

Abstract
No animal models replicate the complexity of human depression. However, a number of behavioral tests in rodents are sensitive to antidepressants and may thus tap important underlying biological factors. Such models may also offer the best opportunity to discover novel treatments. Here, we used several of these models to test the hypothesis that the acid-sensing ion channel-1a (ASIC1a) might be targeted to reduce depression. Genetically disrupting ASIC1a in mice produced antidepressant-like effects in the forced swim test, the tail suspension test, and following unpredictable mild stress. Pharmacologically inhibiting ASIC1a also had antidepressant-like effects in the forced swim test. The effects of ASIC1a disruption in the forced swim test were independent of and additive to those of several commonly used antidepressants. Furthermore, ASIC1a disruption interfered with an important biochemical marker of depression, the ability of stress to reduce BDNF in the hippocampus. Restoring ASIC1a to the amygdala of ASIC1a−/− mice with a viral vector reversed the forced swim test effects, suggesting that the amygdala is a key site of ASIC1a action in depression-related behavior. These data are consistent with clinical studies emphasizing the importance of the amygdala in mood regulation, and suggest that ASIC1a antagonists may effectively combat depression.
Introduction
Depression remains one of the most disabling medical diseases but the molecular pathways underlying depression are poorly understood, and existing treatments are too often ineffective. Although current therapies reduce depression, they fail to resolve symptoms completely in as many as 50% of cases (Berton and Nestler, 2006). Remission rates are even worse for those who have failed initial medication trials (Rush et al., 2006). Thus, novel treatments are critically needed.
The causes of depression and the mechanisms of action of antidepressants remain poorly understood. Nevertheless, several advances provide a foundation for current understanding of neuroanatomical and neurochemical factors in mood regulation. Multiple brain regions have been implicated, including prefrontal cortex, cingulate, striatum, thalamus, and limbic structures (Soares and Mann, 1997). The amygdala is interconnected with many of these regions and well positioned to play a central role in mood regulation. Given that the amygdala is also critical for anxiety (Bechara et al., 1995; Kent and Rauch, 2003; Rauch et al., 2003), it is notable that depression is frequently accompanied by anxiety and panic (Kessler et al., 1998).
A landmark in understanding depression and treatment came with the discovery of the importance of monoamines (Sulser et al., 1962; Schildkraut, 1995). Monoamine depletion precipitates depression in susceptible individuals (Young et al., 1985; Benkelfat et al., 1994) and elevating monoamines is the principle cited mechanism of antidepressant action (Sulser et al., 1962; Berton and Nestler, 2006). Another important advance came with the recognition that stress plays a critical role (Selye, 1955; Kessler, 1997) and that the hypothalamic–pituitary–adrenal (HPA) axis is dysregulated in depression (Carroll et al., 1976; Stokes, 1995). More recently, neurotrophic factors and neuroplasticity have been implicated (Duman, 2002; Nestler et al., 2002). These foundations provide important insight into depression, and have intensified the pursuit of new molecular targets and therapies (Holtzheimer and Nemeroff, 2006).
In this study, we explored the contribution of the acid-sensing ion channel-1a (ASIC1a) in depression-related behavior. ASIC1a is a member of the Degenerin/Epithelial Na+ Channel family (Waldmann et al., 1997; Welsh et al., 2002; Wemmie et al., 2006, 2008). ASIC1a is required for acid-evoked currents in central neurons, where it contributes to synaptic plasticity (Wemmie et al., 2002; Cho and Askwith, 2008) and in the regulation of dendritic spines (Zha et al., 2006). ASIC1a is expressed widely in the central and peripheral nervous systems and is robustly expressed in structures associated with mood including the amygdala, bed nucleus of the stria terminalis, cingulate cortex and nucleus accumbens (Wemmie et al., 2003; Coryell et al., 2007). Consistent with this pattern of localization, previous studies suggested that disrupting ASIC1a attenuates amygdala activity (Wemmie et al., 2003; Coryell et al., 2007) and ASIC1a knock-out mice exhibited deficits in conditioned and unconditioned fear (Wemmie et al., 2003; Coryell et al., 2007). Conversely, overexpressing ASIC1a in transgenic mice increased fear conditioning (Wemmie et al., 2004). The role of ASIC1a in neuron plasticity, its distribution in the limbic circuit, and its effects on anxiety-related behavior, suggested the hypothesis that targeting ASIC1a might reduce depression-related behavior.
Materials and Methods
Sucrose preference and chronic mild stress.
Sucrose preference was assessed in individually housed, stressed and unstressed mice (Pothion et al., 2004). Briefly, two bottles (one 8% sucrose and one water) were placed in each cage for 3 d; 2 d to habituate and 1 d to assess sucrose preference. The locations of the bottles were rotated each 24-h period. At the beginning and end of the final 12-h period both bottles were weighed, and sucrose preference was defined as sucrose solution consumed/total liquid consumed × 100%. Unstressed mice only underwent the 3 d sucrose preference assessment. Stressed mice also underwent 6 d of unpredictable stressors in the following order: restraint (3 h), wetted bedding (12 h), restraint (3 h), 45° angle cage tilt (12 h), paired housing with an unfamiliar mouse (1 h), exposure to a predator odor (1 h) (trimethylthiazoline, Pherotech International). Sucrose preference was then assessed during the final overnight period as described above.
Tail suspension test.
This test was performed by the Tennessee Mouse Genome Consortium (http://www.tnmouse.org/neuromutagenesis/behavioral.html). Mice were suspended by the tail from a ledge ∼60 cm from the ground with adhesive tape. Immobility was defined as the absence of movement and was scored over a 6–min trial.
Forced swim test.
Mice were placed in a 4000 ml beaker filled with 3500 ml of water (25°C) and video taped during a 6 min trial. Immobility time was scored by a blinded experimenter and was defined as absence of motion except that required to keep head above water (Heinrichs and Koob, 2005). Fluoxetine (3 mg/kg, i.p.), desipramine (2 mg/kg, i.p.), and bupropion (2 mg/kg, i.p.) were each delivered 30 min before the forced swim test (FST). P-chlorophenylalanine methyl ester (PCPA; 300 mg/kg, i.p.) was injected daily for 3 d before the FST (Heurteaux et al., 2006). PcTx venom, PcTx1 peptide, and A-317567 were injected by intracerebroventricular cannula (described below).
