

Abstract
The activity of the anticancer drug cisplatin is a consequence of its ability to bind DNA. Platinum adducts bend and unwind DNA duplex, creating recognition sites for nuclear proteins. Following DNA damage recognition, the lesions will either be repaired, facilitating cell viability, or if repair is unsuccessful and the Pt adduct interrupts vital cellular functions, apoptosis will follow. With the use of the benzophenone-modified cisplatin analogue Pt-BP6, 25-bp DNA duplexes containing either a 1,2-d(G*pG*) intrastrand or a 1,3-d(G*pTpG*) intrastrand cross-link were synthesized, where the asterisks designate platinated nucleobases. Proteins having affinity for these platinated DNAs were photo-cross-linked and identified in cervical, testicular, pancreatic and bone cancer cell nuclear extracts. Proteins identified in this manner include the DNA repair factors RPA1, Ku70, Ku80, Msh2, DNA ligase III, PARP-1, and DNA-PKcs, as well as HMG-domain proteins HMGB1, HMGB2, HMGB3, and UBF1. The latter strongly associate with the 1,2-d(G*pG*) adduct and weakly or not at all with the 1,3-d(G*pTpG*) adduct. The nucleotide excision repair protein RPA1 was photo-cross-linked only by the probe containing a 1,3-d(G*pTpG*) intrastrand cross-link. The affinity of PARP-1 for platinum-modified DNA was established using this type of probe for the first time. To ensure that the proteins were not photo-cross-linked because of an affinity for DNA ends, a 90-base dumbbell probe modified with Pt-BP6 was investigated. Photo-cross-linking experiments using this longer probe revealed the same proteins, as well as some additional proteins involved in chromatin remodeling, transcription, or repair. These findings reveal a more complete list of proteins involved in the early step of the mechanism of action of the cisplatin and its close analogue carboplatin.
Keywords: DNA damage, Anticancer Agents, Cisplatin, DNA repair, PARP-1
Introduction
The anticancer activity of cis-diamminedichloroplatinum(II), or cisplatin, was discovered over three decades ago and the compound remains one of the most effective therapeutic agents for testicular germ-cell tumors.[1] Cisplatin and its FDA-approved analogs carboplatin and oxaliplatin are ineffective against certain cancers because of intrinsic or acquired drug resistance.[2] Our research has focused on elucidating the mechanisms of action of this family of compounds with the goal of providing more effective therapies. Four early steps in the cisplatin mechanism have been delineated, namely, cell entry, drug activation, Pt–DNA binding, and protein binding to platinated DNA. The present work focuses on the last of these steps, specifically, the immediate cellular response to platinum-damaged DNA.
Cisplatin preferentially binds to the N7 of guanine bases forming intrastrand and interstrand cross-links with residues in close proximity. The most common lesion is a 1,2-d(G*pG*) adduct in which the platinum is bound to adjacent guanines on the same DNA strand.[3,4] Another common cisplatin-DNA adduct is the 1,3-d(G*pNpG*) cross-link in which the bound guanine residues are separated by one nucleotide. These types of lesions, which comprise over 90% of adducts formed in cisplatin-treated individuals,[2] bend and unwind the duplex.[2,5] DNA distorted by cisplatin is a recognition site for a number of cellular proteins.[6,7] The first such proteins to be identified contain a high-mobility group (HMG) domain and bind specifically to 1,2-d(G*pG*) intrastrand cisplatin-DNA cross-links.[8,9] These proteins include high mobility group box protein 1 (HMGB1), high mobility group box protein 2 (HMGB2), structure-specific recognition protein (SSRP1), and upstream binding factor 1 (UBF1).[8–11]
Following recognition of DNA damage, a series of cellular signaling pathways are activated. These damage-response pathways can result in DNA repair or cell death. Several repair processes have been implicated in response to platinum-damaged DNA. Chief among these is nucleotide excision repair (NER), which removes platinum adducts from DNA[12] and is correlated with cisplatin resistance.[13] In addition, E. coli and human cancer cells deficient in recombination repair pathways are sensitized to cisplatin damage.[14,15] Inhibition of the mismatch repair (MMR) pathway correlates with increased resistance to the drug[16]. Proteins involved in repair processes bind preferentially to cisplatin-DNA adducts, including the Ku70/80 subunits of the mulitprotein complex DNA protein kinase (DNA-PK)[17]. The DNA-PK complex participates in non-homologous end-joining (NHEJ) repair of double-strand breaks (DSB).[18] These findings suggest that the identification of proteins that bind to platinum-modified DNA in cells with varying sensitivities to cisplatin may provide insight into processing of cisplatin adducts in these different contexts.[19]
Studies of the cellular proteins that interact with cisplatin-damaged DNA date back many years. Early work applied electrophoretic mobility shift assays (EMSAs) to demonstrate that xeroderma pigmentosum group E binding factor (XPE-BF) binds preferentially to cisplatin-modified versus unmodified DNA.[20] Using EMSAs to identify proteins with an affinity for platinum-modified DNA would require a separate experiment for each of numerous nuclear proteins. In another study, use of a cisplatin-modified DNA probe to screen a cDNA expression library identified the HMG-domain protein SSRP1.[10,21] Because SSRP1 contains an HMG domain that is homologous to domains in HMGB1 (formerly HMG1), it was hypothesized that the latter would also bind to cisplatin-modified DNA. Following isolation and purification of rat HMGB1, gel retardation assays revealed that the protein binds to the 1,2-d(G*pG*), but not the 1,3-d(G*pG*), intrastrand cross-link of cisplatin.[8] That HMGB1 and HMGB2 have an affinity for cisplatin-damaged DNA was also revealed by southwestern blotting methods.[9,22] Despite these successes, however, neither cDNA library screening nor southwestern blotting methods can gauge protein binding to platinated DNA in the context of multiprotein complexes.
Globally platinated DNA immobilized onto a column treated with nuclear extracts identified Pt–DNA-binding proteins DNA-PK, HMGB1, replication protein A (RPA), and xeroderma pigmentosum group A protein (XPA).[23] This method requires a sensitive adjustment of salt concentration to remove proteins with different affinities for the probe. Also, the use of globally platinated DNA does not distinguish which of many possible Pt–DNA adducts is being recognized.
A superior method for identifying proteins that bind to platinum-modified DNA utilizes a cisplatin analogue capable of forming a covalent bond to capture DNA-damage-response proteins following incubation with nuclear extracts. This method is sensitive to all nuclear proteins that bind to Pt-modified DNA, affording a more complete assessment. Photo-cross-linking provides a convenient way to bind the proteins covalently to the platinated DNA, which can then be isolated for identification.
To achieve this type of photo-cross-linking, we first synthesized a cisplatin analogue containing a photo-reactive azide moiety.[24] Control experiments performed during this study showed that cis-{Pt(NH3)2}2+-d(G*pG*) adduct itself was activated by the 302 nm irradiation required to convert the aryl azide to a nitrene. The aryl azide-modified analogue of cisplatin did not form photo-cross-links more effectively than the cis-{Pt(NH3)2}2+ adduct alone.[24] We also showed that the cis-{Pt(NH3)2}2+ could be more efficiently activated using a laser at 325 or 350 nm to irradiate the sample.[25] The formation of DNA-platinum-protein complexes may be important in the processing of cisplatin cross-links.[26] A limitation of experiments using cis-{Pt(NH3)2}2+ as the cross-linker is that the protein must come into close contact with the platinum atom in order to form a covalent DNA-Pt-protein linkage. This requirement make it difficult to capture DNA-damage recognition proteins that recognize the bulge created on the undamaged DNA strand,[27] where it is unlikely to be close enough to the cis-{Pt(NH3)2}2+ adduct to be photo-cross-linked.
In order to cross-link proteins bound to Pt–DNA adducts more effectively, a photo-reactive benzophenone moiety was tethered to a platinum center. This compound, Pt-BP6, contains a six-carbon linker separating the platinum complex from the benzophenone (Scheme 1), a hexamethylene chain proving to be optimal for efficient protein photo-cross-linking.[28] With an excitation wavelength of 365 nm, benzophenone is an excellent photo-activatable cross-linker for biological applications. The photophore excitation energy is low enough such that it does not cause significant damage to biological samples, and benzophenone moieties are not prohibitively sensitive to background light sources.[29] Cross-linking is initiated by photoexcitation of the benzophenone moiety to form a diradical, which can attack a nearby protein backbone carbon or amino acid side chain to abstract a hydrogen atom. The ketyl and alkyl radicals that form then recombine to generate a covalent C–C bond, a mechanism described in detail elsewhere.[29]
In preliminary work, we prepared a site-specifically platinated duplex containing a single 1,2-d(G*pG*) Pt-BP6 adduct, exposed it to HeLa nuclear extracts, and irradiated the solution to activate the benzophenone moiety. Several photo-cross-linked proteins were observed, two of which were identified as HMGB1 and poly(ADP-ribose) polymerase-1 (PARP-1).[28] Binding to HMGB1 was expected, given the results of much previous work, but PARP-1 had not been known to have an affinity for platinum-damaged DNA. In the present study, we have built upon this initial discovery to increase the scale of the preparative study, which has allowed us to identify the entire family proteins that photo-cross-link to this probe. Photo-cross-linking experiments were then extended to include a 25-bp duplex probe containing a different adduct, the Pt-BP6 1,3-d(G*pTpG*) intrastrand cross-link. We also applied the methodology to investigate nuclear extracts from a variety of cancer cells with varying sensitivities to cisplatin, namely, cervical, testicular, pancreatic, and bone. Because some DNA end-binding proteins, such as those in the Ku family,[30] make contacts deep along the DNA and could potentially be photo-cross-linked without binding to the platinum lesion, and because PARP-1 can bind to many types of DNA constructs, including DNA ends,[31] a longer, 90-base (41-bp) dumbbell DNA probe was also tested (Figure 1). The dumbbell probe contains two loops, or hairpin arms, instead of blunt ends. PARP-1 may bind to these loops, but in so doing only contacts the first four adjacent DNA base pairs.[32] Using the 90-base dumbbell probe, the ability of PARP-1 to bind to platinum-damaged DNA was investigated. As will be described, every protein identified to bind to the photo-activated Pt-BP6-modified 25-bp probe was also cross-linked by the 90-mer, and a number of additional proteins were identified. The results provide a more complete inventory of nuclear proteins that contact the major cisplatin adducts in cancer cells, knowledge of which will facilitate future mechanistic studies.
Figure 1.
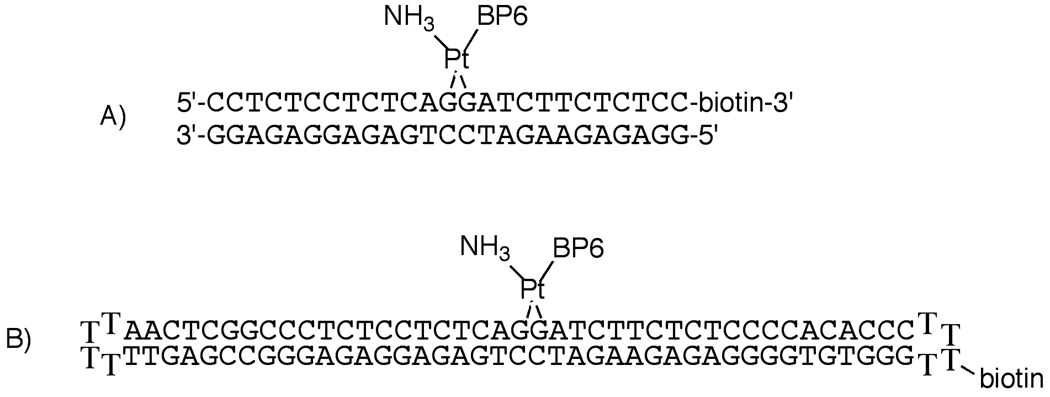
The structures of the DNA probes used in these experiments. In these probes, the benzophenone moiety may be oriented toward the 3'- or 5'-end of the DNA. A mixture of the two possible orientational isomers of PtBP6-modified DNA was used in these experiments. A) The 25-bp duplex probe. B) The 90-base dumbbell probe.
Results
Overview of Results
A 25-bp duplex containing a site-specific 1,2-d(G*pG*) intrastrand cross-link of the photo-active cisplatin analogue Pt-BP6, where the asterisks designate the platinated nucleobases, was used to photo-cross-link proteins with an affinity for platinum-modified DNA, most of which could not be identified in our preliminary study.[28] Cross-linking conditions were optimized here in order to characterize them. A related 25-mer containing a 1,3-d(G*pTpG*) intrastrand adduct of Pt-BP6 was synthesized and characterized. Using this probe, several proteins were photo-cross-linked that differed from those in the set obtained with the 25-mer containing the 1,2-d(G*pG*) intrastrand adduct. A 25-bp duplex probe containing a Pt-BP6 1,2-d(G*pG*)-d(CpT) mismatch was also prepared and used in analytical-scale photo-cross-linking experiments. The probes were applied in photo-cross-linking experiments using nuclear extracts from cancer cell lines with different sensitivities to cisplatin. Finally, a 90-base dumbbell probe was used to ensure that the proteins identified by the 25-bp probe were not photo-cross-linked due to the proximity of the platination site to the DNA ends, for which the proteins might have some affinity. Incorporation of a biotin moiety at the ends or along the hairpin turn in the probes, for their subsequent capture and stringent washing on a streptavidin column, was an important design feature in these experiments.
