-oxygen bond of the epoxide moiety (10–13). Resistance proteins of the FosA-class are metalloenzymes that catalyze the nucleophilic addition of the tri-peptide glutathione to the C1 position of the antibiotic, cleaving the epoxide ring and inactivating the antibiotic (Scheme 1).
Scheme 1.

While various biochemical methods including X-ray crystallography (14), electron paramagnetic resonance spectroscopy (15–17), and kinetic characterization have answered many questions regarding how the Mn(II) and K+ ions and fosfomycin bind to the FosA enzymes, little is known about how the proteins bind GSH and catalyze the addition of the thiol to the antibiotic. Work presented here reports the elucidation of a potential GSH binding site through manual docking of GSH to the model of FosAPA obtained from the crystal structure followed by energy minimization of the docked complex. Mutagenesis of potential binding and catalytic residues followed by kinetic characterization and GSH binding studies provide additional support for the proposed binding site.
EXPERIMENTAL PROCEDURES
Materials
Glutathione was purchased from RPI (Mt. Prospect, IL). Potassium chloride, manganese chloride and thiazolyl blue tetrazolium bromide were purchased from Sigma (St. Louis, MO). Fosfomycin disodium salt was purchased from Fluka (Ronkonkoma, NY). Acetonitrile (HPLC grade) was purchased from Fisher. AccQ-FlourTM Derivatizing Reagent (AQC) was purchased from Waters (Milford, MA).
GSH docking and energy minimization
Glutathione was docked manually in the active site of FosAPA from the PDB coordinate file 1LQP (16) with an interactive molecular graphics package. GSH was initially positioned based on experimental evidence that the thiol is added to C1 of fosfomycin during catalysis (18). A second fosfomycin molecule bound at the entrance of the FosA active site was removed from the crystal structure complex to facilitate the placement of GSH, and side chain conformations for active site residues Q36 and K90 were adjusted using a backbone-dependent rotamer library (19) to eliminate unfavorable steric interactions. Water molecules in or near the active site entrance that overlapped significantly with the docked GSH molecule were removed, but all other water molecules observed in the crystal structure were retained in the model complex.
GSH contains a γ-glutamyl residue that is not well represented by the standard AMBER ff99 force field (20), so suitable geometry and partial charge parameters were developed, using methyl glutamine as a model compound. A geometry-optimized structure for GSH was computed using a Hartree-Fock method with a 3–21G* basis set, and equilibrium bond lengths and angles were taken directly from this structure. All necessary bond, bond angle, and torsion angle force constants were available in the standard potential function data set. Molecular electrostatic potentials were calculated over a grid of points for both GSH and fosfomycin using Hartree-Fock optimized geometries and a 6–31G* basis set. Partial charges for each substrate were then derived by fitting the electrostatic potential to an atom-centered point charge model. Additional parameters for fosfomycin and active site Mn(II) and K+ ions were taken directly from the standard database.
Hydrogen atoms were added to the enzyme-substrate complex using the LeAP module, and the resultant complex was refined with steepest-descent and conjugate-gradient energy minimization to remove remaining unfavorable steric interactions or conformational strain. Energy minimization calculations were performed with a distance-dependent dielectric model, using the AMBER 8.0 package (AMBER 8)(21). All quantum mechanical calculations were performed using Gaussian98 (22). Charge-fitting calculations were done with the RESP package (23).
Generation of Mutants
Site-directed mutagenesis was performed on the FosAPA expression plasmid using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). Oligonucleotide primers (Invitrogen - Carlsbad, CA) were designed to change codon 34 from TGG to GCG (W34A) and CAC (W34H), codon 36 from CAG to AAC (Q36N), codon 39 from TAT to TTT (Y39F), codon 90 from AAG to GCG (K90A), codon 93 from CGC to GCG (R93A), and to insert CGC GTC GAG (RVE) between codons 93 (R) and 94 (S). All mutations were confirmed by DNA sequencing.
