

Abstract
A tandem Diels-Alder/azido Schmidt reaction sequence provides rapid access to the core skeleton shared by several Stemona alkaloids including stenine, neostenine, tuberstemonine, and neotubererostemonine. The discovery and evolution of inter- and intramolecular variations of this process and their applications to total syntheses of (±)–stenine and (±)–neostenine is described. The stereochemical outcome of the reaction depends on both substrate type and reaction condition, enabling the preparation of both (±)–stenine and (±)–neostenine from the same diene/dienophile combination.
Keywords: Total synthesis, Stemona alkaloids, Diels-Alder, azides, Schmidt reaction
Chinese and Japanese traditional medicines have for centuries utilized extracts of stemonaceous plants as remedies for the treatment of respiratory ailments. These extracts and the isolated Stemona alkaloids have been associated with insecticidal, anthelmintic, antitussive and various neurochemical effects, although mechanisms have rarely been identified.1 Recently, interest in these alkaloids was further piqued by the demonstration of effective in vivo activity of two skeletally-related Stemona alkaloids, neostenine 3 and neotuberostemonine 4, against citric acid-induced cough in guinea pig animal models.2 In addition, the Stemona alkaloid tuberstemonine 2 has demonstrated inhibitory activity on excitatory transmission at the crayfish neuromuscular junction.3 The Stemona alkaloids have attracted substantial interest from synthetic chemists partly because of these links to biological activity and partly from their challenging structural complexity. Stenine has been the focus of several successful synthetic efforts4 and has inspired a number of synthetic approaches.5 In addition, tuberostemonine 2 was synthesized by Wipf.6 However the stenine isomer, neostenine 3, had not yet been prepared via total synthesis at the outset of this project.7
A noteworthy challenge in any stenine synthesis is the construction of the B ring, which is fused to three additional rings. In addition, each of its carbon atoms is a stereogenic center. This issue was addressed using an intramolecular Diels–Alder cyclization in three out of the four first-published syntheses of this target (Scheme 1; the stenine numbering system used throughout is that presented in a recent review1f). The first synthesis of stenine by Hart in 1990 not only set the precedent for utilizing a Diels–Alder approach to this target, but also established an iodolactonization/Keck allylation sequence as a solution to the problem of stereoselective ethyl group installation.4a,b Morimoto utilized a chiral oxazoline-based intramolecular Diels–Alder cyclization of 5 to synthesize the naturally occurring enantiomer of stenine.4c-e Padwa applied an impressive Diels–Alder/ring opening/1,2-methylthioshift cascade to append the B and D rings onto an existing 7-membered C ring in a single operation.4g,h Of all the completed syntheses to date, only the route used by Wipf does not employ a Diels–Alder approach for the construction of the cyclohexane ring.4f These workers utilized the selective reduction of a π-allyl palladium complex of the indolone 6, which was readily synthesized from L-tyrosine and converted into the natural enantiomer of stenine in 22 steps.
Scheme 1.

Our own interest in Stemona alkaloid synthesis arose from the recognition that the 7-membered C ring could arise from an intramolecular Schmidt reaction8 of an azide such as 8 (Scheme 2). This step would also form the D ring of stenine while opening the door to constructing 8 via an intramolecular Diels–Alder reaction similar to Hart's synthesis (and sharing the same basic disconnection with Morimoto's route). In this full account, we describe the pursuit of this strategy, which led to the discovery that both the Diels–Alder and the Schmidt reaction steps could be accomplished in a single chemical operation. This not only streamlined the stenine synthesis, but also led to the development of a general synthetic methodology based on this tandem reaction.9 Furthermore, we describe how the reconsideration of possible Diels–Alder routes to cis-1-decalones permitted a second generation, extremely efficient approach to stenine and ultimately to the total synthesis of the antitussive agent neostenine.
Scheme 2.

