

Abstract
Attempts to introduce the highly versatile vinyl group into other organic molecules in a chemo-, regio- and stereoselective fashion via catalytic activation of ethylene provided challenging opportunities to explore new ligand and salt effects in homogeneous catalysis. This review provides a personal account of the development of enantioselective reactions involving ethylene.
Keywords: Carbon-carbon bond formation, hydrovinylation, asymmetric catalysis, ligand effects, nickel, palladium, exo-cyclic stereocontrol, all-carbon quaternary center, diene cyclization
1. Introduction
1.1 The Origins
Unlike most of my peers I started my academic career late after more than a decade in industry. Having benefited from my experience at the highly nurturing and scientifically exciting environment of DuPont Central Research, where I spent my formative years as an independent scientist, I moved to my current position at the Ohio State University in 1995. Just prior to the move, in a highly productive collaboration with two talented colleagues, Dr. Al Casalnuovo and Dr. Tim Ayers, I had just published several papers on the use of readily available carbohydrate-derived ligands in asymmetric catalysis. These studies provided some of the first unequivocal evidence for electronic tuning of an asymmetric catalyst for enhancement of enantioselectivity. The first practical asymmetric hydrocyanations of olefins and a general synthesis of 2-arylpropionic acids including (S)-naproxen followed. Cheap hydrogenation catalysts based on readily available d-sugars (d-glucose and d-N-acetamidoglucose) for the synthesis of D- and L-amino acids were also disclosed. With the intellectual property aspects of these discoveries adequately covered, there was little further interest at DuPont to follow up this research for reasons that had to do more with business than science. So, after moving to Ohio State, I decided to base my first research proposal on what I thought were some exciting initial leads in asymmetric hydrocyanation, a C-C bond-forming reaction of immense potential. In trying to solve the remaining problems of substrate scope and selectivity we were going take a rather empirical approach based on ligand-tuning, an approach that had served us well. In the event, the proposal received mixed reviews and I decided to look elsewhere for a new project, still keeping the focus on the underlying theme of selectivity and efficiency in broadly applicable organic reactions.
In initiating a new project, I was convinced that asymmetric catalysis of C-C bond-forming reactions that involve neutral feedstocks would be a fertile area for research, providing ample opportunities for training graduate and postdoctoral students. After all, Nature makes exquisite use of the most basic of feedstocks, carbon dioxide and water to make many of the molecules that sustain life on earth. Such a project would bring challenges in two prominent areas of modern organic synthesis, activation and stereoselective incorporation of readily available carbon sources for synthesis of valuable intermediates. If successful, this research would add to our repertoire of very powerful synthetic methods with implications for how we make such intermediates in the laboratory and manufacture on larger scale in industry. Under the best of circumstances such processes could even be ‘green’ if we operated under ambient (energy efficient) conditions, used only catalytic amounts of metal and made only the desired products (i.e., high selectivity), thereby avoiding costly separation processes.
In this review I shall attempt to summarize our contributions to the area of heterodimerization of olefins in a more or less chronological order. A review4a we published in 2003 should be consulted for detailed history of early developments, which have been summarized here for the sake of completion. In any comprehensive account of this nature, repetition of some of the already reported results is inevitable; but they are discussed here from a perspective that is often lost in a more traditional narrative of a journal article. You seldom hear about the blind alleys traveled, nor about the ill-conceived conjectures that eventually pay off for the wrong reasons. This update also includes significant results on the HV reactions of dienes, generation of all-carbon quaternary centers and applications to natural product synthesis that involve the reactions of highly functionalized substrates.
1.2 Olefin Dimerization Reactions
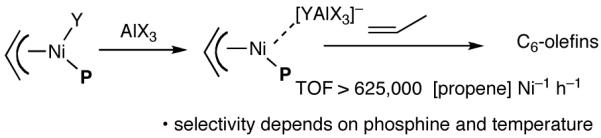
The search for another efficient C-C bond-forming reaction that uses feedstock carbon sources led us to a remarkable review published by Wilke in Angew. Chem. Int. Ed.1 In this paper the author summarized several years of work on allyl metal and metal hydride intermediates carried out at the Max-Plank Institute für Kohlenforschung in Mulheim. Among the many carbon-carbon bond-forming reactions catalyzed by a cationic nickel hydride described in this paper is the homodimerization of propene, which forms the basis of the Dimersol technology (eq 1).2 This reaction is one of the most efficient homogeneous catalyzed C-C bond-forming reactions known outside realm of the single-site olefin polymerization catalysis. The active catalyst, generated from [η3-(allyl)NiX]2, a trivalent phosphorus ligand and a Lewis acid, produces a mixture of C6-olefins from propene with turnover frequencies in excess of 625,000 [propene][Ni]-1[h]-1.2,3 Conspicuously absent in these early studies were applications of such dimerization reactions for the synthesis of fine chemicals, especially functionalized small molecules.4
 |
(1) |
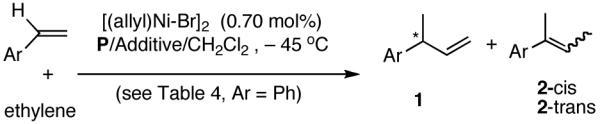
2. Hydrovinylation Reactions
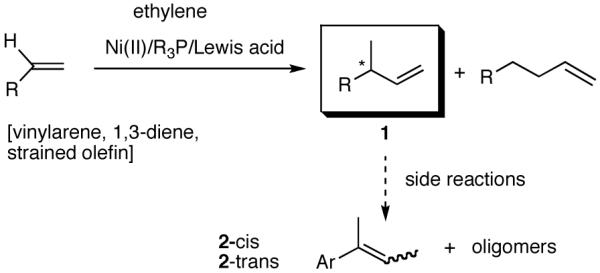
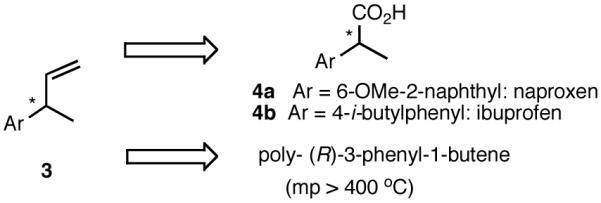
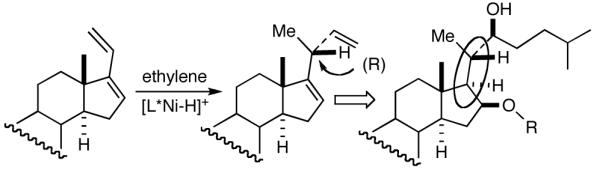
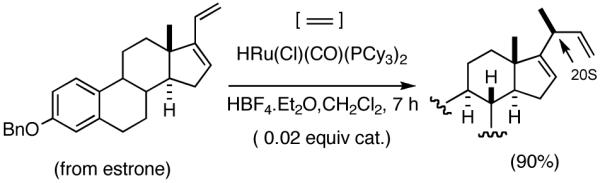
Among the olefin dimerization reactions, the hydrovinylation (HV) reaction, viz., the addition of a vinyl group and a hydrogen across a double bond (eq 2), looked especially promising for fine chemical synthesis if the pesky issues of scope and selectivity could be adequately resolved. Since the branched product 1 (eq 2) is chiral, a regio- and stereoselective version of this reaction, in principle, could provide a variety of olefin-derived products in enantiomerically pure form. For example, enantioselective hydrovinylation of vinylarene derivatives will lead to 3-arylbutenes (3) that can be used for the synthesis of widely used antiinflammatory 2-arylpropionic acids (eq 3).5 One of the hydrovinylation products of styrene, (R)-3-phenyl-1-butene, has been reported to give a very high melting (>400 °C) isotactic polymer under Ziegler conditions.1 Yet another application might be in finding a solution to the long-standing problem of control of exocyclic stereochemistry, an example of which is shown in the context of a steroid-D-ring functionalization via the hydrovinylation of a diene (eq 4). As seen in the steroids, a chiral side chain carrying a methyl group is a very common structural motif in many important natural products, and often this side chain is attached at a stereogenic center of a ring. Classical procedures for the installation of these stereocenters often involve circuitous routes. Further, can the reaction be used for carba-functionalization of strained double bonds as shown in eq 5?
 |
(2) |
 |
(3) |
 |
(4) |
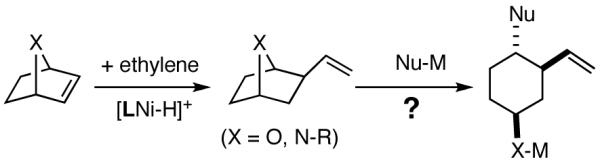 |
(5) |
2.1 A Brief History of Hydrovinylation Reactions
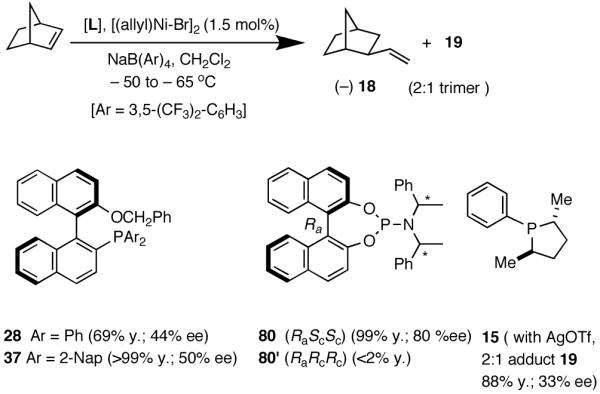
The hydrovinylation reaction has a long history4a dating back to 1965 when Alderson, Jenner and Lindsey (of DuPont Central Research)6a first reported the use of hydrated Rh and Ru chlorides to effect codimerization of ethylene at high pressures (1000 psi) with a variety of olefins including styrene and butadiene. Styrene has served as a prototypical test case for most investigations reported to date. In early studies, in addition to Rh,6 other metals such as Ru6a,7, Co,8 Pd9 and Ni10 were also used, and in most instances the reactions were complicated by isomerization of the initially formed 3-arylbutenes and oligomerization of the starting olefins (eq 2). Notable among the early studies are also the first examples of asymmetric hydrovinylation of 1,3-cyclooctadiene, norbornene and norbornadiene using a combination of [η3-C3H5)NiCl]2/Et3Al2Cl3 and a monoterpene-derived chiral phosphine, even though the selectivities were unacceptably poor.11
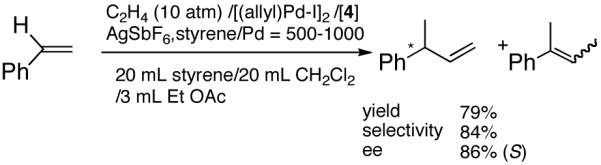
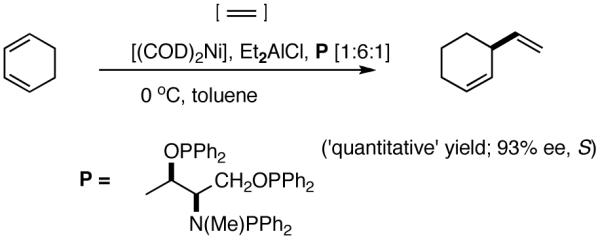
Even though some initial reports9,12 seemed to indicate that the Pd-catalyzed reactions gave mostly linear products and/or extensive isomerization, subsequent studies have shown that use of ligands such as 513 and 614 under carefully chosen reaction conditions, permit the isolation of the branched product. Acceptable yields and best selectivities are achieved under low conversions since isomerization of the primary product is a persistent problem with many of these reactions. Among these ligands, the phosphinite 5 is particularly noteworthy (eq 6).13 With the appropriate counter ion (SbF6-), 3-phenyl-1-butene can be synthesized in a moderate yield and in ee’s up to 86% (S).
 |
(6) |
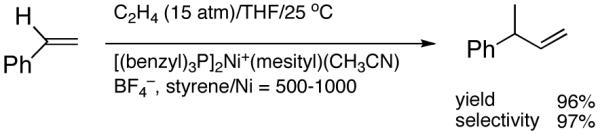 |
(7) |
Recent improvements in the Ni-catalyzed heterodimerization reaction includes the use of [ArNi(PR3)(MeCN)]+ BF4- (Ar = mesityl, R = benzyl) which served as an efficient catalyst for hydrovinylation of styrene (eq 7).15 High turnover numbers (up to 1915 h-1) and selectivities for the 3-arylbutenes can be achieved for a variety of styrenes at 15 bar ethylene pressure. Heteroatom substituents are tolerated, but ring alkylated styrenes give poor yields. The reaction rates fall unacceptably low below 20 °C, and as the temperature is increased isomerization of the initially formed product is seen. Substitution of tribenzylphosphine with cis-myrtanyldiphenylphosphine give high selectivity towards 3-phenylbutene, albeit with a disappointing enantioselectivity (∼7% ee). Since there is an exothermic polymerization of ethylene at the end of the relatively more facile heterodimerization, control of temperature is crucial to get good selectivities under these reaction conditions. Monteiro et al16 reported the use of dicationic nickel complexes ([Ni(CH3CN)6]2+] 2[BF4]-/Ph3P/Et2AlCl) at room temperature and 10 bar pressure of ethylene to get yields of 68 to 87% of various hydrovinylation products. Isomerization of the primary product can be prevented by maintaining a high pressure of ethylene (>10 bar). A unique feature of this catalyst system that is not seen in any other Ni-catalyzed reactions is that chelating phosphines [e.g., diphenylphosphinoethane (dppe) or N,N-dimethyl-1-[2-(diphenylphosphino)ferrocenyl]-ethylamine (dppfa)] do not inhibit the reaction (eq 8). Preparatively useful Ni-catalyzed asymmetric hydrovinylation reactions will be dealt with in greater detail in section 2.3.
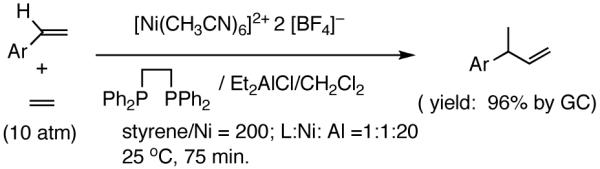 |
(8) |
2.2 Ruthenium- and Cobalt-Catalyzed Hydrovinylation Reactions
While this review is not intended to be exhaustive, two notable results that show considerable promise are worthy of mention before discussing our own contributions in the area of Ni-catalyzed HV reactions. Recently, Yi introduced a combination of (PCy3)2(CO)Ru(Cl)H and HBF4.OEt2 for the HV of styrene.17a With only scanty details reported, the scope and generality of this procedure still remain to be established (eq 9). We found that this reaction can be carried out under 1 atmosphere of ethylene using AgOTf as an additive.17b
 |
(9) |
Vogt reported18 that hydrovinylation of styrene can be accomplished using a Co-chelate under 30 bar ethylene even though conversion and selectivity in an enantioselective version remain poor (eq 10).
 |
(10) |
2.3 Best Practices Prior to 1997. Ni-Catalyzed Hydrovinylation Reactions
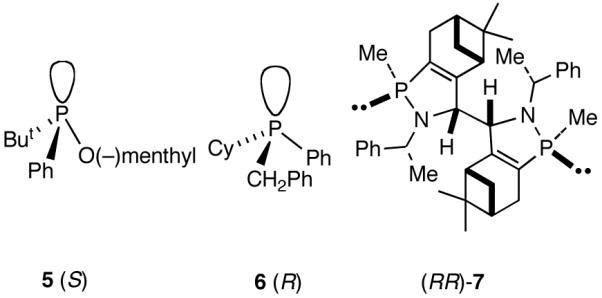
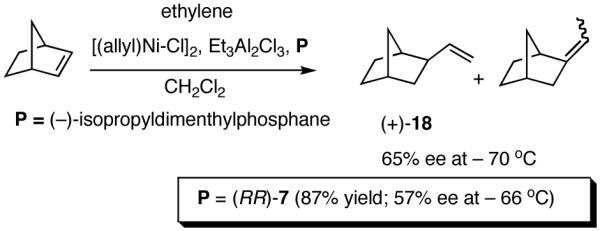
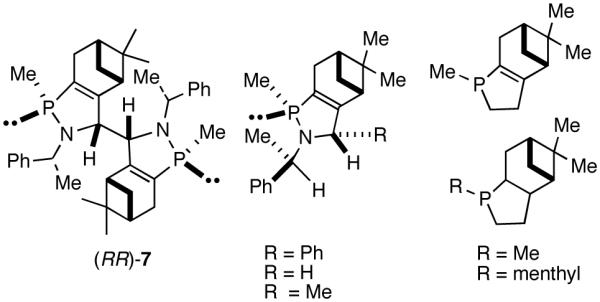
A careful examination of the published research before 1997 when we initiated the new project showed that the best catalyst reported for this reaction was also the one that gave the best enantioselectivity. This was the Wilke system that used [η3-allyl)NiCl]2/[(RR)-7]/Et3Al2Cl3].4c,19 With this catalyst, varying ee’s are obtained depending on the reaction conditions. The azaphospholene (RR)-7 (Figure 1) is a very special ligand for the hydrovinylation of vinylarenes and 1,3-dienes, and the Ni-complexes derived from this ligand were claimed in a patent19 to give unprecedented enantioselectivities for many of the substrates (eq 11 and eq 12). A variety of vinylarenes including 4-chlorostyrene, 4-isobutylstyrene, 2-methylstyrene and 6-methoxy-2-vinylnaphthalene gave very high ee’s in the hydrovinylation reaction. The ligand (RR)-7 is prepared from (-)-(R)-myrtenal and (+)-(R)-1-phenylethylamine in a multistep process.4c One other congener of this compound, the diastereomer (RS)-7 (prepared from (-)-(R)-myrtenal and (-)-(S)-1-phenylethylamine) is much less active and selective for the hydrovinylation of styrene. Monomeric and structurally related versions of this ligand have been prepared4c,21 in an attempt to simplify the synthesis and it has been found that catalytic activity and enantioselectivity invariably fall below useful levels.
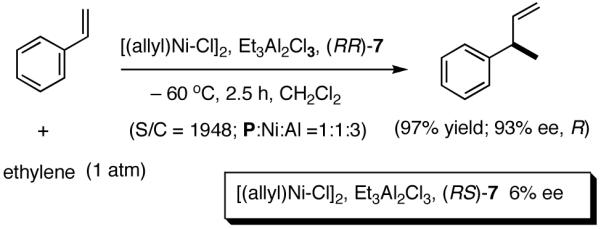 |
(11) |
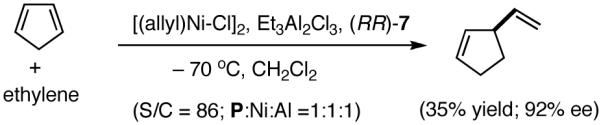 |
(12) |
Figure 1.