Expression and purification of mutants
Mutants were expressed and purified as described for native protein (24). Briefly, for the mutants of FosAPA, bacteria were lysed by sonication followed by centrifugation to pellet the cell debris. The lysate was dialyzed against 20 mM Tris (pH 7.5) prior to loading onto a DEAE-cellulose column equilibrated with this buffer. Protein was eluted with a salt gradient [(20 to 500 mM NaCl in 20 mM Tris (pH 7.5)]. Fractions containing the enzyme were pooled and dialyzed against 20 mM KH2PO4 (pH 6.8). Protein was passed isocratically through a hydroxylapatite column equilibrated with the same buffer. Fractions containing the enzyme were pooled and dialyzed against 20 mM TMA/HEPES (pH 8.0) containing Chelex resin and 5 mM EDTA followed by dialysis against 20 mM TMA/HEPES (pH 8.0). Protein was then demetallated and passed isocratically through an S100 gel filtration column. The protein was then dialyzed in 20 mM HEPES (pH 8.0), concentrated, and stored at −80 (C.
Kinetic analysis of native and mutant enzymes
The kinetic assay for native protein was followed as described (24) to determine kinetic parameters for mutants. Briefly, enzyme was incubated with the appropriate metal(s) prior to addition of thiol and fosfomycin (disodium or TMA salt). Reactions were quenched at a time such that less than 20% of the variable substrate was consumed and derivatized with a fluorescent reagent (AQC) prior to HPLC analysis. Data was fit to a Michaelis-Menton curve to calculate kcat, KM, and kcat/KM. For the K90A and R93A mutations saturation with GSH was not possible for this analysis inasmuch as the maximum practical concentration of GSH in the assay is 100 mM. For these mutants a kcat/KMGSH was calculated by fitting the linear portion of the curve with a straight line (25).
Fluorescence titration of FosAPA with glutathione
A solution of 5 μM FosAPA in the presence of 50 μM MnCl2 and 100 mM KCl was prepared in 20 mM TMA/HEPES (pH 8.0) that had been filtered through a 0.2 μm filter. A solution of buffer containing MnCl2 and KCl was prepared similarly. Only 1 μM enzyme was used for the Q36N, Y39F, S50A, K90A and R93A mutants. GSH was added to both solutions incrementally while monitoring the change in fluorescence. Titration measurements were made in triplicate on a SPEX Fluorolog-3 spectrofluorimeter (Jobin Yvon Horiba, Edison, NJ) in the constant-wavelength mode. Solutions were excited at 275 nm (3 nm slit width), and emission measurements were taken at 340 nm (3 nm slit width) for most mutants. For the K90A and R93A mutants, excitation was at 295 nm (2 nm slit width) and emission was measured at 328 nm (4 nm slit width). This is due to the lower absorbance of GSH at 295 nm versus 275 nm. After correction for fluorescence of the buffer solution, data were averaged and fit to a hyperbola to obtain the apparent Kd.
Determination of the minimum inhibitory concentration (MIC100)
To test whether or not the mutations made in FosAPA affected the ability of the enzyme confer resistance to the antibiotic the mutant expression plasmids were transformed into E. coli and grown on LB plates containing different amounts of the antibiotic. Each plate contained between 0 and 20 mg/mL fosfomycin, 100 μg/mL ampicillin, and 10 μg/mL glucose-6-phosphate. Plates were divided into six sections to accommodate six different samples. For each plate one section was native FosAPA (positive control), another was E. coli transformed with empty vector (pET20, negative control), and the other four were the various mutants. For each spot on the plate, 30 μL of suspension of bacteria (OD600 = 0.03) in LB/Amp media was added and spread onto the plate. Plates were incubated at 37° C for 16 hours. Plates were visualized by spraying them with a solution of 0.5 mg/mL thiazolyl blue tetrazolium bromide.
RESULTS
GSH docking and minimization
A potential GSH binding site was identified in FosAPA through manual molecular docking followed by energy minimization of the docked complex. Favorable intermolecular hydrogen bonding or electrostatic interactions to the docked GSH in the model include R93, K90, S50, Y39, Q36, and W34 as illustrated in Figure 1. Other residues making van der Waals contacts with GSH include Q91, H64, Y62, C48, Y128, and R119. Figure 1 shows the location of GSH in a ribbon diagram of FosAPA as well as a diagram of hydrogen bonding interactions between GSH and the protein. (Figure 1 here)
Figure 1.