Results and Discussion
First Generation Approach: A Formal Synthesis of Stenine
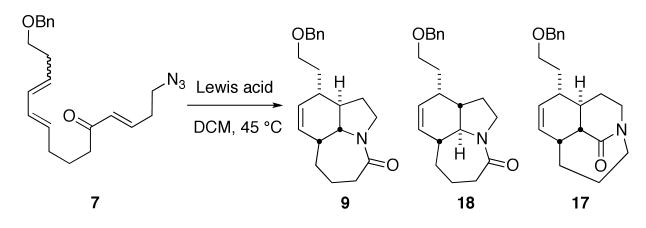
In its original formulation, the total synthesis of stenine was built around the idea of carrying out an intramolecular Diels–Alder reaction on a substrate like 7 (Scheme 2). Although we considered the possibility that this step could be combined with its subsequent intramolecular Schmidt reaction, as both reactions entailed the use of Lewis acid promotion, there was at the time no experimental evidence that such a step would be possible. A key element of this plan was that the intramolecular Diels–Alder reaction would occur via an endo transition state, an outcome for which precedent existed.10
The requisite azido triene 7 was constructed using standard means. The building blocks 13 and 14 were prepared from 1,3-propanediol and 1,5-pentanediol, accordingly (Scheme 3). A modified Julia coupling11 between sulfone 13 and aldehyde 14 afforded 15 as an inseparable 85:15 mixture of isomers at the new double bond. Although it was not possible to completely remove the undesired cis isomer at any step prior to the Diels–Alder, we expected the E, Z diene to be less reactive in the downstream cycloaddition reaction. Removal of the silyl protecting group of 15, followed by Swern oxidation, gave an aldehyde that was treated with the lithium anion of dimethyl methylphosphonate. This provided a β-hydroxyphosphonate that was subsequently oxidized with TPAP/NMO to give the β-oxophosphonate 16. This sequence gave better overall yields than an alternative path that entailed initial conversion to the corresponding carboxylic ester followed by lithiomethyl dimethylphosphonate addition. The resulting β-oxophosphonate 16 was subjected to a Horner-Wadsworth-Emmons reaction12 with 3-azidopropanal13 to afford the triene 7 in 85% yield.
Scheme 3.

Our earliest attempts to carry out a combined Diels–Alder/Schmidt reaction resulted only in the isolation of a single product in modest yield (16–38%). Specifically, treatment of 7 with 1.5 equiv of Et2AlCl led to gas evolution upon heating the reaction to ca. 45 °C. The first product isolated from this reaction had an absorption in the IR spectrum at 1680 cm-1 and a 13C NMR signal at 188 ppm, with no trace of the diagnostic azide IR absorption near 2100 cm-1. These data permitted the assignment of this material as a bridged lactam product (compound 17, Table 1).14 Upon closer examination, the desired fused amide 9 along with a stereoisomer 18 were also found; both of these lactams were considerably more polar than 7 and required more polar chromatography conditions for isolation. Following a series of optimization experiments, we settled on treatment of 7 with 1 equiv of MeAlCl2 in refluxing dichloromethane, which afforded the tricyclic lactam 9 in 43% yield and its bridged and fused isomers 17 and 18 in a combined yield of 36% yield (Table 1). Assuming that only the major component of the 85:15 mixture of 11,12-olefin geometry isomers of 7 reacts, this yield corresponds to an overall conversion of the reactive trans-trans triene isomer to lactam 9 of 51% yield. Only poor yields of the desired lactam were obtained using other non-aluminum-based Lewis acids.
Table 1.
Selected optimization trials for the tandem Diels–Alder/azido Schmidt reaction.
 | ||||||
|---|---|---|---|---|---|---|
| entry | Lewis acid | Lewis acid equiv | 9 yield (%) | 18 yield (%) | 17 yield (%) | combined yield (%) |
| 1 | InCl3 | 1.0 × 2 | 20 | 18 | 22 | 60 |
| 2 | AlMe3 | 1.0 | - | - | - | 0 |
| 3 | Et2AlCl | 0.65 × 2 | 28 | 12 | 20 | 60 |
| 4 | Et2AlCl | 1.0 | 41 | 9 | 28 | 78 |
| 5 | MeAlCl2 | 1.0 | 43 | 12 | 24 | 79 |
The outcomes of the tandem Diels–Alder/Schmidt reaction for triene substrate 7 arise as shown in Scheme 4. Both the desired lactam product 9 and the bridged lactam 17 are obtained from the same Diels–Alder intermediate formed via an endo transition state.10 Following azide addition to carbonyl and assuming antiperiplanar C→N bond migration,15 an intermediate containing an equatorial N2+ group would afford lactam 9, whereas an axially oriented leaving group would give the bridged compound 17. Lactam 18 results from an exo transition state in the Diels–Alder cyclization, followed by the D-ring-forming/C-ring-expansion process. Interestingly, no bridged adduct is formed from the exo Diels–Alder product because the azidohydrin intermediate bearing an axial N2+ group is antiperiplanar to a hydroxyl group instead of a migratable carbon.
Scheme 4.