Whole-cell voltage-clamp recordings.
Cortical neurons were obtained from 1–2 d old pups and cultured for 8–14 d as described (Wemmie et al., 2003; Askwith et al., 2004). Whole-cell voltage-clamp recordings were obtained at 20−23°C using an Axopatch 200B amplifier and Clampex 8.2 (Axon Instruments) sampled at 200 μs interval and filtered at 2 kHz. Extracellular pH was changed with a rapid solution changer (RSC-200; Biologic). Membrane voltage was maintained at −70 mV. Bath solutions contained (in mm): 100 NaCl, 5.4 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 MES, and pH was adjusted with TMA·OH. Patch pipettes (3–5 MΩ) contained (in mm): 10 NaCl, 70 K-gluconate, 10 KCl, 1 MgCl2, 10 EGTA, 25 HEPES, and 3 Na2ATP, adjusted to pH 7.3 with KOH. A-317567 (kindly provided by Drs. Alan Light and Jon Rainier, University of Utah, Salt Lake City, UT) was suspended in 5% DMSO in deionized water for a 10 mm stock and diluted in the bath solutions to a final concentration of 45 μm.
Intracerebroventricular surgeries and injections.
Intracerebroventricular guide cannulae were implanted into the left lateral ventricle of anesthetized mice (0.3 mm caudal, 1.0 mm lateral, 3.0 mm ventral with respect to bregma) (Coryell et al., 2007). Cannulae were fixed to the skull with dental cement and an anchoring screw. Two to four days later, 5 μl PcTx venom (9 ng/μl-24 h before testing), PcTx1 peptide (100 nm-90 min before testing), or A-317567 (1 mm-90 min before testing) in sterile artificial CSF (ACSF) (in mm: NaCl 124, KCL 3, NaH2PO4 1.2, MgSO4 1.2, CaCl2 2, NaHCO3 26) or ACSF alone was injected by hand (over 60 s) using a 10 μl Hamilton syringe and PE10 tubing connected to a 30-gauge stainless steel injector. Mice were returned to the home cage until behavioral testing. Animals with misplaced or plugged cannula were excluded from the analyses.
Corticosterone radioimmunoassay.
Blood was collected via submandibular gland bleed. For stress measurements, collections occurred 30 min following swim stress (10 min), a described by others (Solich et al., 2008). For baseline measurements samples were taken from mice in the home cage. All collections occurred between 10:00–11:00 A.M. Serum was isolated and corticosterone levels were determined using a radioimmunoassay (MP Biomedical).
RNA isolation and real-time PCR.
Immediately following 3–hr restraint stress (Smith et al., 1995), mice were killed, brains were removed and hippocampus was isolated frozen on dried ice and stored at −80°C until analysis. RNA was isolated using the RNAeasy lipid tissue mini kit (Qiagen). RNA (2 μg) was converted to cDNA by reverse transcription with random hexomers. TaqMan Gene expression assay Mm00432069 m1 for the exon 1–2 boundary was used quantify BDNF transcript normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Applied Biosystems). Reactions were performed in triplicate and assessed using the ABI PRISM 7700 fast sequence detection system.
Production of adeno-associated virus vectors.
Viral vectors were produced by the University of Iowa Gene Transfer Vector Core as described previously (Davidson et al., 2000). The vectors were AAV2/1 chimeric viruses with AAV1 capsids and AAV2 ITRs and contained a CMV promoter driving either ASIC1a or enhanced green fluorescent protein (eGFP). To facilitate the screening of AAV-ASIC1a transduced brain regions we inserted an internal ribosomal re-entry site (IRES) followed by eGFP, downstream from ASIC1a in the AAV-ASIC1a vector; however this produced no detectable eGFP expression (data not shown). Therefore, AAV-eGFP with AAV-ASIC1a vectors were injected together in the experimental group (see below). The AAV-eGFP produced no behavioral effects when injected alone (see Fig. 6 B).
Figure 6.
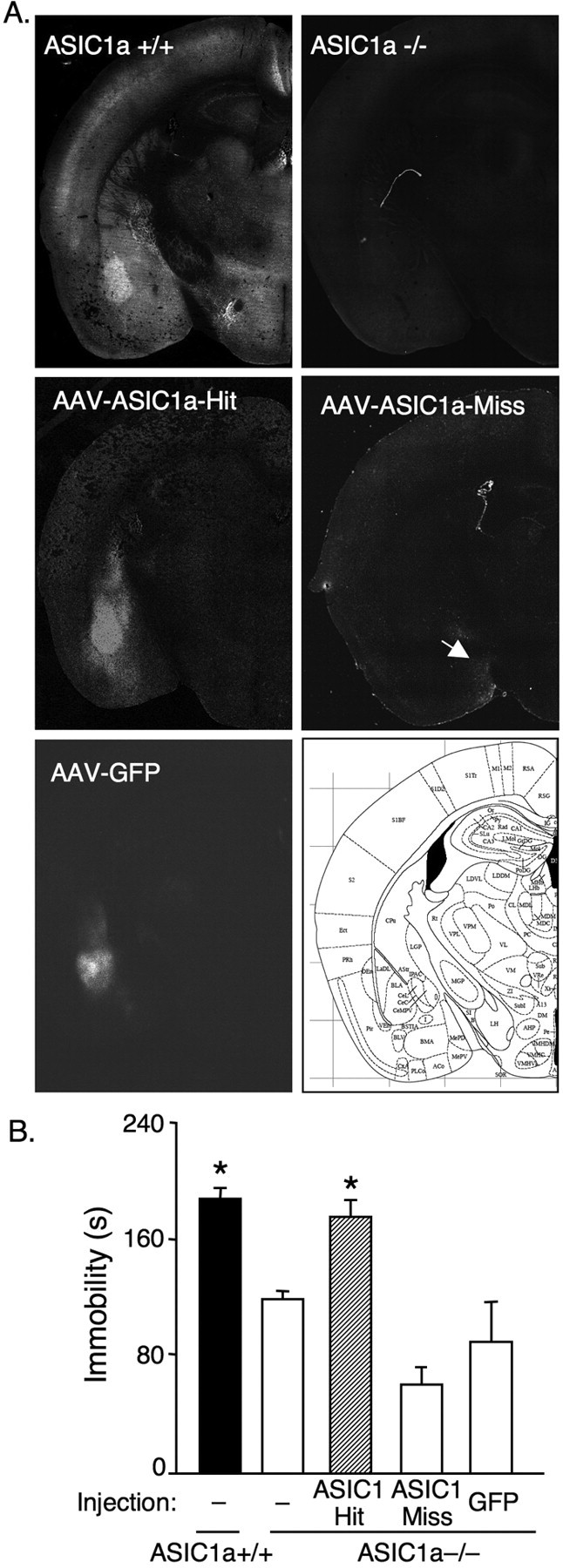
Restoring ASIC1a expression to the basolateral amygdala of ASIC1a−/− mice normalizes depression-related behavior. A, AAV-mediated gene expression. ASIC1 immunofluorescent labeling in an uninjected ASIC1a+/+ mouse, an uninjected ASIC1a−/− mouse, and an ASIC1a−/− mouse injected with AAV-ASIC1a in the basolateral amygdala (AAV-ASIC1a-Hit) and one that missed the basolateral amygdala (AAV-ASIC1a-Miss; arrow indicates viral transduction in the extreme edge of the medial amygdaloid nucleus). Also shown is an ASIC1a−/− mouse with AAV-GFP injected into the BLA. B, Targeting ASIC1a expression to the amygdala of ASIC1a−/− mice with AAV-ASIC1a significantly increased immobility in the forced swim test [uninjected ASIC1a+/+ (n = 11), uninjected ASIC1a−/− (n = 9), AAV-ASIC1a-Hit (n = 7), AAV-ASIC1a-Miss (n = 6), AAV-GFP (n = 6)]. One-way ANOVA revealed a significant effect of group (p < 0.001, F (4,34) = 19.3). Planned contrast testing found that restoring ASIC1a expression to the BLA increased immobility in ASIC1a−/− mice relative to the other ASIC1−/− groups (*p < 0.001), and to levels not significantly different from ASIC1a+/+ mice (p = 0.464). As observed in the earlier experiments, the ASIC1a+/+ mice had significantly increased immobility relative to uninjected ASIC1a−/− mice (*p < 0.001).