Synthesis and characterization of a 25-base-pair 1,2-d(G*pG*) intrastrand cross-linked, biotinylated probe
A 25-base-pair DNA containing a 1,2-d(G*pG*) intrastrand cross-link of Pt-BP6 was synthesized as reported previously[28] The purification procedure did not effect significant separation of the orientational isomers[28,33] of Pt-BP6-modified DNA, and a mixture of the two isomers was therefore used in these studies. Following purification, the collected peaks were dialyzed to remove salt using a membrane with a molecular weight cutoff of 3500. In a typical purification, 5 nmol of platinated DNA was obtained, a 17% yield. The ratio of bound platinum atoms per DNA fragment was determined to be 0.99 ± 0.10. The presence of the desired duplex, with a calculated value of 7430.9 m/z, was confirmed by ESI-MS, which revealed an experimental value of 7432.4 ± 3.7 m/z.
Synthesis and characterization of a 25-base-pair 1,3-d(G*pTpG*) intrastrand cross-linked, biotinylated probe
The reaction product of activated Pt-BP6 with a 25-bp DNA duplex containing a single d(G*pTpG*) site was purified by RP-HPLC (Figure S.1, Supporting Information). Three peaks eluting at 8.5 min, 13.7 min, and 14.3 min were collected and contained 7.6 nmol, 1.4 nmol, and 4.3 nmol of DNA, respectively, corresponding to yields of 27%, 5%, and 15%.
The first product, eluting at 8.5 min, was unplatinated starting material. Peaks 2 and 3 contained 0.90 ± 0.16 and 0.96 ± 0.09 Pt atoms per DNA strand, suggesting that they are not fully resolved bands corresponding to 5’- and 3’-orientational isomers[33] of a singly platinated 1,3-d(G*pTpG*) adduct. This behavior was also observed for the 1,2-d(G*pG*) probe (see above). ESI-MS analysis (data not shown) confirmed the absence of the two chloride ligands of Pt-BP6, which are replaced by Pt–N7 guanine bonds in the product. For a singly platinated DNA complex, the calculated m/z is 7956.3. For peaks 2 and 3, the experimental values are 7955.7 ± 0.6 m/z and 7955.8 ± 0.7, respectively, establishing that the platinum atom is bound in a bifunctional manner, with neither chloride nor another buffer-derived ligand remaining in the coordination sphere. Nuclease digestion analyses of the platinated probe (Figure S.2, Supporting Information) confirm that the platinum atom is bound to guanine bases. For products 2 and 3, peaks corresponding to guanosine had significantly diminished intensity in the assay. The peaks for the other three bases were not affected by platination (Figure S.2, Supporting Information).
25-Base-pair duplex probes
Radiolabeled 25-bp duplex probes were prepared in approximately 70% yield, an estimated value since UV-vis measurements were not taken on the radioactive material. The double-stranded constructs could be well separated from single-stranded material by native PAGE, and the ImageQuant gel analysis program used to determine their purity indicated it to be approximately 100% (Figure S.3, Supporting Information).
For preparative-scale photo-cross-linking experiments, 25-bp duplex probes were prepared without a radiolabel. Following purification, 455 pmol of the unplatinated 25-bp duplex, 379 pmol of the 25-bp duplex containing a 1,2-d(G*pG*) intrastrand Pt-BP6 cross-link, and 331 pmol of the corresponding 1,3-d(G*pTpG*) Pt-BP6 adduct were isolated. These values correspond to yields of 91%, 76% and 66%, respectively.
Preparative-scale photo-cross-linking of a 25-bp duplex containing a 1,2-d(G*pG*) intrastrand Pt-BP6 adduct in HeLa nuclear extracts
The Pt-BP6-modified DNA was irradiated in the presence of HeLa nuclear extracts to generate covalent bonds between the benzophenone moiety of the platinum compound and any proteins in the vicinity of the damage site. The DNA-to-protein ratio was adjusted to be the same for both preparative-scale and analytical-scale experiments. After immobilization of the biotinylated DNA-platinum-protein conjugates on streptavidin-coated magnetic beads, the complexes were washed stringently with high salt and SDS-containing buffers to remove any non-covalently bound proteins. SDS-PAGE analysis was performed on a slab gel to obtain better separation of proteins. An analytical-scale photo-cross-linking experiment was run in parallel to the preparative-scale experiment as a guide for excising DNA-platinum-protein complexes, as shown in Figure 2B, lane 1. Mass spectral analysis of the preparative-scale photo-cross-linking experiment identified several proteins in each of the six major bands on the analytical gel (Table 1), including the DNA repair proteins PARP-1, DNA ligase III, Msh2, and all three members of the DNA-PK heterotrimer, as well as HMG-domain proteins HMGB1, HMGB2, HMGB3 and UBF1.
Figure 2.
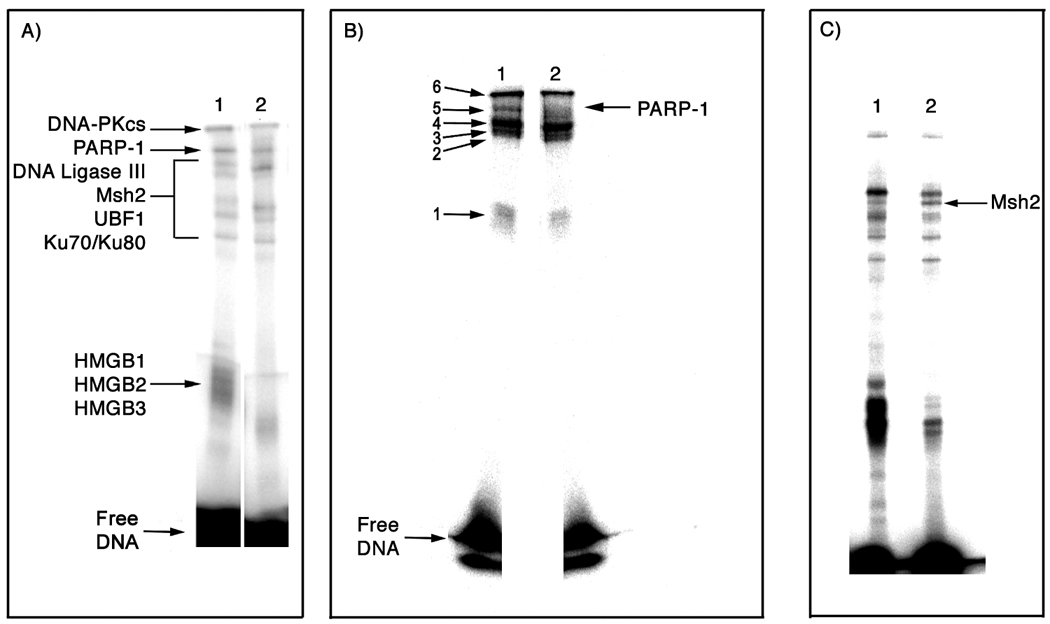
Analytical-scale photo-cross-linking results. A) 10% SDS-PAGE analysis of photo-cross-linking of HeLa nuclear extracts using a 25-bp duplex containing a 1,2-d(G*pG*) (lane 1) or 1,3-d(G*pTpG*) (lane 2) cross-link of Pt-BP6. B) 12% SDS-PAGE analysis of photo-cross-linking using a 25-bp duplex containing a 1,2-d(G*pG*) cross-link of Pt-BP6 incubated with HeLa nuclear extracts (lane 1) or nuclear extracts from HeLa cells in which PARP-1 has been silenced using RNAi (lane 2). Lane 1 was used as a guide to excise six bands from preparative scale photo-cross-linking experiments for analysis by trypsin digest-coupled LC-MS/MS. C) 10% SDS-PAGE analysis of photo-cross-linking of NTera2 nuclear extracts using a 25-bp duplex containing a 1,2-d(G*pG*) cross-link of Pt-BP6 (lane 1) or a 25-bp duplex containing a mismatched 1,2-d(G*pG*)/d(CpT) cross-link of Pt-BP6 (lane 2).
Table 1.
Proteins identified by preparative-scale photo-cross-linking of a 25-bp duplex containing a 1,2-d(G*pG*) intrastrand Pt-BP6 adduct.
| Band[a] | Protein[b] | Probability[c] | MW (Da)[d] |
Unique Peptides[e] |
Protein Function |
|---|---|---|---|---|---|
| 1 | HMGB1 | 3.50E-10 | 24878 | 5 | HMGB1 shields cisplatin-DNA adducts from excision repair[40] |
| HMGB2 | 3.12E-07 | 24018 | 5 | Analysis of the NCI library of cancer cell lines reveals a correlation between cells expressing high levels of HMGB1 and cisplatin sensitivity[100,101] |
|
| HMGB3 | 4.14E-08 | 22980 | 3 | HMGB1, HMGB2 and HMGB3 are chromatin architectural proteins[36] | |
| 2 | Ku70 | 4.47E-11 | 69829 | 17 | Ku70 and Ku80 are members of the DNA-PK complex involved in nonhomologous end-joining repair of double-strand breaks[102] |
| Ku80 | 2.50E-09 | 82652 | 5 | ||
| PARP-1 | 7.01E-11 | 113012 | 4 | See text | |
| 3 | Ku80 | 4.44E-15 | 82652 | 37 | |
| Ku70 | 1.11E-15 | 69829 | 22 | ||
| PARP-1 | 1.35E-10 | 113012 | 9 | ||
| 4 | Ku80 | 2.22E-15 | 82652 | 30 | |
| Ku70 | 1.26E-12 | 69829 | 27 | ||
| UBF1 | 7.96E-07 | 89350 | 3 | HMG-domain protein responsible for initiation of RNA polymerase I[11] | |
| PARP-1 | 9.88E-10 | 113012 | 14 | ||
| DNA-PKcs | 4.01E-11 | 468786 | 13 | ||
| DNA ligase III | 3.91E-08 | 112834 | 6 | See text | |
| Msh2 | 8.53E-06 | 104676 | 4 | Protein involved in the recognition of base pair mismatches[42] | |
| 5 | PARP-1 | 2.63E-13 | 113012 | 42 | |
| DNA-PKcs | 4.26E-11 | 468786 | 29 | Member of the DNA-PK complex involved in nonhomologous end-joining repair of double-strand breaks[102] |
|
| DNA ligase III | 8.10E-14 | 112834 | 11 | ||
| Msh2 | 7.57E-12 | 104676 | 5 | ||
| 6 | DNA-PKcs | 8.73E-15 | 468786 | 146 |
The LC-MS/MS analysis was carried out on six gel slices corresponding to the bands indicated in Figure 2B, Lane 1. The proteins found in each band are listed here.
Proteins identified as described in the Experimental Section.
Probability of the peptide identification being an artifact of background signal from the LC-MS/MS instrument. For example, a probability of 1.0E-4 indicates that there is a 0.01% chance that the identified peptide is not actually present in the sample. The values listed correspond to the peptide most likely to be present for each protein.
Masses listed are those of the full-length proteins.
The number of peptides identified for each protein. Peptides differing by only one amino acid were not considered to be unique.
A control lane containing unplatinated DNA was included in the preparative scale photo-cross-linking experiment. Proteins identified in this lane, as well as proteins for which the molecular weight was inconsistent with the band location, were excluded from the list in Table 1, which reports the results of three repeats of preparative-scale photo-cross-linking experiments. In the table, the “Band” column refers to the relative mobility of the protein-platinum-DNA complex through the gel (Figure 2B, lane 1), with band one being the fastest migrating species. The “Unique Peptides” column reports the number of individual peptide fragments matching those expected from the known protein sequence. Peptides differing by only one amino acid were not considered to be unique. The “Probability” column assigns a number to each protein based on the likelihood of a coincidental match of the best unique peptide for each protein. For example, three unique peptides were identified that correspond to the protein UBF1. One of these peptides, RQLQEERPELSESELTRL, produced MS/MS data with a probability of 7.96 × 10−7, meaning that there is a 0.0000796% probability that this identification is an artifact due to background. Peptides with probabilities of 1.0 × 10−3 or higher were excluded.
Analytical-scale photo-cross-linking of a 25-bp duplex containing a 1,3-d(G*pTpG*) intrastrand adduct
Following purification, the 25-bp probe containing a 1,3-d(G*pTpG*) Pt-BP6 intrastrand adduct was used in photo-cross-linking experiments (Figure 2A, lane 2). For this probe, only a weak band was present near the position where bands arising from HMGB1, HMGB2, and HMGB3 cross-linked to the 1,2-d(G*pG*) adduct appear, indicating that these proteins are bound weakly, if at all. The higher molecular weight bands were similar, although not identical, to those of the 1,2-d(G*pG*) crosslink probe study. Proteins photo-cross-linked by the 1,3-d(G*pTpG*) probe were identified by preparative-scale experiments.
Preparative-scale photo-cross-linking of the 1,3-d(G*pTpG*) intrastrand adduct of Pt-BP6 using HeLa cell nuclear extracts
Preparative scale photo-cross-linking experiments were carried out using the 25-bp duplex containing a 1,3-d(G*pTpG*) Pt-BP6 intrastrand cross-link in a manner similar to that described for the 1,2-d(G*pG*) adduct. In this study, a desthiobiotin-modified probe was required in order to avoid reaction between the platinum atom and the sulfur-containing biotin moiety. The desthiobiotin-tethered duplex has comparable affinity for streptavidin-coated magnetic beads as the biotin-tethered duplex originally employed (data not shown). The cross-linking experiments identified several proteins that bind to this construct (Table 2). Unlike the probe containing a 1,2-d(G*pG*) Pt-BP6 cross-link, this probe exhibited only weak affinity for HMGB1 and HMGB3, and no affinity at all for HMGB2 or UBF1. The probe formed photo-cross-links to DNA repair proteins PARP-1, DNA ligase III, Msh2, all three members of the DNA-PK heterotrimer, and RPA1, which was not photo-cross-linked by the 1,2-d(G*pG*) adduct. The significance of the protein probability values is discussed above. Fragment mass spectra of proteins identified by only one peptide are available in Fig. S.6 of Supporting Information.