We briefly attempted to improve the overall conversion of triene to lactam by deliberately carrying out the Diels–Alder and the Schmidt reactions separately (Scheme 5). The former step could be nicely optimized, but deprotection of the hydroxy group with PPTS led to epimerization of the cis-decalone 20 to the undesired trans isomer. This was not an issue at all in the combined reaction, suggesting another advantage in using the domino procedure. While this work was in progress, Jung and coworkers reported the successful synthesis of an advanced intermediate of stenine using a similar Diels–Alder reaction to that in Scheme 4, followed by a four-step Beckmann rearrangement/N-alkylation sequence to form the BCD ring skeleton of stenine.5d
Scheme 5.

The formal synthesis of stenine was finished as shown in Scheme 6. Removal of the benzyl ether from 9, oxidation of the resulting hydroxy group, and iodolactonization gave butyrolactone 23 in 80% yield from 9. Keck allylation16 followed by methylation of the lactone proceeded stereoselectively to provide the Hart intermediate 24 in 67% yield over two steps. Cleanly carrying out the methylation of the lactone required a little work. Initial attempts using up to 2 equiv of LDA to deprotonate the lactone resulted in mainly unreacted starting material. The ability of LHMDS to deprotonate lactones is documented,17 however allowing this enolate to react with 10 equiv of MeI for 1.5-2.0 h at -78 °C afforded predominantly the dimethylated derivative. Finally it was determined that treatment of the lactone 23 with 1.8 equiv of LHMDS at -78 °C, followed by the addition of 10 equiv of MeI at the same temperature for 40 min afforded exclusively the monomethylated product 24 in 72% yield for the alkylation step and completed the formal synthesis of stenine.18 In Hart's synthesis of stenine,4a,b 24 was carried on to the stenine 1 in four steps and 63% yield. Overall, this first generation formal synthesis, including the final four steps as reported by Hart,4a,b would require 21 steps from commercially-available material and afford stenine in 7.2% overall yield. This would constitute the highest overall yield for known syntheses at the time (range 0.9–3.0%) but the route was longer than the shortest known route at the time (Padwa's; 16 steps4g,h).
Scheme 6.