Surgery and viral injection.
Mice were anesthetized and placed in a stereotaxic frame (Kopf). Viral vector injectate was either a combination of 70% AAV-ASIC1a (titer 2.28 × 1013 viral genomes (vg)/ml) and 30% AAV-eGFP (titer 1.78 × 1014 vg/ml) or AAV-eGFP alone. Virus (0.5 μl, same for both groups) was infused using a 10 μl Hamilton syringe with a 33 gauge blunt tipped needle and a microinjector pump at a rate of 0.2 μl/min; the needle rested in position for 5 min postinjection. The injection coordinates were 1.5 mm posterior to bregma, 3.5 mm lateral to the midline, and 4.5 mm the from pia surface. Following injection, incisions were sutured and topical anesthetic was applied to the wound (Bupivi-caine, 0.5%). Mice recovered for 14 d before behavioral analysis.
Tissue processing and target verification.
Following behavioral analysis, brains were freshly frozen on dry ice and embedded in OCT tissue freezing medium. Coronal slices (10 μm) were cut and mounted on slides using the CryoJane cryosectioning system (Electron Microscopy Systems). Slides were then either thawed for immediate eGFP detection and imaging or processed for immunohistochemical detection of ASIC1a as described below. Slides were coverslipped with Vectashield (Vector Laboratories) and visualized with a Zeiss epifluorescence microscope. Images were captured at 10× magnification and compiled using the tiling function to create composite images of coronal hemisections. Borders for the basolateral amygdala (BLA) were defined based on the anatomical landmarks in Paxinos (Paxinos and Franklin, 2001). Correctly targeted injections (hits) were defined as having fluorescence above background within bilateral BLA. Off target injections (misses) were defined as having fluorescence above background in the temporal lobe but not in both BLA.
ASIC1 immunohistochemistry.
Coronal slices (10 μm) of fresh frozen brain were cut, mounted on slides using the CryoJane sectioning protocol (Electron Microscopy Sciences), and postfixed in phosphate buffered saline (PBS) with 4% formaldehyde and 4% sucrose for 10 min, followed by 0.25% Triton X-100 in PBS for 5 min at 23°C, and then incubated in affinity purified ASIC1a antibody (Wemmie et al., 2003) (1:50) for 24 h at 4°C. Following PBS wash, sections were incubated with Cy3-coupled anti-rabbit IgG (Jackson ImmunoResearch Labs) 1:200 for 1 h, 23°C. Slices were visualized with a Zeiss epifluorescence microcope (LSM 510 Meta). Images were captured at 10× magnification and compiled using the tiling software function (Zeiss) to create composite image of coronal hemi-sections.
Statistical analysis.
Values are expressed as mean ± SEM. t test was used to assess significance in experiments comparing 2-groups. For experiments comprised of multiple factors a two-way ANOVA, with test for interaction, was used (SPSS). One-way ANOVA was used for single factor experiments involving more than two groups. For ANOVA results, planned contrast testing was used to test relationships hypothesized a priori between groups. p values (two-tailed) <0.05 were considered significant.
Results
ASIC1a disruption reduces depression-related behavior
In rodents, unpredictable chronic mild stress decreases the normal preference for sucrose-containing water versus regular drinking water. This might parallel anhedonia in depression or it might reflect differences in carbohydrate craving in depressed patients. Importantly, most currently available antidepressants block this effect in rats and mice (Berton and Nestler, 2006). To explore the possible role of ASIC1a in depression-related behavior, we tested the effects of ASIC1a on sucrose preference before and following chronic mild stress. Under basal conditions, ASIC1a disruption had no effect on the normally strong preference for 8% sucrose (Fig. 1 A). Unpredictable stress reduced sucrose preference in both ASIC1a+/+ and ASIC1a−/− mice. However, the loss of ASIC1a attenuated the sucrose preference reduction caused by unpredictable stress.
Figure 1.