Table 2.
Proteins identified by preparative-scale photo-cross-linking of a 25-bp duplex containing a 1,3-d(G*pTpG*) intrastrand Pt-BP6 adduct.
| Band[a] | Protein[a] | Probability[a] | MW (Da) | Unique Peptides[a] |
Protein Function |
|---|---|---|---|---|---|
| 1 | HMGB1 | 2.57E-04 | 24878 | 1 | See Table 1 for descriptions of proteins that are also listed here. |
| HMGB3 | 4.97E-04 | 22980 | 1 | ||
| 2 | PARP-1 | 1.61E-07 | 113012 | 2 | |
| RPA1 | 2.13E-06 | 68095 | 1 | ||
| Ku70 | 1.30E-05 | 69799 | 1 | ||
| 3 | Ku80 | 7.72E-13 | 82652 | 13 | |
| PARP-1 | 1.20E-08 | 113012 | 5 | ||
| RPA1 | 8.47E-07 | 68095 | 1 | RPA1 is involved in DNA-damage recognition of NER. It has a 2-fold greater affinity for cisplatin 1,3-d(G*pNpG*) vs. 1,2-d(G*pG*) intrastrand adducts[39] |
|
| DNA ligase III | 1.36E-06 | 112834 | 4 | ||
| Msh2 | 1.51E-05 | 104676 | 3 | ||
| Ku70 | 2.21E-05 | 69799 | 3 | ||
| 4 | PARP-1 | 8.47E-10 | 113012 | 14 | |
| DNA ligase III | 8.51E-07 | 112834 | 6 | ||
| 5 | PARP-1 | 2.44E-07 | 113012 | 7 | |
| DNA ligase III | 4.46E-07 | 112834 | 3 | ||
| 6 | DNA-PKcs | 4.94E-09 | 468786 | 8 |
See Table 1 for heading descriptions.
Effect of shRNA-induced RNAi on PARP-1 expression
Figure 3A illustrated the results of western blot experiments, using an antibody against PARP-1, on HeLa nuclear extracts. Cells expressing the shRNA (lane 3) had a lower level of PARP-1 than the control HeLa cells (lane 1). The DNA sequence from which this shRNA is transcribed is 5’-CCAACTCCTACTACAAGC-TTTCTCGAGAAAGCTTGTAGTAGGAGTTGGTTTTTG-3’. Cells expressing the shRNA were sorted into single clones for selection of those containing minimal PARP-1 levels. As indicated in Figure 3B, clone #11 (lane 5) exhibited no detectable PARP-1 by western blot analysis. This clone was used in all subsequent experiments.
Figure 3.

A) Western blot analysis of HeLa cell nuclear extracts transfected with plasmids containing shRNA for silencing of the PARP-1 gene (lanes 2 and 3). The extract of untransfected HeLa cells (lane 1) serves as an experimental control. B) Western blot analysis of single clones of HeLa cells transfected with the plasmid for RNAi of PARP-1. The nuclear extracts of clones #4, #5, #11, and #13 were loaded in lanes 3 to 6, respectively. Untransfected HeLa cells (lane 1) and transfected but unsorted cells (lane 2) were used as controls.
Analytical-scale photo-cross-linking using nuclear extracts from cells with PARP-1 silenced
Nuclear extracts from HeLa cells in which PARP-1 was silenced by RNAi were used in photo-cross-linking experiments with the probe containing a 1,2-d(G*pG*) intrastrand Pt-BP6 adduct (Figure 2B). The band identified as PARP-1 by preparative-scale photo-cross-linking was completely abolished when using nuclear extracts from PARP-1 silenced cells.
Analytical-scale photo-cross-linking of a 25-bp duplex containing a 1,2-d(G*pG*) intrastrand adduct at a GT mismatched base pair
A 25-bp duplex containing a 1,2-d(G*pG*) intrastrand cross-link at a mismatched base pair was synthesized and purified. The duplex was constructed such that the platinated guanine on the 3’-side of the top strand was mismatched against a thymine residue on the bottom strand. Photo-cross-linking experiments were performed using this probe and NTera2 nuclear extracts (Figure 2C). The main result was formation of a band containing the protein Msh2, which appears with much greater intensity in lane 2. The increased affinity of Msh2 for the compound lesion diminishes the binding of other proteins that would otherwise recognize the platinated duplex. In particular, HMG-domain proteins and PARP-1 are photo-cross-linked less effectively, and proteins that comprise the DNA-PK heterotrimer are also somewhat competed.
Analytical-scale photo-cross-linking using nuclear extracts from cancer cell lines of different origin
Nuclear extracts from cervical (HeLa), bone (U2OS), testicular (NTera2), and pancreatic (BxPC3) cancer cells were used in photo-cross-linking experiments with both the 1,2-d(G*pG*) and 1,3-d(G*pTpG*) adducts of Pt-BP6 (Figure 4). The proteins cross-linked to each probe appear to be the same for nuclear extracts from the four different cell lines, but with varying quantities. The most dramatic difference occurs for PARP-1, the band for which is strong using nuclear extracts from NTera2 cells, but very weak for nuclear extracts from BxPC3 cells.
Figure 4.
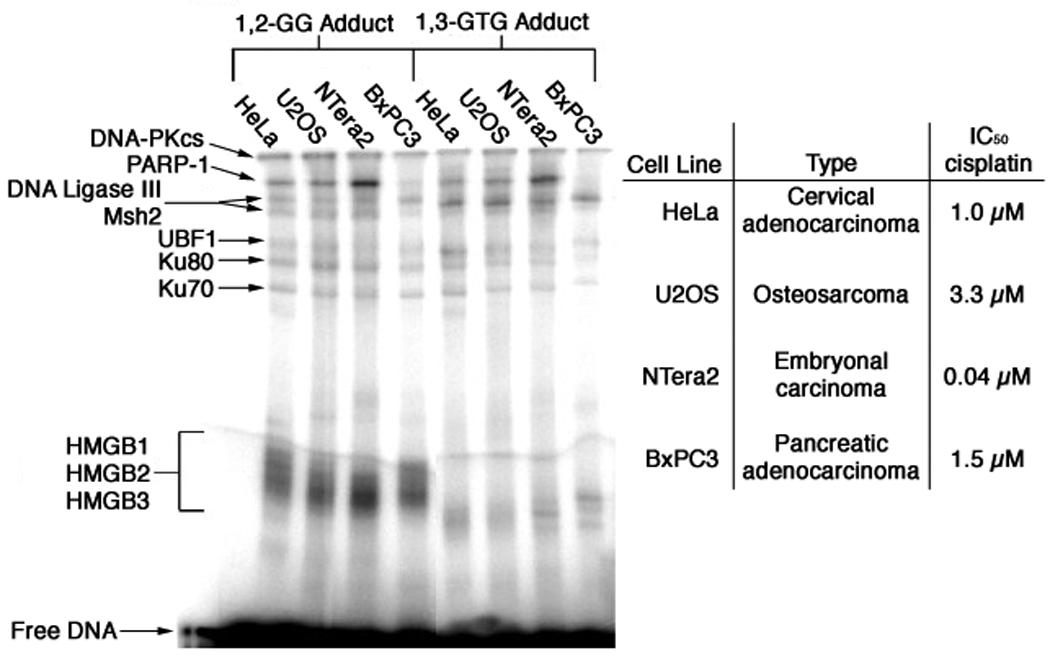
Photo-cross-linking of 25-bp Pt-BP6 probe with nuclear extracts from various cell lines.
Western blot analysis of PARP-1 in HeLa and NTera2 cells
Western blot analyses of HeLa and NTera2 nuclear extracts reveal that PARP-1 expression is significantly higher in the latter (Figure 5). This result indicates that the greater amount of photo-cross-linking to PARP-1 in NTera2 nuclear extracts is a consequence of its higher expression level.
Figure 5.

Western blot analysis of PARP-1 expression using HeLa (lane 1) and NTera2 (lane 2) nuclear extracts.
Synthesis of a site-specifically platinated 90-base probe
A biotinylated 65-base oligonucleotide was prepared on the DNA synthesizer and purified by 6% urea-PAGE. A total of 12.4 nmol was collected. The 65-base DNA and a Pt-BP6-modified 25-base DNA were phosphorylated, annealed, and ligated to make a 90-base dumbbell probe (Figure S.4, Supporting Information), the formation of which was confirmed by 6% urea-PAGE analysis (Figure 6A). The 90-base probe was isolated by 6% urea-PAGE and visualized by audioradiography (Figure 6B). The dumbbell was resynthesized on a preparative scale probe and visualized by UV shadowing, which revealed a band migrating the same distance through the gel as the radiolabeled 90-base DNA product. The DNA was excised from the gel and isolated by the crush and soak method. Following extraction from the gel, 630 pmol of the platinated DNA was obtained.
Figure 6.

The 90-base DNA was synthesized from 25-base and 65-base DNA fragments. The 90-base product was purified A) and the purity determined B) by 6% urea-PAGE.
Characterization of the site-specifically platinated 90-base probe
To determine whether the excised DNA contained the desired 90-base probe, the preparative-scale sample was analyzed by nuclease digestion. The analysis was altered from that used for the 25-base DNA by addition of an initial step involving exonuclease III, T7 exonuclease, and mung bean nuclease. This step is necessary to digest the loops and duplex portions of the DNA prior to incubation with the single-stranded nuclease S1. HPLC analysis revealed a peak in addition to those of the four nucleobases with a retention time of 19.5 min (Figure S.5, Supporting Information). The ratios of optical bands at 280 nm and 260 nm for cytosine, thymine, guanine, and adenine are expected to be 1.03, 0.66, 0.78 and 0.16, respectively.[34] Experimentally, these ratios were 1.06, 0.66, 0.79 and 0.20, in good agreement with the theoretical values. For the peak at 19.5 min, this ratio was 2.14, indicating that the peak is unlikely to contain a DNA base. RP-HPLC analysis of the buffers and enzymes alone, without DNA, indicated that this peak is an artifact and not a product of digestion of the 90-base DNA probe (Figure S.5A, Supporting Information).
The C:G:T:A ratios of the 65-base DNA, 25-base DNA, and desired 90-base dumbbell product are 13:23:15:14, 12:2:9:2; and 25:25:24:16, respectively. For the unplatinated probe (Figure S.5B, Supporting Information), the experimental ratio was 23.1:26.5:23.7:15.7 and, for the platinated probe, (Figure S.5C, Supporting Information) it was 24.1:25.9:23.6:15:4. These values compare well with the expected ratios for a 90-base probe, indicating that the DNA was properly constructed. To determine whether the DNA was in the completely ligated dumbbell form, a radiolabeled sample was digested with mung bean nuclease (MBN). The expected product would contain 41 bases (Figure 7A), in agreement with the experimental result (Figures 7B,C). MBN digestion of a 90-base DNA that is only singly ligated produced a radiolabeled DNA fragment of 33 bases (Figure 7C).
Figure 7.
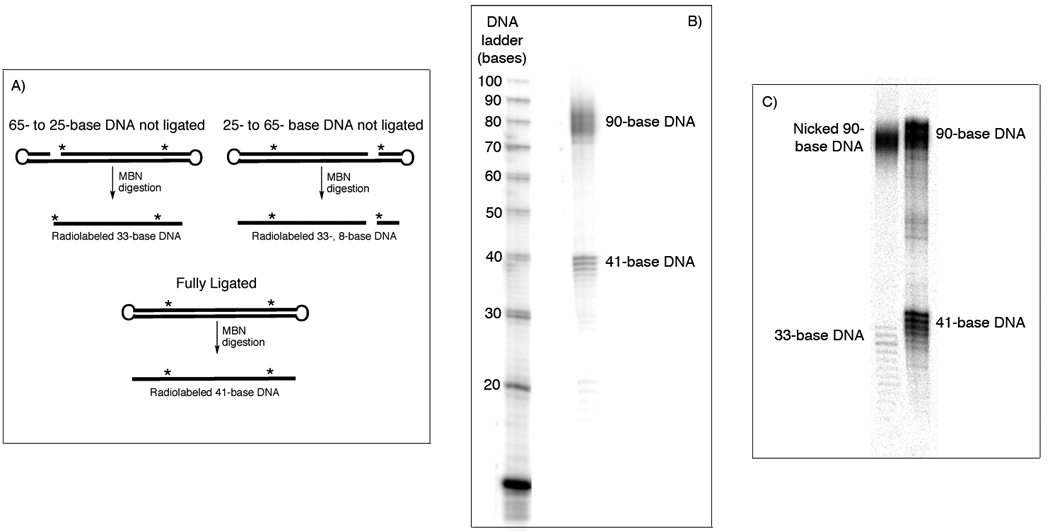
The purified 90-base DNA was analyzed by mung bean nuclease (MBN) digestion. A) This enzyme will cleave the looped ends of the DNA, which will produce a different product for singly- or doubly-ligated material. B) Digestion of the 90-base probe indicates that a fully ligated DNA was isolated. C) A control experiment using a singly ligated DNA demonstrates the 33-base product produced after MBN digestion of this type of DNA.
Analytical-scale photo-cross-linking of the platinated 90-base probe
Photo-cross-linking using a radiolabeled 90-base dumbbell probe containing a site-specific 1,2-d(G*pG*) adduct of Pt-BP6 and nuclear extracts from various cancer cell lines yielded several specific protein-platinum-DNA complexes. The results are presented in Figure 8.
Figure 8.