Second-Generation Synthesis of (±)-Stenine
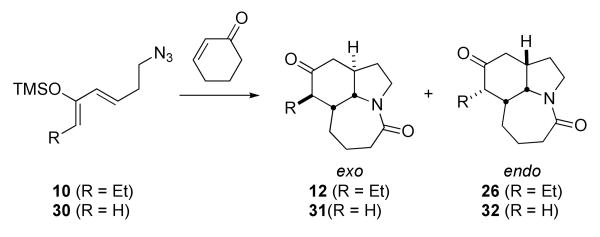
Having completed this synthetic effort, our attention turned to the further development of the tandem Diels–Alder/intramolecular Schmidt reaction. In particular, we learned that the sequence could nicely accommodate intermolecular Diels–Alder reactions.9 Also, the 2003 report that neostenine exhibited strong antitussive activity in a guinea pig model2a provided strong motivation to revisit the problem of Stemona alkaloid synthesis in general, with an eye toward practical routes that would be amenable to analogue synthesis. The possibility of using an intermolecular Diels–Alder/intramolecular Schmidt sequence was especially attractive because it would require many fewer steps in starting material preparation than our first-generation route. We accordingly contemplated a Diels–Alder disconnection between C-9/C-10 and C-9a/C-1, which retrosynthetically leads to cyclohex-2-en-1-one and silyloxydiene 10 as starting materials (Scheme 2). This route would additionally allow early incorporation of the ethyl side chain, obviating the need for a multistep removal of the terminal ethylene from an allylated precursor (i.e., 24→1, Scheme 6).
These tenets were rapidly verified and an interesting stereochemical situation revealed through the experiments shown in Scheme 7. Thus, the known19 Horner-Wadsworth-Emmons reagent 25 was prepared in 99% yield from commercially-available dimethyl methylphosphonate and butyryl chloride. Olefination of 3-azidopropanal13b (available in a single step from acrolein and HN3) afforded an enone that was readily converted to the corresponding trimethylsilyloxy diene 10. Treatment of cyclohexenone with SnCl4 and diene 10 afforded a ca. 3:1 ratio of Diels–Alder/Schmidt adducts 12 and 26, with the former compound – arising from an exo-selective Diels–Alder step – predominating.
Scheme 7.

This quick success was gratifying as it permitted the preparation of a key intermediate containing three rings and four stereocenters in the targeted compound in only four steps from very simple starting materials. However, the stereochemical outcome of this sequence determines which ultimate target can be obtained using it, as shown in Scheme 8. Thus, the exo approach observed here maps the four centers obtained in compound 12 nicely onto stenine, whereas the alternative endo reaction would find utility in a synthesis of the isomeric neostenine 3, pending a successful epimerization of the ethyl group along the way. Predominant exo selectivity in Diels–Alder reactions of cyclic dienophiles has been previously noted by Corey and coworkers20 and is here likely due to significant steric interactions between one of the γ protons of the cyclohexenone with the incoming nucleophilic silyl enol ether.
Scheme 8.

The completion of the synthesis is shown in Scheme 9. All of the additional stereocenters were generated by highly selective substrate-directed reactions: axially directed alkylation and reduction reactions on purified 12 afforded compounds 27 and 28, respectively. An X-ray crystal structure of lactone 28 verified the stereostructure shown (see Supporting Information). The installation of the final methyl group and removal of the lactam carbonyl were carried out as previously established. Thus, alkylation of the lactone 28 proceeded smoothly to give the known oxostenine 29; reduction via the thiolactam as reported by others afforded stenine 1.4a,b,f-h,6 The spectrum of the natural product thus prepared fully matched those of the literature values. Overall, the total synthesis was accomplished in nine steps from commercially-available reagents and 14% overall yield.21
Scheme 9.