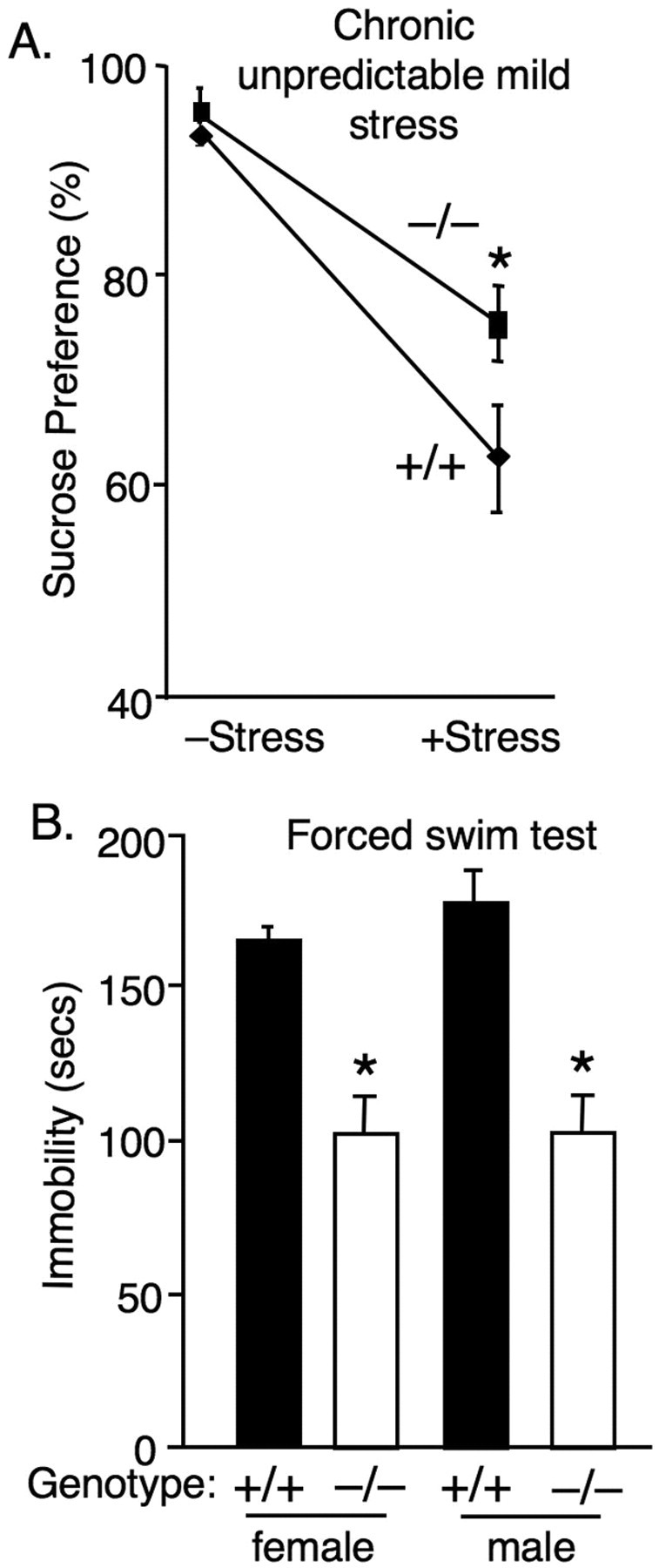
Loss of ASIC1a produces antidepressant-like effects. A, ASIC1a disruption attenuated the effect of unpredictable chronic mild stress (CMS) on sucrose preference. Two-way ANOVA with interaction revealed a significant overall model (p < 0.001, F (3,33) = 398) and a significant stress by genotype interaction [p < 0.01, F (1,33) = 6.39; ASIC1a+/+ CMS– (n = 14), ASIC1a−/− CMS– (n = 12), ASIC1a+/+ CMS + (n = 6), ASIC1a−/− CMS+ (n = 5)]. Planned contrast testing also revealed a significantly greater sucrose preference in the ASIC1a−/− mice in the stressed condition (*p = 0.003, t (33) = 3.22). B, ASIC1a disruption reduced immobility in the FST [*p < 0.001, t (47) = 7.30; ASIC1a+/+ (n = 28), ASIC1a−/− (n = 21)]. ASIC1a disruption produced similar effects in female and in male mice in the FST; two-way ANOVA found no effect of gender and no gender by genotype interaction (F <1).
The tail suspension test and the forced swim test (Cryan and Holmes, 2005; Cryan and Slattery, 2007) also exhibit a high degree of validity for predicting antidepressant effects in people. While medications targeting primarily anxiety such as benzodiazepines produce little effect on the forced swim test (Porsolt et al., 1977). In addition to their strong predictive validity, these tests might mirror the helplessness and despair of depression. The tail suspension test probes whether mice resist when being suspended by the tail or how readily they give up and hang motionlessly. The forced swim test assesses resistance or immobility in an inescapable pool of water (Cryan and Holmes, 2005; Cryan and Slattery, 2007). We examined ASIC1a+/+ and ASIC1a−/− mice in these tests and found that consistent with an antidepressant-like effect, the ASIC1a−/− mice had significantly less immobility in the tail suspension test (p = 0.006, t (18) = 3.13; n = 10 per group) and in the forced swim test (Fig. 1 B). Similar effects were observed in both male and female mice (Fig. 1 B). Previous studies suggest that animal size, locomotor activity and swimming are normal in the ASIC1a-null mice (Wemmie et al., 2002; Coryell et al., 2007), so these factors are unlikely to explain the antidepressant-like behaviors.
ASIC1a inhibitors have antidepressant effects
To assess whether the antidepressant-like effects of ASIC1a disruption depend on the chronic loss of ASIC1a and possibly a product of differential brain development, we tested the effects of acutely administering ASIC1a inhibitors. Previous studies showed that venom from the tarantula Psalmopoeus cambridgei contains a peptide PcTx1 that reversibly blocks ASIC1a-mediated currents (Escoubas et al., 2000) and produces effects in mice similar to ASIC1a gene disruption (Xiong et al., 2004; Coryell et al., 2007). Therefore, we tested the hypotheses that (1) PcTx1-containing venom and (2) purified PcTx1 peptide would produce antidepressant effects in the forced swim test. We chose to deliver the antagonists to the lateral cerebral ventricle (intracerebroventricularly) to bypass the blood–brain-barrier and to test the CNS as the likely site of action. Relative to vehicle-injected controls, both the venom and purified PcTx1 peptide significantly reduced immobility time (Fig. 2 A,B). As a control for specificity, we also tested the effects of PcTx venom and purified peptide in ASIC1a−/− mice, and found no effect.