A. Ribbon diagram of FosAPA with GSH shown in the docked position (Mn(II) in purple and K+ in dark blue). Residues that contribute electrostatic or hydrogen bonding interactions with GSH are shown in yellow while residues that are in van der Waals contact are shown in light blue. B. Diagram of residues participating in potential hydrogen-bonding or electrostatic interactions with GSH. The numbers represent the distances in Å. The position of the thiol group of GSH relative to Y39 and fosfomycin C1 is indicated. The distance between sulfur of GSH and C1 of fosfomycin is 3.5 Å, which is a favorable van der Walls contact.
Kinetic analysis of native and mutant enzymes
The ionization of GSH during catalysis often involves electrophilic assistance by an active site tyrosine or serine residue in the canonical GSH transferases (26). Although S50 was a preliminary candidate for playing this role in FosAPA, the S50A mutation had little effect on catalysis (Table 1) but has a relatively large effect on the KdGSH. Results from the energy-minimized docked enzyme-substrate complex indicated that the hydroxyl group of Y39 is in a good position to assist in the ionization GSH, being 3.5 Å from the unionized thiol of GSH and the C1 group of fosfomycin, respectively. The ionized thiol was not represented explicitly in our calculations (see Methods). The Y39F mutant displayed a 13-fold reduction in turnover and a 50-fold decrease in catalytic efficiency, kcat/KMGSH (Table 1).
Table 1.
Kinetic and binding parameters for the native and mutant enzymes.a
| Enzyme | kcat (s-1)b | kcat/KMGSH (M−1s−1) | KdGSH (mM) | kcat/KMfos (M−1s−1) | MIC100 (mg/mL) |
|---|---|---|---|---|---|
| FosAPA | 180 ± 6 | (4.1 ± 0.8) × 104 | 0.13 ± 0.03 | (9 ± 1) × 105 | > 20 |
| FosAPA - K+ | ~10 | 62 ± 2 | 39 ± 3 | (1.4 ± 0.3) × 103 | n.d. |
| FosAPA-K-loopc | ~14 | 26.6 ± 0.5 | 39 ± 2 | (1.0 ± 0.2) × 104 | 2 |
| W34A | 32 ± 3 | (2.0 ± 0.4) × 102 | n.d. | (2.1 ± 0.5) × 104 | < 1 |
| W34H | 30 ± 4 | (5.4 ± 1.1) × 103 | 0.1 ± 0.02 | (1.1 ± 0.3) × 104 | > 20 |
| Q36N | 109 ± 3 | (1.4 ± 0.1) × 104 | 32 ± 3 | n.d. | 4 |
| Y39F | 14 ± 2 | (9 ± 1) × 102 | 21 ± 1 | (5 ± 3) × 105 | 1 |
| S50A | 134 ± 2 | (6.2 ± 0.4) × 104 | 60 ± 3 | (1.8 ± 0.9) × 106 | 2 |
| K90A | ~30 | (3.4 ± 0.2) × 102 | 22 ± 3 | (2.4 ± 0.9) × 104 | 2 |
| R93A | ~30 | (1.9 ± 0.1) × 102 | 23 ± 3 | (3 ± 1) × 104 | 2 |
n.d. – value not determined.
Numbers preceded by a tilde (~) are estimated from Lineweaver-Burke Linear Regression analysis.
Three-residue insertion mutant that disrupts the K+-binding loop (see Figure 3).
Results from the molecular docking indicated that the side chains of W34, Q36, K90, and R93 are within hydrogen bonding or favorable electrostatic distance of various carbonyl and carboxylate oxygens of the substrate. The mutation of W34, K90, and R93 to alanine all resulted in a significant decrease in kcat and kcat/KM, analogous to that seen in the Y39F mutant (Table 1).
Fluorescence titration of enzyme with GSH
Titration of FosAPA with GSH as monitored by intrinsic protein fluorescence revealed that the protein has an equilibrium dissociation constant similar to the unrelated canonical GSH transferases (Table 1, Figure 2) (26). In contrast, the KdGSH for the Q36N, Y39F, S50A, K90A and R93A mutants increased between 150 and 400-fold as compared to the native enzyme (Table 1, Figure 2).