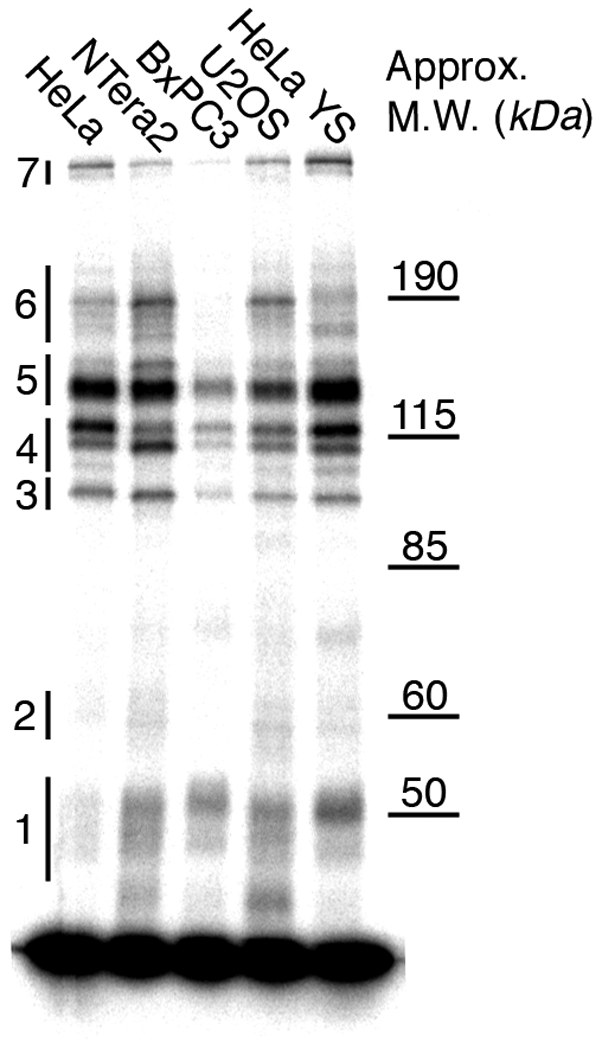
Photo-cross-linking of the 90-base DNA with several cancer cell nuclear extracts. The bands used for preparative-scale experiments are indicated.
Preparative-scale photo-cross-linking of the platinated 90-base probe
Preparative-scale photo-cross-linking experiments using the 90-base dumbbell probe and HeLa nuclear extracts led to the identification of many nuclear proteins with an affinity for this platinated DNA (Table 3). All of the proteins identified using the 25-bp probe were detected with the 90-base probe. The additional proteins identified were the transcription factor YB-1, mismatch repair proteins Msh3 and Msh6, excision repair proteins DDB1, RFC1 and RFC2, and the chromatin remodeling proteins SMARCA3, DNA topoisomerase I, protein polybromo-1 (PB-1), SMARCA4, SMARCC2, and FACT subunit SPT16. Fragment mass spectra of proteins identified by only one peptide are available in Fig. S.6 of Supporting Information.
Table 3.
Proteins identified by preparative-scale photo-cross-linking of a 90-base DNA containing a 1,2-d(G*pG*) intrastrand Pt-BP6 adduct.
| Band[a] | Protein[a] | Probability[a] | M.W. (Da) | Unique Peptides[a] |
Comments |
|---|---|---|---|---|---|
| 1 | HMGB1 | 2.36E-05 | 24878 | 1 | See Table 1 for descriptions of proteins that are also listed here. |
| HMGB2 | 3.80E-08 | 24018 | 1 | ||
| HMGB3 | 8.64E-08 | 22980 | 1 | ||
| 2 | YB-1 | 8.32E-07 | 35902 | 1 | Y-box binding protein-1 (YB-1) is a transcription factor containing a cold-shock domain (CSD) that binds to cisplatin-modified DNA[70] |
| RFC2 | 3.31E-05 | 39132 | 2 | Involved in intermediate steps of nucleotide excision repair[72] | |
| 3 | Ku70 | 7.29E-08 | 69799 | 11 | |
| 4 | Msh2 | 1.70E-07 | 112834 | 9 | |
| Ku70 | 3.72E-07 | 69799 | 8 | ||
| Ku80 | 3.89E-07 | 82652 | 3 | ||
| UBF1 | 7.21E-07 | 89350 | 1 | ||
| Msh3 | 5.72E-06 | 127376 | 2 | Protein involved in the recognition of base pair mismatches[42] | |
| DNA topo I | 1.05E-05 | 90668 | 3 | Chromatin remodeling protein involved in relaxing superhelical DNA[85] | |
| 5 | PARP-1 | 1.61E-11 | 113012 | 13 | |
| RFC1 | 2.67E-10 | 128175 | 4 | Binds to double-stranded DNA and interacts with proliferating cell nuclear antigen (PCNA) to initiate transcription[72] |
|
| PB-1 | 2.02E-09 | 192823 | 5 | SWI/SNF chromatin remodeling protein containing an HMG-domain[90] | |
| Msh3 | 4.04E-06 | 127376 | 3 | ||
| SMARCA4 | 2.14E-09 | 184529 | 4 | SWI/SNF chromatin remodeling protein[89] | |
| SMARCA3 | 1.31E-08 | 113856 | 2 | SWI/SNF chromatin remodeling protein[89] | |
| DNA Ligase III | 1.91E-08 | 112834 | 10 | ||
| DDB1 | 7.71E-05 | 126887 | 2 | Nucleotide excision repair protein that binds to cisplatin-modified DNA[2,20,66,71] | |
| 6 | PARP-1 | 6.32E-08 | 113012 | 7 | |
| SMARCC2 | 2.69E-06 | 132796 | 2 | SWI/SNF chromatin remodeling protein[89] | |
| DNA Ligase III | 7.68E-07 | 112834 | 5 | ||
| Msh6 | 2.44E-07 | 152688 | 1 | Protein involved in the recognition of base pair mismatches[42] | |
| Msh2 | 2.75E-06 | 104676 | 3 | ||
| SPT16 | 4.93E-05 | 119838 | 2 | FACT complex subunit that increases the affinity of the HMG-domain protein SSRP1 for cisplatin-modified DNA[79] |
|
| 7 | DNA-PKcs | 1.48E-08 | 468786 | 15 |
See Table 1 for heading descriptions.
90-base probe repair assay
The 90-base probe remains intact following exposure to nuclear extracts from various cell lines both before and after UV irradiation (Figure S.7, Supporting Information). Gel electrophoretic analysis of DNA exposed to the extracts revealed no difference from that for untreated DNA. The lane labeled “U2OS + UV” in Fig. S.7 indicates that exposure to UV light also does not effect the probe. If the dumbbell ends were severed from the probe, as occurs following incubation with mung bean nuclease (Figure 7B), a digestion product of 41 bases would be present. If the platinum adduct were excised by NER proteins, a DNA fragment of 28–32 bases would appear in the gel.
Discussion
Overview of the methodology
Since the discovery that cisplatin targets DNA, much effort has been expended to identify proteins with an affinity for the platinated duplex. In the present study, we prepared and characterized site-specifically modified 25-bp duplexes containing a 1,2-d(G*pG*) or 1,3-d(G*pTpG*) cross-link as well as a 90-base dumbbell probe containing a 1,2-d(G*pG*) intrastrand cross-link, all bearing a pendant photo-activatable benzophenone cross-linking agent. Upon irradiation, these probes form covalent bonds between the platinated DNA and any proteins in a nuclear extract for which they have an affinity. Isolation of the DNA-platinum-protein complexes followed by trypsin digestion and mass spectrometric analysis allowed the proteins to be identified. Unlike southwestern blotting and other techniques, the present approach exposes the platinated DNA to multiprotein complexes. The method requires proteins to compete for binding to the probe, thus providing a more realistic assessment of the cellular milieu. Although cisplatin itself can form DNA-protein cross-links, in order to form such a covalent bond the protein would have to be in very close proximity to the adduct.[24,35] Pt-BP6 probes utilize a photo-reactive moiety with a six-carbon atom linker chain, allowing for a large number of DNA-bound proteins to be captured. Our probes also can carry site-specific platinum adducts such that proteins having an affinity for a particular chemical cross-link can be analyzed.
Identification of the proteins
Many of the proteins identified in the photo-cross-linking experiments conducted in this study (Table 1–Table 3) appear in more than one band. This phenomenon occurs for a number of reasons. During photo-cross-linking and subsequent work-up steps, the samples were kept at 4°C whenever possible. Despite these efforts, some protein degradation is inevitable. Degraded proteins travel down the gel more rapidly and the fragments appear in faster eluting bands. The nature of trypsin-digest coupled mass spectrometric analysis does not distinguish between full-length proteins and protein fragments. For example, a small amount of DNA-PKcs, a protein that is clearly identified in the most slowly eluting band (Figure 2B, lane 1), also occurs to a lesser degree in the three bands running just below it in the gel. Another common problem with trypsin-digest coupled mass spectrometry is a phenomenon known as carry-over. The instrumentation is extremely sensitive, able to detect attamole quantities of peptides, and very small amounts of protein from one run can be carried over into the next.
Even with 2 mg of nuclear extracts used for photo-cross-linking, only the band containing DNA-PKcs is distinctly visible by Coomassie staining of the gels. Since the analytical-scale photo-cross-linking must be used as a guide to excise bands that run extremely close together on the gel, some overlap of excised bands is inevitable. The methodology was able to isolate and identify a band containing DNA-PKcs and one containing HMGB1, HMGB2 and HMGB3. As described below, the presence of PARP-1 cross-linked to our probe was identified by its absence in experiments using nuclear extracts from HeLa cells in which the protein was silenced with RNAi (Figure 2B). Also, the identity of Msh2 in a band was verified by photo-cross-linking with a DNA duplex containing a combination of a platinum adduct and a DNA mismatch (Figure 2C). The identities of Ku70, Ku80, and UBF1 proteins in the remaining bands are consistent with their molecular weights. For the 90-base dumbbell probe, it was possible to identify proteins present in the various bands, despite their large number (Table 3). Results with this probe clearly demonstrate that proteins photo-cross-linked to the 25-bp duplex probe are not a consequence of their affinity for DNA ends.
HMG-domain proteins bind with greater affinity to 1,2-d(G*pG*) cross-links
A significant difference between the Pt 1,2-d(G*pG*) and 1,3-d(G*pTpG*) intrastrand cross-links is that the former are better recognized by HMG-domain proteins.[8] These chromosomal proteins bind to distorted DNA structures such as four-way junctions and induce further bending of the duplex.[36] The present results indicate that, in cell extracts, HMGB1 and HMGB3 bind weakly to the 1,3-d(G*pG*) adduct, whereas HMGB1, HMGB2, HMGB3, and the HMG-domain protein UBF1 all bind strongly to the 1,2-d(G*pG*) cross-link (Figure 2A, Table 1 and Table 2). This finding supports previous work utilizing electrophoretic mobility shift assays (EMSAs) of purified HMGB1, which reveal that the protein binds to the 1,2-d(G*pG*) but not the 1,3-d(G*pTpG*) adduct.[8] Here we provide conclusive evidence that this behavior occurs in a nuclear milieu, where many other proteins can compete for binding to the platinum cross-link.
The data provided here also establish for the first time an affinity of HMGB3 for platinum-modified DNA. The absence of prior studies linking HMGB3 to the processing of platinum-DNA adducts most likely stems from the fact that this protein is usually expressed during embryonic development and not in adult tissues.[37] One proteomic analysis of platinum resistant versus sensitive ovarian tumors indicates that HMGB3 is expressed to a greater degree in resistant cells, although not dramatically.[38]
Nucleotide excision repair proteins bind to Pt-BP6-modified DNA
Replication protein A1 (RPA1) is a component of the nucleotide excision repair apparatus, the major machinery for removing cisplatin adducts from DNA.[2] We find this protein to be photo-cross-linked only to the 1,3-d(G*pTpG*) intrastrand adduct of Pt-BP6. EMSA studies using purified RPA1 indicate that the protein has a 1.5–2-fold stronger affinity for the 1,3-d(G*pTpG*) than the 1,2-d(G*pG*) cisplatin cross-link.[39] The 1,3-d(G*pTpG*) adduct is repaired more efficiently by NER than the 1,2-d(G*pG*) adduct, and repair of the latter is further inhibited, exclusively for the 1,2-d(G*pG*) cross-link, by the binding of HMG-domain proteins.[40] The affinity of HMG-domain proteins in nuclear extracts for the 1,2-d(G*pG*) intrastrand cross-link has been established in the present experiments, as discussed earlier. Our results indicate that RPA1 binds with greater affinity to the 1,3-d(G*pTpG*) intrastrand cross-link of Pt-BP6 in the nuclear milieu, where other proteins such as HMGB1 are available to compete for binding. That RPA1 does not bind to the 1,2-d(G*pG*) intrastrand adduct of Pt-BP6 most likely reflects lower affinity for the lesion in addition to competition for binding from HMG-domain proteins.
Nucleotide excision repair of cisplatin adducts results in excised DNA fragments of approximately 28–32 base pairs,[40] which is longer than the 25-bp probe. Use of a longer DNA probe modified with Pt-BP6 should therefore increase the ability of nucleotide excision repair proteins to bind and become activated, providing a more complete account of proteins that bind to platinated DNA. Photo-cross-linking studies using the longer 90-base dumbbell probe are discussed below.
Mismatch repair recognition complexes bind to platinum-damaged DNA
The binding of mismatch repair proteins to cisplatin-damaged DNA is relevant to the anti-proliferative effects of the drug and is discussed in detail in the literature.[16,41–46] The affinity of MMR protein Msh2 for various constructs of cisplatin-modified DNA has been demonstrated.[41,47] Msh2 forms a complex with either Msh3 or Msh6 when it binds to DNA containing mismatches. These two distinct mismatch repair recognition complexes have different affinities for certain types of mismatched bases and nucleotide insertions or deletions.[48] The current study establishes that both protein complexes Msh2/Msh3 and Msh2/Msh6 will bind to platinum-modified DNA in nuclear extracts (Table 3).