Figure 2.

ASIC1a antagonists produce antidepressant-like effects in ASIC1a+/+ but not in ASIC1a−/− mice. A, Psalmopoeus cambridgei venom (5 μl, 9 ng/μl, i.c.v.) containing the ASIC1a antagonist PcTx1 significantly decreased immobility in the FST in ASIC1a+/+ mice [venom (n = 11), vehicle (n = 10)], but not in ASIC1a−/− mice [venom (n = 9), vehicle (n = 9)]. Two-way ANOVA with interaction revealed a significant overall model (p < 0.001, F (3,35) = 12.29), and a significant venom by genotype interaction (p < 0.01, F (1,35) = 10.69). Planned contrast tests found a significant effect of venom in ASIC1a+/+ mice (*p < 0.001, t (35) = 4.74), but not in ASIC1a−/− mice (p = 0.946, t (35) = 0.069). B, Similar to P. cambridgei venom, purified PcTx1 peptide (5 μl, 100 nm, i.c.v.) reduced immobility in ASIC1a+/+ mice [PcTx1 (n = 10), vehicle (n = 4)], but not in ASIC1a−/− mice [PcTx1 (n = 6), vehicle (n = 5)]. Two-way ANOVA found similarly significant effects of the PcTx1 by genotype interaction. Planned contrast tests revealed a significant effect of PcTx1 peptide in ASIC1a+/+ mice (*p = 0.003, t (21) = 3.33) but not in ASIC1a −/− mice (p = 0.626, t (21) = 0.494). C, The ASIC1a antagonist A-317567 (45 μm) reversibly blocked acid-evoked currents in cultured mouse cortical neurons, and (D) A-317567 (5 μl, 1 mm, i.c.v.) reduced FST immobility in ASIC1a+/+ mice [A-317567 (n = 12), vehicle (n = 16)], but not in ASIC1a−/− mice [A-317567 (n = 8), vehicle (n = 8)]. Two-way ANOVA with interaction revealed a significant overall model (p < 0.001, F (3,40) = 14.69), and a significant drug by genotype interaction (p < 0.05, F (1,40) = 5.07). Planned contrast tests found a significant effect of A-317567 in ASIC1a+/+ mice (*p < 0.01, t (40) = 2.92), but not in ASIC1a−/− mice (p = 0.549, t (40) = –0. 61).
We also tested a small molecule inhibitor, A-317567, an amiloride-like compound (Dubé et al., 2005). Previously, A-317567 blocked ASIC-like currents in cultured dorsal root ganglion neurons (Dubé et al., 2005). Here we found that A-317567 also blocked acid-evoked currents in cultured cortical neurons (Fig. 2 C). A-317567 reversibly blocked currents evoked by pH 6, which are mediated by ASIC1a-containing channels (Wemmie et al., 2002; Askwith et al., 2004). To test whether this small molecule inhibitor also produced antidepressant effects by blocking ASIC1a, we administered A-317567 (5 μl, 1 mm, i.c.v.) to ASIC1a+/+ and ASIC1a−/− mice and assessed behavior in the forced swim test. Similar to the antidepressants, A-317567 reduced immobility in ASIC1a+/+ mice (Fig. 2 D). However no effect was observed in the ASIC1a−/− mice. Thus, with acute administration ASIC antagonists produce ASIC1a-dependent antidepressant-like effects.
Monoamine reuptake inhibitors do not require ASIC1a for antidepressant effects
Considering whether current antidepressants and ASIC1a inhibitors act through related mechanisms, we explored whether monoamine reuptake inhibitors depend on ASIC1a; if so, then they should have no effect or a diminished effect in the ASIC1a-null mice. We tested 3 antidepressants; fluoxetine (3 mg/kg, i.p.), desipramine (2 mg/kg, i.p.), and bupropion (2 mg/kg, i.p.), which target the serotonin, norepinephrine, and dopamine systems respectively. Each drug was delivered 30 min before the forced swim test. As expected, each of these antidepressants reduced immobility in ASIC1a+/+ mice (Fig. 3 A–C). Moreover, they also reduced immobility in the ASIC1a−/− mice. Because some bupropion doses increase locomotor activity (David et al., 2003) and might confound forced swim mobility, we tested 2 mg/kg, intraperitoneal bupropion on activity in the open field for 30 min as described (Coryell et al., 2007). Consistent with others' results (David et al., 2003), this bupropion dose did not significantly increase activity (ASIC1a+/+ saline = 8024 ± 726 (mean beam breaks ± SEM), bupropion = 8130 ± 460; ASIC1a−/−saline = 6631 ± 743, bupropion = 6873 ± 800; n = 3–4 per group). Two-way ANOVA indicated no drug effect (F <1), no genotype effect (F (1,10) = 3.78, p > 0.05), and no interaction (F <1). Thus, the antidepressant actions of these medications are unlikely to depend on ASIC1a.
Figure 3.

Monoaminergic antidepressants produce effects independent of ASIC1a. A, The serotonin reuptake inhibitor fluoxetine decreased immobility in the forced swim test both in ASIC1a+/+ mice [drug (n = 6), vehicle (n = 5)] and in ASIC1a−/− mice [drug (n = 10), vehicle (n = 8)]. Two-way ANOVA revealed significant effects of fluoxetine (p < 0.001, F (1,25) = 18.6) and genotype (p < 0.001, F (1,25) = 115) but not an interaction (F <1). Planned contrast tests revealed a significant effect of fluoxetine in both genotypes (*p < 0.01). B, Similarly, the norepinephrine reuptake inhibitor desipramine decreased immobility both in ASIC1a+/+ mice [drug (n = 6), vehicle (n = 11)] and in ASIC1a−/− mice [drug (n = 5), vehicle (n = 5)]. Two-way ANOVA revealed a significant effect of desipramine (p < 0.01, F (1,23) = 10.9) and genotype (p < 0.05, F (1,23) = 6.37) but not an interaction (F <1). Planned contrast tests revealed a significant effect of desipramine in both genotypes (*p < 0.05). C, The dopaminergic antidepressant bupropion also decreased immobility in ASIC1a+/+ mice [drug (n = 6), vehicle (n = 4)] and in ASIC1a−/− mice [drug (n = 7), vehicle (n = 8)]. Two-way ANOVA revealed a significant effect of bupropion (p < 0.001, F (1,21) = 22.3), but not genotype (p > 0.05, F (1,21) = 2.2) and no interaction (p > 0.05, F (1,21) = 2.1). Planned contrasts revealed significant effects of bupropion in both genotypes (*p < 0.05).