Assorted Ligands Useful for Asymmetric Hydrovinylation
2.4 Mechanism of Ni-Catalyzed Hydrovinylation of Vinylarenes
In the absence of meaningful mechanistic work, we started our research with a working hypothesis for the mechanism of the reaction.22 Even though much of the early studies of hydrovinylation of styrene are characterized by lack of any selectivity, many of them provide significant mechanistic insights into the reaction. For example, kinetic and solvent effect studies of hydrovinylation with NiX2/AlEt3/BF3.OEt2/P(OPh)310 e,f,g provided some early indications of the [Ni-H]+ coordination to a styrene and subsequent addition. Deactivating effect of a solvent was found to increase in the order CH2Cl2, PhF, PhCl, PhMe, PhNO2, Et2O, consistent with inhibitory effect of a coordinating Lewis base. Studies of D-distribution in the product when the hydrovinylation was carried out with D2C=CD2 provided further evidence for the involvement of a cationic nickel-hydride intermediate.10f Even though a catalytically active Ln[Ni-H]+ has not been isolated, its generation and inter-1 and intramolecular23 additions have been documented. Since these early studies, Brookhart and DiRenzo have provided more details of their mechanistic study of closely related Pd-catalyzed codimerization of styrene and ethylene.24 Based on all the available evidence and our own initial observations (vide infra), a hypothetical mechanism, shown in Scheme 1, can be proposed for this reaction.
Scheme 1.

Proposed Mechanism for the Hydrovinylation of Styrene
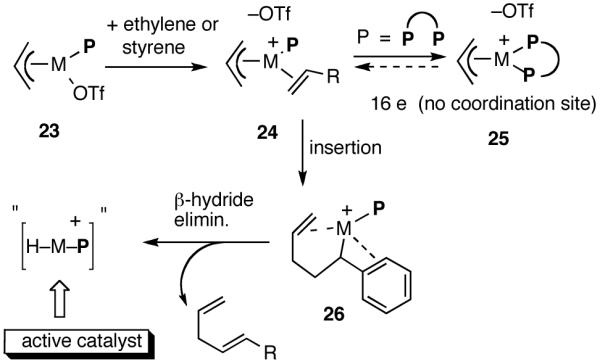
Functional equivalent of a catalyst can be represented by 11, a cationic metal hydride intermediate associated with a weakly coordinated counteranion, and a phosphine. This species is formed by the Lewis acid-assisted dissociation of the Ni-X bond from the 16-electron phosphine complex 8, coordination of ethylene (or styrene) to form 10, insertion into the allyl Ni-bond followed by subsequent β-hydride elimination. Several crystal structures of complexes related to Ni-allyl compounds 8 and 9 are known with Cy3P [X = MeAlCl3],11b P(menthyl)(Me)(But) [X = Cl]25a and P(menthyl)2(Me)[X = Me].25b Addition of the metal hydride to the vinylarene would lead to the benzyl complex 13, which is shown as a 16-electron η3-structure. Ligand substitution with ethylene leads to 14. At higher concentrations of ethylene and styrene this species could serve as a catalyst resting state. Strong evidence for such a situation has been provided by Brookhart and Direnzo24 in mechanistically related [(allyl)Pd(Cy3P)]+ [BARF]- mediated dimerization of styrene. Insertion of ethylene followed by β-hydride elimination from 15 regenerates the metal hydride catalyst and the product 1. A number of anecdotal observations reported in the literature and some made during our studies study can be accommodated by this mechanism.
Diminished reactivity of electron-deficient vinylarenes might arise from low rate of metal-hydride addition (11 -> 13).
Apparent poor reactivity of substrates carrying heteroatoms when R2AlX-type Lewis acids are employed could be the result of the coordination of these atoms to aluminum.
Deactivating effects of the coordinating solvents.
Isomerization of the initially formed 3-aryl-1-butene to 2-aryl-2-butens (1 -> 2) could be mediated by the metal hydride via sequential addition-elimination reactions.
Total inhibition of the reaction by chelating phosphines (vide infra).
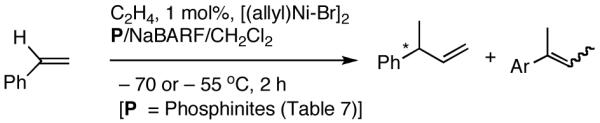
2.5 A New Protocol for Hydrovinylation of Vinylarenes Amenable to Asymmetric Catalysis
We have already alluded to the fact that among the earlier developments, only the Wilke’s azaphospholane ligand (RR)-71,19 gave satisfactory yield and selectivity for this potentially important reaction (eq 11 and 12). Subsequent work has shown that the protocols using this ligand is possibly of limited value for the development of a broadly applicable hydrovinylation reaction. At the outset of our work we speculated that the scope and selectivity of hydrovinylation could be increased significantly by eliminating the trouble-some Lewis acids from the Wilke system. In its place we would use a silver salt whose weakly coordinating anion can be easily replaced from the coordination sphere of Ni by an olefin prior to the insertion step (10 --> 11--> 12 in Scheme 1). Further, we expected the phosphine ligand to play a crucial role in dictating the selectivity of the reaction, as was apparent from some of the seminal ligand tuning studies that had been carried out by the Wilke group.1,4b As for the effect of the counter anion (Y in Scheme 1), the situation appeared uncertain, as it was known that the selectivity varied considerably with the nature of the ligand. For example, coordinating anions (Et2AlCl2-, OTf-, BF4-) give higher ee’s with ligand (RR)-7,4c but an opposite effect is observed with highly basic ligands like (menthyl)2PPri, where the best anions are the highly dissociating ones like SbF6- and PF6-.4b
 |
(13) |
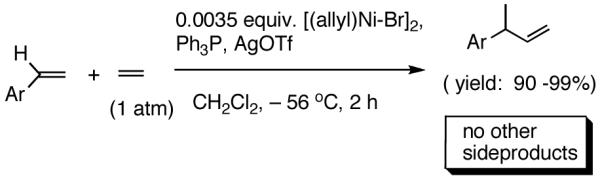
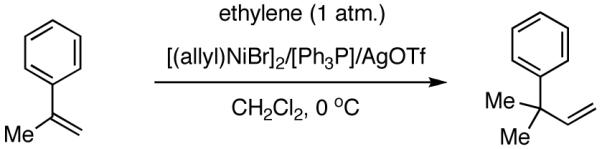
I still recall the day this project was assigned to Dr. Nobu Nomura, an exceptionally bright and hard-working postdoctoral fellow who came from Nagoya University (to which he has since returned as Faculty), with a warning of many of the risks that might lie ahead. Nobu proceeded to methodically investigate the effects of variations of ligands, counter ions and other parameters on the course of the hydrovinylation of styrene. After an extensive effort he discovered a new protocol (eq 13) for this highly demanding reaction.22 During these investigations Nobu encountered every conceivable problem imaginable in trying to react two alkenes to get a third alkene as the major product in a coupling reaction. These included oligomerization of styrene, polymerization of ethylene, isomerization of the initially formed 3-phenylbutene, precipitation of the metal (Ni or Pd) or complete lack of reactivity, depending on the phosphine, the silver salt, solvent and temperature. However, several reactions gave just enough encouraging results26 to feed his persistence. In the end, a reliable protocol that gave unprecedented chemical yield and selectivity in the hydrovinylations of a series of substituted vinylarenes was arrived at. This involved the use of a combination of [(allyl)NiBr]2, triphenylphosphine, and weakly coordinating counter anion, triflate (OTf-) as the precatalyst (eq 13 and Table 1). Typically, the reaction is carried out under 1 atmosphere of ethylene at -56 °C in methylene chloride as the solvent, using 0.007 equiv. of the catalyst. Under these conditions no oligomerization of ethylene or styrene or rearrangement of the initially formed product was detected. In sharp contrast to the previously observed diminished reactivity for vinylarenes with Lewis basic centers, no such limitations are apparent under the new conditions (entries 2, 5, 9, Table 1). Derivatives such as 4-isobutylstyrene, 3-fluoro-4-phenylstyrene, 2-methoxy-6-vinylnaphthalene and 3-benzoylstyrene - all potential precursors of important antiinflammatory agents - give excellent yields of the hydrovinylation products. Hydrovinylation product of 3- and 4-bromostyrenes (entries 3 and 8) are other potentially important intermediates that can be transformed into useful products through organometallic cross-coupling reactions. As expected, the use of a number of chelating bis-phosphines, aminophosphines and 1,2-bis-diarylphosphinitites give no products under otherwise identical conditions. These include 1,3-bis-diphenylphosphinopropane (DPPP), 2,2′-bis-diphenylphosphino-1,1′-binaphthyl (BINAP), [2,2,-dimethyl-1,3-dioxolane-4,5-diylbismethylene]bisdiphenyl-phosphine (DIOP), N-(tert-butoxycarbonyl)-4-(diphenylphosphino)-2-[(diphenylphosphino]methyl)pyrrolidine (BPPM). Methyl substitution at α- or β- carbons of styrene also leads to poor yields (21% and 49% respectively).
Table 1.
Hydrovinylation of Vinylarenes (Eq 13)
| entry | vinylarene | % yielda | conditionsb |
|---|---|---|---|
| 1. | styrene | >95 (99) | (i) |
| 2. | 4-methoxystyrene | >95 (98) | (i) |
| 3. | 4-bromostyrene | >95 (98) | (i) |
| 4. | 2-vinylnaphthalene | (99) | (i) |
| 5. | 6-MeO-2-vinylnaphthalene (MVN) | (90) (97) |
(i), (ii) |
| 6. | 4-i-Bu-styrene | >90 (99) >97 (99) |
(i) (ii) |
| 7. | 3-F-4-C6H5-styrene | (88) | (i) |
| 8. | 3-bromostyrene | (99) | (i) |
| 9. | 3-Ph-C(O)-styrene | (99) | (i) |
| 10. | 3-Me-styrene | (>99) | (i) |
| 11. | 4-Me-styrene | (>99) | (i) |
| 12. | 2-Cl-styrene | 30 | (i) |
| 13. | 4-Cl-styrene | (>99) | (i) |
isolated yield; selectivity >98 in all cases. In brackets are yields based on gas chromatography.
(i) 0.007 equiv. cat., CH2Cl2/-55 °C/2 h. (ii) (R)-MOP/Ar4B- Na+/ CH2Cl2/-56 °C/2 h (see section 4.1, New Ligands for Asymmetric Hydrovinylation’)
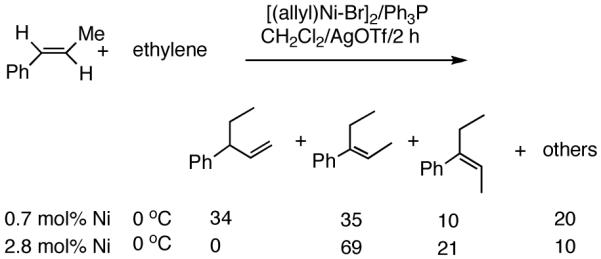
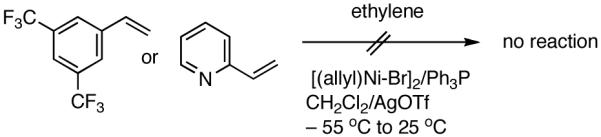
At higher temperatures (-20 °C) up to 54% yield of the primary product is formed from E-β-methyl styrene with contamination from isomerization (eq 14). Z-Stilbene gives a mixture of olefinic products in low yields. Not unexpectedly, the recovered stilbene is a mixture of Z- and E- isomers, providing further support for a [(L)Ni-H]+ intermediate in these reactions. Other related substrates that fail to undergo the hydrovinylation reaction under a variety of conditions include 3,5-bis-trifluromethylstyrene, 2-vinylpyridine and N-vinylcarbazole. While the electron-deficient nature of the styrene may retard Ni-coordination, the lack of reactivity of vinylpyridine may have its origin in the formation of stable intermediates assisted by the pyridine nitrogen.
 |
(14) |
 |
(15) |
2.6 Heterodimerization of Styrene with Other Olefins Including Propene.27
Unlike heterodimerization reactions of ethylene, no synthetically useful heterodimerization reaction using propene was known before our work. We find that propene reacts with styrene and substituted styrenes under conditions slightly modified from what was previously described for ethylene giving excellent yields of the expected products (eq 16, Table 2). The reaction with propene proceeds at a higher temperature (-15 °C to 10 °C vs. -56 °C for ethylene), especially in the case of the more electron-deficient styrene derivatives.27 As expected, a mixture of regioisomeric products (with propene-C1 addition to the benzylic position, 16, as the major one) is obtained.
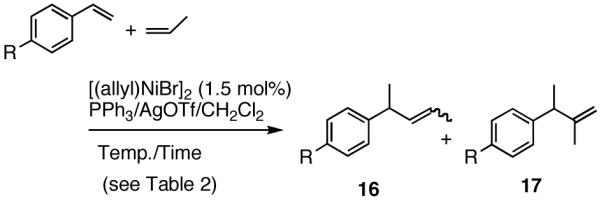 |
(16) |
Table 2.
Heterodimerization of Propene and Vinylarenes (Eq 16)
| no | R | temp. °C | time (min) | yielda | 16:17 |
|---|---|---|---|---|---|
| 1 | i-Bu | -15 | 15 | 96 | 3:1 |
| 2 | OMe | -15 | 60 | 86 | 4:1 |
| 3 | Cl | 0 | 15 | 94 | 4:1 |
| 4 | Br | 0 | 10 | 95 | 4:1 |
| 5 | OAc | -40 | 30 | 98b | 5:1 |
| 6 | PhC(O)-c | 10 | 15 | 94 | 4:1 |
| 7 | NTs2c | 10 | 25 | 92 | 2:1 |
| 8 | MVN,c,d | -5 | 60 | 88 | 10:1 |
Isolated yield
1.4 Mol% Ni used
3 Mol% Ni used
2-Methoxy-6-vinylnaphthalene
2.7 Other Heterodimerization Reactions26
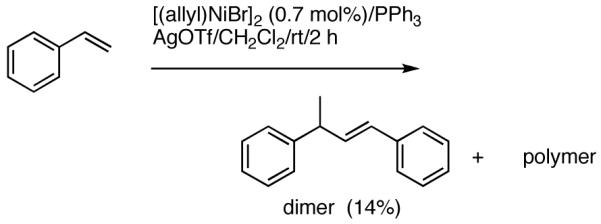
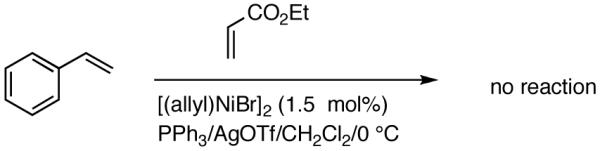
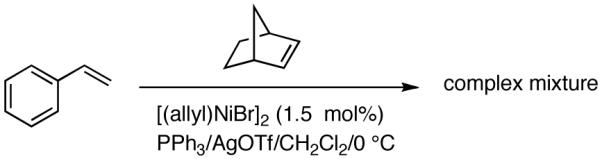
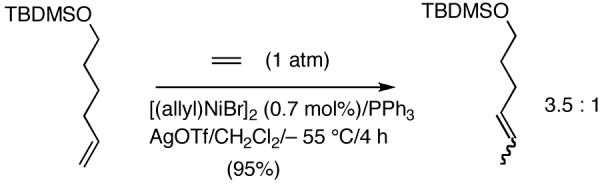
Reaction of styrene alone with [(allyl)2NiBr]2 and Ph3P at room temperature in the presence of AgOTf leads to the formation of 14% styrene dimer along with extensive polymerization (eq 17). Attempts to effect heterodimerization of styrene and cylohexene or ethyl vinyl ether also lead to polymer formation. Varying amounts of styrene dimer can be detected in gas chromatography (eq 18, 19) under these conditions. Codimerization of styrene and ethyl acrylate does not proceed under the standard hydrovinylation conditions (eq 20) using Ph3P and AgOTf, whereas with norbornene a complex mixture of hydrocarbons is obtained (eq 21). Treatment of a typical terminal olefin, 1-tert-butyldimethylsiloxy-5-hexene with ethylene under hydrovinylation conditions leads to clean isomerization of the double bond to give a mixture of Z- and E 1-tert-butyldimethylsiloxy 4-hexenes (eq 22).
 |
(17) |
 |
(18) |
 |
(19) |
 |
(20) |
 |
(21) |
 |
(22) |
2.8 Hydrovinylation of Norbornene28
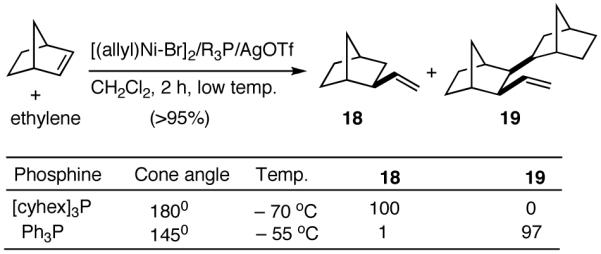
Like differences in electronic properties and size between two olefins, ring strain is another differentiating factor that could be exploited to effect a selective heterodimerization. We find that the protocol using [(allyl)NiBr]2/phosphine/AgOTf works equally well for the heterodimerization of norbornene and ethylene (eq 23), the course of the reaction being dependent on the phosphine that is employed. Tricyclohexylphosphine gives the expected 1:1 adduct (18) in nearly quantitative yield, whereas triphenylphosphine gives a 2:1 adduct (19) between norbornene and ethylene. For further identification (vide infra under Asymmetric Hydrovinylations), the trimer was converted into the alcohol 20. This remarkable selectivity is presumably related to the cone angles of the two phosphines and the relative reactivities of the two olefins. It is conceivable that norbornene is more reactive than ethylene and thus undergoes a fast initial dimerization, when a smaller phosphine (Ph3P) is used (Scheme 2). The initially formed σ-nickel complex 21, for stereoelectronic reasons, cannot undergo β-hydride elimination, and hence react with another olefin, ethylene, giving finally the 2:1 adduct 19. With a bulky phosphine, only addition to ethylene is feasible giving the 1:1 adduct.
 |
(23) |
Scheme 2.