Figure 2.

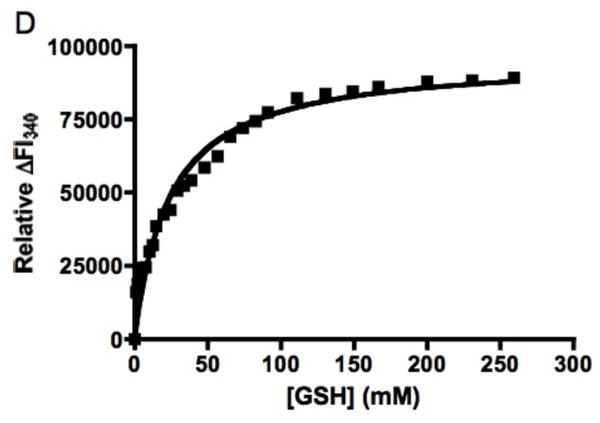
A. Fluorescence spectra of the FosAPA•Mn(II)•K+ complex in the absence (—) and presence (---) of GSH upon excitation at 275 nm. B. Fluorescence spectra of the R93AFosAPA•Mn(II)•K+ complex in the absence (—) and presence (---) of GSH upon excitation at 295 nm. C. Relative change in intrinsic protein fluorescence for FosAPA•Mn(II)•K+ at 340 nm upon addition of GSH. The solid line is the fit of the data to a rectangular hyperbola with a KdGSH = 0.13 ± 0.03 mM. D. Relative change in intrinsic protein fluorescence for R93AFosAPA•Mn(II)•K+ at 340 nm upon addition of GSH. The solid line is the fit of the data to a rectangular hyperbola with a KdGSH = 23 ± 3 mM.
The two cationic residues K90 and R93 reside at the base of the K+-binding loop as is illustrated in Figure 3. The FosA enzymes are activated up to 100-fold by K+ or other monovalent cations. It is interesting to note that the catalytic efficiency of the enzyme and its ability to bind GSH are compromised in the absence of K+ to an extent that is comparable to either the K90A or R93A mutants (Table 1). To further investigate this observation the connection between the K90/R93 GSH binding residues and the residues involved in the binding of K+ was disrupted by a three-residue insertion (Figure 3). The catalytic and binding properties were quite similar to the behavior of the enzyme in the absence of K+.
FIGURE 3.

Sequence alignment of the FosAPA and FosATN enzymes near the K-loop showing the position of the three-residue insertion to disrupt the register between K90/R93 and the K+-binding loop. Residues involved in the binding of GSH, K+ and the insertion are shown in red, green and blue, respectively.
The role of W34 in the enzyme is more complex. The W34A mutant displayed a 30-fold decrease in kcat/KMGSH (Table 1), which would be consistent with a role in the binding of GSH as suggested by the model. However the protein precipitated during the fluorescence titration experiments at relatively low [GSH], a fact that precluded a determination of KdGSH and suggested that the mutant was unstable in the presence of GSH. In contrast, the W34H mutant, featuring an imidazole NH group that has potential to serve as a hydrogen-bond donor to GSH was quite stable and showed near-native levels of enzyme activity and GSH binding affinity. Two of the other mutants also displayed curious behavior. Both the S50A and Q36N mutants exhibited elevated dissociations constants for GSH but appeared to maintain the catalytic efficiency of the native enzyme.
The mutation of residues in the GSH binding site had, in general, a smaller effect on the efficiency of the enzyme as judged by kcat/KMfos. This result is consistent with the fact that residues thought to be involved in GSH binding, not the recognition of fosfomycin, were chosen for analysis. The largest deficit in catalytic efficiency with respect to kcat/KMfos (600-fold) was recorded in the absence of K+ ion, a fact that may suggest multiple roles of K+ in catalysis.