Serotonin depletion does not alter the antidepressant effects of ASIC1a inhibition
Selective serotonin reuptake inhibitors (SSRIs) are the most widely used antidepressants; therefore we wanted to further test whether the behavioral effects of ASIC1a blockade in ASIC1a+/+ mice depend on serotonin signaling. We depleted serotonin with the tryptophan hydroxylase inhibitor P-chlorophenylalanine (PCPA). Though PCPA does not increase depression-like behavior, it blocks the antidepressant effects of fluoxetine and other serotonin reuptake inhibitors in the forced swim test and in other animal models of depression (Heurteaux et al., 2006). We reasoned that if serotonin is necessary for the antidepressant effect of ASIC1a blockers, then like fluoxetine, ASIC1a inhibitors should be sensitive to serotonin depletion. Mice were given intraperitoneal injections of PCPA (300 mg/kg, daily, 3 d) or vehicle control (saline, daily, 3 d). We then injected fluoxetine (3 mg/kg, i.p.) or PcTx (5 μl, 9 ng/ml, i.c.v.) and antidepressant activity was assessed. Consistent with previous reports, PCPA prevented the fluoxetine effects in the forced swim test (Fig. 4 A). In contrast, PCPA had no effect on the antidepressant-like responses to PcTx1 or ASIC1a disruption (Fig. 4 B). These data suggest that PcTx1 and ASIC1a disruption produce antidepressant effects through mechanisms different from fluoxetine.
Figure 4.
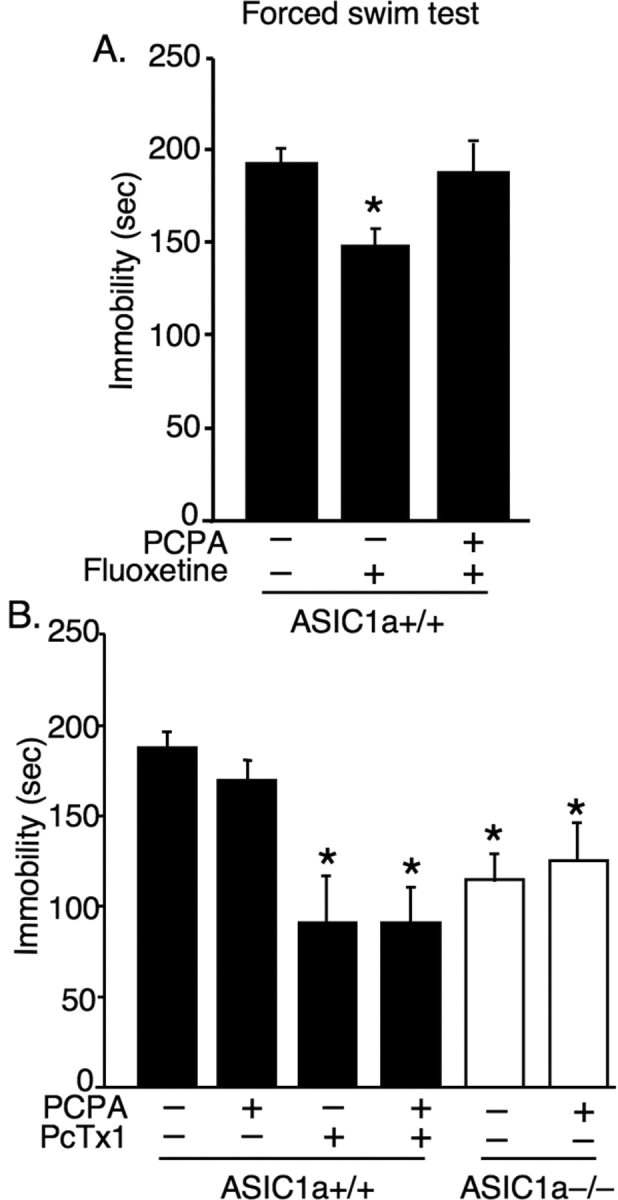
In ASIC1a+/+ mice, the antidepressant-like effects of ASIC1a antagonist occur independent of serotonin. A, Depleting serotonin with the tyrosine hydroxylase inhibitor PCPA eliminated fluoxetine effects in the forced swim test [untreated (n = 15), +fluoxetine (n = 6), +fluoxetine +PCPA (n = 3)]. One-way ANOVA revealed a significant overall effect (p < 0.05, F (2,21) = 4.50). Planned contrast tests showed that fluoxetine decreased immobility compared with untreated mice (*p = 0.009, t (21) = 2.89). However, mice treated with both fluoxetine and PCPA did not differ significantly from untreated controls (p = 0.884, t (21) = –0.141). B, ASIC1a antagonist PcTx1 continued to produce antidepressant-like effects following serotonin depletion [untreated (n = 11), +PCPA (n = 9), +PcTx1 (n = 4), +PcTx1 +PCPA (n = 8)]. One-way ANOVA revealed a significant overall effect (p < 0.001, F (3,28) = 8.60). Planned contrast tests revealed a significant reduction in immobility in both PcTx1-treated groups compared with untreated controls (*p < 0.001) and in both ASIC1a−/− groups relative to untreated ASIC1a+/+ controls (*p < 0.05). The PCPA-treated ASIC1a−/− mice did not differ from untreated ASIC1a−/− mice (p = 0.59).
Stress-evoked corticosterone responses are normal in the ASIC1a−/− mice
Stress stimulates corticosterone release from the adrenal glands. Furthermore, HPA axis dysfunction has long been implicated in the etiology of depression (Selye, 1955; Carroll et al., 1976; Stokes, 1995). Therefore we assessed serum corticosterone levels in ASIC1a+/+ and ASIC1a−/− mice at baseline and following swim stress. We found that stress similarly stimulated corticosterone release in both genotypes (Fig. 5 A), suggesting that ASIC1a disruption does not attenuate the corticosterone response.