Previous work using EMSAs indicates that the mismatch repair protein, Msh2, has a greater affinity for a 1,2-d(G*pG*) cisplatin cross-link in which the unplatinated DNA strand contains a mismatched thymine opposite the 3’-guanosine.[41] This result nicely correlates with the finding of the present study, in which the cross-linking efficiency is enhanced in the mismatched probe (Figure 2C). The increased affinity of Msh2 for DNA containing the mismatched platinum cross-link is accompanied by diminished photo-cross-linking to the other proteins. If a DNA polymerase were able to bypass a cisplatin adduct, the resulting product would incorporate a mismatched base[49] and become a better binding site for mismatch repair proteins, inducing futile attempts to repair the compound lesion and eventually signal apoptosis.[14]
Are Pt-BP6 adducts a reasonable facsimile for those of cisplatin?
Our photo-cross-linking results demonstrate that the Pt-BP6-modified probe mimics cisplatin-modified DNA. Many of the proteins cross-linked by Pt-BP6 have been previously associated with cisplatin-damaged DNA. This finding indicates that cis-{Pt(NH3)(N-(6-aminohexyl)-4-benzophenonamide)}2+-DNA cross-links are processed in a manner identical to those of cis-{Pt(NH3)2}2+. The probe containing a 1,2-d(G*pG*) intrastrand adduct forms photo-cross-links to several HMG-domain proteins whereas the probe containing a 1,3-d(G*pTpG*) intrastrand Pt-BP6 adduct, like that for cisplatin, bound these proteins weakly or not at all. Also, only the 1,3-d(G*pTpG*) probe formed photo-cross-links to the nucleotide excision repair protein, RPA1 (Table 1 and Table 2). The probe containing a compound lesion of a 1,2-d(G*pG*) adduct of Pt-BP6 and a C·T mismatch photo-cross-linked more strongly to the mismatch repair protein Msh2 (Figure 2C). These results agree with work from the literature using cisplatin-DNA probes in EMSAs with purified proteins.[8,39,41]
The fact that the ammine/alkyl amine platinum complex Pt-BP6 behaves like cisplatin is relevant to the mechanism of the orally available platinum(IV) compound cis,trans,cis-[Pt(NH3)(cyclohexylamine)(acetato)2Cl2], satraplatin, which is has undergone clinical trials for hormone resistant prostate cancer. Satraplatin is reduced to platinum(II) in blood, and in cells the cis-{Pt(NH3)(cyclohexylamine)}2+ cation is expected to be the DNA-binding moiety.[33,50] DNA adducts formed by satraplatin are apparently not recognized by mismatch repair proteins.[44] Our present results suggest that satraplatin-DNA adducts, which contain a pendant cyclohexylamine structurally similar to the benzophenone arm of the BP6 ligand, may be processed by the cell in a manner identical to that of cisplatin.
Photo-cross-linking results are not strongly dependent on the composition of nuclear extracts from different cancer cell lines
The photo-cross-linking experiments were extended to include nuclear extracts from a variety of cancer cell lines having variable sensitivities to cisplatin (Figure 4). The proteins photo-cross-linked by the platinated probe are generally consistent among all cell lines tested, although their relative amounts differ. The fact that extracts from a variety of cells exhibit an identical array of platinated DNA-binding proteins is consistent with recent work indicating that the success of oxaliplatin, by comparison to cisplatin or carboplatin, to treat colorectal cancer is a consequence of increased cellular uptake[51] and not differential processing of the Pt-DNA adducts. The one notable difference in the present work is the extent of photo-cross-linking of PARP-1. The amount of photo-cross-linking of the protein between the different extracts varies greatly, with the greatest levels appearing in extracts from NTera2 testicular cancer cells (Figure 4, Lane 3). The consequences of PARP-1 binding to platinum-modified DNA are discussed below. Photo-cross-linking with nuclear extracts from the same cell lines using the 90-base Pt-BP6 probe (Figure 8) showed similar results. Fewer proteins are present in BxPC3 nuclear extracts that bind to platinated DNA than in similar extracts from the other cells. Unlike experiments using the 25-bp probe (Figure 2B), photo-cross-linking studies with the 90-base probe using nuclear extracts in which PARP-1 is silenced did not reveal a resolved band containing this protein (Figure 8). This finding is most likely due to overlap between the PARP-1-platinum-DNA complex and other proteins having a similar molecular weight (Table 3).
In summary, the results from the present photo-cross-linking experiments reveal that recognition of platinum-damaged DNA is largely conserved between nuclear extracts from cell lines with differing sensitivities to cisplatin and that DNA damage recognition is not likely to be the sole reason for the variable response of the cells to cisplatin treatment.
PARP-1 binds to platinum-damaged DNA
Photo-cross-linking experiments identified poly(ADP-ribose) polymerase-1 (PARP-1) as a key protein that binds to both the 1,2-d(G*pG*) and 1,3-d(G*pTpG*) intrastrand Pt-BP6 cross-links. Using nuclear extracts from HeLa cells in which PARP-1 was silenced by RNAi, we confirmed the identity of the band containing PARP-1 with the 25-bp duplex probe (Figure 2B). PARP-1 has been associated with base excision repair[52] and a backup system of nonhomologous end joining (NHEJ); this latter process repairs double strand breaks when the DNA-PK heterotrimer is compromised.[53,54] In both of these activities, PARP-1 recruits DNA ligase III to the damage site,[52] which we propose to be the main reason for DNA ligase III photo-cross-linking by our probes.
PARP-1 catalyzes the addition of poly(ADP-ribose) (PAR) polymers onto acceptor proteins in a reaction that consumes NAD+.[31] PAR polymers created by PARP proteins can signal the release of apoptosis-inducing factor (AIF) from mitochondria, which initiates apoptosis.[55] PARP activity depletes cellular reservoirs of NAD+, leading to a shutdown of glycolysis. Since cancer cells rely on glycolysis for ATP production, the cells will die by necrosis.[56] The formation of PAR polymers on nuclear proteins is important, since each unit of the polymer contains two negatively charged phosphate moieties, which electrostatically repel the DNA backbone in the vicinity of a PAR-modified protein.[57] A major target of PARP-1 activity is PARP-1 itself, which leads to dissociation of the enzyme from DNA. PARP-1 can also can modify histone proteins, relaxing their DNA interactions, which may facilitate repair.[31]
Given the sensitivity of NTera2 cells to cisplatin, the high levels of PARP-1 expression in this context (Figure 5) suggest a role for this protein in mediating the anticancer activity of the drug. PARP-1 is commonly mutated in germ cells, specific variants being Val762Ala and Lys940Arg, two residues in the catalytic domain of the protein.[58] Compromised activity of PARP-1 by these mutations may be the reason that such high levels of the protein are present in this cell line. PARP inhibitors have been used to sensitize cells to cisplatin and carboplatin,[59,60] although there are conflicting results that indicate no effect.[61] On balance, the data suggest that PARP-1 may facilitate repair of Pt–DNA adducts, since its inhibition sensitizes cells to cisplatin. The anticancer potential of PARP inhibitors and platinum drugs in combination therapies is currently being investigated in phase I and II clinical trials.[62]
Given this information, the identification of PARP-1 as a protein that binds to platinum-modified DNA is a significant discovery. Since the protein also recognizes double strand breaks, we investigated the possibility that it binds to the ends of our 25-mer probe rather than the platinum cross-link by preparing a site-specifically platinated 90-base dumbbell probe. PARP-1 can bind to the hairpin loops of dumbbell DNA, but in so doing only makes contact with the four adjacent base pairs.[32] In our probe, the platination site is 19 base pairs from the loops (Figure 1B) making it very unlikely that PARP-1 would be photo-cross-linked without binding at the site of platination. Photo-cross-linking experiments using the 90-base dumbbell probe confirmed that every protein identified using the 25-bp probe, including PARP-1, binds at the platinum site and not the DNA ends (Table 3). Several proteins were identified using this longer probe that were not found with the 25-bp duplex probe as discussed below.
The DNA-PK heterotrimer binds to the 90-base dumbbell probe
Experiments using the 90-base probe eliminated the possibility that photo-cross-linking was a consequence of protein binding bound to DNA ends except for Ku70 and Ku80 (Table 3), which interact with DNA double-strand breaks as well as loops[63] to form a complex that translocates along the DNA duplex.[64] DNA-bound Ku proteins also recruit DNA-PKcs,[65] and the Ku family are also known bind to cisplatin-modified DNA.[17] Investigating the specificity of the Ku proteins for platinum-modified DNA remains a challenge owing to these properties and, from the present studies, we are unable to conclude definitively that their capture in the photo-cross-linking studies is solely the consequence of platinum lesion recognition.
Proteins involved in nucleotide excision repair bind to the 90-base DNA probe
The DNA damage binding protein-1 (DDB-1), which is the 142-kDa subunit of the complex DDB, originally known as xeroderma pigmentosum complementation group E protein binding factor (XPE-BF), was photo-cross-linked by the 90-base probe. The cisplatin-modified DNA-binding ability of this protein has been demonstrated by using EMSAs with extracts from XPE cells, which contain mutated DDB-1.[20,66] This protein stimulates nucleotide excision repair (NER) of UV-damaged DNA,[67] although it is apparently not necessary for repair of a cisplatin 1,3-d(G*pTpG*) intrastrand adduct.[68] The role of DDB-1 in cisplatin-damaged DNA repair is not well understood; it may be one of the first NER proteins to bind to damaged DNA.[69]
The transcription factor YB-1, which was photo-cross-linked by the 90-base probe, binds to cisplatin-modified DNA.[70] The consequences of this binding may derive from the interaction of YB-1 with proliferating cell nuclear antigen (PCNA), a protein necessary for NER.[2,71] Another PCNA-interacting protein photo-cross-linked by our 90-mer probe is replication factor C (RFC).[69] Five RFC proteins are involved in DNA replication and repair.[72] RFC1 binds to double-stranded DNA and interacts with PCNA to initiate transcription.[72] A role for RFC1 in DNA repair is supported by experiments demonstrating that the protein is necessary to promote cell survival following cisplatin or UV exposure.[73] The protein RFC2 was also photo-cross-linked. RFC2 binds to damaged DNA downstream of damage recognition in the nucleotide excision repair pathway following excision of the damaged bases.[74,75] It may be responsible for keeping the damage response activated until repair is completed.[76]
Since RFC2 has been implicated in binding to damaged DNA after excision of a lesion, the integrity of the 90-base probe following incubation with nuclear extracts was investigated. After a two-hour period, the DNA was analyzed by PAGE, which indicated that Pt-BP6 was not excised (Figure S.7, Supporting Information). This result proves that RFC2 binds to platinum-damaged DNA prior to excision of the lesion. Our experiments also demonstrate that the dumbbell DNA structure is stable in nuclear extracts, consistent with other studies of this type of DNA structure.[77] The stability of dumbbell DNA structures in biological media has led to an investigation of their therapeutic potential as decoys for transcription factors.[78]
The photo-cross-linking of SPT16 gives structural information about the binding of the FACT complex to platinum-modified DNA
The SPT16 subunit of the FACT (FAcilitates Chromatin Transcription) complex increases the affinity of its other subunit, the HMG-domain protein SSRP1, for cisplatin-modified DNA.[79] Structural analysis of the HMG-domains of HMGB1 indicate that they domains bind to the widened minor groove of cisplatin-modified DNA in a hydrophobic notch opposite the lesion.[80–82] The photo-cross-linking of SPT16 but not SSRP1 suggests that the latter interacts mainly with DNA across from the platinum cross-link, that that SPT16 is directed toward the adduct. The photo-cross-linking of SPT16 demonstrates that it binds to platinum-modified DNA in the context of the nuclear milieu and is relevant to the processing of platinum adducts.
Chromatin remodeling proteins bind to platinum-modified 90-base dumbbell DNA
DNA topoisomerase I (topo I) binds to closed circular DNA and DNA-histone complexes containing cisplatin adducts.[83,84] Topo I relaxes superhelical DNA by severing one strand, crossing the unbroken strand through the break, and then re-ligating the broken strand. This function is necessary for transcription.[85] The stalling of topo I at DNA damage sites can interfere with repair, an effect blocked by poly(ADP-ribosyl)ation of topo I by PARP-1 and PARP-2, which dissociates the protein from DNA.[86] The combination of cisplatin and topo I inhibitors, such as camptothecin, has improved response in clinical trials.[87] The synergistic cell-killing by these two drugs is not well understood, but the effect only occurs in cells with a functional homologous recombination repair pathway.[88] The ability of topo I to bind non-nucleosomal platinum-modified DNA has not been established, but the current study demonstrates that the interaction will occur in nuclear extracts.
The SWI/SNF family proteins SMARCA3, SMARCA4, SMARCC2, and protein polybromo-1 (PB-1) were photo-cross-linked by the 90-base dumbbell probe. The acronym SMARC stands for SWI/SNF-related matrix-associated actin-dependent regulators of chromatin. These proteins are responsible for chromatin remodeling, which allows the transcription machinery to processes DNA that is tightly wrapped around histones.[89] One member of this family, PB-1, contains an HMG-domain,[90] which may bind to the platinum site and recruit the other SWI/SNF proteins. The affinity of PB-1 and the SWI/SNF family of proteins for platinum-modified DNA has not been demonstrated prior to the present work. The ability of these proteins to bind to platinum-modified DNA may inhibit excision of the adduct by NER proteins or hijack them from their native functions.