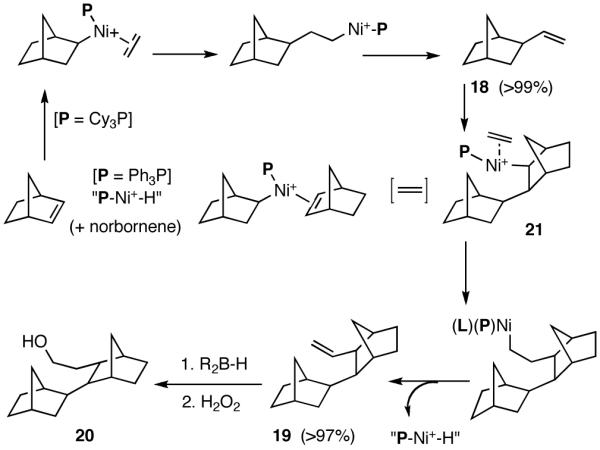
Ligand Dependence in the Hydrovinylation of Norbornene
3. Nickel-Catalyzed Enantioselective Hydrovinylation Reactions
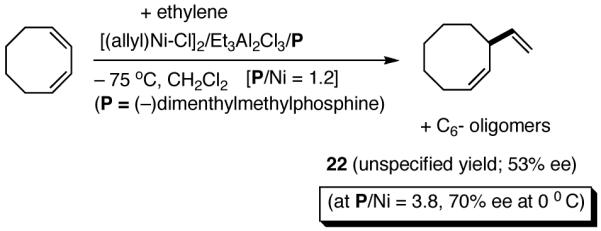
Asymmetric hydrovinylation of 1,3-cyclooctadiene (eq 24) using (-)-isopropyldimenthylphosphine as a ligand is one of the first examples11a of an asymmetric carbon-carbon bond-forming reaction ever reported, even though the selectivity was unacceptably low (up to 70% ee, unspecified yield) by today’s standards. Under somewhat similar conditions, norbornene (eq 25) and norbornadiene give the corresponding 2-exo-vinyl products in 65% ee (at -70 °C) and 78% ee (-65 °C) respectively.11b Depending on the temperature, varying degrees of isomerization to the ethylidene derivatives (E and Z) are observed in both cases. The full details of reaction conditions and characterization of products in these and many other early hydrovinylations are difficult to locate.
 |
(24) |
 |
(25) |
3.1 Azaphospholene Ligands
Before the recent resurgence of activity in this area, the best ligand for the hydrovinylation of vinylarenes (eq 11), cyclopentadiene (eq 12) and norbornene (eq 25), has been the azaphospholene [(RR)-7, (Figure 3)], used in conjunction with an (allyl)nickel halide dimer and a Lewis acid like Et3Al2Cl3.4c,19 Attempts to modify the azaphospholene ligand (Figure 2) suggest that this class of compounds is of a narrow scope, and possibly of limited value for the development of a broadly applicable hydrovinylation reaction, especially for a practical enantioselective version. The use of pyrophoric aluminum alkyl Lewis acids is another major limitation of this protocol, especially if the scope of the reaction is to be expanded to heteroatom-containing substrates.
Figure 3.
Chelating Ligands Examined for the Ni-catalyzed Hydrovinylation Reactions
Figure 2.
Phospholene Ligands for Asymmetric Hydrovinylation
3.2 Aminophosphine/Phosphinite (AMPP) Ligands
A nickel complex prepared in situ from an aminophosphine/phosphinite ligand derived from (2S3R)-threonine is an efficient catalyst for the hydrovinylation of 1,3-cyclohexadiene (eq 26).29 Other aminoalcohol-derived AMPP ligands gave lower selectivities.
 |
(26) |
3.3 Use of Chelated Phosphines
If the proposed mechanism (Scheme 1) has any validity, there is only one ligand per metal in the catalytically active species. A number of studies have indicated that the Ni-catalyzed hydrovinylation reaction might be inhibited by chelating phosphines even when the reactions are carried out under widely different conditions.4b,15a Nonetheless we investigated the viability of different classes of chelating phosphines under the newly discovered protocol (eq 13). A list of ligands and some typical reaction conditions tested are listed in Figure 3. Careful examination of the crude reaction products by gas chromatography and NMR spectroscopy revealed that no C-C coupling products (oligomers, hetero- or homodimers) were formed under these reaction conditions even when the reaction is run at higher temperatures.
In retrospect, the conspicuous lack of activity of chelated phosphines in the hydrovinylation is not surprising. As shown in Scheme 3, the generation of the active catalyst is possible only if the OTf in the initially formed complex 24 is efficiently displaced by one of the olefins (ethylene or styrene), and this event is followed by an insertion (e. g., to form 26) and a β-hydride elimination. A strongly chelating bis-phosphine would either prevent the formation of 24 or effectively reduce its concentration such that the insertion pathway is no longer available.
Scheme 3.
Why a Chelating Ligand Maybe Unsuitable for Hydrovinylation of a Vinylarene
4. Synergistic Relation Between Hemilabile Ligands and Counteranions30
4.1 New Ligands for the Asymmetric Hydrovinylation Reaction. 2-Diphenylphosphino-2′-alkoxy-1,1′-binaphthyl (MOP) Ligands
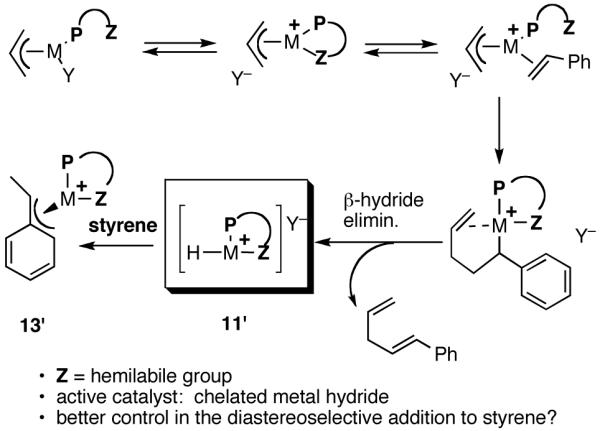
Considering the requirement of an open coordination site for ethylene in the critical steps of the reaction (Scheme 1), we wondered whether a monophosphine that also carried a hemilabile group31 might have an advantage, since such a group can stabilize the putative cationic intermediates by internal coordination. The argument goes that the weakly coordinated group could be displaced by the olefin at the appropriate stage. In addition, such coordination might lead to a chelated metal hydride (11′ = [P∼Z-Ni-H]+) with better diastereoselective discrimination (Scheme 4) in the key addition to the prochiral faces of the olefin (for example, in the formation of the η3-benzyl-Ni- intermediate 13′) [11′ and 13′ corresponds to 11 and 13 in the general mechanism shown in Scheme 1]. Making the reasonable assumption that all the subsequent steps proceed with retention of configuration, it can be argued that the enantioselectivity is determined at the stage of the metal hydride addition.
Scheme 4.
Use of a Chelated Metal Hydride: Better Diastereoselectivity?
A number of ‘hemilabile’ groups including carboxylate (anionic), ester carbonyl, triarylphosphonoyl, sulfur (from a thiophene) moiety have been investigated in a variety of reactions including codimerization of ethylene and styrene,32a oligomerization of ethylene,32a-d and ethylene/CO oligomerization.32e,f Since our eventual goal was to develop an asymmetric version of the hydrovinylation reaction, we decided to explore the use of a hemilabile ligand in the context of a chiral ligand. In the absence of any clear lead, an ether-oxygen was chosen as the hemilabile group in the first ligands we investigated. This choice was not entirely arbitrary since phosphino-ether systems have been extensively investigated,31 starting with the initial o-diphenylphosphinoanisole, which was the first hemilabile ligand to be so named.31a
In the event, (R)-2-diphenylphosphino-2′-methoxy-1,1′-binaphthyl (27, MOP)33 in which the methoxy moiety would play the role of the hemilabile ligand, was chosen for the initial study. The BINAP structural motif was considered especially attractive since it allowed considerable flexibility in ligand tuning including variations of the 2′-substituents, which would allow further explorations of the hemilabile ligand concept.
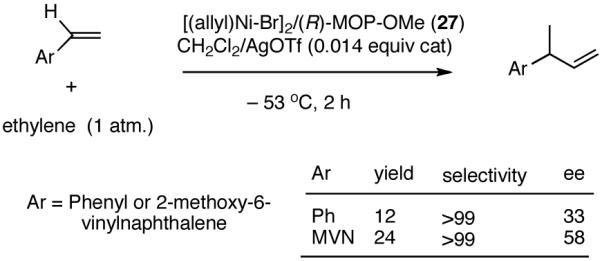 |
(27) |
Hydrovinylation of styrene and 2-methoxy-6-vinylnaphthalene (MVN) were carried out using the MOP ligand under the standard protocol described earlier (eq 13) using AgOTf and the results are shown in eq 27. A highly selective reaction ensues yielding the expected product albeit in disappointingly low conversion (% yield: 12 and 24) and enantioselectivity (% ee: 33 and 58). The conversions were of special concern since nearly quantitative reactions were routinely observed in reactions reported earlier (Table 1). Even though the exact origin of the diminished activity of a Ni-catalyst with a hemilabile ligand under these conditions remained unknown, for further development of the reaction we relied on the following rationale (Scheme 5).
Scheme 5.
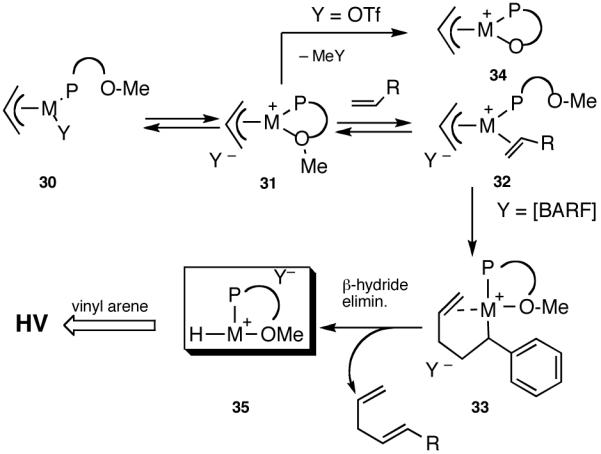
Effect of Conterions on NiII[MOP]-Mediated Hydrovinylation
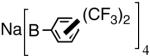
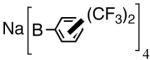
The initially formed complex 30 could be in equilibrium with a chelated complex 31. The generation of the catalyst is possible only if the hemilabile ligand is successfully displaced by an olefin to form 32. The relative concentrations of 30, 31 and 32 thus become an important factor in the catalyst turnover. Low concentrations of the catalytically competent species 32 and/or side reactions, which remove the catalyst (for example, by methylation of the triflate to give catalytically inactive34 34) may account for the poor reactivity under these reaction conditions. Support for this conjecture comes from the fact that upon replacement of the triflate by a totally dissociated, non-nucleophilic counteranion, tetrakis-[3,5bis-(trifluoromethyl)phenyl] borate (B[(3,5-(CF3)2C6H3)]4, BARF)35 the activity of the catalyst system is completely restored. The primary products from 4-isobutylstyrene and MVN are formed in more than 95% yields with enantioselectivities (ee) of 40% and 62% (eq 28) respectively.
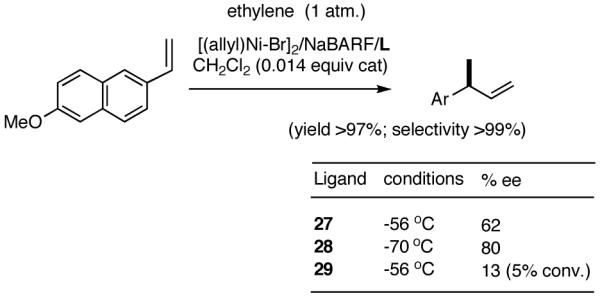 |
(28) |
Further studies revealed that a minor modification in the ligand structure (change of OMe to OCH2Ph, 28) improved the ee for MVN to 80% when the reaction is carried out at -70 °C. Styrene gave a disappointing 20% ee. The weakly coordinating O-alkyl groups in these ligands appear to be crucial for the success of the reaction since yield and enantioselectivity for the ligand with an ethyl group (29) in the place of the methoxy group are only 13% and 3% ee respectively with BARF as the counteranion.
4.2 Effect of Hemilabile Groups
To the best of our knowledge, this was the first time explicitly planned use of hemi-labile coordination to amplify enantioselectivity of a chemical reaction was reported. Therefore we decided to take a closer look at the effect of various groups at the 2′-position of the binaphthyl scaffold on the hydrovinylation reaction. Table 3 lists the results of reactions of 6-methoxy-2-vinylnaphthalene done under the standard protocol (eq 28) using different MOP-type ligands with different 2′-substituents.
Table 3.
Effect of BINAP 2′-Substituents on Hydrovinylation of 2-Methoxy-6-vinylnaphthalenea
| entry | ligandb | (2′ group) | yield (%) | %ee |
|---|---|---|---|---|
| 1. | (R)-BINAP | Ph2P | 0 | - |
| 2. | 27 | OCH3 | >98 | 62 |
| 3. | 36 | i-Pr | 69 | 70 |
| 4. | 28 | OCH2Ph | 97 | 73c |
| 5. | 28 | OCH2Ph | 93 | 80d |
| 6 | 38 (aRR) | OC(H)(Ph)(CH3) | >98 | 71 |
| 7. | 39 (aRS) | OC(H)(Ph)(CH3) | 79 | 65 |
| 8. | 29 | CH2CH3 | 12 | <3 |
| 9. | 40 | OC(O)CH3 | 0 | - |
| 10. | 41 | P(O)Ph2 | 0 | - |
| 11. | 42 | OMe | 94 | 63 |
| 12. | 43 | OMe | 93 | 63 |
| 13. | 44 | OMe | 99 | 81 |
Increasing the steric bulk of the 2′-O-alkyl substituent has little effect on the enantioselectivity of the MVN reaction, but the yield of the product is reduced. Thus O-i-Pr derivative 36 under identical conditions gave 69% yield and 70% ee. For MVN, a benzyloxy analog of MOP (28) gave 80% ee when the reaction was carried out at -70 °C. Evidence of the involvement of hemilabile oxygen may also be inferred from different activities of catalysts prepared from BINAP derivatives with (R)- and (S)-phenethyl ether side-chains (38 and 39). While the former gave an excellent yield (>98%, 71% ee) of the product, the latter gave only 79% (65% ee) yield. In an attempt to probe the effect of the hemilabile ligand, we prepared the 2′-ethyl analog 29 and tested this ligand under both sets of conditions, viz., using AgOTf and NaBARF as additives. For the hydrovinylation of MVN using BARF counteranion 12% yield and 3% ee of the product were obtained, whereas AgOTf gave less than 2% conversion. If the hemilabile ligation is important, one should expect different reactivities from ligands with varying donor properties.31,32 Allyl(Ni) complexes of 2′-acetoxy (40) and diphenylphosphosphoryl (41) analogs failed to produce any hydrovinylation products under the standard reaction conditions (entries 9 and 10). Phosphinoxide is known to be a strongly coordinating group32a and it is not surprising if the catalyst generation is prevented due to the inability of an olefin to displace this group. As for the acetoxy derivative 35, carbonyl oxygen is known to be a strongly coordinating atom as compared to an ether-oxygen in a variety of metal complexes.36 A limited effort made to modify the diaryl substituents of MOP led to no significant improvements in the HV of styrene.26,37
4.3 Solvent and Salt Effects26
As expected from the proposed mechanism, the reaction shows pronounced solvent effects. Under conditions described in equation 27 (-55 °C, 0.7 mol% [(allyl)NiBr]2, NaBARF, 2 h), the following yields and enantioselectivities were observed for the solvents indicated; CH2Cl2 (97, 73); ether (87, 77); toluene (88, 74); THF (0, 0). Tetrahydrofuran is a strongly coordinating solvent and it is no surprise that under these conditions no hydrovinylation is observed. The experiments using styrene also showed for the first time that other dissociated silver salts (AgSbF6 and AgNTf2) could effectively replace NaBARF in these reactions.
4.4 Electronic Effects
Finally, electronic effect of ligands on the hydrovinylation selectivity was examined by comparison of ee’s obtained using ligands 42 and 43 with that from 27 (Table 3, entries 2, 11 and 12). In sharp contrast to the Ni(0)-catalyzed hydrocyanation, Rh(I)-catalyzed hydrogenation or the Pd(0)-catalyzed allylation,38 ligand electronic properties appear to have little effect on hydrovinylation; in each case the chemical yield and ee were almost identical. Note that mechanistically the most significant difference between these reactions and hydrovinylation is that there is no change in the oxidation state of the metal in the catalytic cycle of the hydrovinylation reaction. Nickel(II) with its ligands plays the role of a complex Lewis acid!
4.5 Other Protocols for Ni-catalyzed Hydrovinylation Reactions
During the course of these investigations we have uncovered a number of other viable procedures for this exacting reaction. Thus a catalyst prepared from allyl 2-diphenylphosphinobenzoate 45 and Ni(COD)2 or the corresponding potassium salt of the acid (48) and allyl nickel bromide (Scheme 6) shows very good activity and excellent selectivity in the hydrovinylation reactions of styrene when activated with (C6F5)3B40 (eq 29). Structurally related catalysts have been used for oligomerization of ethylene.32a-c,40 These novel methods (Scheme 6) for the preparation of the neutral carboxylate complexes (e.g., 46) from the allyl ester or the acid might find other applications.
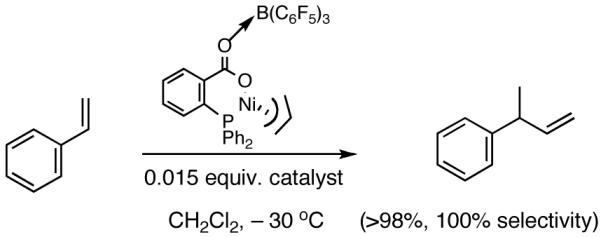 |
(29) |
Scheme 6.
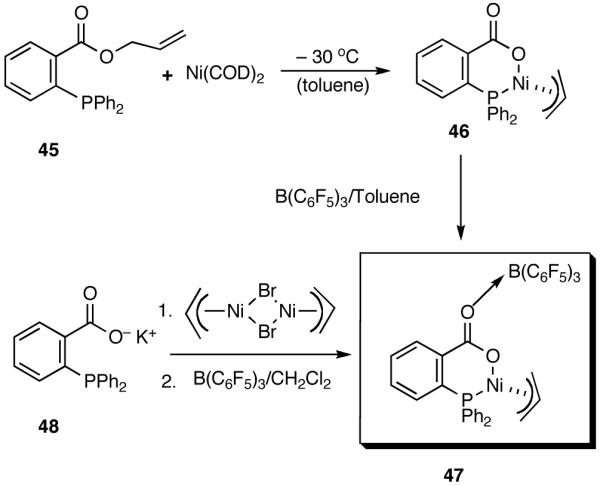
A Single Component Catalyst for Hydrovinylation of Styrene
4.6 A Model for the Asymmetric Induction in HV Reactions Catalyzed by (R)-MOP-Ni(allyl)-BARF
Even though the details of the mechanism of asymmetric HV including the nature of the turnover limiting and enantioselectivity determining steps remain unknown, a useful, working model for the transition-state maybe constructed based on reasonable assumptions derived from experimental observations. In this connection, we regarded the absence of electronic effects, which could complicate simple steric arguments with some consolation. Maybe we do not have to worry about inscrutable reactivity differences between diastereomeric intermediates. If that is the case, the first stereo-differentiating step could be used to build a model. This would be the addition of a chelated metal hydride through one of the four possible square planar Ni(II) complexes (49-52) shown in Figure 5. In the preferred intermediate/transition state, the olefin will be coordinated trans to the PAr2 (sterically less encumbered compared to the corresponding cis-olefin/PAr2 structures) and the metal-hydride addition will take place from the re-face of the olefin (e. g., through the transition state B in Figure 6), eventually leading to the observed major product. In this orientation, the interaction between the hydrogen ortho to the OR group of the ligand and the aromatic moiety of the vinylarene is minimized as the distance between the Ni-atom and the benzylic carbon is reduced during the bond-formation. Such interaction would retard addition to the si-face. In partial support of this argument, the observed ee for a bulky vinylarene (2-methoxy-3-vinylnaphthalene) is significantly higher than that for simple styrene derivatives (80% vs <30%) under identical conditions. Further in the hydrovinylation of styrene and 4-methylstyrene, a 3′-methyl-substituted MOP-derivative (44, see Figure 4) gave significantly higher enantioselectivity compared to the 3′-unsubstituted ligand 60% ee vs (<25% ee).37 It is expected that a 3′-susbstituent in MOP would destabilize the transition state A leading to the si-face addition.