Figure 5.

ASIC1a−/− mice have normal stress-evoked corticosterone responses, but BDNF reduction depends on ASIC1a. A, Swimming-induced stress significantly increased corticosterone levels in both genotypes [ASIC1a+/+ unstressed (n = 12), ASIC1a+/+ stressed (n = 13), ASIC1a−/− unstressed (n = 11), ASIC1a−/− stressed (n = 12)]. Two-way ANOVA revealed a significant effect of stress (p < 0.001, F (1,44) = 146), but not genotype (F <1), and no interaction (F <1). Stress significantly increased corticosterone in both genotypes (*p < 0.001). B, C, Restraint stress reduced BDNF mRNA in the hippocampus of ASIC1a+/+ mice [unstressed (n = 7), stressed (n = 7)] but not in ASIC1a−/− mice [unstressed (n = 8), stressed (n = 7)]. BDNF mRNA was assessed by real-time PCR. BDNF detection threshold normalized to GAPDH (Δ Ct) (B) and BDNF levels are plotted as fold change in stressed mice relative to baseline (C). Two-way ANOVA with interaction revealed a significant overall model (p < 0.05, F (3,25) = 4.10), and a significant stress by genotype interaction (p < 0.05, F (1,25) = 7.11). Planned contrast tests found a significant effect of stress in ASIC1a+/+ mice (*p = 0.002), but not in ASIC1a−/− mice (p = 0.826).
Stress-evoked BDNF reduction depends on ASIC1a
Brain derived neurotrophic factor (BDNF) plays a prominent role in current theories of depression (Dranovsky and Hen, 2006; Sahay et al., 2007). Stress alters the expression of BDNF in several brain regions including the nucleus accumbens and amygdala (Aguilar-Valles et al., 2005; Berton et al., 2006), and BDNF in the hippocampus has emerged as an important biological marker of depression; stress reduces BDNF in the hippocampus and antidepressants block the effect (Smith et al., 1995; Duman et al., 1997). Therefore, to explore whether ASIC1a disruption altered BDNF expression or affected the stress response, we measured BDNF mRNA levels in the hippocampus by quantitative PCR (qPCR) at baseline and following restraint stress. Consistent with findings by others (Duman and Monteggia, 2006), restraining wild-type mice reduced BDNF expression in the hippocampus (Fig. 5 B,C). In the ASIC1a−/− mice, BDNF levels were slightly lower at baseline (Fig. 5 B). Strikingly, stress did not reduce BDNF levels in the ASIC1a-null mice. Thus, like antidepressant therapies, ASIC1a disruption prevented the ability of stress to lower BDNF in the hippocampus.
Restoring ASIC1a in the amygdala eliminates the antidepressant-like phenotype in ASIC1a-null mice
Because ASIC1a is robustly expressed in the amygdala and the amygdala has been implicated in human depression (Soares and Mann, 1997; Drevets, 2000), we wondered to what degree ASIC1a activity in the amygdala might contribute to depression-related behavior. We developed an adeno-associated virus (AAV) vector to target ASIC1a expression to the amygdala of ASIC1a-null mice (Coryell et al., 2008) and assessed the effect in forced swim test (Fig. 6 A,B). Restoring ASIC1a expression in the basolateral amygdala increased immobility in the ASIC1a−/− mice to wild-type levels (Fig. 6 B). In contrast, AAV-eGFP injections and AAV-ASIC1a injections that missed the basolateral amygdala had no effect. Although, this technique is not precise enough to tell us exactly what cells are responsible, these results suggest that the amygdala is a key site of ASIC1a action in this depression-related behavior.
Discussion
Loss or inhibition of ASIC1a produced antidepressant-like effects in multiple depression-related behaviors. Although the relationship between these behavioral models and the pathophysiology of depression are not well elucidated, the strong predictive validity of these models suggests that ASIC1a antagonists may have similar antidepressant effects in humans (Cryan and Slattery, 2007). The antidepressant-like effect was observed whether we manipulated ASIC1a genetically or with peptide and small molecule inhibitors, suggesting that both chronically and acutely interrupting ASIC1a produce similar effects. Strengthening the likelihood that the antidepressant effects of PcTx1 and A-317567 were mediated through ASIC1a, these ASIC1a antagonists produced no effect in the ASIC1a-null mice.
Despite extensive investigation, the mechanism of action of current antidepressant therapies and the pathophysiological causes of depression remain elusive. Thus it is difficult to place ASIC1a in relevant pathways. However, identifying ASIC1a as a possible target in depression raises the intriguing possibility that ASIC1a might modulate depression through novel mechanisms. Here, we explored three possible pathways by which ASIC1a might affect depression-related behavior: (1) HPA axis function, (2) BDNF regulation, and (3) monoamine signaling. Stress-evoked HPA activation measured by corticosterone release appeared normal in the ASIC1a-null mice. However, BDNF reduction in the hippocampus during stress depended on ASIC1a. This raises an intriguing mechanism that may help explain how ASIC1a contributes to depression-related behaviors and how inhibiting ASIC1a attenuates effects of stress. Emotional stress precipitates and can worsen depression, and it is well established that stress reduces BDNF mRNA in the hippocampus (Martinowich et al., 2007). Conversely, current antidepressants, including electroconvulsive therapy, reverse the stress-mediated BDNF decreases (Duman and Monteggia, 2006; Ploski et al., 2006), suggesting a compelling role for hippocampal BDNF in depression and in treatment. Recently it has been suggested that stress alters BDNF in other brain regions and that this may play a critical role in depression (Aguilar-Valles et al., 2005; Berton et al., 2006); it will be interesting to learn whether ASIC1a is involved in these changes. In addition, the association between ASIC1a and BDNF suggests a novel way by which ASIC1a might exert some of its other diverse effects. Our previous studies implicated ASIC1a in synaptic plasticity (Wemmie et al., 2002), dendrite morphology (Zha et al., 2006) and in neurodegenerative processes (Xiong et al., 2004; Friese et al., 2007), all of which are affected by BDNF.