In vivo efficacy of FosA and its mutants
A biological test of the efficacy of the enzyme is how well the protein performs in conferring resistance to fosfomycin in E. coli. The bacterium is quite sensitive to fosfomycin with an MIC100 of <0.025 mg/mL in agar (13). The MIC100 values observed for most mutant proteins described here exhibit marked decreases in their ability to confer resistance to fosfomycin when the expression plasmids are transformed into E. coli. The greatest changes are seen in the W34A, Y39F, K90A, and R93A mutants and the K-loop mutant (Table 1). These five mutants displayed MIC values of less than 1 mg/mL for the W34A and Y39F mutants and less than 2 mg/mL for the K90A and R93A mutants. There is a loose correlation between the mutant kcat/KMGSH and the value of the MIC. Typically, mutants with a kcat/KMGSH less than 103 M−1s−1 have very low MIC values (< 2 mg/mL), while those above 103 M−1s−1 have much greater MIC values (usually as high as native FosAPA, 20 mg/mL). There are two exceptions to this. The Q36N and S50A mutants have respectable values of kcat/KMGSH that are ≥ 104 M−1s−1, but relatively low MIC values of ≤ 4 mg/mL). Both of these mutants have high turnover numbers but also high KdGSH.
DISCUSSION
Model of the GSH binding site
The model of the GSH binding site generated by manual docking and energy minimization of the docked complex suggests that there are six residues that contribute hydrogen-bonding or ionic interaction with GSH and six others that are within van der Waals contact of the substrate. The quality of the model was assessed experimentally by comparison of the kinetic, binding and biological data of the mutants predicted to be involved in ionic or hydrogen-bonding interactions with GSH. To initiate the discussion it is useful to point out a recent paper that utilized targeted saturation mutagenesis of FosAPA to determine essential, near essential and non-essential residues for fosfomycin resistance in E. coli (27). The 24-targeted residues in this particular study were selected based on their proximity to the active site as revealed by the published crystal structure of the protein (14). Of the 12 residues identified in the model of the GSH binding site presented here, ten were among the test set for the saturation mutagenesis study and seven of the ten were determined to be either “essential” (W34, Y39, H64, R119) or “near essential” (K90, C48, Y128). So, the structural model for the GSH binding site appears to be largely, though not completely, consistent with the previous saturation mutagenesis studies (27).
Activation of GSH
The FosA proteins contain several tyrosine residues near the active site that could serve to facilitate the ionization of GSH during catalysis. Two of these, Y62 and Y100, are conserved throughout the fosfomycin resistance protein family and have previously been implicated in the binding of fosfomycin (28). In addition to the strictly conserved tyrosine residues, Y39 seemed a good candidate for participation in the ionization of the thiol of GSH due to its position proximal to the thiol group of GSH in the docked model. In addition, this residue is conserved in the FosB group of resistance proteins that utilize L-cysteine rather that GSH (29). Available data from canonical GSH transferases indicates that hydrogen bonding to the thiol can lower the pKa of the thiol (26). This interaction is often assisted by secondary interactions with other hydrogen-bonding partners. The potential hydrogen-bonding interaction between the hydroxyl groups of Y39 and Y128, conserved in the FosA proteins, is consistent with a second-sphere interaction that assists in the deprotonation of the thiol of GSH via Y39. It is interesting to note that Y128 was identified as a “near essential” residue in the targeted saturation mutagenesis study that is cited above (27). A 13-fold reduction in activity was observed for the Y39F mutant. While this reduction is not as great as seen in equivalent mutants of the canonical GSH transferases, the activation of the oxirane ring of fosfomycin by the Mn(II) center increases the electrophilicity of the oxirane carbon and could therefore decrease the influence of thiolate anion formation on catalysis (30).