Figure 5.
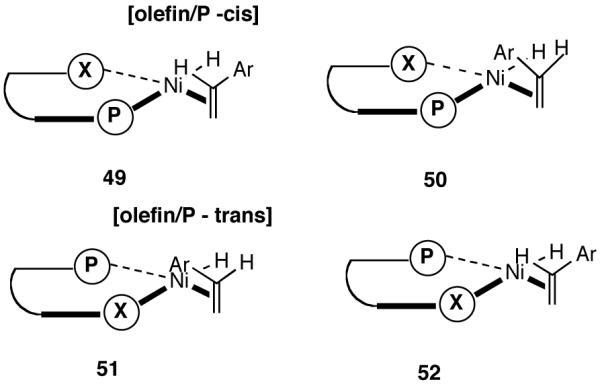
Possible Modes of Olefin Complexation to [Ni(II)-H]+. X Represents the Hemilabile Coordination Site
Figure 6.

A Model for Asymmetric Induction in the HV of a Vinylarene Using an R-MOP-Ni-Complex
Figure 4.

2,2′-Disubstituted-1,1′-Binaphtthyl Ligands for Asymmetric Hydrovinylation
4.7 De Novo Design of an Asymmetric Ligand. 1-(2-X-Aryl)-2,5-dialkylphospholanes (X = Hemilabile Group)
Our search for an in-house catalyst for the Ni-catalyzed asymmetric HV followed a minimalist approach that was based on the following requirements for the ligand: (i) a source of chirality, in the form a chiral P atom or a chiral scaffolding; (ii) an appropriately placed group, capable of forming a kinetically labile chelate. With regard to the second item, one could try heteroatoms of various donor abilities or operate on the size of the chelate ring to modulate the critical hemilabile properties of the group X.
One example that fits the design criteria outlined above is the phospholane 53 shown in Figure 7, and the proposed model for asymmetric induction is depicted in Figure 8. Note that the cis-P/olefin complex might appear to prefer re-face addition (61a). There is no such discernable preference for the trans-P/olefin complex 62. Our conjecture, admittedly without much rationale, was that additional elements of chirality near the hemilabile atom might increase selectivity, even though the exact nature of such control may have to be learned by further experimentation.
Figure 7.
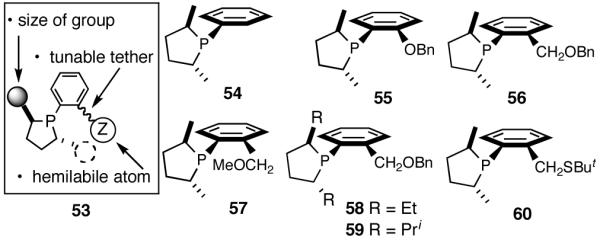
Elements of a Minimalist Ligand for Asymmetric Hydrovinylation and Some Examples
Figure 8.
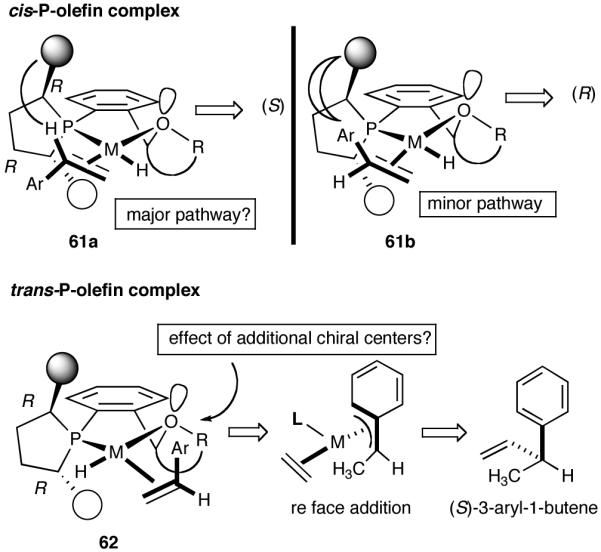
A Working Model for Asymmetric Induction in (Phospholane)Ni(II) Catalysis
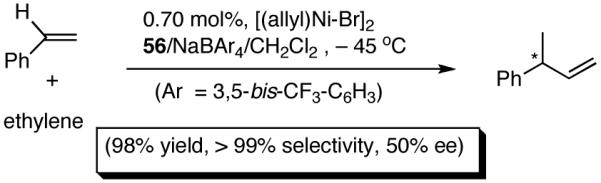
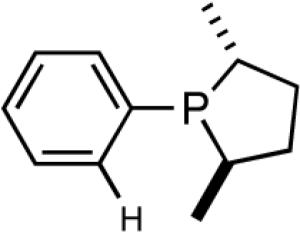
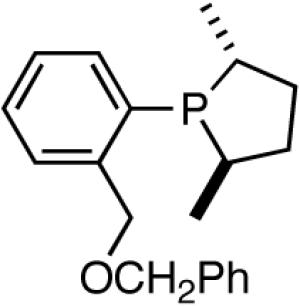
A series of simple phospholane derivatives 54-60 (Figure 7) were prepared and tested for asymmetric HV.41 We started with the 1-arylphospholano-ligand 54, and a close analog, 55, with a potential hemilabile group at the ortho-position. While we find 54 to be an excellent ligand for the Ni-catalyzed hydrovinylation of vinylarenes, especially with OTf as the counterion (Table 4, entry 1), 55 leads to significant isomerization of the initially formed product 1 (to 2, eq 30) under the standard reaction conditions even at -55 °C. One of the principal differences between 55 and the versatile MOP ligand (28), we conjecture, is the placement of the hemilabile alkoxy group with respect to the phosphorus. In 55 it is on the β∼-carbon and in 28 it is on the δ-carbon, resulting in a 5-vs 7-memebered Ni-chelate intermediate in the respective cases. This might have attendant consequences with respect to reversibility of the Ni-H addition, once the product 1 is formed. In order to probe the effect of the relative positioning of the hemilabile group, the o-benzyloxymethyl analog 56 was prepared, and most gratifyingly, this ligand proved to be one of the best for highly selective hydrovinylation reactions (eq 31). No trace of isomerization products (2) was detected under optimum conditions!
Table 4.
Effect of Counterions on the Hydrovinylation of Styrene Using ‘Hemilabile’ Phospholane Ligands
| entry | additive | (%yield; %ee) | remarks | |
|---|---|---|---|---|
| 54 | 56 | |||
| 1 | AgOTf | 94 (37) | <4 (-) | 37 %ee (S) with 54 |
| 2 | AgClO4 | 95 (low) | <2 (-) | 29% isom. with 54 |
| 3 | AgNTf2 | <2 | 48 | 47 %ee (S), 9% isom. with 56 |
| 4 | AgSbF6 | <2 | 94 | 48 %ee (S) with 56 |
| 5 | NaBAr4 | <2 | 97 | 50 %ee (S) with 56 |
| 6 | NaBAr4 | <2 | 97 | 42 %ee (S) with 57 |
The results of hydrovinylation of styrene41 using 54 and 56 shown in Table 4 deserve some comments. For the simplest phospholane ligand 54, with no possibility of
 |
(30) |
 |
(31) |
hemilabile coordination, the reaction does not proceed unless a weakly-coordinating anion such a OTf is used (entries 1 and 2). Incidentally, with ClO4- significant isomerization of the primary product is observed when this ligand is used. Additives such as AgBF4, NaBPh4, AgNTf2, AgSbF6 and Na(B[3,5-(CF3)2-C6H3)]4) used in conjunction with 54 (Table 4, column 3) gave practically no reaction under the standard conditions (entries 3-5), mostly because of immediate precipitation of Ni(0) from the solution. In sharp contrast, for ligand 56 (or 57) with the o-alkoxymethylphenyl substituent, best results were obtained with non-coordinating counter anions BARF- and SbF6- (entries 4-6). Catalyst solution containing these combinations also appeared to be remarkably stable for at least two days at room temperature, as judged by 31P NMR. Not surprisingly, AgOTf, AgClO4, AgBF4 were found to be ineffective with ligands 56 and 57. Some support for the hemilabile coordination has been obtained by NMR spectroscopy.41
Increasing the size of the 2,5-substituents on the phospholane improves the enantioselectivity. Thus the diethyl derivative 58 (Figure 7) gave 63% and 67% ee’s for styrene and 4-i-butylstyrene in highly selective reactions. For 4-i-butylstyrene, a precursor for ibuprofen, this represented one of the highest overall selectivities recorded at the time we reported these ligands. The 2,5-diisopropylphospholane (59) appears to be too bulky to effect the hydrovinylation reaction. Even at 25 °C most of the starting material was recovered.
Finally the ligand 60 (Figure 7) with an o-CH2SBut group in place of the CH2OR substituents does not give any reaction. The sulfur atom in this ligand is likely to be a strong donor.
Based on the working model for asymmetric induction in this reaction (Figure 8), we decided to examine the effect of introducing additional elements of chirality at the hemilabile position (see structure 62, Figure 8). We prepared a series of P-(2-X-aryl)-2,5-dialkylphospholanes (X = dioxan-2-yl or dioxalan-2-yl, Table 5) via modification of a procedure we had published earlier (Scheme 7).41,42
Table 5.
Synergistic Effects of Hemilabile Coordination and Counterions in the Hydrovinylation of Styrene
| ligand | additive (MX) | yield/selectivity |
|---|---|---|
 |
AgOTf | 94 (37% ee) |
 |
<4 | |
 |
AgOTf | <2 |
 |
97 (50% ee) |
Scheme 7.
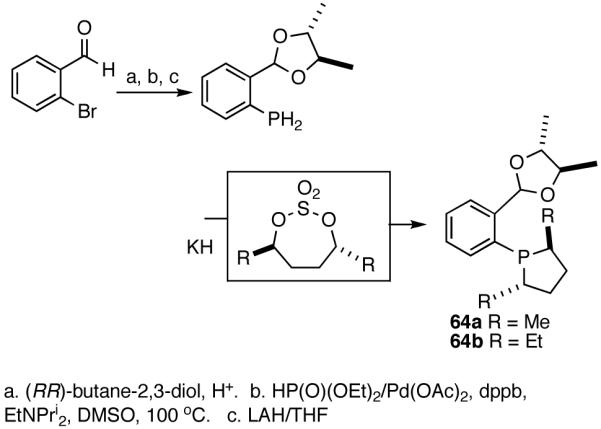
Synthesis of a Prototypical Phospholane/Acetal Ligand 64
In scouting experiments the hydrovinylation reaction was carried out using 0.007 equivalents of Ni and the phosphine ligand in an atmosphere of ethylene at - 55 °C and the results are tabulated in Table 6. The acetalcontaining phospholanes (63-67), in general, are excellent ligands for asymmetric hydrovinylation, giving quantitative yields and selectivities (for the 3-arylbutenes) of the expected products.
Table 6.
Asymmetric Hydrovinylation of 4-Isobutylstyrene with Phospholane Ligands
| entry | ligand | conv. | sel(%)a | %ee |
|---|---|---|---|---|
| 1. | 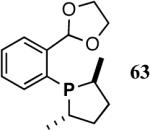 |
>99.5 | >99.5 | 85 (R) |
| 2. | 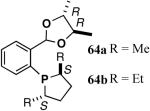 |
>99.5 83 |
>99.5 >99.5 |
91 (R) 88 (R) |
| 3. | 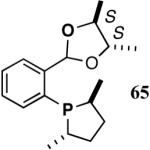 |
>99.0 | 90b | 71 (R) |
| 4. | 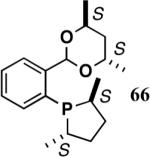 |
>99.5 | >99.5 | 85 (R) |
| 5. | 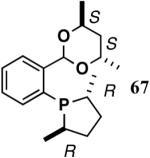 |
>99.5 | >99.5 | 90 (S) |
selectivity for 3-arylbutene.
rest cis- and trans-2-arylbutenes
The ligand 63, with an achiral acetal appendage, gives 85% ee in the asymmetric hydrovinylation of 4-i-butylstyrene (entry 1, Table 6). The combination of (SS)-2,5-dimethylphospholane and acetal derived from (RR)-2,3-butanediol (64a) gives the best selectivity (91% ee, entry 2). Increasing the size of the phospholane 2,5-substituents from Me to Et (64b) appear to have little effect on ee, but significantly, the rate of the reaction is slower (entry 2). A change in configuration of the 4,5-carbons of the 1,3-dioxalane (65, entry 3) leads to onset of isomerization of the primary product (up to 10%). Significant deterioration of the enantioselectivity (71% ee) is also observed. Structurally analogous ligands 66 and 67 with 1,3-dioxane side-chain behave in a similar fashion. In this case, as expected, the (RR)-phospholane/(SS)-dioxane combination (67) gives the best results (entry 5). An examination of the results from entries 1-5 shows that the stereoselectivity of the reaction is dictated by the chirality of the phospholane ring, with the (RR)-phospholane favoring (S)-3-arylbutene, in accordance with the proposed model.
Use of the ligand 64a in hydrovinylation of other vinylarenes gave the following ee’s under the typical reaction conditions (0.70 mol% Ni/-55 °C, >99.5% yield, unless specified otherwise): styrene (88); 4-methylstyrene (86); 4-bromostyrene (71); 4-methoxystyrene (73); 2-methoxy-6-vinylnaphthalene (86, 73% yield.). Except for 4-bromostyrene, at the time of this Communication, these were among the highest ee’s reported for the asymmetric hydrovinylation of these substrates.
 |
(32) |
Finally, efficiency of the catalyst for the reaction was examined using ligand 64a. In a reaction carried out with 4-i-butylstyrene/[Ni(II)/64a] ratio of 1428 (0.07 mol% catalyst) a yield of 86% (rest, starting material) was realized (eq 32).
4.8 Diarylphosphinite Ligands
Even though the initial studies with the MOP (eq 27) and 1-aryl-2,5-dialkylphospholane (eq 32) ligands provided a number of useful parameters such as the effect of hemilabile coordination and counteranions to improve the efficiency and selectivity of the catalyst system, the enantioselectivity in the hydrovinylation of styrene derivatives remained modest. In continued efforts to improve the enantioselectivity we recently screened a large number of ligands and found that easily accessible diarylphosphinites serve as excellent ligands for this exacting reaction.43a,28b Sugar phosphinites are a class of easily synthesized ligands we used before with remarkable success in other asymmetric reactions such as hydrocyanation,38c,f,g hydrogenation38d,h,i and allylation reactions.38j They are readily amenable to steric and electronic tuning; a highly desirable attribute for ligands for asymmetric catalysis. The results of hydrovinylation of styrene using these ligands are shown in Table 7.
Table 7. Hydrovinylation of Styrene Using Diarylphosphinite Ligandsa.
| entry | ligand | Ar | conv./y.b | sel.c | %eed |
|---|---|---|---|---|---|
| 1. | 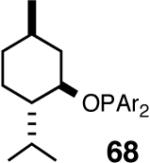 |
B | 99/86 | 86 | >5 |
| 2. | 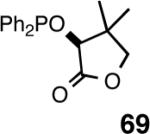 |
- | --/87 | 99 | 6 |
| 3. | 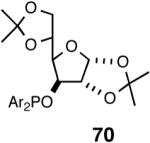 |
B | 68/68 | 99 | 29 (R) |
| 4. | 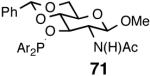 |
A | 62/62 | 99 | 32 (S) |
| 5. | 71 | B | 35/35 | 99 | 28 (S) |
| 6. | 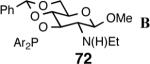 |
B | 0 | -- | -- |
| 7. | 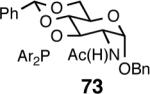 |
A | 97/93 | 96 | 9 (S) |
| 8. | 73 | B | 93/93 | 99 | 45 (S) |
| 9. | 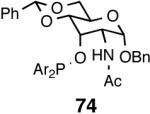 |
 |
|||
| 10. | 74 | B | 42/42 | 99 | 62 (S) |
| 11. | 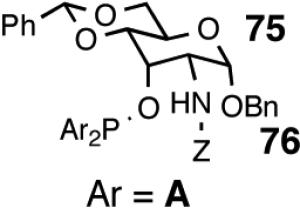 |
Z = CF3CO | 99/40 | 40e | 87 (S) |
| 12. | Z = PhCO | 99/23 | 23e | 82 (S) | |
See eq 33. A Ar = 3,5-Me2-C6H3; B Ar = 3,5-(CF3)2-C6H3.
isolated yield.
% of 3-phenyl-1-butene.
by HPLC.
conv. >99%
Principally, bis-(3,5-dimethylphenyl)- and bis- (3,5-di-trifluoromethylphenyl)- phosphinites were chosen for this study. In general, outstanding selectivity for 3-phenyl-1-butene is observed with variety of phosphinites. Whether a 3,5-bis-CH3-C6H3-substituent or a 3,5-bis-CF3-C6H3-substituent on phosphorus is better depends on the configuration of the carbon to which is attached the diaryl phosphinite moiety. In the gluco-series (entry 7/8) the CF3-aromatic substituent is better, where as in the allo-series (entry 9/10) the CH3-aromatic substituent is better. The allo-configuration for the ligand (entries 9,10) is clearly superior compared to the gluco-derivative (entries 7, 8) for higher enantioselectivity. Finally, the acyl group on nitrogen showed a pronounced effect on the selectivity of the reaction (entries 11 and 12). Whereas the acetyl substituent on nitrogen gives consistently high selectivity (most of the time >99%) for the desired product, alkyl groups inhibit the reaction (entry 6). The N-COCF3 and N-COPh derivatives promote concomitant isomerization of the initially formed 3-phenyl-1-butene to a mixture of 2-phenyl-2-butenes under the reaction conditions, reducing the selectivity for the former to 40% and 23% respectively (entries 11 and 12). Remarkably the highest ee for styrene (87%) in this series is observed for the N-C(O)CF3 derivative.
 |
(33) |
 |
(34) |
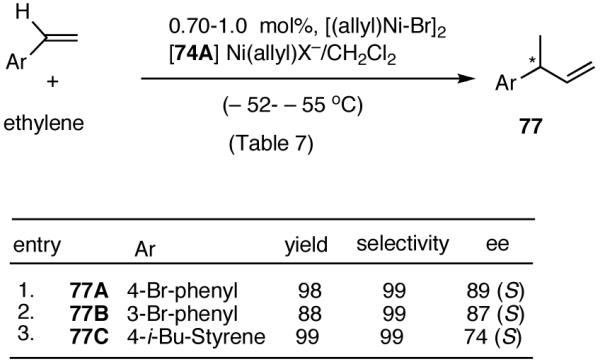
In overall yield and selectivity, the diarylphosphinite 74A is one of the best ligands for the Ni-catalyzed asymmetric hydrovinylation of styrene (Table 7, entry 9). Most gratifyingly, ligand 74 A is also one of the best ligands for the hydrovinylation of other derivatives such as 4-bromostyrene, 3-bromostyrene and 4-i-butylstyrene (eq 34). In the case of 4-bromostyrene up to 98% isolated yield (>99% selectivity for the desired product) with 89% enantiomeric excess is obtained. Selectivities for 74A and other related ligands in the hydrovinylation of 4-bromostyrene are shown in Table 8.