Pt-BP6-modified DNA is able to photo-cross-link proteins with low cellular abundance
The binding of proteins to DNA is an equilibrium process, but photo-cross-linking creates a covalent bond, which prevents proteins from dissociation. Due to this covalent linkage, any protein with an affinity for platinum-modified DNA will be photo-cross-linked by the probe. As few as 500 zeptomoles of trypsin-digested peptides can be detected by the Thermo Electron Model LTQ Ion Trap mass spectrometer used in our experiments.[91] In practice, the instrument used in these studies will detect sub-femtomole amounts of peptide in complex mixtures,[92] corresponding to fewer than 6.02 × 108 molecules. In our preparative-scale photo-cross-linking experiments, we used 2 mg of HeLa nuclear extracts. These extracts are collected from five 175 cm2 cell culture plates, each containing approximately 1.8 × 107 cells. If a protein were expressed at a level of only 10 copies per cell, there would be 9 × 108 copies of that protein present in our extracts. Although such poorly expressed proteins would have to compete for binding with other more abundant ones, photo-cross-linking will create an irreversible covalent bond between any protein having an affinity for the platinum-modified DNA and the probe. These considerations indicate that proteins expressed at a level as low as ten copies per cell should be detectable by our methods.
Several highly abundant proteins present at upwards of 105 molecules per cell were identified using Pt-BP6-modified DNA, including PARP-1[93] and the Ku proteins.[94] Other proteins identified are not nearly as abundant, such as the MMR proteins Msh2, Msh3, and Msh6, as well as the SWI/SNF family proteins, for which there are only 100–200 molecules per cell,[95,96] and DDB1, which is present at 8,000 molecules per cell.[97] The expression of HMGB3 in adult tissues was undetectable by RT-PCR,[37] but was identified using our methodology. The identification of these proteins demonstrates that these photo-cross-linking experiments provide a very sensitive method for detection of platinum-damaged DNA-binding proteins.
Conclusion
DNA duplexes site-specifically platinated with a cisplatin analog containing an amino ligand bearing a pendant photo-activatable moiety, Pt-BP6, were prepared to capture proteins with an affinity for Pt-modified DNA. Differences in photo-cross-linking between a 25-bp probe containing a 1,2-d(G*pG*) or 1,3-d(G*pTpG*) intrastrand cross-link reflect preferences previously reported for DNA adducts of cisplatin studied in isolation. For example, HMG-domain proteins strongly favor the 1,2-d(G*pG*) intrastrand cross-link whereas the nucleotide excision repair protein RPA1 prefers the 1,3-d(G*pTpG*) adduct. Similarly, a duplex containing the 1,2-d(G*pG*) adduct of Pt-BP6 carrying a mismatched base opposite the platination site has a greater affinity for the mismatch repair protein, Msh2. These results indicate that the Pt-BP6-DNA lesion is processed in manner analogous to cisplatin-DNA lesions and that recognition of damaged DNA is effective in the context of the nuclear environment. The Pt-BP6–DNA probe allowed us to identify PARP-1 binding specificity to Pt-modified DNA. PARP-1 is currently a subject of much scrutiny by cancer biologists, and the discovery that it binds to platinum-modified DNA warrants further attention. This protein is differentially expressed among the sampling of cancer cell lines routinely investigated in our laboratory and high levels of expression in testicular cancer cells lead to greater photo-cross-linking. Because of the affinity of some of the identified proteins for DNA ends, photo-cross-linking experiments were conducted with a site-specifically platinated 90-base dumbbell probe, in which the capture of most proteins bound to DNA ends is highly unlikely. Photo-cross-linking experiments with the 90-base probe identified several previously unrecognized platinum-DNA damage recognition proteins. Included are YB-1, DDB1, as well as several other proteins involved in nucleotide excision repair and chromatin remodeling. A numerical analysis reveals that this technology should be able to detect proteins expressed at very low levels that bind to platinum-DNA damage in cells. The ability of these proteins to bind to platinum-modified DNA suggests that further studies should be carried out to define the role of these proteins in the processing of DNA damage. The effect on cisplatin sensitivity of silencing or chemical inhibition of these proteins is also warranted.
Experimental Section
Materials
Solvents and chemical reagents were purchased from commercial sources. Potassium tetrachloroplatinate(II) was provided by Engelhard. DNA strands were synthesized on an Applied Biosystems ABI 392 DNA/RNA synthesizer using solvents and reagents supplied by Glen Research. Enzymes were purchased from New England BioLabs and Promega. UV-vis spectra were obtained on an HP 8453 UV-visible spectrometer. Platinum analyses were performed by flameless atomic absorption spectroscopy on a Perkin-Elmer AAnalyst 300 system. Analytical and preparative HPLC work was performed either on an HP Waters or an Agilent 1200 HPLC system. UV irradiation was conducted in a Stratagene Stratalinker UV Crosslinker. Protein digestion and analyses were performed by trypsin digestion-coupled LC-MS/MS analysis at the MIT Biopolymers facility. Antibodies used in western blot experiments were purchased from Axxora Life Sciences.
Synthesis of a 1,2-d(G*pG*) intrastrand Pt-BP6-modified 25-bp DNA
A 25-base-pair DNA duplex, site-specifically platinated with Pt-BP6, was synthesized as reported previously.[28] Briefly, cis-ammine(BP6)dichloroplatinum(II) (Scheme 1B) was activated by treatment with AgNO3 and allowed to react in a 2:1 molar ratio with a 25-base single stranded DNA containing one d(G*pG*) site, 5’-CCTCTCCTCTCAGGATCTTCTCTCC-biotin-TEG-3’. The singly-platinated 25-bp was then isolated by reverse-phase HPLC and characterized by UV-visible absorbance, and atomic absorption spectroscopy, and by ESI-MS.
Synthesis of a 1,3-d(G*pTpG*) intrastrand Pt-BP6-modified 25-base single stranded DNA
In a method similar to that used to prepare a DNA strand containing the 1,2-d(G*pG*) adduct, cis-ammine(BP6)dichloroplatinum(II) was activated with AgNO3. To a solution of a 25-base DNA (35 nmol) containing one d(G*pTpG*) site, 5’-CCTCTCCTCTCGTGATCTTCTCTCC-desthiobiotin-TEG-3’, in 10 mM NaH2PO4 (pH 6.3) was added activated Pt-BP6 (70 nmol). The reaction was incubated overnight at 37 °C with mixing. The singly-platinated 25-base DNA was then isolated by reverse-phase HPLC on an Agilent C18 300-SB column. The method used was 0 – 12% B over 5 min, followed by 12–25% over 25 min, where solvent A contained 95:5 water:acetonitrile, 0.1% trifluoroacetic acid, and solvent B contained 90:10 acetonitrile:water, 0.1% trifluoroacetic acid. The collected fractions were neutralized with dilute sodium hydroxide and lyophilized. The samples were then reconstituted in water and dialyzed to remove salt.
Characterization of 1,3-d(G*pTpG*) intrastrand Pt-BP6-modified 25-base DNA probes
Each collected peak was characterized by UV-visible absorbance and atomic absorption spectroscopy to establish a 1:1 ratio of platinum per DNA. The collected peaks were characterized by ESI-MS to confirm that both chloride ligands were replaced by Pt–DNA bonds. Platination of the guanine bases was verified by enzymatic digestion of the DNA. The digests consisted of 100 mM NaOAc (20 µL, pH 5.2), 100 mM ZnCl2 (1 µL), nuclease P1 (10 µL), and DNA (150 pmol). The total reaction volumes were increased to 100 µL with ddH2O. The solutions were mixed thoroughly and incubated at 37 °C overnight. Next, 1.5 M Tris-HCl (5 µL, pH 8.8) and calf intestinal phosphatase (1µL, CIP) were added. After a 4-h incubation at 37 °C, the samples were centrifuged at 5000 rpm for 5 min. The supernatants were analyzed by RP-HPLC using a Supelcosil LC-18-S column. The method was 5 – 15% B over 30 min, 15 – 80% B over 10 min, and 80% B for 5 min, where solvent A was 100 mM NaOAc, pH 5.2 and solvent B was methanol.
Synthesis of Pt-BP6-modified duplex 25-bp probes
For analytical-scale photo-cross-linking experiments, each platinated top strand (50 pmol) was annealed to an equimolar amount of the complementary bottom strand and radiolabeled by standard T4 PNK phosphorylation procedures. The duplex 25-bp was then isolated by 15% native PAGE using audioradiography to locate the radiolabeled DNA, which was subsequently extracted from the gel. The final product was then analyzed by 15% native PAGE. For preparative scale photo-cross-linking experiments, each platinated or unplatinated top strand (500 pmol or 1 nmol) was annealed to an equimolar amount of the complementary bottom strand. The double-stranded DNA was purified by 15% native PAGE and visualized by UV shadowing. Following excision of the desired band, the DNA was extracted from the gel and dialyzed to remove salt. The concentration of the DNA was then determined by UV-vis spectroscopy.
Design and synthesis of a 25-bp duplex containing a mismatched 1,2-d(G*pG*) intrastrand adduct of Pt-BP6
A single-stranded 25-base DNA of the sequence 5’-GGAGAGAAGATCTTGAGAGGAGAGG-3’ was prepared on a DNA synthesizer using standard phosphoramidite methods. The DNA was heated to 55 °C for 14 h and then purified by 12% denaturing PAGE. The DNA was located using UV shadowing and then extracted from the gel and dialyzed as described above. This DNA was then annealed to a complementary 25-base DNA containing a 1,2-d(G*pG*) intrastrand adduct of Pt-BP6 and the duplex was purified and used for analytical-scale photo-cross-linking experiments as described above.
Synthesis of a biotinylated 65-base DNA
DNA having the sequence 5’-CCACACCCTTTTGGGTGTGGGGAGAGAAGATCCTG-AGAGGAGAGGGCCGAGTT(biotin-T)TTTAACTCGGC-3’ was prepared at a 1 µmol scale. Biotin-T represents a commercially available biotin-modified thymine phosphoramidite (Glen Research). The DNA was purified by 6% urea-PAGE and visualized by UV shadowing and excised. The DNA was extracted from the gel and dialyzed versus water using dialysis tubing with a molecular weight cutoff of 3500 (Pierce).
Synthesis of site-specifically modified 90-base probe
For analytical-scale experiments, 100 pmol each of 25-Pt-BP6 and 65-biotin were radiolabeled using T4 PNK (New England Biolabs) and a mixture of unradiolabeled and γ-32P-ATP. The radiolabeled DNA was annealed in NEBuffer 3 (100 µL, New England BioLabs, 50 mM Tris-HCl pH 7.9, 100 mM NaCl, 10 mM MgCl2, 1 mM DTT) from 90 °C to 16 °C over 4 h. The DNA was ligated by the addition of T4 DNA ligase reaction buffer (100 µL; 50 mM Tris-HCl, 10 mM MgCl2, 1 mM ATP, 10 mM DTT pH 7.5, 400 units T4 DNA ligase) and incubated at 16 °C overnight. The ligated DNA was analyzed and then purified by 6% urea-PAGE. The bands were isolated by audioradiography and extracted from the gel. The DNA was then re-annealed and ethanol precipitated. For preparative-scale experiments, 2 nmol 65-biotin and 1 nmol each of platinated and unplatinated 25-base DNA were phosphorylated using T4 PNK and ATP (10 mM). The DNA was annealed and ligated in a method analogous to that used for the analytical-scale probes. The ligated DNA was purified by 6% urea-PAGE and the band was isolated by UV shadowing. The DNA was extracted from the gel and then reannealed and ethanol precipitated.
Characterization of site-specifically modified 90-base probe
To ensure that the collected product contained one 65-base DNA and one 25-base DNA, the preparative-scale probe was analyzed by nuclease digestion. Each digestion was performed with purified DNA (75 pmol). The DNA was digested with exonuclease III, T7 exonuclease, and mung bean nuclease for 1 h at 30 °C in NEBuffer 4 (60 µL; 50 mM potassium acetate, 20 mM tris-acetate, 10 mM magnesium acetate, 1 mM DTT pH 7.9). To the solution was then added 10X S1 nuclease buffer (10 µL; 500 mM sodium acetate pH 4.5, 2.8 M NaCl, 45 mM ZnSO4) and nuclease S1 (10 µL, Promega). The solutions were mixed thoroughly and incubated at 37 °C overnight. Next, 1.5 M Tris-HCl (5 µL, pH 8.8) and calf intestinal phosphatase (1 µL, CIP) were added. After a 4-h incubation at 37 °C, the samples were centrifuged at 5000 rpm for 5 min. The supernatants were analyzed by RP-HPLC using a Supelcosil LC-18-S column. The eluant was a gradient of 5 – 15% B over 30 min, 15 – 80% B over 10 min, and 80% B for 5 min, where solvent A was 100 mM NaOAc, pH 5.2 and solvent B was methanol. To examine whether the DNA was fully ligated, the analytical-scale probe was digested with mung bean nuclease, which cleaves the looped ends. The purity of the DNA was first analyzed by 6% urea-PAGE. As a control, a singly ligated 90-base probe was synthesized on the DNA synthesizer, purified, and radiolabeled as above. In an eppendorf tube, DNA (1 pmol) was incubated in MBN reaction buffer (50 mM sodium acetate, 150 mM NaCl, 1 mM ZnSO4, 5 units mung bean nuclease, pH 5.0) for 30 min at room temperature. The reactions were analyzed by 8% or 12% urea-PAGE.
Cell culture
Cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37 °C under 5% CO2. Once growth reached ~80% confluence, cells were trypsinized and passed or collected. Cells between passages 4 and 18 were collected and used for preparing nuclear extracts. Testicular (NTera2), cervical (HeLa), bone (U2OS), pancreatic (BxPC3), and HeLa cells in which PARP-1 was silenced by RNAi were used for these experiments.