Stereochemical Divergence of the Diels–Alder/Schmidt Reaction: Syntheses of Neostenine and 13-Epineostenine
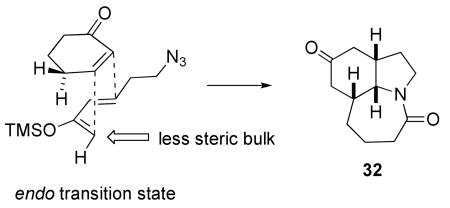
The analysis in Scheme 8 indicated that modification of the stereochemical outcome of the Diels–Alder/Schmidt reaction from exo to endo selectivity could lead to a viable neostenine intermediate. Accordingly, we investigated the effect of changing diene structure and Lewis acid on the stereochemistry of the key step (Table 2). As used above, SnCl4 gave the best overall result, yielding Diels–Alder/Schmidt product in 70% yield as a 3:1 mixture of exo and endo isomers. Predominant endo stereochemistry could be obtained in two ways. First, the use of a less strongly coordinating Lewis acid such as BF3•OEt2 gave exclusively endo Diels–Alder product in modest yield, albeit as a mixture of isomers at the ethyl group (resulting from partial epimerization). Alternatively, the silyloxy diene 30, lacking the ethyl side chain, also afforded predominantly the endo lactam 32.
Table 2.
Stereochemical studies of the Diels–Alder/Schmidt sequence.
 | ||||
|---|---|---|---|---|
| entry | diene | Lewis acid | product(s) | yield (exo/endo ratio) |
| 1 | 10 | SnCl4 | 12, 26 | 70% (3:1) |
| 2 | 10 | BF3•OEt2 | 26 | 55% (endo only; mixture of ethyl epimers) |
| 3 | 30 | SnCl4 | 31, 32 | 82% (1:3.4) |
It is likely that the removal of the ethyl group from the diene affords predominantly endo product due to the easing of steric interactions between the diene and the cyclohexenone (eq 1). A dependence of stereochemistry on diene substitution was previously registered by Corey and coworkers, who analyzed it in the context of changes in diene size and conformation.20 The reason for the switch in stereochemistry due to the use of BF3•OEt2 is less clear, although it could be due to a lengthening of the bond forming between the β carbon of the enone and the silyl enol ether in the transition state leading to 26.
 |
(1) |
The endo-selective tandem Diels–Alder/Schmidt reaction was scaled up as shown in Scheme 10. Addition of 1.5 equiv of silyloxy diene 10 to a mixture of cyclohex-2-en-1-one and 1 equiv BF3•OEt2 at -78 °C, followed by the further addition of 1.5 equiv of BF3•OEt2 after warming to ca. -30 °C provided the tricyclic lactams 26a,b as a mixture of ethyl epimers in 43% yield, the major component being the expected isomer 26a (Scheme 7, α ethyl isomer). Under these conditions, we also isolated an azide-containing mixture to which we assigned structure 33 as the major component, based mainly on the 13C NMR spectrum of the mixture (diagnostic ketone peaks at 212.1 and 213.0 ppm). Treatment of a CH2Cl2 solution of this mixture with TiCl4 afforded an additional quantity (12% yield based on 10) of the tricyclic lactams 26a,b. Attempts to improve the one-pot yield of 26a,b through longer reaction times, heating of the reaction to reflux or the addition of one equivalent of TiCl4 to the reaction mixture after allowing to stir at room temperature all gave lower isolated yields of 26a,b. MM2 calculations suggested that 26a, containing a pseudoaxial methyl group, would be 4.4 kcal/mol higher in energy relative to the desired, equatorial isomer 26b. Thus, the keto lactam mixture readily converged onto 26b upon base treatment.
Scheme 10.

Ketone 26b was regio- and stereospecifically alkylated with ethyl bromoacetate to give ketoester 34 (Scheme 11). Initial attempts to reduce 34 with NaBH4 gave an inseparable mixture of lactone products in 54% yield (35a and 35b, ratios not rigorously determined). A screen of reducing reagents revealed that the combination of CeCl3 and L-Selectride gave a complex mixture of products that contained a pair of chemical shifts in the 13C NMR spectrum at 97.7 and 98.4 ppm. Based on these downfield signals we proposed that the mixture contained the diastereomeric lactols 36 shown in Scheme 11. This was confirmed by TPAP oxidation of the mixture, which led smoothly to a single lactone 35a.
Scheme 11.