The coupling of GSH and K+ binding
In the model of the GSH binding site, other residues forming intermolecular hydrogen bonds to the docked substrate include R93, K90, S50, Q36, and W34 (Fig. 1). Of the residues that interact with GSH, K90 and R93 are particularly interesting as they both are proposed to ion-pair with the terminal carboxyl of GSH, a feature that is common in many GSH binding proteins. In addition, these residues are located near the K-loop of the enzyme (Figure 4) suggesting that K+-binding and the binding of GSH may be coupled. Mutation of either of these residues, the absence of a monovalent cation, or the mutational disruption of the K-loop results in a substantial decrease in kcat/KMGSH, KdGSH, and the ability of the enzyme to confer resistance to fosfomycin. Although the binding of K+ to the enzyme is known to be required for full activity, the exact role of K+ in catalysis has remained elusive. The crystal structure of the enzyme (14) suggested that the potassium ion served to orient the phosphonate of fosfomycin in the active site. The results presented here indicate that the role of K+ and the K-loop may be more complex and perhaps also involves the proper orientation of K90 and R93 to assist in the binding of GSH. These results indicate that both K90 and R93 are crucial for fully functional enzyme. This is in contrast to the previous observation that R93 is a “non-essential” residue (27).
FIGURE 4.

Stereo-view of the interaction of GSH with the K-loop of FosAPA. The Mn(II) and K+-ions are shown as purple and orange spheres, respectively.
The W34A mutant was deficient in catalysis and the ability to confer resistance. It also tended to precipitate during titrations with GSH suggesting that the mutant protein was unstable. The results with the W34A mutant are consistent with the previous identification of W34 as an “essential” residue (27). However, the W34H mutant exhibited good catalytic properties and conferred substantial resistance to fosfomycin in E. coli, suggesting that W34, while important, is not absolutely essential.
Although the Q36N mutant exhibited kcat/KMGSH close to that of the native enzyme it had a greatly elevated KdGSH and conferred only a modest degree resistance in E. coli. This result is consistent with the proposed role of the side-chain of Q36 in binding GSH (Figure 1). This residue was not targeted for saturation mutagenesis in the previous study (27), but appears to be essential or near-essential for biological function based on the results of the present study.
The role of the hydroxyl group of S50 in catalysis and the biological function of the enzyme remains unclear. The computational model indicates that the side-chain hydroxyl group is within hydrogen-bonding distance of the γ-glutamyl-carbonyl of GSH. Indeed, the S50A mutant has a very elevated KdGSH. However, the enzyme is as catalytically efficient as the native enzyme, but is deficient in fosfomycin resistance in the agar assay used in this work. Interestingly, S50 was designated as a “non-essential” residue in the saturation mutagenesis study (27). The reasons for this apparent discrepancy may be the differences in agar assays used in each study. The assay used in this work included glucose-6-phosphate, which enhances the uptake of the antibiotic.
Correlation of the kinetics, binding data, and biological activity
There is a reasonable correlation of the kcat/KMGSH and the KdGSH with the ability of most of the mutants to confer resistance to fosfomycin in E. coli. However the correlation is far from perfect. This fact reflects the complexity of the interactions necessary for an efficient enzyme including the binding of Mn(II), K+ and fosfomycin. There are also obvious differences in the conditions for the in vitro assays for catalytic activity and the in vivo conditions for the antibiotic assay. It is true that, in general, the effects of the mutations on kcat/KMGSH and KdGSH are more pronounced than their effects on kcat/KMfos. This is not unexpected since the mutations were designed to target residues proposed to be involved in the binding of GSH and not fosfomycin.
Conclusions
The kinetic and functional properties of site-specific mutants and previous work on targeted saturation mutagenesis (27) lends solid experimental support to a computationally derived GSH binding site. The results also indicate that the binding of K+ to the K-loop may also be important for the orientation of the K90 and R93 residues that are necessary for effective binding of GSH.
Supplementary Material
Footnotes
This work was supported by Grants R01 AI042756, T32 ES007028, and P30 ES000267 from the National Institutes of Health.