Table 8.
Asymmetric Hydrovinylation of 4-Bromostyrene Using Phosphinite Ligandsa
| entry | ligand (counterion) | yield (%) | selectivityb | %eec |
|---|---|---|---|---|
| 1. | 74A (SbF6) | 98 | >99 | 89 (S) |
| 2. | 74A (BARF) | 94 | 94 | 89 (S) |
| 3. | 74A (BF4) | 24 | >99 | 86 (S) |
| 4. | 74A (OTf) | 70 | >99 | 74 (S) |
| 5. | 74B (BARF) | 19 | >99 | 43 (S) |
| 6. | 70A (BARF) | 88 | >99 | 13 (S) |
| 7. | 70B (BARF) | 41 | >99 | 47 (S) |
1 atm. ethylene, 1 mol% [(allyl)NiP]+ X-, CH2Cl2,- 55 °C, 2 h.
for 3-aryl-1-butene; >99% means no other hydrocarbon products were observed.
determined by HPLC.
A study of the effect of the counteranion on this reaction (Table 8) shows that SbF6 is marginally better than BARF (entry 2), whereas BF4 and OTf appear to be inferior (entries 3 and 4).
The enantiomeric excess of 3-(4-bromophenyl)-1-butene, 77A, from which other 2-arylpropionic acids could be prepared by crosscoupling chemistry is ∼ 89%. For example, Kumada coupling of 77A and i-BuMgBr in the presence of 1 mol% of (dppe)NiCl2 gave 77C. Subsequent ozonolysis and oxidation of the resulting aldehyde gave ibuprofen, whose configuration and enantiomeric excess were established by conversion to the known (-)-menthyl esters. Gas chromatograpic analysis of these esters using chirasil-L-val column revealed baseline separation, with a diastereomeric excess of 89% for the (R)-ibuprofen ester. This establishes the overall selectivity and the absolute configuration of the primary product (S) of hydrovinylation of 4-bromostyrene.
The hydrovinylation of 3-bromostyrene using 74A as a ligand gives the corresponding 3-aryl-1-butene in 88% yield and 87% enantioselectivity (eq 34, entry 2).
Finally, studies with 4-i-butylstyrene (eq 34, entry 3) serve as a reminder that a single ligand is unlikely to have broad applicability, and further fine-tuning maybe needed before practical levels of asymmetric induction can be achieved for individual substrates.
4.9 Phosphite Ligands
Binaphthol-derived phosphites prepared from carbohydrate diols are also competent ligands28b,43 for the hydrovinylation of styrene under conditions described in eq 34, using BARF as a counter ion. The yield and enantioselectivity for styrene hydrovinylation are modest and appear to be dictated by the configuration of the BINAP unit rather than the carbohydrate backbone.
4.10 Phosphoramidite Ligands
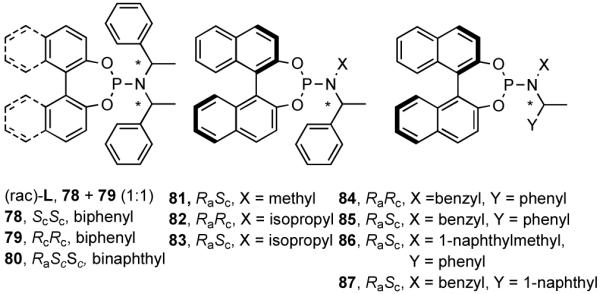
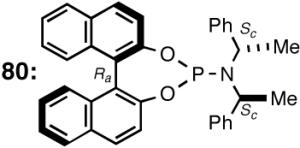
Phosphoramidites, originally introduced by Feringa44 for the asymmetric Cu-catalyzed conjugate addition of dialkylzinc reagents to enones, are among the most versatile and tunable ligands for C-C and C-H bond-forming reactions.45 Phosphoramidites were introduced for hydrovinylation of vinylarenes by Leitner46 and, later, for norbornene by our group28 under the conditions we originally prescribed for these reactions.22 For several vinylarenes including 4-bromostyrene46 and norbornene28 highest efficiencies and selectivities were recorded. However, 4-isobutylstyrene (precursor for ibuprofen) gave only 28% conversion and 68% ee. To expand the scope of the reaction we undertook a systematic study of ligand tuning using the phosphoramidites derived from 1.1′-binaphthol, 1,1′-biphenol and a variety of α-methylarylamines. These studies resulted in the highest enantioselectivities reported to date for HV of a broad spectrum of vinylarenes.47 A partial list of ligands examined in this study are shown in Figure 9.47b
Figure 9.
Selected Phosphoramidite Ligands Used for Hydrovinylation
The feasibility of ligand control in hydrovinylation was initially investigated using p-methoxystyrene, an electron-rich model substrate that consistently had given one of the poorest reactions among vinylarenes tested previously. We started these investigations using a modified protocol (eq 31) that had originally been developed for MOP and phospholane ligands.
Among the ligands examined,47 in addition to the original Feringa ligand 80, two others stand out. The ligand 78 (or its enantiomer 79), which has only a lowly biphenyl backbone instead of a chiral binaphthyl unit and is significantly cheaper, still yields similar selectivities and conversions. The ligand 87, in which the (S)-N-α-methylbenzyl groups are replaced with an achiral benzyl and a chiral (S)-α-methyl-1-naphthyl group is by far the best ligand for this exacting reaction48 yielding nearly quantitative yield and selectivity (Table 8). Surprisingly, ligands prepared from achiral dibenzylamine and enantiopure 2,2′-binaphthol (not shown) gave no conversion.
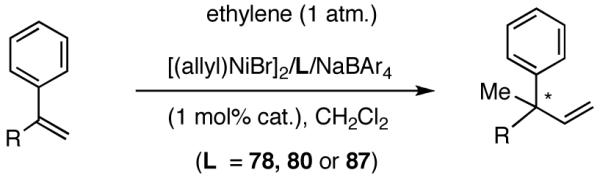
Once the best ligands were identified, the studies were extended to several vinylarenes and the results are tabulated in Table 9. The enantioselectivities observed for the 3-arylbutenes 89-92, which are precursors for arylpropionic acids ibuprofen, naproxen, flurbiprofen and fenoprofen (entries 2-5), represent the highest overall selectivities reported todate for any viable intermediates for these important compounds.49 In one case Craig Smith, who has been involved wih the development of the phosphoramidite ligands, has shown that HV of 4-isobutylstyrene can be accomplished with 0.00014 equiv catalyst (substrate /catalyst ratio = 7142) in 4.67 h at 0 °C. For the biphenyl-derived ligands 78 and 79, the configuration of the amine determines the sense of asymmetric induction. With the S-chiral moiety in the amine portion of the ligand, the product configuration in all cases is also S. As seen in entries 1-5, the lack of axial chirality in the ligand leads to little erosion of ee, suggesting that for simple substrates a more elaborate (and expensive) binaphthol-based phosphoramidite is not necessary to achieve high stereoselectivity. In all cases examined, 87 yielded the best results in terms of overall yield and selectivity. To the best of our knowledge this is a novel ligand.
Table 9.
Asymmetric Hydrovinylation of Vinylarenes Using Finely Tuned Phosphoramidesa
| no. | product | lig. | convn/yield | sel. | ee(%)e/conf |
|---|---|---|---|---|---|
| 1. |  |
78 | >99/82 | >99 | 95, S |
| 80 | >99/79 | >99 | 95, S | ||
| 87 | >99/77 | >99 | 97, S | ||
| 2. | 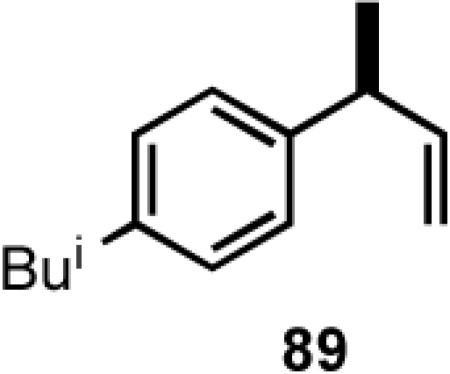 |
78 | >99/97 | 98 | 90, S |
| 80 | >99/98 | 99 | 90, S | ||
| 87 | 97/97 | >99 | 96, S | ||
| 3. | 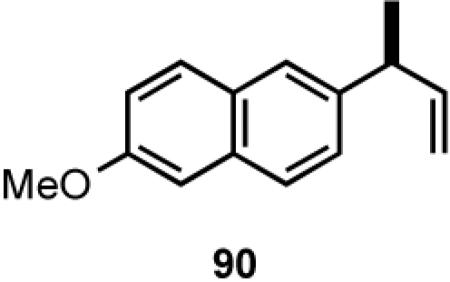 |
78 | >99/93 | >99 | 90, S |
| 80 | >99/94 | <99 | 95, S | ||
| 87 | >99/89 | >99 | 99, S | ||
| 4. | 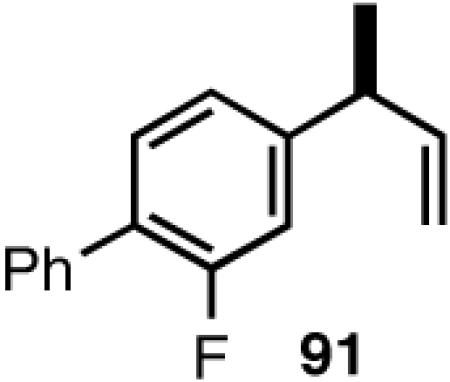 |
78 | >99/90 | >99 | 80, S |
| 80 | >99/93 | >99 | 86, S | ||
| 87 | >99/92 | >99 | 97, S | ||
| 5. | 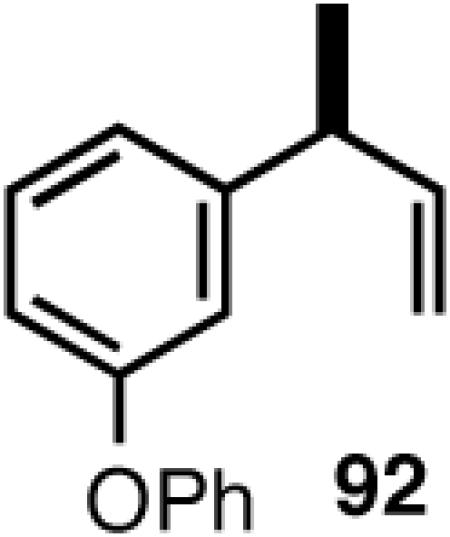 |
78 | >99/91 | >99 | 95, S |
| 80 | >99/96 | >99 | 97, S | ||
| 87 | >99/92 | >99 | 97, S |
See reference 47 for details and a complete set of ligands
5. Generation of All-Carbon Quaternary Centers55
The search for new methods for stereoselective generation of all carbon-quaternary centers is a subject of considerable topical interest.50 Several important pharmaceutically relevant compounds, among them, analgesic (-)-eptazocine,51 protein kinase C activator lyngbyatoxin,52 cognitive enhancing agent (-)-phenserine,53 and serotonin antagonist LY42696554 contain all-carbon quaternary centers at the benzylic position. Hydrovinylation of 2-aryl-1-alkenes26 generate a quaternary center at the benzylic positions (eq 35) and introduces a highly versatile latent functionality in the form of a vinyl group. The resulting intermediates could be quite valuable for further synthetic elaboration. An asymmetric variant of this reaction is shown in eq 36.55
 |
(35) |
 |
(36) |
In scouting studies using 2-phenyl-1-butene (93, Table 10) as substrate, catalysts derived from the MOP ligand (Figure 4, 28) show no reactivity while those derived from phospholane ligand 56 (Figure 7), which gave high ee’s and turnover numbers in the hydrovinylation of a number of styrene derivatives42 and 1,3-dienes (vide infra),58 show only moderate reactivity under similar conditions.55 Among the chiral ligands examined, the phosphoramidite 78,47 80,55,56 and 8747 were found to provide the best results. These ligands, when treated with [(allyl)NiBr]2 followed by NaBARF gave a very active pre-catalyst that effects the hydrovinylation of 1-ethylstyrene at low temperature with as little as 1 mol% of catalyst to give a nearly quantitative reaction.56 Under these conditions, no oligomerization product is detected, as judged by careful GC analysis and 1H NMR spectroscopy. The yields and selectivities are highly reproducible, and as expected, best selectivity is observed at low temperatures. They are independent of the catalyst loading or extent of reaction, clearly indicating the total absence of non-selective reactions.
Table 10.
Asymmetric Hydrovinylation of 2-Aryl-1-alkenes. Generation of All-Carbon Quaternary Centersa
| entry | vinylarene | T (°C)/t (h) | conv./yield | sel. | ee (%) |
|---|---|---|---|---|---|
| 1. |  |
-70/4 | >99/>95 | >99 | >95 |
| 2. |  |
-69/12 | >99/>90 | >99 | 90 |
| 3. |  |
-70/11 | >94/>90 | >95 | 90 |
| 4. |  |
24/20 | 61/60 | >97 | >95 |
| 5. |  |
-70/8 | >98/93 | >96 | >50 |
| 6. |  |
-70/14 | >99/>98 | >99 | 93 |
| 7. |  |
-70/4 | >98/70 | 71 | >95 |
Results of asymmetric hydrovinylation of several 2-aryl-1-alkenes under the optimal conditions are tabulated in Table 9. While substrates 93 and 94 gave excellent selectivity for the formation of the expected product, the 4-chloro derivative 95 gave up to 5% isomerization of the starting olefin (entry 3). A similar minor side reaction was also observed for the substrates 97 and 99. An isopropyl group at the 1-position of the styrene (96) retards the reaction (entry 4), and it is best accomplished at 24 °C with 10 mol% catalyst. Even though the yield of the reaction is only moderate, very high ee (∼97%) was observed for the isolated product. The 2-naphthyl derivative 98 gave excellent yield (>98%) and selectivity (>99%) for the expected product. The tetralin derivative 99 represents a different class of substrates that under-went the hydrovinylation reaction giving >95% ee. Significant isomerization (∼30%) of the starting material to an endocyclic olefin is a major detraction of this otherwise useful reaction.
Compounds (e.g., 100b) structurally related to the HV product 100a from 99 have been synthesized previously via intramolecular asymmetric Heck reactions (∼93% ee),51 stoichiometric oxazoline directed alkylation (∼99% ee),57a and enzyme-catalyzed desymmetrization of a chiral malonate (97% ee).57b By comparison, the asymmetric hydrovinylation route is significantly shorter, and operationally simpler.
Among the other olefins 101-103, only the acyclic diene 103 undergoes hydrovinylation, and the product 104 is formed in nearly racemic form, contaminated with product of ethylene addition at the benzylic position.
6. Asymmetric Hydrovinylation of 1,3-Dienes58
Although asymmetric hydrovinylation of 1,3-cyclooctadiene (eq 24), is one of the earliest reported metal-catalyzed asymmetric C-C bond-forming reactions,11a,59 no satisfactory solution to the problem of hydrovinylation of 1,3-dienes had emerged until 2006.4 Both the Wilke conditions19 using the azaphospholene ligand (RR)-7 (Figure 2 and eq 12), and the use of a catalyst from aminophosphine phosphinite/Ni(COD)2/Et2AlCl,60 reported for 1,3-cyclohexadiene (eq 26), are limited either by the esoteric nature of the azaphospholene ligand, which permits no structural simplifications,21 and/or by the constraints imposed by the need for a strong Lewis acid like EtAlCl2. The isomerization of the product 1,4-diene at higher conversion could be one of the limitations of a recently reported non-asymmetric Ru-catalyzed reaction (eq 37).61 Asymmetric version of this reaction remained largely unexplored until our work.
 |
(37) |
We wondered whether the beneficial effects of the synergistic effects between ligands and counter ions could be applied to develop a viable Ni-catalyzed hydrovinylation of 1,3-dienes. An asymmetric version of this reaction would be especially attractive for 1-vinylcycloalkenes, since the product 1,4-dienes would allow control of absolute and relative configurations of the side-chains and of other stereogenic centers on the ring, a common feature in many important natural products, including steroid D-rings, serrulatanes and psuedopterosins (Scheme 8).58
Scheme 8.
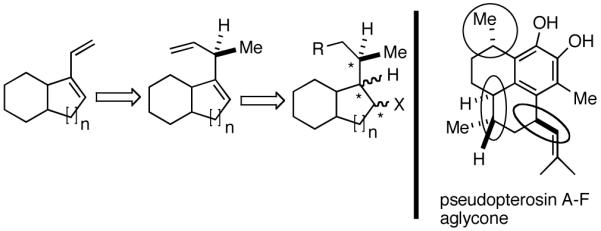
Hydrovinylation of 1,3-Dienes and Control of Exocyclic Stereochemistry
Our studies58 started with an examination of hydrovinylation of cyclohexa-1,3-diene (106) and 4-t-butyl-1-vinylcyclohexene (107), using the procedure we successfully employed for the hydrovinylation of vinylarenes [eq 13: Ph3P/[(allyl)NiBr]2/AgOTf, 0.07 equiv. Ni, low temp., CH2Cl2, 1 atm ethylene]. It soon became apparent that under these conditions, 1,3-dienes were much less reactive compared to the vinylarenes, and higher temperatures (∼25 °C) were needed for the reaction.
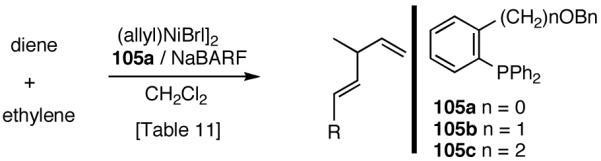 |
(38) |
We decided to explore new protocols for this potentially useful reaction by systematically examining the use of the hemilabile ligand effects41 using 107 (Table 11) as a substrate and ligands 105a∼c as ligands (eq 38). These studies revealed that the best ligand for this reaction was 2-benzyloxyphenyldiphenylphosphine (105a). Thus, 0.14 mol% of a catalyst generated from 105a, allyl-nickel bromide dimer and NnBARF effects the reaction of 107 with ethylene (1 atm) to give a quantitative yield of the product 116, as a mixture of two diastereomers (eq 38 and Table 11, entry 2). This product is formed with exquisite regioselectivity (1,2-addition at the less hindered olefin). The racemic, axially chiral olefin 107 gave a nearly ∼2:1 mixture of diastereomers. The results of hydrovinylation of other typical dienes are shown in Table 11. In general, excellent yields (>97%) and selectivities (>95%) are observed for the hydrovinylation of both cyclic and acyclic dienes (entries 1, 2, 5-9) under 1 atmosphere of ethylene. Lack of selectivity is seen only for 1-vinylcyclohexene (108, entry 3) and 1-vinylcyclopentene 109 (entry 4), which gave a mixture of 1,2- and 1,4-addition products.