Neostenine was prepared by the methylenation/hydrogenation sequence shown in Scheme 12. Greene and coworkers have developed a two-step sequence for the methylenation of esters and lactones involving initial formation of an α-carboxylic acid followed by condensation with formaldehyde and subsequent decarboxylation.22 We found that this sequence gave better overall yields to the more common Eschenmosher salt alkylation method.23 The α-carboxylic acid intermediate was not purified, but used directly in the condensation/decarboxylation sequence to provide methylene lactone 37 in 49% isolated yield. Hydrogenation over Adams catalyst in methanol/acetic acid (1:1 mixture) gave a single methylated lactone 38 in 91% yield, while hydrogenation over palladium on carbon produced a mixture containing traces of the epimeric methyl isomer (compound 39; see Scheme 13). Selective thioamide formation was achieved using a P2S10 method developed by Curphy,24 which gave a more easily-purified reaction mixture than Lawesson's reagent. Reduction of the thioamide with Raney nickel proceeded smoothly to give racemic neostenine 3 in 93% yield. Spectral comparison of the synthetic material to reported values confirmed its identity.25 X-ray crystallography of our synthetic material unambiguously showed it to be identical to the reported structure of the natural product. Overall, the total synthesis was accomplished in thirteen steps from commercially-available reagents and 10% overall yield.
Scheme 12.

Scheme 13.

Since one goal of our global program is the preparation of stenine analogues for biological screening, lactone 35a was converted to the methyl epimer of neostenine via the three-step sequence shown in Scheme 13. Alkylation with LiHMDS and methyl iodide provided the single methyl lactone 39 in 89% yield. As expected, this alkylation provided the C-13 epimer of neostenine due to approach of the alkylating agent form the convex face formed by the cis-fused AB ring junction. Compound 39 was converted to 13-epineostenine 40 analogous to the method used above24 for the reduction of the lactam carbonyl to a tertiary amine. The high-resolution mass spectrum of 40 and its 1H and 13C NMR spectra were consistent with a stereoisomer of the natural product, which we have assigned as the previously unknown 13-epineostenine.
Conclusion
The productive relationship between synthetic methodology development and the total synthesis of natural products has been much discussed and is well illustrated in the work described in this paper. The first-generation total synthesis of stenine demonstrated the first combination of an intramolecular Diels–Alder reaction and an intramolecular Schmidt reaction. This discovery spurred on additional development of this useful synthetic method, described elsewhere,9 which in turn led us to unusually efficient syntheses of two natural products (stenine and neostenine) and one novel analogue (13-epineostenine). Although not discussed here at all, even an unanticipated side-product of the main reaction, bridged bicyclic lactam 17, led to a separate research project directed toward the understanding of the unusual chemical properties of this previously unknown class of “twisted amides”.26
These projects required the optimization of different stereochemical outcomes of the intermolecular Diels–Alder/intramolecular Schmidt domino reaction, a very useful aspect of the sequence that we are currently using in a broader program to discover fully synthetic analogues of these alkaloids. In addition, the brevity of these routes have provided sufficient quantities of stenine congeners that will be used to investigate the as yet unknown mode of action of the observed antitussive activity of some of these alkaloids. This work will be published in due course.
Supplementary Material
Figure 1.