Abbreviations used: AMBER, Assisted Model Building with Energy Refinement; AQC, AccQ-FluorTM fluorescent derivatizing reagent; DEAE, (diethylamino)ethyl resin; EDTA, ethylenediaminetetraacetic acid; FosA, fosfomycin resistance protein A; FosB, fosfomycin resistance protein B; GSH, glutathione; HA, hydroxylapetite resin, HEPES, N-(2-hydroxethyl)piperazine-N′-ethanesulfonic acid; MIC, minimum inhibitory concentration; MOPS, 3-(N-morpholino)propanesulfonic acid; MurA, UPD-N-acetylglucosamine 3-enolpyruval transferase; RESP, restrained electrostatic potential; TMA, trimethylamine.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Walsh C. Where will new antibiotics come from? Nat Rev Microbiol. 2003;1:65–70. doi: 10.1038/nrmicro727. [DOI] [PubMed] [Google Scholar]
- 2.Hall BG. Innovation - Anti-infectives - Predicting the evolution of antibiotic resistance genes. Nat Rev Microbiol. 2004;2:430–435. doi: 10.1038/nrmicro888. [DOI] [PubMed] [Google Scholar]
- 3.Levy SB, Marshall B. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med. 2004;10:S122–S129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 4.Hendlin D, Stapley EO, Jackson M, Wallick H, Miller AK, Wolf FJ, Miller TW, Chaiet L, Kahan FM, Foltz EL, Woodruff HB. Phosphonomycin, a new antibiotic produced by strains of Streptomyces. Science. 1969;166:122–123. doi: 10.1126/science.166.3901.122. [DOI] [PubMed] [Google Scholar]
- 5.Christensen B, Leanza WJ, Beattie TR, Patchett AA, Arison BH, Ormond RE, Kuehl FA, Alberssc G, Jardetzk O. Phosphonomycin - Structure and synthesis. Science. 1969;166:123–124. doi: 10.1126/science.166.3901.123. [DOI] [PubMed] [Google Scholar]
- 6.Kahan FM, Kahan JS, Cassidy PJ, Kropp H. Mechanism of action of fosfomycin (phosphonomycin) Ann N Y Acad Sci. 1974;235:364–386. doi: 10.1111/j.1749-6632.1974.tb43277.x. [DOI] [PubMed] [Google Scholar]
- 7.Kim DH, Lees WJ, Kempsell KE, Lane WS, Duncan K, Walsh CT. Characterization of a Cys115 to Asp substitution in the Escherichia coli cell wall biosynthetic enzyme UDP-GlcNAc enolpyruvyl transferase (MurA) that confers resistance to inactivation by the antibiotic fosfomycin. Biochemistry. 1996;35:4923–4928. doi: 10.1021/bi952937w. [DOI] [PubMed] [Google Scholar]
- 8.Kadner RJ, Winkler HH. Isolation and characterization of mutations affecting the transport of hexose phosphates in Escherichia coli. J Bacteriol. 1973;113:895–900. doi: 10.1128/jb.113.2.895-900.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsuruoka T, Yamada Y. Characterization of spontaneous fosfomycin-resistant cells of E. coli in vitro. J Antiobiot. 1975;28:906–911. doi: 10.7164/antibiotics.28.906. [DOI] [PubMed] [Google Scholar]
- 10.Garcialobo JM, Ortiz JM. Tn2921, A Transposon Encoding Fosfomycin Resistance. J Bacteriol. 1982;151:477–479. doi: 10.1128/jb.151.1.477-479.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arca P, Rico M, Brana AF, Villar CJ, Hardisson C, Suarez JE. Formation of an adduct between fosfomycin and glutathione - A new mechanism of antibiotic-resistance in bacteria. Antimicrob Agents Chemother. 1988;32:1552–1556. doi: 10.1128/aac.32.10.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suarez JE, Mendoza MC. Plasmid-encoded fosfomycin resistance. Antimicrob Agents Chemother. 1991;35:791–795. doi: 10.1128/aac.35.5.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bernat BA, Laughlin LT, Armstrong RN. Fosfomycin resistance protein (FosA) is a manganese metalloglutathione transferase related to glyoxalase I and the extradiol dioxygenases. Biochemistry. 1997;36:3050–3055. doi: 10.1021/bi963172a. [DOI] [PubMed] [Google Scholar]
- 14.Rife CL, Pharris RE, Newcomer ME, Armstrong RN. Crystal structure of a genomically encoded fosfomycin resistance protein (FosA) at 1.19 angstrom resolution by MAD phasing off the L-III edge of Tl. J Am Chem Soc. 2002;124:11001–11003. doi: 10.1021/ja026879v. [DOI] [PubMed] [Google Scholar]