Table 11.
Hydrovinylation of 1,3-Dienes
| entry | diene | conv. | regiosel. | product(s) |
|---|---|---|---|---|
| 1. | 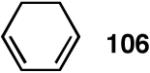 |
94 | >99 | 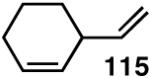 |
| 2. | 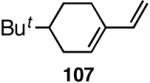 |
>99 | 98 | 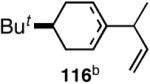 |
| 3. | 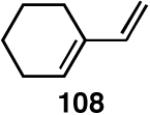 |
>99 | 68 | 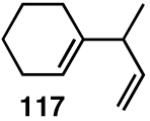 |
| 4. | 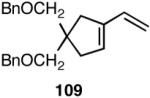 |
>99 | 72 | 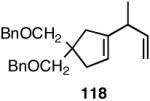 |
| 5. | 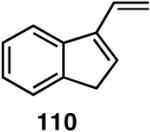 |
>99 | >99 |  |
| 6. | 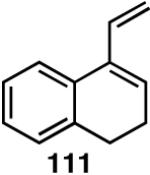 |
>99 | 98 |  |
| 7. | 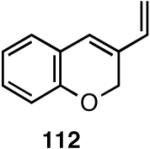 |
∼97 | >99 | 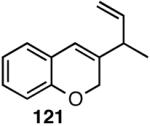 |
| 8. | 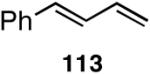 |
>99 | 95 | 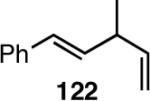 |
| 9. | 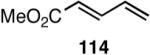 |
99 | 98 | 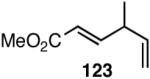 |
see reference 58 for details.
mixture of two diastereomers (∼2:1)
Table 12 shows asymmetric hydrovinyaltion of 1,3-dienes. Thus hydrovinylation of 110, 111 and 112 under our standard conditions (eq 38) using the phospholane 64a42 or the phosphoramidite ligand 80 gave exceptionally high yields, regio- and enantioselectivities for these cyclic dienes. Acyclic diene 113 under these conditions gave low selectivity even with the phosphoramidite 80. However a structurally related ligand derived from biphenol (78, Figure 9) gave up to 84% ee.47 The high selectivity for acyclic diene is noteworthy since this is a class of challenging substrates for asymmetric transformations.61b, 63
Table 12.
Asymmetric Hydrovinylation of 1,3-Dienes
| Ligand: | 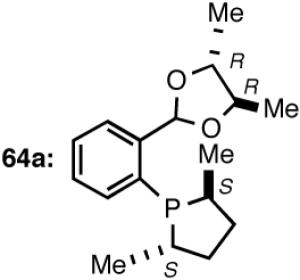 |
 |
||
|---|---|---|---|---|
| diene | conv. % (sel. %) | %ee (config.) | conv. % (sel. %) | %ee (config.) |
| 110 | >99 (>99) | 85 (R) | >99 (>99) | 96 (S) |
| 111 | >99 (97) | 93 (R) | >99 (>99) | >99 (S) |
| 112 | >99 (99) | 38 (R) | >99 (>99) | 95 (S) |
| 113 | 88 (>99) | <5% | >99 (∼96) | 77 (?) |
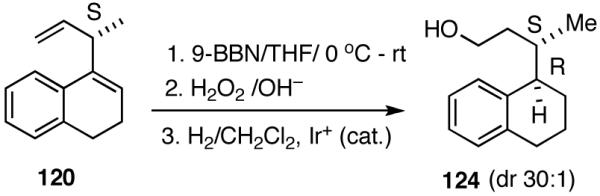
A number of different strategies can be envisioned for controlling the configuration of the ring carbon to which the side-chain is attached.62 One example is shown in eq 39. The hydroboration (9-BBN, H2O2) of (S)-120 followed by directed hydrogenation using Crabtree’s catalyst, ([(COD)(Cy3P)Ir(py)]+ PF6-), gives a reduced product (124, dr 30:1) with very high stereoselectivity.
 |
(39) |
7. Asymmetric Hydrovinylation of Norbornene
We have already alluded to the initial results on hydrovinylation of norbornene as one of the first metal-catalyzed asymmetric C-C bond-forming reactions (eq 25) and the remarkable dependence of the reaction on the cone angle of the phosphine employed (eq 23).11b,19 The results obtained with the new ligands are shown in eq 40 and Table 13.28
 |
(40) |
Table 13.
Asymmetric Hydrovinylation of Norbornenea
| Entry | Ligand | Additive | 18 (%) | 19 (%) | %ee |
|---|---|---|---|---|---|
| 1. | 28 | NaBARF | 69 | 0 | 44 |
| 2. | 28 | AgSbF6 | >99 | 0 | 50 |
| 3. | 28 | AgNTf2 | >99 | 0 | 50 |
| 4. | 37 | AgSbF6 | >99 | 0 | 44 |
| 5. | 80 (RaScSc) | NaBARF | >99 | 0 | 80 |
| 6. | 80 (RaScSc) | AgOTf | 20 | 0 | -- |
| 7. | 80 (RaScSc) | AgSbF6 | 0 | 99 | 34 (19) |
| 8. | 80′ (RaRcRc) | NaBARF | <2% | 0 | -- |
| 9. | 15 | AgOTf | 11 | 88 | 33 (19) |
Ozonolysis of 18 followed by oxidation of the resulting aldehyde gave norbonane-2-carboxylic acid, the enantiomers of which were converted into esters of (S)-methyl mandelate by the standard procedure using DCC. The absolute configuration of these diastereomers had been fully established before.64 As expected, phosphines with large cone angles (28 and 37, 80) give exclusively the 1:1 adduct in nearly quantitative yield and modest enantioselectivity (entries 1-5). Note the use of highly dissociated counteranions in these reaction. No trace of the 2:1 adduct 19 is observed under these conditions. The selectivity with the phosphoramidite ligands (entries 5-8) depends on both the counteranion and the nature of the secondary amine appendage. Whereas the (RaScSc)-isomer is a good ligand (entry 5), the corresponding (RaRcRc)-diastereomer 80′ gives less than 2% of the product (entry 8). Suprisingly, for the ligand 80 (RaScSc), the counter anion determines whether 1:1 or 1:2 adduct is produced. With NaBARF only 1:1 adduct is produced (entry 5), whereas AgSbF6 (which we have successfully used in place of NaBARF in some early hydrovinylation experiements43a), now gives exclusively the 2:1 adduct 19 in nearly quantitative yield (entry 7)! Phospholane 15 gives mostly the 2:1 adduct (entry 9). A modest enantioselectivity of 33% has been observed for this product as determined by the Mosher ester method.28 As we have documented before, the use of AgOTf as an additive is important for the ligands like 15 with no hemilabile side-chain. Chelating ligands inhibit the reaction under the typical conditions reported here.
8. Applications of Asymmetric Hydrovinylation Reactions
8.1 (S)- or (R)-2-Arylpropionic Acids
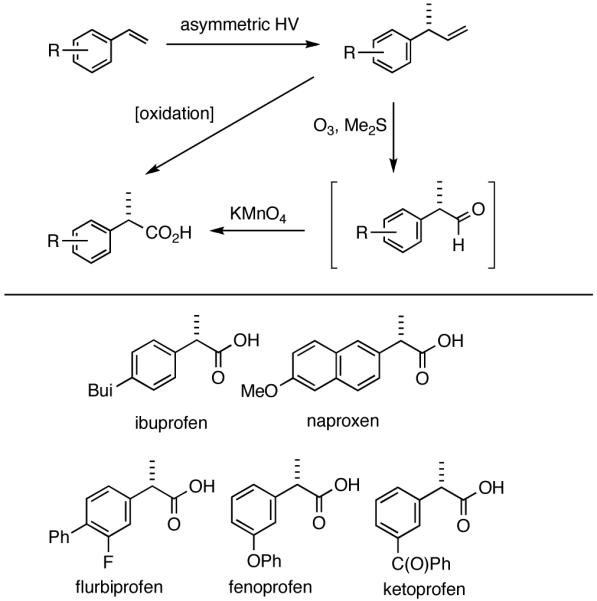
2-Arylpropionic acids are the most widely used non-steroidal antiinflammatory agents (NSAID).65 Naproxen, 2-(6-methoxy-2-naphthyl)-2-propionic acid, which is the only NSAID currently sold in enantiomerically pure form (S) is resolved by a classical resolution.66 Most members of this important class of compounds can in principle be synthesized by oxidative cleavage of the double bond of the hydrovinylation products of vinylarenes (Scheme 9). With our recent syntheses of various 3-arylbutenes of very high enantiomeric purity (>96% ee)47 this becomes a viable route. Thus Table 9 shows highly enantioselective syntheses of compounds 89, 90, 91 and 92, precursors of ibuprofen, naproxen, flurbiprofen and fenoprofen respectively, via hydrovinylation of the appropriate vinylarene using the ligand 87.66 We have since carried out the HV of 3-bromostyrene in very high ee and the product from this reaction has been converted into ketoprofen via 125.67
Scheme 9.
A General Synthesis of 2-Arylpropionic Acids
Oxidative cleavage by ozone of the double bond in the HV products followed by further oxidation of the resulting aldehydes by KMnO4 or NaClO2 give ibuprofen (from 89) and flurbiprofen (from 91) in acceptable yield without any racemization at the intermediate aldehyde stage (Table 14). More electron-rich naproxen substrate 90 was best oxidized with NaIO4 and KMnO4. These conditions also gave the best yields for the oxidation of the ketoprofen precursor 3-(3-bromophenyl)-1-butene. Likewise, the fenoprofen precursor 125 was obtained using RuCl3/NaIO4 from the corresponding 3-arylbutene. In each case the ee of the final product was confirmed by chiral stationary phase gas chromatography of the (L)-menthyl esters.28b,43a
Table 14.
Synthesis of 2-Arylpropionic Acids via Oxidationa
| entry | product | oxidant | yield | %ee/conf. |
|---|---|---|---|---|
| 1 | Ibuprofen | O3 | 98 | 98 (S) |
| 2. | Naproxen | KMnO4/NalO4 | 67 | >97 (S) |
| 3. | Flurbiprofen | O3 | 95 | 98 (S) |
| 4. | Fenoprofen | RuCl3/NalO4 | 91 | >99 (S) |
| 5. |  |
KMnO4/NalO4 | 93 | 91 (S) |
Enantiomers of compounds shown in Table 9 used for the syntheses
8.2 (R)-α-Curcumene (127) and (R)-ar-Turmerone (131)68
Several important classes of natural products, among them, bisabolanes, heliannanes, serrulatanes and pseudopterosins are characterized by a benzylic chiral center, often carrying a methyl group at this position.69 Diverse biological activities exhibited by these compounds include antiinflammatory, antiviral and antimycobacterial properties and they have attracted considerable attention from synthetic chemists. No less than 12 non-racemic syntheses of the simplest member of this class of compounds, (R)-(-)-α-curcumene (127) are known. (R)-(-)-α-curcumene and related (R)-(-)-ar-turmerone (131) are the constituents of a large number of essential oils and it has been amply demonstrated that intermediates for their synthesis could in principle be used for a number of other bisabolane and other related terpenes.69a
In spite of their rather simple structures, the stereo-center at the benzylic position poses a significant challenge in the asymmetric synthesis of even curcumene.70 Arguably, the ‘shortest (incidentally, also the most recent) route’ starts with citronellal and involves 6 steps and multiple chromatographic separations to produce curcumene in 28% overall yield.71 An exceptionally short synthesis based on asymmetric hydrovinylation of 4-methylstyrene is shown in Scheme 10. This synthesis starts with hydrovinylation of 4-methylstyrene. In the racemic series, the hydrovinylation of 4-methylstyrene can be achieved in nearly quantitative yield and >99% selectivity for the desired 3-arylbutene using ethylene at 1 atm and catalytic amounts of [(allyl)NiBr]2, Ph3P and AgOTf (eq. 13). Chiral ligands like MOP derivative 28, sugar-derived diarylphosphinite 74A and binaphthol-derived phosphoramidite 80 which gave high ee’s for other styrene derivatives, gave unacceptably low ee’s for 4-alkylstyrenes. However, the Ni(II)-complex from 1-aryl-2,5-dialkylphospholane ligand 64a gave greater than >99% yield of 126 with an er of (R:S) 93:7 (Scheme 10).68 Treatment of compound 126 with 9-BBN in THF, followed by the addition of Pd(PPh3)4 (5 mol%), K3PO4 (1.5 equiv.), 2-methyl-1-bromopropene (2 equiv.) and dioxane and stirring at 60 °C afforded (-)-α-curcumene (127) as a colorless oil in 55% overall yield in three steps from 4-methylstyrene (Scheme 10).
Scheme 10.
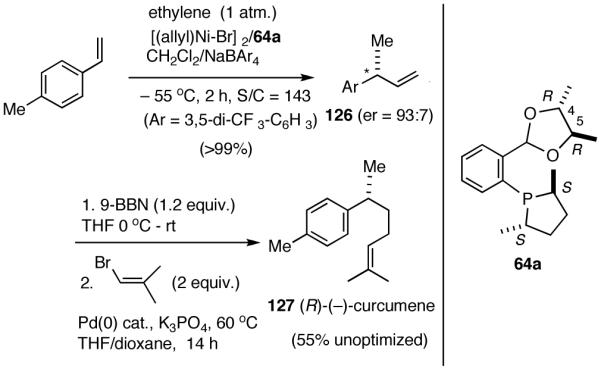
Synthesis of α-Curcumene from 4-Methylstyrene
Synthesis of (R)-(-)-ar-turmerone is accomplished starting with the 3-arylbutene 126. The olefin 126 is subjected to hydroboration with disiamylborane in THF, followed by oxidation with hydrogen peroxide to give alcohol 128 in 84% isolated yield in two steps (Scheme 11). Swern oxidation of alcohol 128 gives aldehyde 129 in 90% yield. Treatment of aldehyde 129 with 2-methyl-1-propenylmagnesium bromide in THF at -78 °C gave a diastereomeric mixture (at C9: 6:5) of alcohol(s) 130 in 78% isolated yield. Swern oxidation of alcohol 130 gave (R)-(-)-ar-turmerone 131 in 44% yield.
Scheme 11.
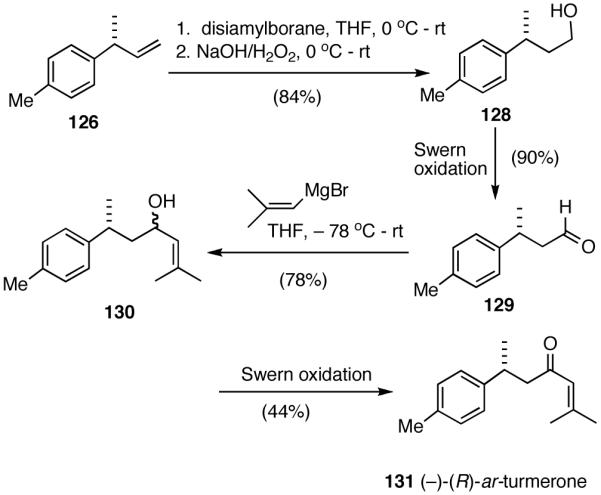
Synthesis of (-)-(R)-ar-Turmerone
8.3 Control of the Configuration of Steroidal D-Ring Side Chain72
Several creative solutions to the problem of installation of stereogenic centers on the steroid D-ring and on the side chains (see Figure 10: 132-134) have been developed over the years, even though no broadly applicable methods that use readily available precursors have emerged.73 The problem is especially acute for the synthesis of the unnatural 20(S)-epimers. Consider for example, precursor 133 for calcitriol analogs with exocyclic C20(S)-configuration, which have been shown to have significant biological activity.74 These molecules are currently prepared by circuitous routes that involve the equilibration of the aldehyde 134, obtained from vitamin D2 and subsequent reactions of the minor isomer isolated from the mixture.75
Figure 10.
Steroids with Functionalized D-Rings
Hydrovinylation of a steroid-derived diene 135 (Figure 11, eq 41) has already been used in a Ru-catalyzed hydrovinylation to prepare 20(S)-compound in a highly stereoselective fashion.61a We wondered whether both the 20(S)- and 20(R)- compounds could be prepared by overcoming the inherent selectivity of the steroid nucleus by the use of enantiomerically pure ligands in a Ni-catalyzed reaction.
 |
(41) |
Figure 11.
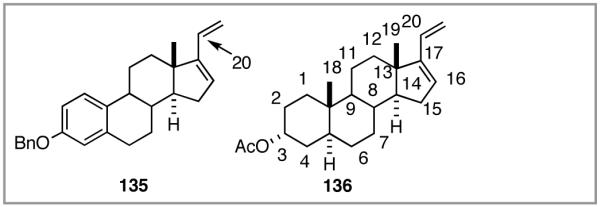
Prototypical Steroidal Dienes for Hydrovinylation
We explored the Ni-catalyzed diene hydrovinylation of two prototypical steroidal dienes 135 and 136 (Figure 11) using the ligands shown in Figure 12. Several ligands we had successfully employed for hydrovinylaton of vinylarenes and dienes either did not react [Figure 12: Ph3P, MOP (27), 105b, 105c] or gave mixtures (105a, 64a) of stereo- and regioisomers (eq 42).
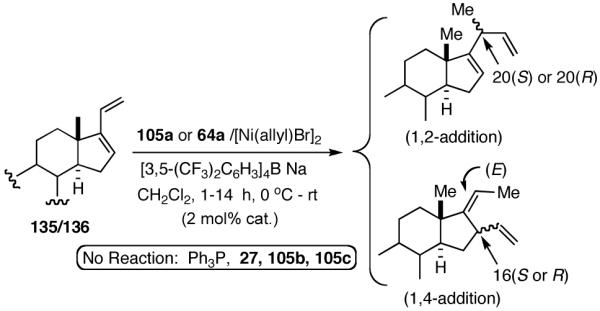 |
(42) |
Figure 12.
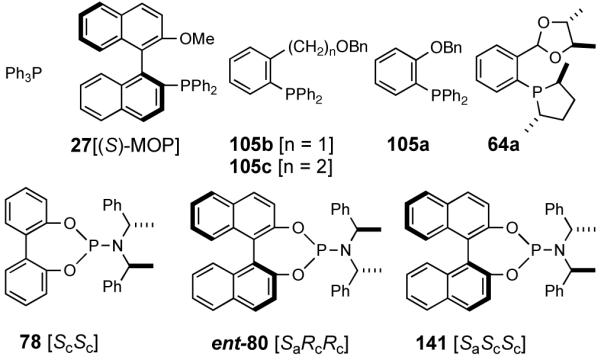
Ligands for HV of Steroidal Dienes
We anticipate this lack of selectivity to be a recurring problem in the context of this and other future synthetic objectives in which hydrovinylations of key chiral intermediates will be involved. It is entirely conceivable that the inherent diastereoselectivity in such substrates could be low, or even opposite to what would be desired. Thus, from a synthetic perspective, either the enhancement of the inherent selectivity or overriding such an outcome with the use of a tunable asymmetric catalyst became a highly desirable goal. Looking for a general solution to this problem, we decided to examine the selectivity of the hydrovinylation reactions using fine-tuned phosphoramidites that served us well in other situations. The results are shown in eq 43 and 44.