Selected Stemona alkaloids.
Acknowledgments
We acknowledge Professor Ge Lin for generously providing detailed NMR spectra of neostenine for comparison. We thank the National Institute of General Medical Sciences (GM-49093 and PO50-GM069663) for financial support, Benjamin Neuenswander for HPLC-MS, David Vander Velde and Sarah Neuenswander for NMR assistance and Douglas Powell and Victor Day for X-ray crystallography. J.E.G. gratefully acknowledges the Madison A. and Lila Self Fellowship Program for its support.
Footnotes
Supporting Information Available. Experimental details and characterization data for all new compounds, including X-ray structures (CIF files) of 3 and 28. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For reviews see: Götz M, Edwards OE. In: The Alkaloids. Manske RHF, editor. Vol. 9. Academic Press: New York; 1967. pp. 545–551.Götz M, Strunz GM. In: Alkaloids. Wiesner K, editor. Vol. 9. Butterworth; London: 1973. pp. 143–160.Lin WH, Ye Y, Xu RS. J Nat Prod. 1992;55:571–576.Xu RS. Stud Nat Prod Chem. 2000;21:729–772.Pilli RA, Ferreira de Oliveira MdC. Nat Prod Rep. 2000;17:117–127. doi: 10.1039/a902437i.Greger H. Planta Med. 2006;72:99–113. doi: 10.1055/s-2005-916258.Xu YT, Hon PM, Jiang RW, Cheng L, Li SH, Chan YP, Xu HX, Shaw PC, But PPH. J Ethnopharmacology. 2006;108:46–53. doi: 10.1016/j.jep.2006.04.022.
- 2.(a) Chung HS, Hon PM, Lin G, But PPH, Dong H. Planta Med. 2003;69:914–920. doi: 10.1055/s-2003-45100. [DOI] [PubMed] [Google Scholar]; (b) Leung PHH, Zhang L, Zuo Z, Lin G. Planta Med. 2006;72:211–216. doi: 10.1055/s-2005-916195. [DOI] [PubMed] [Google Scholar]
- 3.Shinozaki H, Ishida M. Brain Res. 1985;334:33–40. doi: 10.1016/0006-8993(85)90564-5. [DOI] [PubMed] [Google Scholar]
- 4.(a) Chen CY, Hart D. J Org Chem. 1990;55:6236–6240. [Google Scholar]; (b) Chen CY, Hart D. J Org Chem. 1993;58:3840–3849. [Google Scholar]; (c) Morimoto Y, Iwahashi M, Nishida K, Hayashi Y, Shirahama H. Angew Chem. 1996;108:968–970. [Google Scholar]; (d) Morimoto Y, Iwahashi M, Nishida K, Hayashi Y, Shirahama H. Angew Chem Int Ed Engl. 1996;35:904–906. [Google Scholar]; (e) Morimoto Y, Iwahashi M, Kinoshita T, Nishida K. Chem Eur J. 2001;7:4107–4116. doi: 10.1002/1521-3765(20011001)7:19<4107::aid-chem4107>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]; (f) Wipf P, Kim Y, Goldstein DM. J Am Chem Soc. 1995;117:11106–11112. [Google Scholar]; (g) Ginn JD, Padwa A. Org Lett. 2002;4:1515–1517. doi: 10.1021/ol025746b. [DOI] [PubMed] [Google Scholar]; (h) Padwa A, Ginn JD. J Org Chem. 2005;70:5197–5206. doi: 10.1021/jo050515e. [DOI] [PubMed] [Google Scholar]
- 5.(a) Morimoto Y, Nishida K, Hayashi Y, Shirahama H. Tetrahedron Lett. 1993;34:5773–5776. [Google Scholar]; (b) Morimoto Y, Iwahashi M. Synlett. 1995:1221–1222. [Google Scholar]; (c) Goldstein DM, Wipf P. Tetrahedron Lett. 1996;37:739–42. [Google Scholar]; (d) Jung SH, Lee JE, Joo HJ, Kim SH, Koh HY. Bull Korean Chem Soc. 2000;21:159–160. [Google Scholar]; (e) Booker-Milburn KI, Hirst P, Charmant JPH, Taylor LHJ. Angew Chem Int Ed Engl. 2003;42:1642–1644. doi: 10.1002/anie.200250507. [DOI] [PubMed] [Google Scholar]; (f) Zhu L, Lauchli R, Loo M, Shea KJ. Org Let. 2007;9:2269–2271. doi: 10.1021/ol070397c. [DOI] [PubMed] [Google Scholar]