- 15.Bernat BA, Laughlin LT, Armstrong RN. Elucidation of a monovalent cation dependence and characterization of the divalent cation binding site of the fosfomycin resistance protein (FosA) Biochemistry. 1999;38:7462–7469. doi: 10.1021/bi990391y. [DOI] [PubMed] [Google Scholar]
- 16.Smoukov SK, Telser J, Bernat BA, Rife CL, Armstrong RN, Hoffman BM. EPR study of substrate binding to the Mn(II) active site of the bacterial antibiotic resistance enzyme FosA: A better way to examine Mn(II) J Am Chem Soc. 2002;124:2318–2326. doi: 10.1021/ja012480f. [DOI] [PubMed] [Google Scholar]
- 17.Walsby CJ, Telser J, Rigsby RE, Armstrong RN, Hoffman BM. Enzyme control of small-molecule coordination in FosA as revealed by P-31 pulsed ENDOR and ESE-EPR. J Am Chem Soc. 2005;127:8310–8319. doi: 10.1021/ja044094e. [DOI] [PubMed] [Google Scholar]
- 18.Bernat BA, Laughlin LT, Armstrong RN. Regiochemical and stereochemical course of the reaction catalyzed by the fosfomycin resistance protein, FosA. J Org Chem. 1998;63:3778–3780. [Google Scholar]
- 19.Dunbrack RL, Karplus M. Backbone-Dependent Rotamer Library For Proteins - Application To Side-Chain Prediction. J Mol Biol. 1993;230:543–574. doi: 10.1006/jmbi.1993.1170. [DOI] [PubMed] [Google Scholar]
- 20.Wang J, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J Comput Chem. 2000;21:1049–1074. [Google Scholar]
- 21.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA. AMBER. Vol. 8. University of California; San Francisco: 2004. [Google Scholar]
- 22.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA, Jr, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Baboul AG, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Gonzalez C, Head-Gordon M, Replogle ES, Pople JA. Gaussian 98. Gaussian, Inc; Pittsburgh, PA: 1998. Revision A.9. [Google Scholar]
- 23.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A well-behaved electrostatic potential based method using charge restrains for determining atom-centered charges: the RESP model. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 24.Rigsby RE, Fillgrove KL, Beihoffer LA, Armstrong RN. Fosfomycin Resistance Proteins: A Nexus of Glutathione Transferases and Epoxide Hydrolases in a Metalloenzyme Superfamily. Methods Enzymol. 2005;401:367–379. doi: 10.1016/S0076-6879(05)01023-2. [DOI] [PubMed] [Google Scholar]
- 25.Copeland Robert A. Enzymes. Vol. 104. Wiley-VCH; New York: 2000. p. 136. [Google Scholar]
- 26.Armstrong RN. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem Res Toxicol. 1997;10:2–18. doi: 10.1021/tx960072x. [DOI] [PubMed] [Google Scholar]
- 27.Beharry Z, Palzkill T. Functional analysis of active site residues of the fosfomycin resistance enzyme FosA from Pseudomonas aeruginosa. J Biol Chem. 2005;280:17786–17791. doi: 10.1074/jbc.M501052200. [DOI] [PubMed] [Google Scholar]
- 28.Rigsby RE, Rife CL, Fillgrove KL, Newcomer ME, Armstrong RN. Phosphonoformate: A minimal transition state analogue inhibitor of the fosfomycin resistance protein, FosA. Biochemistry. 2004;43:13666–13673. doi: 10.1021/bi048767h. [DOI] [PubMed] [Google Scholar]
- 29.Cao M, Bernat BA, Wang Z, Armstrong RN, Helmann JD. FosB, a cysteine-dependent fosfomycin resistance protein under the control of σw, an extracytoplasmic function σ factor in Bacillus subtilis. J Bacteriol. 2001;183:2380–2383. doi: 10.1128/JB.183.7.2380-2383.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu S, Zhang P, Ji X, Johnson WW, Gilliland GL, Armstrong RN. Contribution of Tyrosine 6 to the Catalytic Mechanism of Isoenzyme 3-3 of Glutathione S-Transferase. J Biol Chem. 1992;267:4296–4299. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.