Short hairpin RNA (shRNA)-induced RNAi on PARP-1 in HeLa cells
We designed several DNA sequences that transcribe to shRNAs for silencing PARP-1 in HeLa cells. These DNA constructs were cloned into the plasmid vector pcDNA3.1-Zeo (-) -U6. This plasmid, derived from pcDNA3.1-Zeo (-) (Invitrogen), has the human U6 promoter inserted upstream of the cloning site to drive shRNA transcription. The transfected HeLa cells were selected in DMEM with Zeocin (100 µg/ml). After Zeocin selection, cells were collected by trypsin digestion and their nuclear extracts were prepared as previously reported.[28] The total protein concentrations of the extracts were estimated by bicinchoninic acid solution (BCA) assay. For each sample, extracts with 20 µg of total protein were analyzed by western blot. With the use of a MoFlo high-performance cell sorter (DakoCytomation), HeLa cells transfected with the plasmid were sorted into wells of two 96-well plates. The instrument set up allows only 0.1 µl of diluted culture with no more than one cell being added to each well. The single clones were incubated at 37 °C for two weeks. During this time, the medium was changed every 4 d. These cells were collected by trypsin digestion and grown in 6-well plates. The nuclear extracts of survived clones were analyzed by western blot to evaluate the expression level of PARP-1.
Nuclear extraction
Nuclear extracts were prepared as reported previously.[28] In a typical extraction, a cell pellet from 5 × 175 cm2 plates was used. Nuclear extracts were quantified by using the BCA assay and stored at -80 °C. Between 0.5 – 1 mg of protein were obtained from each 175-cm2 plate. For preparative scale photo-cross-linking experiments, 4 mg of nuclear extracts were required and could be obtained from 4–8 × 175 cm2 plates…
Analytical scale photo-cross-linking
Established conditions for photo-cross-linking were used.[28] Briefly, radiolabeled DNA (1 pmol) was incubated with nuclear extracts (20 µg) in binding buffer (20 µL; 10 mM Tris-HCl pH 7.5, 10 mM MgCl2, 50 mM KCl, 1 mM EDTA, 0.05% NP-40, 0.2 µg/mL BSA). The reaction was incubated on ice under UV irradiation for 2 h. Irradiation was performed by using NEC FL8BL 8W bulbs, which provide a maximum at 360 nm with minimal output at 300 nm (http://www.nelt.co.jp/english/products/safl/). The latter wavelength could potentially labilize the platinum-DNA bonds. The samples were placed at a distance of 4 cm from the lamps. The reactions were analyzed by 10% SDS-PAGE with a 4% stacking gel for the 25-bp probe or 7.5% SDS-PAGE with a 4% stacking gel for the 90-base probe. The gel was dried and visualized using an Amersham Biosciences phosphorimager.
Preparative scale photo-cross-linking
To unradiolabeled DNA (100 pmol) in binding buffer (100 µL) was added HeLa nuclear extract (2 mg). The reaction mixtures were irradiated for 2 h on ice. To each reaction mixture was then added streptavidin-coated magnetic beads (5 mg, Dynabeads, Invitrogen), and the solutions were incubated with mixing for 20 min at 4 °C. To the solution was then added 2X elution buffer (400 µL; 50 mM Tris-HCl, 10 mM DTT, 1% SDS, pH 7.9) and the mixture was incubated with agitation for 20 min at 4 °C. The supernatant was then removed from the beads using a magnet. The beads were washed four times with high salt buffer (5 mM Tris-HCl, 500 µM EDTA, 1 M NaCl, pH 7.5), each time removing the supernatant using the magnet. The beads were then washed five times with 1X elution buffer, again each time removing the supernatant using the magnet. After the fifth elution buffer wash was removed, 2X SDS-PAGE loading buffer (40 µL; 100 mM Tris-HCl, 20 mM DTT, 2% SDS, 30% glycerol, 0.1% bromophenol blue, pH 7.9) was added to the solution. The solution was heated to 90 °C for 10 min to dissociate the biotinylated DNA from the beads and the supernatant was quickly removed using the magnet. The reactions were resolved by 10% SDS-PAGE with a 4% stacking gel for the 25-bp probe or 7.5% SDS-PAGE with a 4% stacking gel for the 90-base probe. Following electrophoresis, the gel was removed from the plates and the lanes containing the preparative scale photo-cross-linking products were separated from the rest of the gel using a blade. These lanes were then soaked in Coomassie Blue R-250 for 1 h and destained in 10% methanol, 7% glacial acetic acid, 83% ddH2O 2 times for 1 h each. The bands were located using the results from the radioactive analytical scale cross-linking experiment as a guide. Molecular weight ladders were run on both gels, such that the locations of each band on the gel could be precisely estimated prior to excision. Bands were excised from lanes containing Pt-BP6-platinated DNA as well as unplatinated DNA. The excised bands were analyzed by trypsin digest-coupled LC-MS/MS. The peptide fragments were then processed by SEQUEST protein analysis software to identify the parent proteins.[98] The proteins present in the lane containing the unplatinated DNA were eliminated from the list of those found in the lane with the Pt-BP6-platinated DNA. Proteins were accepted only if they occurred in two repetitions of the cross-linking experiments. In accordance with suggested guidelines for the identification of proteins by trypsin digest-coupled mass spectrometry,[99] the proteins identified were only accepted if meeting the criteria of low probability of coincidental identification (P < 1.0 × 10−3) and/or multiple unique matching peptides found. “Unique peptides” were defined as having entirely different sequences from one another; two peptides differing only by a modified amino acid were scored as being identical. Mass spectra from proteins identified by a single peptide match with good probability are available in the Supporting Information. Almost all of the proteins found in these experiments were identified by three or more peptides and two repetitions, and these results are presented with great confidence.
90-base probe repair assay
DNA (1 pmol) was incubated in binding buffer (20 µL) with nuclear extracts (20 µg) from HeLa, NTera2, BxPC3 U2OS, and HeLa YS cells. The reactions were incubated on ice for 2 h. A sample containing U2OS nuclear extracts was exposed to UV irradiation for 2 h to assess whether irradiation effects the stability of the DNA. The samples were phenol extracted and ethanol precipitated to remove proteins and concentrate the DNA. The samples were analyzed by 8% urea-PAGE.
Scheme 1.
A) Structure of cis-diamminedichloroplatinum(II), or cisplatin. B) Structure of the photoactive cisplatin analogue, Pt-BP6.
Acknowledgements
We thank Richard Cook, Alla Leshinsky, Rick Schiavoni, and Dr. Ioannis Papayannopoulos from the MIT Biopolymers and Proteomics laboratories for their assistance with trypsin digest-coupled mass spectrometry experiments and for helpful discussions. We acknowledge Carl Novina and Derek Dykxhoorn for constructing plasmid vector pcDNA3.1-Zeo (-) -U6 used for silencing PARP-1 in HeLa cells. We also thank members of our laboratory for many helpful suggestions and comments on the manuscript.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Horwich A, Shipley J, Huddart R. Lancet. 2006;367:754–765. doi: 10.1016/S0140-6736(06)68305-0. [DOI] [PubMed] [Google Scholar]
- 2.Jamieson ER, Lippard SJ. Chem. Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 3.Cohen GL, Ledner JA, Bauer WR, Ushay HM, Caravana C, Lippard SJ. J. Am. Chem. Soc. 1980;102:2487–2488. [Google Scholar]
- 4.Fitchtinger-Schepman AMJ, van der Veer JL, den Hartog JHJ, Lohman PHM, Reedijk J. Biochemistry. 1985;24:707–713. doi: 10.1021/bi00324a025. [DOI] [PubMed] [Google Scholar]
- 5.Rice JA, Crothers DM, Pinto AL, Lippard SJ. Proc. Natl. Acad. Sci. U.S.A. 1988;85:4158–4161. doi: 10.1073/pnas.85.12.4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jung Y, Lippard SJ. Chem. Rev. 2007;107:1387–1407. doi: 10.1021/cr068207j. [DOI] [PubMed] [Google Scholar]
- 7.Zlatanova J, Yaneva J, Leuba SH. FASEB J. 1998;12:791–799. doi: 10.1096/fasebj.12.10.791. [DOI] [PubMed] [Google Scholar]
- 8.Pil PM, Lippard SJ. Science. 1992;256:234–237. doi: 10.1126/science.1566071. [DOI] [PubMed] [Google Scholar]
- 9.Hughes EN, Engelsberg BN, Billings PC. J. Biol. Chem. 1992;267:13520–13527. [PubMed] [Google Scholar]
- 10.Toney JH, Donahue BA, Kellett PJ, Bruhn SL, Essigmann JM, Lippard SJ. Proc. Natl. Acad. Sci. U.S.A. 1989;86:8328–8332. doi: 10.1073/pnas.86.21.8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Treiber DK, Zhai X, Jantzen H-M, Essigmann JM. Proc. Natl. Acad. Sci. U.S.A. 1994;91:5672–5676. doi: 10.1073/pnas.91.12.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chu G. J. Biol. Chem. 1994;269:787–790. [PubMed] [Google Scholar]
- 13.Saldivar JS, Wu X, Follen M, Gershenson D. Gynecol. Oncol. 2007;107:S56–S71. doi: 10.1016/j.ygyno.2007.07.043. [DOI] [PubMed] [Google Scholar]
- 14.Zdraveski ZZ, Mello JA, Marinus MG, Essigmann JM. Chem. Biol. 2000;7:39–50. doi: 10.1016/s1074-5521(00)00064-8. [DOI] [PubMed] [Google Scholar]
- 15.Chan N, Koritzinsky M, Zhao H, Bindra R, Glazer PM, Powell S, Belmaaza A, Wouters B, Bristow RG. Cancer Res. 2008;68:605–614. doi: 10.1158/0008-5472.CAN-07-5472. [DOI] [PubMed] [Google Scholar]
- 16.Lin X, Kim H-K, Howell SB. J. Inorg. Biochem. 1999;77:89–93. doi: 10.1016/s0162-0134(99)00134-8. [DOI] [PubMed] [Google Scholar]
- 17.Turchi JJ, Henkels K. J. Biol. Chem. 1996;271:13861–13867. doi: 10.1074/jbc.271.23.13861. [DOI] [PubMed] [Google Scholar]
- 18.Jackson SP, Jeggo PA. Trends Biochem. Sci. 1995;20:412–415. doi: 10.1016/s0968-0004(00)89090-8. [DOI] [PubMed] [Google Scholar]
- 19.Chaney SG, Sancar A. J. Natl. Cancer Inst. 1996;88:1346–1360. doi: 10.1093/jnci/88.19.1346. [DOI] [PubMed] [Google Scholar]
- 20.Chu G, Chang E. Science. 1988;242:564–567. doi: 10.1126/science.3175673. [DOI] [PubMed] [Google Scholar]
- 21.Bruhn SL, Pil PM, Essigmann JM, Housman DE, Lippard SJ. Proc. Natl. Acad. Sci. U.S.A. 1992;89:2307–2311. doi: 10.1073/pnas.89.6.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.CC-K Chao. Mutat. Res. 1991;264:59–66. doi: 10.1016/0165-7992(91)90046-7. [DOI] [PubMed] [Google Scholar]
- 23.Turchi JJ, Henkels KM, Hermanson IL, Patrick SM. J. Inorg. Biochem. 1999;77:83–87. doi: 10.1016/s0162-0134(99)00145-2. [DOI] [PubMed] [Google Scholar]
- 24.Kane SA, Lippard SJ. Biochemistry. 1996;35:2180–2188. doi: 10.1021/bi952240a. [DOI] [PubMed] [Google Scholar]
- 25.Mikata Y, He Q, Lippard SJ. Biochemistry. 2001;40:7533–7541. doi: 10.1021/bi010318j. [DOI] [PubMed] [Google Scholar]
- 26.Chálová K, Brabec V, Kašpárková J. Nucleic Acids Res. 2007;35:1812–1821. doi: 10.1093/nar/gkm032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buterin T, Meyer C, Giese B, Naegeli H. Chem. Biol. 2005;12:913–922. doi: 10.1016/j.chembiol.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 28.Zhang CX, Chang PV, Lippard SJ. J. Am. Chem. Soc. 2004;126:6536–6537. doi: 10.1021/ja049533o. [DOI] [PubMed] [Google Scholar]
- 29.Dormán G, Prestwich GD. Biochemistry. 1994;33:5661–5673. doi: 10.1021/bi00185a001. [DOI] [PubMed] [Google Scholar]