- 6.(a) Wipf P, Spencer SR, Takahash H. J Am Chem Soc. 2002;124:14848–14849. doi: 10.1021/ja028603t. [DOI] [PubMed] [Google Scholar]; (b) Wipf P, Spencer SR. J Am Chem Soc. 2005;127:225–235. doi: 10.1021/ja044280k. [DOI] [PubMed] [Google Scholar]
- 7.Professor Kevin Booker-Milburn and coworkers have recently completed an independent synthesis of neostenine (personal communication).
- 8.(a) Aubé J, Milligan GL. J Am Chem Soc. 1991;113:8965–8966. [Google Scholar]; (b) Aubé J, Milligan GL, Mossman CJ. J Org Chem. 1992;57:1635–1637. [Google Scholar]; (c) Milligan GL, Mossman CJ, Aubé J. J Am Chem Soc. 1995;117:10449–10459. [Google Scholar]; (d) Desai P, Schildknegt K, Agrios KA, Mossman CJ, Milligan GL, Aubé J. J Am Chem Soc. 2000;122:7226–7232. [Google Scholar]
- 9.Zeng Y, Reddy S, Hirt E, Aubé J. Org Lett. 2004;6:4993–4995. doi: 10.1021/ol047809r. [DOI] [PubMed] [Google Scholar]
- 10.(a) Gras JL, Bertrand M. Tetrahedron Lett. 1979:4549–4552. [Google Scholar]; (b) Coe JW, Roush WR. J Org Chem. 1989;54:915–930. [Google Scholar]
- 11.(a) Blakemore PR, Cole WJ, Kocienski PJ, Morley A. Synlett. 1998:26–28. [Google Scholar]; (b) Kocienski PJ, Bell PR, Blakemore PR. Synlett. 2000:365–366. [Google Scholar]
- Paterson I, Yeung KS, Smaill JB. Synlett. 1993:774–776. [Google Scholar]
- 13.(a) Boyer JH. J Am Chem Soc. 1951;73:5248–5252. [Google Scholar]; (b) Ma Y. Heteroatom Chem. 2002;13:307–309. [Google Scholar]
- 14.For a review of bridged amides, see: Greenberg A. In: Structure and Reactivity. Liebman JF, Greenberg A, editors. VCH; New York: 1988. pp. 139–178.
- 15.Kishi Y, Goodman RM. J Am Chem Soc. 1998;120:9392–9393. and references therein. [Google Scholar]
- 16.Yates JB, Keck GE. J Am Chem Soc. 1982;104:5829–5830. and references therein. [Google Scholar]
- 17.Rathke MW. J Am Chem Soc. 1970;92:3222–3223. [Google Scholar]
- 18.Golden JE, Aubé J. Angew Chem Int Ed Engl. 2002;41:4316–4318. doi: 10.1002/1521-3773(20021115)41:22<4316::AID-ANIE4316>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 19.Khatri NA, Schmitthenner HF, Shringarpure J, Weinreb SM. J Am Chem Soc. 1981;103:6387–6393. [Google Scholar]
- 20.Ge M, Stoltz BM, Corey EJ. Org Lett. 2000;2:1927–1929. doi: 10.1021/ol0060026. [DOI] [PubMed] [Google Scholar]
- 21.Zeng Y, Aubé J. J Am Chem Soc. 2005;127:15712–15713. doi: 10.1021/ja055629m. [DOI] [PubMed] [Google Scholar]
- 22.Murta MM, de Azevedo MBM, Greene AE. Synth Commun. 1993;23:495–503. [Google Scholar]
- 23.For reviews on the synthesis of α-methylene lactones, see: Grieco PA. Synthesis. 1975:67–82.Hoffman HMR, Rabe J. Angew Chem Int Ed Engl. 1985;24:94–110.Petragnani N, Ferraz HMC, Silva GVJ. Synthesis. 1986:157–183.
- 24.Curphey TJ. J Org Chem. 2002;67:6461–6473. doi: 10.1021/jo0256742. [DOI] [PubMed] [Google Scholar]
- 25.The originally reported spectrum of neostenine contains a typographical error. The 13C NMR chemical shift originally reported by Lin and coworkers2a at 39.79 ppm actually appears at 37.97 ppm. We thank Professor Lin for kindly confirming this and for providing the NMR spectra of naturally occurring neostenine.
- 26.Lei Y, Wrobleski AD, Golden JE, Powell DR, Aubé J. J Am Chem Soc. 2005;127:4552–4553. doi: 10.1021/ja050214m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.