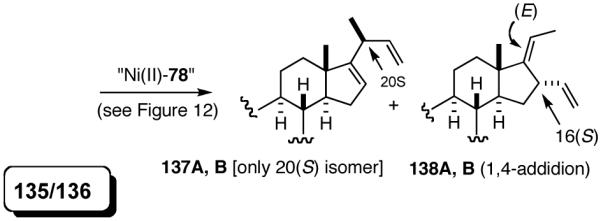 |
(43) |
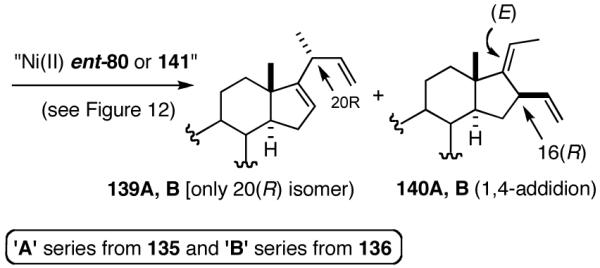 |
(44) |
Preparatively, the most useful reactions involve the use of ligands 78 and ent-80, which give the 20(S) or the 20(R) compound respectively along with minor amounts of a 1,4-adduct (eq 43 and 44). The highly stereoselective formation of the otherwise scarce C20(S)-isomer uncontaminated with the corresponding (R)-epimer is particularly noteworthy. The stereochemistry (α-vinyl appendage at C16) in the 1,4-adduct(s) 138A is deduced from the fact that this is the only other product formed concomitant with the 20(S) compound 137A. It is reasonable to assume that these two compounds originate from the same allyl-Ni intermediate arising from the α-face addition of the cationic Ni-H to the starting diene (Scheme 12). Same arguments hold for the formation of 139A and 140A except the reaction starts with β-face addition of the metal hydride.
Scheme 12.
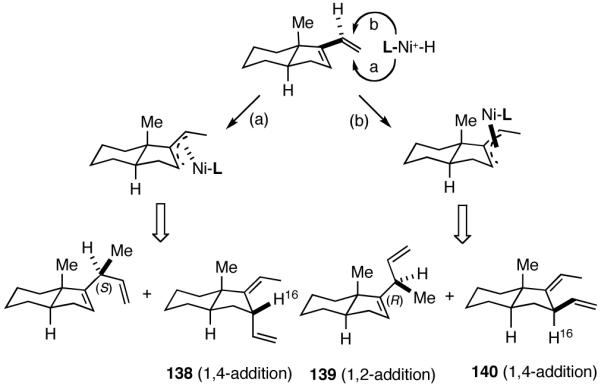
Origin of 1,2- and 1,4-Adducts in Hydrovinylation Reactions of Steroidal Dienes
The steroid D-ring can be elaborated in a myriad of ways using the diene functionality in the adducts. For example, selective hydroboration of the mono-substituted olefin in 137A derived from estrone gives an alcohol (142A) which could serve as a precursor for more advanced intermediates. Catalytic hydrogenation of this alcohol gives a single product, 143A. The endocyclic π-bond (C16-C17) in 142A will also be a useful handle for oxygenation of the D-ring, a key feature in many important steroidal glycosides, including potent anticancer agent OSW-1.73k Compounds in the series B, derived from 3-epiandrosterone can also be prepared by similar routes.
8.4 Intramolecular Reactions: Synthesis of Carbocyclic and Heterocyclic Compounds79
The dimerization reaction can be applied for the synthesis of cyclic compounds if the reaction is carried out in an intramolecular fashion. In this context, the Pd-catalyzed cyclization of eneynes, which, in principle could involve a [LnPd-H]+ intermediate, is a well known reaction.76 However, relatively little attention has been paid to the corresponding cyclization of α,ω-dienes using late metal catalysts.77 Except for a few isolated reports,78 Pd and Ni-catalyzed reactions have not been explored for the synthesis of carbocyclic compounds until our initial report.79,80 One of earliest examples is shown in eq 45.78d We find that the conditions developed for the hydrovinylation of vinylarenes22 can be applied for the efficient cyclization of α,ω-dienes (eq 46, 47).79 The ease of synthesis of starting materials and the diminished Lewis acidity of these metals (vis-à-vis early transition metals77) should make this process especially attractive for substrates that contain heteroatoms. As illustrated in equations 48 and 49, with unsymmetrical dienes, there is also the possibility of very good regiochemical control. An enantioselective version of this reaction has also been reported.80a
 |
(45) |
 |
(46) |
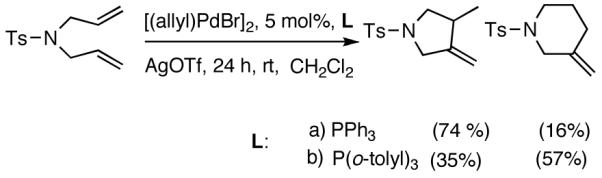 |
(47) |
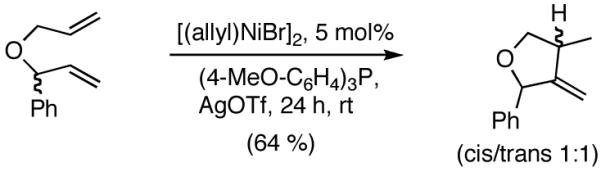 |
(48) |
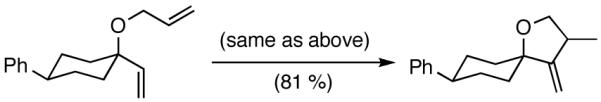 |
(49) |
8.5 Large Scale Synthesis
A patent claims Ni-catalyzed asymmetric hydrovinylation of styrene at -60 °C on a 8.26 kg (79.6 mol) scale using the azaphospholene ligand (RR)-7.19 The low yield (41%) and moderate enantioselectivity (87% ee) suggest that further developmental efforts are needed before the reaction can be practiced on a manufacturing scale for the synthesis of pharmaceutical intermediates such as 3-aryl-1-butenes. Several recent discoveries including new protocols, and the use of highly tunable ligands brighten the prospect of developing a practical process. For example, hydrovinylations of several 2-arylpropionic acid precursors have been carried out on a laboratory scale using the ligand 87 (Figure 9) in 90-98% yield and ee’s >96%.47,48,67 In the case of ibuprofen, substrate:catalyst ratio of 7142 (0.014 mol%) has been realized. A detailed procedure for a 50 mmol-scale hydrovinylation was published recently in Org. Synth.56
9. Summary and Future Prospects
The heterodimerization of olefins has great potential as a selective carbon-carbon bond forming reaction when the two olefins involved have different reactivities. With ethylene as one of the reactants, this difference could have its origin in size and electronic factors (e. g. vinylarenes, dienes) or in the higher reactivity of a partner due to inherent strain in the molecule (e. g., norbornene, norbornadiene). Demonstrated examples validate the claim that very high turnover frequency and exquisite selectivity for the desired product can be realized in many reactions. The reaction conditions are tolerant to a wide spectrum of common organic functional groups. The reaction has been shown to proceed under catalysis of Ni, Pd, Co and Ru, and a number of tunable ligand systems for these metals have been identified. With further improvements in ligand design and reaction engineering, expansion of the scope and selectivity of asymmetric hydrovinylation can be expected in the near future. Applications in complex molecule synthesis can also be anticipated.
Scheme 13.
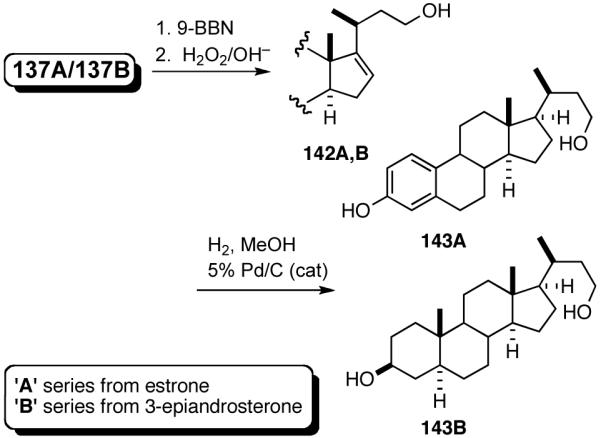
Steroid D-ring Functionalization
Acknowledgment
Financial assistance for this research by US National Science Foundation (CHE-0610349) and the National Institutes of Health (General Medical Sciences, R01 GM075107) is gratefully acknowledged. Further support for various research programs in our group including some aspects of hydrovinylation comes from by US Environmental Protection Agency, the Petroleum Research Fund of the American Chemical Society and the Ohio State University.
References
- (1).Wilke G. Angew. Chem. Int. Ed. Engl. 1988;27:185. [Google Scholar]
- (2).Chauvin Y, Olivier H. Dimerization and Codimerization. In: Cornils B, Herrmann WA, editors. Applied Homogeneous Catalysis with Organometallic Compounds. Vol. 1. VCH; New York: 1996. pp. 258–268. [Google Scholar]
- (3).Bogdanov’c B, Spliethoff B, Wilke G. Angew. Chem. Int. Ed. 1980;19:622. [Google Scholar]
- (4).For a summary of early results, see:RajanBabu TV. Chem. Rev. 2003;103:2845. doi: 10.1021/cr020040g.Other pertinent reviews:Bogdanov’c B. Adv. Organomet. Chem. 1979;17:105.Jolly PW, Wilke G. Hydrovinylation. In: Cornils B, Herrmann WA, editors. Applied Homogeneous Catalysis with Organometallic Compounds. Vol. 2. VCH; New York: 1996. pp. 1024–1048.RajanBabu TV, Nomura N, Jin J, Radetich B, Park H, Nandi M. Chem. Eur. J. 1999;5:1963.Gooßen LJ. Angew. Chem. Int. Ed. 2002;41:3775.
- (5).(a) Rieu J-P, Boucherle A, Cousse H, Mouzin G. Tetrahedron. 1986;42:4095. [Google Scholar]; (b) Sonawane HR, Bellur NS, Ahuja JR, Kulkarni DG. Tetrahedron: Asymmetry. 1992;3:163. [Google Scholar]; (c) Stahly GP, Starrett RM. In: Chirality in Industry II. Collins AN, Sheldrake GN, Crosby J, editors. Wiley; Chichester: 1997. p. 19. [Google Scholar]
- (6).(a) Alderson T, Jenner EL, Lindsey RV. J. Am. Chem. Soc. 1965;87:5638. [Google Scholar]; (b) Umezaki H, Fujiwara Y, Sawara K, Teranishi S. Bull. Chem. Soc. Jpn. 1973;46:2230. [Google Scholar]; (c) Dzhemilev UM, Gubaidullin LY, Tolstikov GA. Bull. Acad. Sci. USSR. 1976:2009. [Google Scholar]
- (7).Use of Ru: (a) see ref. 6(a).Umezaki H, Fujiwara Y, Sawara K, Teranishi S. Bull. Chem. Soc. Jpn. 1973;46:2230.
- (8).Early use of Co:Pu LS, Yamamoto A, Ikeda S. J. Am. Chem. Soc. 1968;90:7170.Pillai SM, Tembe GL, Ravindranathan M. J. Mol. Catal. 1993;84:77.
- (9).Early studies with Pd catalysts:Barlow MG, Bryant MJ, Haszeldine RN, Mackie AG. J. Organomet. Chem. 1970;21:215.Kawamoto K, Tatani A, Imanaka T, Teranishi S. Bull. Chem. Soc. Jpn. 1971;44:1239.For related studies see also:Drent E. 5,227,561. US Patent. 1993 Chem. Abstr. 1994, 120, 31520.Nozima H, Kawata N, Nakamura Y, Maruya K, Mizoroki T, Ozaki A. Chem. Lett. 1973:1163.
- (10).Kawata N, Maruya K, Mizoroki T, Ozaki A. Bull. Chem. Soc. Jap. 1971;44:3217.Kawata N, Maruya K, Mizoroki T, Ozaki A. Bull. Chem. Soc. Jap. 1974;47:413.(c) See also:Kawakami K, Kawata N, Maruya K, Mizoroki T, Ozaki A. J. Catal. 1975;39:134.(d) ref. 6c.Azizov AG, Mamedaliev GA, Aliev SM, Aliev VS. Azerb. Khim. Zh. 1978:3. Chem. Abstr. 1979, 90, 6002.Azizov AG, Mamedaliev GA, Aliev SM, Aliev VS. Azerb. Khim. Zh. 1979:3. Chem. Abstr. 1980, 93, 203573.Mamedaliev GA, Azizov AG. Pol. J. (Japan) 1985;17:1075.
- (11).Bogdanov’c B, Henc B, Meister B, Pauling H, Wilke G. Angew. Chem. Int. Ed. 1972;11:1023.Bogdanov’c B, Henc B, Lösler A, Meister B, Pauling H, Wilke G. Angew. Chem. Int. Ed. 1973;12:954.Among asymmetric metal-catalyzed carbon-carbon bond-forming reactions, only Nozaki’s Cu(II)-catalyzed cyclopropanation of styrene with ethyl diazoacetate predates this discovery. See,Nozaki H, Moriuti S, Takaya H, Noyori R. Tetrahedron Lett. 1966:5239.
- (12).Britovsek GJP, Keim W, Mecking S, Sainz D, Wagner T. J. Chem. Soc., Chem. Commun. 1993:1632.Britovsek GJP, Cavell KJ, Keim W. J. Mol. Catal. A, Chemical. 1996;110:77.(c) For a related dendrimeric Pd-catalyst see,Hovestad NJ, Eggeling EB, Heidbüchel HJ, Jastrzebski JTBH, Kragl U, Keim W, Vogt D, van Koten G. Angew. Chem. Int. Ed. 1999;38:1655. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1655::AID-ANIE1655>3.0.CO;2-2.For a full report,Eggeling EB, Hovestad NJ, Jastrzebski TBH, Vogt D, van Kotten G. J. Org. Chem. 2000;65:8857. doi: 10.1021/jo000433k.Shi W-J, Xie J-H, Zhou Q-L. Tetrahedron: Asymmetry. 2005;16:705.
- (13).Bayersdörfer R, Ganter B, Englert U, Keim W, Vogt D. J. Organomet. Chem. 1998;552:187. [Google Scholar]
- (14).Albert J, Cadena M, Granell J, Muller G, Ordinas JI, Panyella D, Puerta C, Sanudo C, Valerga P. Organometallics. 1999;18:3511.For use of other highly basic phosphines see also:Albert J, Bosque R, Cadena JM, Delgado S, Granell J, Muller G, Ordinas JI, Bardia MF, Solans X. Chem. Eu. J. 2002;8:2279. doi: 10.1002/1521-3765(20020517)8:10<2279::AID-CHEM2279>3.0.CO;2-#.Englert U, Haerter R, Vasen D, Salzer A, Eggeling EB, Vogt D. Organometallics. 1999;18:4390.
- (15).(a) Ceder R, Muller G, Ordinas JI. J. Mol. Catal. 1994;92:127. [Google Scholar]; (b) Muller G, Ordinas JI. J. Mol. Catal., A: Chem. 1997;125:97. [Google Scholar]
- (16).(a) Monteiro AL, Seferin M, Dupont J, Souza RF. Tetrahedron Lett. 1996;37:1157. [Google Scholar]; (b) Fassina V, Ramminger C, Seferin M, Monteiro AL. Tetrahedron. 2000;56:7403. [Google Scholar]
- (17).Yi CS, He Z, Lee DW. Organometallics. 2001;20:802.RajanBabu TV, Nomura N, Jin J, Nandi M, Park H, Sun X. J. Org. Chem. 2003;68:8431. doi: 10.1021/jo035171b.(c) These results have since been confirmed in another more recent publication.Sanchez RP, Jr., Connell BT. Organometallics. 2008;27:2902.
- (18).Grutters MMP, Müller C, Vogt D. J. Am. Chem. Soc. 2006;128:7414. doi: 10.1021/ja058095y.(b) For a related reaction, see:Hilt G, Lüers S. Synthesis. 2002:609.
- (19).Wilke G, Monkiewicz J, Kuhn H. Preparation of optically active azaphospholenes and their use in catalysis for asymmetric codimerization of olefins. 4912274. US Patent. 1990 Chem. Abstr. 1991, 114, 43172.
- (20).Use of the Wilke catalyst system in scCO2 has since been reported. See:Wegner A, Leitner W. J. Chem. Soc., Chem. Commun. 1999:1583.Bösmann A, Franciò G, Janssen E, Solinas M, Leitner W, Wasserscheid P. Angew. Chem. Int. Ed. 2001;40:2697.
- (21).Angermund K, Eckerle A, Lutz F. Z. Naturforsch., B: Chem. Sci. 1995;50:488. [Google Scholar]
- (22).Nomura N, Jin J, Park H, RajanBabu TV. J. Am. Chem. Soc. 1998;120:459. [Google Scholar]
- (23).Muller U, Keim W, Krüger C, Betz P. Angew. Chem. Int. Ed. 1989;28:1011. [Google Scholar]
- (24).DiRenzo GM. Ph. D. Thesis. University of North Carolina; 1997. Mechanistic Studies of Catalytic Olefin Dimerization Reactions Using Electrophilic η3-allylPalladium(II) Complexes.We thank Professor Brookhart and Dr. DiRenzo for a copy of this dissertation.
- (25).(a) Brandes H, Goddard R, Jolly PW, Krüger C, Mynott R, Wilke G. Z. Naturforsch. 1984;39B:1139. [Google Scholar]; (b) Barnett BL, Krüger C. J. Organomet. Chem. 1974;77:407. [Google Scholar]
- (26).Ref. 17(b).
- (27).Jin J, RajanBabu TV. Tetrahedron. 2000;56:2145. [Google Scholar]
- (28).(a) Kumareswaran R, Nandi N, RajanBabu TV. Org. Lett. 2003;5:4345. doi: 10.1021/ol0356284. [DOI] [PubMed] [Google Scholar]; (b) Park H, Kumareswaran R, RajanBabu TV. Tetrahedron Symposium-in Print. 2005;61:6352. [Google Scholar]
- (29).Buono G, Siv C, Peiffer G, Triantaphylides C, Denis P, Mortreux A, Petit F. J. Org. Chem. 1985;50:1781. P. [Google Scholar]
- (30).Nandi M, Jin J, RajanBabu TV. J. Am. Chem. Soc. 1999;121:9899. [Google Scholar]
- (31).For a discussion of hemilabile ligands see:Jeffrey JC, Rauchfuss TB. Inorg. Chem. 1979;18:2658.Bader A, Lindner E. Coord. Chem. Rev. 1991;108:27. A.Slone CS, Weinberger DA, Mirkin CA. The Transition Metal Coordination Chemistry of Hemilabile Ligands. In: Karlin KD, editor. Progress in Inorganic Chemistry. Vol. 48. John Wiley; New York: 1999. pp. 233–350.