- 30.Walker JR, Corpina RA, Goldberg J. Nature. 2001;412:607–614. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- 31.Kim MY, Zhang T, Kraus WL. Genes Dev. 2005;19:1951–1967. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- 32.Potaman VN, Shlyakhtenko LS, Oussatcheva EA, Lyubchenko YL, Soldatenkov VA. J. Mol. Biol. 2005;348:609–615. doi: 10.1016/j.jmb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 33.Hartwig JF, Lippard SJ. J. Am. Chem. Soc. 1992;114:5646–5654. [Google Scholar]
- 34.Andrus A, Kuimelis RG. Unit 10.3: Overview of Purification and Analysis of Syntheic Nucleic Acids. In: Beaucage SL, editor. Current Protocols in Nucleic Acid Chemistry. New York City: John Wiley & Sons; 2000. pp. 10.13.11–10.13.16. [DOI] [PubMed] [Google Scholar]
- 35.Ciccarelli RB, Solomon MJ, Varshavsky A, Lippard SJ. Biochemistry. 1985;24:7533–7540. doi: 10.1021/bi00347a005. [DOI] [PubMed] [Google Scholar]
- 36.Thomas JO, Travers AA. Trends Biochem. Sci. 2001;26:167–174. doi: 10.1016/s0968-0004(01)01801-1. [DOI] [PubMed] [Google Scholar]
- 37.Vaccari T, Beltrame M, Ferrari S, Bianchi ME. Genomics. 1998;49:247–252. doi: 10.1006/geno.1998.5214. [DOI] [PubMed] [Google Scholar]
- 38.Helleman J, Jansen MPHM, Span PN, van Staveren IL, Massuger LFAG, Meijer-van Gelder ME, Sweep FCGJ, Ewing PC, van der Burg MEL, Stoter G, Nooter K, Berns EMJJ. Int. J. Cancer. 2006;118:1963–1971. doi: 10.1002/ijc.21599. [DOI] [PubMed] [Google Scholar]
- 39.Patrick SM, Turchi JJ. J. Biol. Chem. 1999;274:14972–14978. doi: 10.1074/jbc.274.21.14972. [DOI] [PubMed] [Google Scholar]
- 40.Huang JC, Zamble DB, Reardon JT, Lippard SJ, Sancar A. Proc. Natl. Acad. Sci. U.S.A. 1994;91:10394–10398. doi: 10.1073/pnas.91.22.10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fourrier L, Brooks P, Malinge J-M. J. Biol. Chem. 2003;278:21267–21275. doi: 10.1074/jbc.M301390200. [DOI] [PubMed] [Google Scholar]
- 42.Schofield MJ, Hsieh P. Annu. Rev. Microbiol. 2003;57:579–608. doi: 10.1146/annurev.micro.57.030502.090847. [DOI] [PubMed] [Google Scholar]
- 43.Fink D, Zheng H, Nebel S, Norris PS, Aebi S, Lin T-P, Nehme A, Christen RD, Haas M, Macleod CL, Howell SB. Cancer Res. 1997;57:1841–1845. [PubMed] [Google Scholar]
- 44.Fink D, Nebel S, Aebi S, Zheng H, Cenni B, Nehmé A, Christen RD, Howell SB. Cancer Res. 1996;56:4881–4886. [PubMed] [Google Scholar]
- 45.Meyers M, Hwang A, Wagner MW, Boothman DA. Environ. Mol. Mutagen. 2004;44:249–264. doi: 10.1002/em.20056. [DOI] [PubMed] [Google Scholar]
- 46.Chao EC, Lipkin SM. Nucleic Acids Res. 2006;34:840–852. doi: 10.1093/nar/gkj489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Duckett DR, Drummond JT, Murchie AIH, Reardon JT, Sancar A, Lilley DMJ, Modrich P. Proc. Natl. Acad. Sci. U.S.A. 1996;93:6443–6447. doi: 10.1073/pnas.93.13.6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Acharya S, Wilson T, Gradia S, Kane MF, Guerrette S, Marsischky GT, Kolodner R, Fishel R. Biochemistry. 1996;93:13629–13624. doi: 10.1073/pnas.93.24.13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoffmann J-S, Pillaire M-J, Garcia-Estefania D, Lapalu S, Villani G. J. Biol. Chem. 1996;271:15386–15392. doi: 10.1074/jbc.271.26.15386. [DOI] [PubMed] [Google Scholar]
- 50.Carr JL, Tingle MD, McKeage MJ. Cancer Chemother. Pharmacol. 2002;50:9–15. doi: 10.1007/s00280-002-0462-2. [DOI] [PubMed] [Google Scholar]
- 51.Zhang S, Lovejoy KS, Shima JE, Lagpacan LL, Shu Y, Lapuk A, Chen Y, Komori T, Gray JW, Chen X, Lippard SJ, Giacomini KM. Cancer Res. 2006;66:8847–8857. doi: 10.1158/0008-5472.CAN-06-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bouchard VJ, Rouleau M, Poirier GG. Exp. Hematol. 2003;31:446–454. doi: 10.1016/s0301-472x(03)00083-3. [DOI] [PubMed] [Google Scholar]
- 53.Audebert M, Salles B, Calsou P. J. Biol. Chem. 2004;279:55117–55126. doi: 10.1074/jbc.M404524200. [DOI] [PubMed] [Google Scholar]
- 54.Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. Nucleic Acids Res. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu S-W, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL. Proc. Natl. Acad. Sci. U.S.A. 2006;103:18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Amaravadi RK, Thompson CB. Clin. Cancer Res. 2007;13:7271–7279. doi: 10.1158/1078-0432.CCR-07-1595. [DOI] [PubMed] [Google Scholar]
- 57.Schreiber V, Dantzer F, Amé J-C, de Murcia G. Nat. Rev. Mol. Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 58.Shiokawa M, Masutani M, Fujihara H, Ueki K, Nishikawa R, Sugimura T, Kubo H, Nakagama H. Jpn. J. Clin. Oncol. 2005;35:97–102. doi: 10.1093/jjco/hyi028. [DOI] [PubMed] [Google Scholar]
- 59.Miknyoczki SJ, Jones-Bolin S, Pritchard S, Hunter K, Zhao H, Wan W, Ator M, Bihovsky R, Hudkins R, Chatterjee S, Klein-Szanto A, Dionne C, Ruggeri B. Mol. Cancer. Ther. 2003;2:371–382. [PubMed] [Google Scholar]
- 60.Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, Bontcheva-Diaz VD, Cox BF, DeWeese TL, Dillehay LE, Ferguson DC, Ghoreishi-Haack NS, Grimm DR, Guan R, Han EK, Holley-Shanks RR, Hristov B, Idler KB, Jarvis K, Johnson EF, Kleinberg LR, Klinghofer V, Lasko LM, Liu X, Marsh KC, McGonigal TP, Meulbroek JA, Olson AM, Palma JP, Rodriguez LE, Shi Y, Stavropoulos JA, Tsurutani AC, Zhu G-D, Rosenberg SH, Giranda VL, Frost DJ. Clin. Cancer Res. 2007;13:2728–2739. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- 61.Bernges F, Zeller WJ. J. Cancer Res. Clin. Oncol. 1996;122:665–670. doi: 10.1007/BF01209029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. Nat. Rev. Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 63.Arosio D, Cui S, Ortega C, Chovanec M, Marco SD, Baldini G, Falaschi A, Vindigni A. J. Biol. Chem. 2002;277:9741–9748. doi: 10.1074/jbc.M111916200. [DOI] [PubMed] [Google Scholar]
- 64.Turchi JJ, Henkels KM, Zhou Y. Nucleic Acids Res. 2000;28:4634–4641. doi: 10.1093/nar/28.23.4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pawelczak KS, Andrews BJ, Turchi JJ. Nucleic Acids Res. 2005;33:152–161. doi: 10.1093/nar/gki157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chu G, Chang E. Proc. Natl. Acad. Sci. U.S.A. 1990;87:3324–3327. doi: 10.1073/pnas.87.9.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wakasugi M, Kawashima A, Morioka H, Linn S, Sancar A, Mori T, Nikaido O, Matsunaga T. J. Biol. Chem. 2002;277:1637–1640. doi: 10.1074/jbc.C100610200. [DOI] [PubMed] [Google Scholar]
- 68.Moggs JG, Yarema KJ, Essigmann JM, Wood RD. J. Biol. Chem. 1996;271:7177–7186. doi: 10.1074/jbc.271.12.7177. [DOI] [PubMed] [Google Scholar]
- 69.Gillet LCJ, Schärer OD. Chem. Rev. 2006;106:253–276. doi: 10.1021/cr040483f. [DOI] [PubMed] [Google Scholar]
- 70.Ise T, Nagatani G, Imamura T, Kato K, Takano H, Nomoto M, Izumi H, Ohmori H, Okamoto T, Ohga T, Uchiumi T, Kuwano M, Kohno K. Cancer Res. 1999;59:342–346. [PubMed] [Google Scholar]
- 71.Kartalou M, Essigmann JM. Mutat. Res. 2001;478:23–43. doi: 10.1016/s0027-5107(01)00141-5. [DOI] [PubMed] [Google Scholar]
- 72.Mossi R, Hübscher U. Eur. J. Biochem. 1998;254:209–216. [PubMed] [Google Scholar]
- 73.Pennaneach V, Salles-Passador I, Munshi A, Brickner H, Regazzoni K, Dick F, Dyson N, Chen T-T, Wang JYJ, Fotedar R, Fotedar A. Mol. Cell. 2001;7:715–727. doi: 10.1016/s1097-2765(01)00217-9. [DOI] [PubMed] [Google Scholar]
- 74.Rechkunova NI, Maltseva EA, Lavrik OI. Mol. Biol. 2008;42:20–26. [Google Scholar]
- 75.Friedberg EC. Nat. Rev. Cancer. 2001;1:22–33. doi: 10.1038/35094000. [DOI] [PubMed] [Google Scholar]
- 76.Zhou B-BS, Elledge S. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 77.Chu BCF, Orgel LE. Nucleic Acids Res. 1992;20:5857–5858. doi: 10.1093/nar/20.21.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hosoya T, Takeuchi H, Kanesaka Y, Yamakawa H, Miyano-Kurosaki N, Takai K, Yamamoto N, Takaku H. FEBS Lett. 1999;461:136–140. doi: 10.1016/s0014-5793(99)01450-7. [DOI] [PubMed] [Google Scholar]
- 79.Yarnell AT, Oh S, Reinberg D, Lippard SJ. J. Biol. Chem. 2001;278:25736–25741. doi: 10.1074/jbc.M101208200. [DOI] [PubMed] [Google Scholar]
- 80.He Q, Ohndorf U-M, Lippard SJ. Biochemistry. 2000;39:14426–14435. doi: 10.1021/bi001700j. [DOI] [PubMed] [Google Scholar]
- 81.Takahara PM, Rosenzweig AC, Frederick CA, Lippard SJ. Nature. 1995;377:649–652. doi: 10.1038/377649a0. [DOI] [PubMed] [Google Scholar]
- 82.Ohndorf U-M, Rould MA, He Q, Pabo CO, Lippard SJ. Nature. 1999;399:708–712. doi: 10.1038/21460. [DOI] [PubMed] [Google Scholar]
- 83.Kobayashi S, Furukawa M, Dohi C, Hamashima H, Arai T, Tanaka A. Chem. Pharm. Bull. 1999;47:783–790. doi: 10.1248/cpb.47.783. [DOI] [PubMed] [Google Scholar]
- 84.Kobayashi S, Nakagawa S, Uehara N, Hamashima H, Tanaka A. Nucleic Acids Symp. Ser. 2002;2:283–284. doi: 10.1093/nass/2.1.283. [DOI] [PubMed] [Google Scholar]
- 85.Wang JC. Ann. Rev. Biochem. 1996;65:635–692. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- 86.Malanga M, Althaus FR. J. Biol. Chem. 2004;279:5244–5248. doi: 10.1074/jbc.C300437200. [DOI] [PubMed] [Google Scholar]
- 87.Gelderblom HAJ, de Jonge MJA, Sparreboom A, Verweij J. Invest. New Drugs. 1999;17:401–415. doi: 10.1023/a:1006394610219. [DOI] [PubMed] [Google Scholar]
- 88.van Waardenburg RCAM, de Jong LA, van Delft F, van Eijndhoven MAJ, Bohlander M, Bjornsti M-A, Brouwer J, Schellens JHM. Mol. Cancer Ther. 2004;3:393–402. [PubMed] [Google Scholar]
- 89.Saha A, Wittmeyer J, Cairns BR. Nat. Rev. Mol. Cell Biol. 2006;7:437–447. doi: 10.1038/nrm1945. [DOI] [PubMed] [Google Scholar]
- 90.Štros M, Launholt D, Grasser KD. Cell Mol. Life Sci. 2007;64:2590–2606. doi: 10.1007/s00018-007-7162-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mayya V, Rezaul K, Cong Y-S, Han D. Mol. Cell. Proteomics. 2005;4:214–223. doi: 10.1074/mcp.T400015-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Papayannopoulos IA. Personal Communication [Google Scholar]
- 93.D'Amours D, Desnoyers S, D'Silva I, Poirier GG. Biochem. J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- 94.Lieber MR, Ma Y, Pannicke U, Schwarz K. Nat. Rev. Mol. Cell Biol. 2003;4:712–720. doi: 10.1038/nrm1202. [DOI] [PubMed] [Google Scholar]
- 95.Feng G, Tsui H-CT, Winkler ME. J. Bacteriol. 1996;178:2388–2396. doi: 10.1128/jb.178.8.2388-2396.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cairns BR, Lorch Y, Li Y, Zhang M, Lacomis L, Erdjument-Bromage H, Tempst P, Du J, Laurent B, Kornberg RD. Cell. 1996;87:1249–1260. doi: 10.1016/s0092-8674(00)81820-6. [DOI] [PubMed] [Google Scholar]
- 97.Precious BL, Carlos TS, Goodbourn S, Randall RE. Virology. 2007;368:114–121. doi: 10.1016/j.virol.2007.06.024. [DOI] [PubMed] [Google Scholar]
- 98.MacCoss MJ, Wu CC, Yates JRI. Anal. Chem. 2002;74:5593–5599. doi: 10.1021/ac025826t. [DOI] [PubMed] [Google Scholar]
- 99.Carr S, Aebersold R, Baldwin M, Burlingame A, Clauser K, Nesvizhskii A. Mol. Cell. Proteomics. 2004;3:531–533. doi: 10.1074/mcp.T400006-MCP200. [DOI] [PubMed] [Google Scholar]
- 100.Robert J. Personal Communication. 2007 [Google Scholar]
- 101.Robert J, Laurand A, Meynard D, Le Morvan V. In Platinum drugs and DNA repair. Lessons from the NCI-60 panel and clinical correlates. 10th International Symposium on Platinum Coordination Compounds in Cancer Chemotherapy; Italy: Verona; 2007. [Google Scholar]
- 102.Collis SJ, DeWeese TL, Jeggo PA, Parker AR. Oncogene. 2005;24:949–961. doi: 10.1038/sj.onc.1208332. [DOI] [PubMed] [Google Scholar]