- (32).(a) Meking S, Keim W. Organometallics. 1996;15:2650. [Google Scholar]; (b) Keim W. Angew. Chem. Int. Ed. 1990;29:235. [Google Scholar]; (c) Bonnet MC, Dahan F, Ecke A, Keim W, Schulz RP, Tkatchenko I. Chem. Commun. 1994:615. [Google Scholar]; (d) Keim W, Maas H, Mecking S. Z. Naturforsch. 1995;50B:430. [Google Scholar]; (e) Britovsek GJP, Keim W, Mecking S, Sainz D, Wagner T. J. Chem. Soc., Chem. Commun. 1993:1632. [Google Scholar]; (f) Britovsek GJP, Cavell KJ, Keim W. J. Mol. Catal. A, Chemical. 1996;110:77. [Google Scholar]
- (33).Uozumi Y, Tanahashi A, Lee S-Y, Hayashi T. J. Org. Chem. 1993;58:1945. [Google Scholar]
- (34).For a structurally related, unreactive, neutral Pd-complex, see, ref 32 (f).
- (35).Nishida H, Takada N, Yoshimura M, Sonoda T, Kobayashi H. Bull. Chem. Soc. Jpn. 1984;57:2600.Brookhart M, Grant B, Volpe A. Organometallics. 1992;11:3920.For the use of [Ar4B]- in related reactions see:DiRenzo GM, White PS, Brookhart M. J. Am. Chem. Soc. 1996;118:6225.
- (36).Braunstein P, Chauvin Y, Nähring J, DeCian A, Fischer J, Tiripicchio A, Ugozzoli F. Organometallics. 1996;15:5551. [Google Scholar]
- (37).Saha B, RajanBabu TV. J. Org. Chem. 2007;72:2357. doi: 10.1021/jo062044h. [DOI] [PubMed] [Google Scholar]
- (38).Examination of electronic effects has become a common practice in asymmetric catalysis. For some of the early examples of electronic tuning of asymmetric catalysts, see:Inoguchi K, Sakuraba S, Achiwa K. Synlett. 1992:169.Jacobsen EN, Zhang W, Guler ML. J. Am. Chem. Soc. 1991;113:6703.RajanBabu TV, Casalnuovo AL. J. Am. Chem. Soc. 1992;114:6265.RajanBabu TV, Ayers TA, Casalnuovo AL. J. Am. Chem. Soc. 1994;116:4101.Schnyder A, Hintermann L, Togni A. Angew. Chem. Int. Ed. 1995;34:931.Other examples from our work:Casalnuovo AL, RajanBabu TV, Ayers TA, Warren TH. J. Am. Chem. Soc. 1994;116:9869.RajanBabu TV, Casalnuovo AL. J. Am. Chem. Soc. 1996;118:6325.RajanBabu TV, Ayers TA, Halliday GA, You KK, Calabrese JC. J. Org. Chem. 1997;62:6012.RajanBabu TV, Radetich B, You KK, Ayers TA, Casalnuovo AL, Calabrese JC. J. Org. Chem. 1999;64:3429. doi: 10.1021/jo9901182.Clyne DS, Mermet-Bouvier YC, Nomura N, RajanBabu TV. J. Org. Chem. 1999;64:7601.Yan Y, RajanBabu TV. Org. Lett. 2000;2:4137. doi: 10.1021/ol006591f.
- (39).Ref. 37
- (40).Komon ZJA, Bu X, Bazan GC. J. Am. Chem. Soc. 2000;122:1830. [Google Scholar]
- (41).Ref. 30.
- (42).Zhang A, RajanBabu TV. Org. Lett. 2004;6:1515. doi: 10.1021/ol0495063. [DOI] [PubMed] [Google Scholar]
- (43).Park H, RajanBabu TV. J. Am. Chem. Soc. 2002;124:734. doi: 10.1021/ja0172013.For another report of the use of a phosphinite, in Pd-catalyzed hydrovinylation, see:Bayersdörfer R, Ganter B, Englert U, Keim W, Vogt D. J. Organomet. Chem. 1998;552:187.
- (44).(a) Feringa BL. Acc. Chem. Res. 2000;33:346. doi: 10.1021/ar990084k. [DOI] [PubMed] [Google Scholar]; (b) Arnold L,A, Imbos R, Mandoli A, de Vries AHM, Naasz R, Feringa BL. Tetrahedron. 2000;56:2865. [Google Scholar]
- (45).For representative examples of the use of finely tuned phosphoramidites from various research groups, see:Alexakis A, Polet D, Rosset S, March S. J. Org. Chem. 2004;69:5660. doi: 10.1021/jo049359m.Bernsmann H, van den Berg M, Hoen R, Minnaard AJ, Mehler G, Reetz MT, De Vries JG, Feringa BL. J. Org. Chem. 2005;70:943. doi: 10.1021/jo048374o.Streiff S, Welter C, Schelwies M, Lipowsky G, Miller N, Helmchen G. Chem. Commun. 2005:2957. doi: 10.1039/b503713a.Leitner A, Shekhar S, Pouy MJ, Hartwig JF. J. Am. Chem. Soc. 2005;127:15506. doi: 10.1021/ja054331t.Yu RT, Rovis T. J. Am. Chem. Soc. 2006;128:12370. doi: 10.1021/ja064868m.Du H, Yuan W, Zhao B, Shi Y. J. Am. Chem. Soc. 2007;129:11688. doi: 10.1021/ja074698t.
- (46).Franció G, Faraone F, Leitner W. J. Am. Chem. Soc. 2002;124:736. doi: 10.1021/ja012099v.For a computational study of the phosphoramidite ligand system, see:Hölscher M, Franció G, Leitner W. Organometallics. 2004;23:5606.
- (47).Smith CR, RajanBabu TV. Org. Lett. 2008;10:1657. doi: 10.1021/ol800395m.(b) For a scalable procedure for the synthesis of phosphoramidites see,Smith CR, Mans DJ, RajanBabu TV. Org. Synth. 2008;85:238.
- (48).To the best of our knowledge, the ligand 87 has not been described in the literature. For a complete list of phosphoramidites used in this study, see reference 47 and the corresponding Supporting Information.
- (49).Only naproxen is currently sold in enantiomerically pure form. For a review of practical aspects of the synthesis of 2-arylpropionic acids, see:Stahly GP, Starrett RM. In: Chirality in Industry II. Collins AN, Sheldrake GN, Crosby J, editors. Wiley; Chichester: 1997. p. 19.
- (50).For reviews, and history of the problem, see:Douglas CJ, Overman LE. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5363. doi: 10.1073/pnas.0307113101.and references cited therein.Denissova I, Barriault L. Tetrahedron. 2003;59:10105.Corey EJ, Guzman-Perez A. Angew. Chem., Int. Ed. 1998;37:388. doi: 10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V.Romo D, Meyers AI. Tetrahedron. 1991;47:9503.Martin SF. Tetrahedron. 1980;36:419.
- (51).Takemoto T, Sodeoka M, Sasai H, Shibasaki M. J. Am. Chem. Soc. 1993;115:8477. corrections: ibid. J. Am. Chem. Soc.1994, 116, 11207. [Google Scholar]
- (52).For a leading reference, see:Edwards DJ, Gerwick WH. J. Am. Chem. Soc. 2004;126:11432. doi: 10.1021/ja047876g.
- (53).Huang A, Kodanko JJ, Overman LE. J. Am. Chem. Soc. 2004;126:14043. doi: 10.1021/ja046690e. [DOI] [PubMed] [Google Scholar]
- (54).Denmark SE, Fu J. Org. Lett. 2002;4:1951. doi: 10.1021/ol025971t. [DOI] [PubMed] [Google Scholar]
- (55).Zhang A, RajanBabu TV. J. Am. Chem. Soc. 2006;128:5620. doi: 10.1021/ja060999b.(b) For the use of another phosphoramidite, see:Shi W-J, Zhang Q, Xie J-H, Zhu S-F, Hou G-H, Zhou Q-L. J. Am. Chem. Soc. 2006;128:2780. doi: 10.1021/ja057654y.
- (56).For a detailed procedure of this and other related hydrovinylation reactions, see:Smith CR, Zhang A, Mans DJ, RajanBabu TV. Org. Synth. 2008;85:248. doi: 10.15227/orgsyn.085.0248.
- (57).(a) Hulme A,N, Henry SS, Meyers AI. J. Org. Chem. 1995;60:1265. [Google Scholar]; (b) Fadel A, Arzel P. Tetrahedron: Asymmetry. 1997;8:371. [Google Scholar]
- (58).Zhang A, RajnBabu TV. J. Am. Chem. Soc. 2006;128:54.(b) Dan Mans in our group has since completed the synthesis of a number pseudopterosin aglycones by applying back-to-back enantioselective hydrovinylations of vinylarenes and dienes. This work will be reported in due course.
- (59).For an early example (non-asymmetric) of codimerization of ethylene and dienes, see:Su ACL. Adv. Organomet. Chem. 1979;17:269.Peiffer G, Cochet X, Petit F. Bull. Soc. Chim. Fr. II. 1979:415.
- (60).Buono G, Siv C, Peiffer G, Triantaphylides C, Denis A, Mortreux A, Petit F. J. Org. Chem. 1985;50:1781. [Google Scholar]
- (61).(a) He Z, Yi CS, Donaldson WA. Org. Lett. 2003;5:1567. doi: 10.1021/ol030031+. [DOI] [PubMed] [Google Scholar]; (b) He Z, Yi CS, Donaldson WA. Synlett. 2004:1312. [Google Scholar]
- (62).See ref 58 for an extensive listing of references.
- (63).Except for Diels-Alder reactions, asymmetric catalyzed C-C bond-forming reactions of acyclic 1,3-dienes give only moderate regio- and enatio-selectivities. See for example, (a) cyclopropanation:Doyle M. In: Catalytic Asymmetric Synthesis. Ojima I, editor. Wiley-VCH; New York: 2000. (b) ene reaction:Terada M, Mikami K. J. Chem. Soc., Chem. Commun. 1995:2391.(c) hydroformylation:Horiuchi T, Ohta T, Shirakawa E, Nozaki K, Takaya H. Tetrahedron. 1997;53:7795. doi: 10.1021/jo9624051.(d) hydrocyanation:Saha B, RajanBabu TV. Org. Lett. 2006;8:4657. doi: 10.1021/ol062002f.
- (64).Hodgson M, Parker D, Taylor RJ, Ferguson G. Organometallics. 1988;7:1761. [Google Scholar]
- (65).For a review of synthesis of 2-arylpropionic acids, see:Rieu J-P, Boucherle A, Cousse H, Mouzin G. Tetrahedron. 1986;42:4095.Sonawane HR, Bellur NS, Ahuja JR, Kulkarni DG. Tetrahedron: Asymmetry. 1992;3:163.Stahly GP, Starrett RM. In: Chirality in Industry II. Collins AN, Sheldrake GN, Crosby J, editors. Wiley; Chichester: 1997. p. 19.
- (66).For best asymmetric routes todate, Naproxen via Ru-catalyzed asymmetric hydrogenation of 2-arylacrylic acids:Ohta T, Takaya H, Kitamura M, Nagai K, Noyori R. J. Org. Chem. 1987;52:3174.(98% ee). Ni-catalyzed asymmetric hydrocyanation:RajanBabu TV, Casalnuovo AL. J. Am. Chem. Soc. 1996;118:6325.and references cited therein. (95% ee). Ibuprofen via Ru-catalyzed hydrogenation:Uemura T, Zhang X, Matsumura K, Sayo N, Kumobayashi H, Ohta T, Nozaki K, Takaya H. J. Org. Chem. 1996;61:5510. doi: 10.1021/jo961689m.(97% ee). Rh-catalyzed asymmetric hydroformylation:Nozaki K, Sakai N, Nanno T, Higashijima T, Mano S, Horiuchi T, Takaya H. J. Am. Chem. Soc. 1997;119:4413.(92% ee). Hydrovinylation: ref. 42 (91%). (e) Flurbiprofen via dynamic kinetic resolution:Norinder J, Bogár K, Kaupp L, Backvall J-E. Org. Lett. 2007;9:5095. doi: 10.1021/ol702261t.No useful catalytic asymmetric methods are known for other (S)-2-arylpropionic acid precursors.
- (67).Unpublished results. Smith, C. R.; and RajanBabu, T. V. (2008).
- (68).Zhang A, RajanBabu TV. Org. Lett. 2004;6:3159. doi: 10.1021/ol048790v. [DOI] [PubMed] [Google Scholar]
- (69).For recent references see the following and others cited therein. (a) Bisabolanes:Hagiwara H, Okabe T, Ono H, Kamat VP, Hoshi T, Suzuki T, Ando M. J. Chem. Soc., Perkin Trans. 1. 2002:895.Vyvyan JR, Loitz C, Looper RE, Mattingly CS, Peterson EA, Staben ST. J. Org. Chem. 2004;69:2461. doi: 10.1021/jo035778s.(c) Heliannanes:Kishuku H, Shindo M, Shishido K. Chem. Commun. 2003:350. doi: 10.1039/b211227b.(d) Pseudopterosins:Look SA, Fenical W, Jacobs RS, Clardy J. Proc. Natl. Acad. Sci. 1986;83:6238. doi: 10.1073/pnas.83.17.6238.Johnson TW, Corey EJ. J. Am. Chem. Soc. 2003;125:13486. doi: 10.1021/ja0378916.Harrowven DC, Tyte MJ. Tetrahedron Lett. 2004;45:2089.(g) Serrulatanes:Rodriguez A, Ramirez C. J. Nat. Prod. 2001;64:100. doi: 10.1021/np000196g.Dehmel F, Lex J, Schmalz H-G. Org. Lett. 2002;4:3915. doi: 10.1021/ol026827a.
- (70).See reference 68 for a comparison of various methods of for the synthesis of curcumene.
- (71).Hagiwara H, Okabe T, Ono H, Kamat VP, Hoshi T, Suzuki T, Ando M. J. Chem. Soc., Perkin Trans. 1. 2002:895. [Google Scholar]
- (72).Saha B, Smith CR, RajanBabu TV. J. Am. Chem. Soc. 2008;130:9000. doi: 10.1021/ja711475f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).(a) For a leading reference, discussion and citations of earlier work, see:Guevel A-C, Hart DJ. J. Org. Chem. 1996;61:465. doi: 10.1021/jo951531m.and references cited therein. (b) For a pedagogical view of this problem and a historical account of syntheses of erythro- and threo-juvabiones, see:Carey FA, Sundberg RJ. Advanced Organic Chemistry. Ed. 2 Vol. 2. Kulwer; New York: 2001. pp. 848–859.Trost BM, Verhoeven TR. J. Am. Chem. Soc. 1976;98:630. doi: 10.1021/ja00418a063.Snider BB. Acc. Chem. Soc. 1980;13:426.Snider BB, Deutsch EA. J. Org. Chem. 1983;48:1823.Wulkovich PM, Barcelos A, Sereno JF, Baggiolini EG, Hennesey BM, Uskokovic MR. Tetrahedron. 1984;40:2283.Andersen NH, Hadley SW, Kelly JD, Bacon ER. J. Org. Chem. 1985;50:4144.Mikami K, Kawamoto K, Nakai T. Tetrahedron Lett. 1985;26:5799.Mikami K, Loh T-P, Nakai T. J. Chem. Soc., Chem. Commun. 1988:1430.Houston TA, Tonaka Y, Koreeda M. J. Org. Chem. 1993;58:4287.(k) For recent references, dealing with side-chain of the anticancer natural product OSW-1, see:Yu W, Jin Z. J. Am. Chem. Soc. 2002;124:6576. doi: 10.1021/ja012119t.
- (74).Honzawa S, Suhara Y, Nihei K, Saito N, Kishimoto S, Fujishima T, Kurihara M, Sugiura T, Waku K, Takayamaa H, Kittaka A. Bioorg. Med. Chem. Lett. 2003;13:3503. doi: 10.1016/s0960-894x(03)00739-x.and references cited therein. (b) For leading references, see,Kabat MM, Garofalo LM, Daniewski AJ, Hutchings SD, Liu W, Okabe M, Radinov R, Zhou Y. J. Org. Chem. 2001;66:6141. doi: 10.1021/jo015788y.
- (75).Fujishima T, Konno K, Nakagawa K, Kurobe M, Okano T, Takayama H. Bioorg. Med. Chem. 2000;8:123. doi: 10.1016/s0968-0896(99)00262-x.Fernández B, Pérez JAM, Granja JR, Castedo l., Mourino A. J. Org. Chem. 1992;57:3173.See also:Fernández C, Gómez G, Lago C, Momán E, Fall Y. Synlett. 2005:2163.Hijikuro I, Doi T, Takahashi T. J. Am. Chem. Soc. 2001;123:3716. doi: 10.1021/ja003976k.(e) For a review of vitamin D chemistry:Dai H, Posner GH. Synthesis. 1994:1383.Posner GH, Lee JK, White MC, Hutchings RH, Dai H, Kachinski JL, Dolan P, Kensler TW. J. Org. Chem. 1997;62:3299. doi: 10.1021/jo970049w.
- (76).Trost BM. Acc. Chem. Res. 1990;23:34.and references cited therein.
- (77).For the use of early transition metals see:Nugent WA, Taber DF. J. Am. Chem. Soc. 1989;111:6435.Piers WE, Shapiro PJ, Bunel EE, Bercaw JE. Synlett. 1990:74.Knight KS, Waymouth RM. J. Am. Chem. Soc. 1991;113:6268.Molander GA, Hoberg JO. J. Am. Chem. Soc. 1992;114:3123.Negishi E, Takahashi T. Acc. Chem. Res. 1994;27:124.Dzhemilev UM. Tetrahedron. 1995;51:4333.Christoffers J, Bergman RG. J. Am. Chem. Soc. 1996;118:4715.Thiele S, Erker G. Chem. Ber./ Rec. 1997;130:201.Yamaura Y, Hyakutake M, Mori M. J. Am. Chem. Soc. 1997;119:7615.and references cited therein.
- (78).Bright A, Malone JF, Nicholson JK, Powell J, Shaw BL. J. Chem. Soc., Chem. Commun. 1971:712.(b) ref. 4b.Grigg R, Malone JF, Mitchell TRB, Ramasubbu A, Scott RM. J. Chem. Soc., Perkin Trans I. 1984:1745.Behr A, Freudenberg U, Keim W. J. Mol. Catal. 1986;35:9.
- (79).Radetich B, RajanBabu TV. J. Am. Chem. Soc. 1998;120:8007. [Google Scholar]
- (80).α,ω-Diene cyclization and related reactions have since received a lot of attention. For example, see:Bóing C, Franciò G, Leitner W. Chem. Commun. 2005:1456. doi: 10.1039/b417333c.Lloyd-Jones GC. Org. Biomol. Chem. 2003;1:215. doi: 10.1039/b209175p.Bothe U, Rudbeck HC, Tanner D, Johannsen J. Chem. Soc., Perkin Trans. 1. 2001:3305.Perch NS, Pei T, Widenhoefer RA. J. Org. Chem. 2000;65:3836. doi: 10.1021/jo0003192.