The mechanisms by which stress lowers hippocampal BDNF are complex and thus remain elusive. Up to nine distinct promoters drive the expression of multiple gene products and spliced isoforms with epigenetic mechanisms also playing an important role (Tsankova et al., 2006; Martinowich et al., 2007; Lubin et al., 2008; Tabuchi, 2008). Pathways that are both corticosterone-dependent and independent have been implicated in BDNF regulation by stress (Kessler, 1997; Berton and Nestler, 2006). The corticosterone-independent processes observed in adrenalectomized animals are particularly ambiguous (Hansson et al., 2006). The normal corticosterone response in the ASIC1a-null mice observed here may point to a role for ASIC1a in corticosterone-independent mechanisms.
Because most antidepressants target monoamine signaling, we explored whether ASIC1a loss or inhibition produced effects through monoamine pathways. We found that depleting serotonin, the most common treatment target, did nothing to lessen the antidepressant action of ASIC1a inhibition. Moreover, inhibitors of serotonin, norepinephrine, and dopamine reuptake produced antidepressant-like effects independent of ASIC1a. ASIC1a interruption produced effects that were additive to those of monoamine reuptake inhibitors, suggesting that ASIC1a inhibitors might produce added benefit when combined with these currently available therapies. By suggesting a different mechanism of action, these observations further suggest that ASIC1a inhibitors might be effective in people who do not respond to monoamine reuptake inhibitors or who cannot tolerate the side effects.
It has long been speculated that amygdala hyperactivity is a key component in the pathogenesis of depression (Drevets et al., 1992) in that the amygdala is thought to be critical for vigilance and negative emotion (Davis and Whalen, 2001). Several studies have detected changes in amygdala volume (Frodl et al., 2002; Lange and Irle, 2004) and functional imaging has revealed increases in amygdala activity in depressed patients. Increased amygdala blood flow and glucose metabolism correlate with greater illness severity and recurrence (Drevets et al., 2008). The role of the amygdala in these rodent models of depression-related behavior is less well established; however the ability of viral vectors driving ASIC1a expression in the amygdala to restore forced swim behavior to normal levels in the ASIC1a-null mice suggests that this structure plays a key role. Other amygdala manipulations have similarly produced antidepressant-like effects in the forced swim test in rats (Duncan et al., 1986; Salomé et al., 2006). Consistent with the amygdala as a site of ASIC1a action, our previous studies suggest c-Fos expression in the BLA is impaired by ASIC1a disruption (Coryell et al., 2007, 2008). Thus, ASIC1a inhibitors may combat depression by reducing amygdala activity. Because of the importance of the amygdala in negative emotions and fear, we speculate that reducing amygdala activity reduces the emotional burden of stressors like the forced swim test; thus reducing helplessness and other stress-evoked depression-related behaviors. The involvement of an acid-activated channel suggests that endogenous pH fluctuations may occur in the amygdala during emotional stress.
This is the first study exploring ASIC1a inhibition on depression-related behavior. However, a previous genetic study in humans investigated the possible link between polymorphisms in the ASIC1a-encoding gene and major depression and anxiety disorders (Hettema et al., 2008). That study found a possible association between a specific haplotype of the ASIC1a-containing locus and major depression (463 cases), although the data were only marginally significant (p = 0.045, uncorrected for multiple tests). Nevertheless, our observations suggest additional studies might be in order to further investigate the possible association between ASIC1a and the causes of human depression.
Footnotes
Support was provided by National Institutes of Mental Health Grants F31MH076414-03 (M.W.C.) and U01-MH61971 with subcontract (M.N.C.), National Alliance for Research on Schizophrenia and Depression Young Investigator Award, and Department of Veterans Affairs Merit Review Award (J.A.W.).
References
- Aguilar-Valles A, Sánchez E, de Gortari P, Balderas I, Ramírez-Amaya V, Bermúdez-Rattoni F, Joseph-Bravo P. Analysis of the stress response in rats trained in the water-maze: differential expression of corticotropin-releasing hormone, CRH-R1, glucocorticoid receptors and brain-derived neurotrophic factor in limbic regions. Neuroendocrinology. 2005;82:306–319. doi: 10.1159/000093129. [DOI] [PubMed] [Google Scholar]
- Askwith CC, Wemmie JA, Price MP, Rokhlina T, Welsh MJ. ASIC2 modulates ASIC1 H+-activated currents in hippocampal neurons. J Biol Chem. 2004;279:18296–18305. doi: 10.1074/jbc.M312145200. [DOI] [PubMed] [Google Scholar]
- Bechara A, Tranel D, Damasio H, Adolphs R, Rockland C, Damasio AR. Double dissociation of conditioning and declarative knowledge relative to the amygdala and hippocampus in humans. Science. 1995;269:1115–1118. doi: 10.1126/science.7652558. [DOI] [PubMed] [Google Scholar]
- Benkelfat C, Ellenbogen MA, Dean P, Palmour RM, Young SN. Mood-lowering effect of tryptophan depletion. Enhanced susceptibility in young men at genetic risk for major affective disorders. Arch Gen Psychiatry. 1994;51:687–697. doi: 10.1001/archpsyc.1994.03950090019003. [DOI] [PubMed] [Google Scholar]
- Berton O, Nestler EJ. New approaches to antidepressant drug discovery: beyond monoamines. Nat Rev Neurosci. 2006;7:137–151. doi: 10.1038/nrn1846. [DOI] [PubMed] [Google Scholar]
- Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, Monteggia LM, Self DW, Nestler EJ. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- Carroll BJ, Curtis GC, Mendels J. Cerebrospinal fluid and plasma free cortisol concentrations in depression. Psychol Med. 1976;6:235–244. doi: 10.1017/s0033291700013775. [DOI] [PubMed] [Google Scholar]
- Cho JH, Askwith CC. Presynaptic release probability is increased in hippocampal neurons from ASIC1 knockout mice. J Neurophysiol. 2008;99:426–441. doi: 10.1152/jn.00940.2007. [DOI] [PubMed] [Google Scholar]
- Coryell MW, Ziemann AE, Westmoreland PJ, Haenfler JM, Kurjakovic Z, Zha XM, Price M, Schnizler MK, Wemmie JA. Targeting ASIC1a reduces innate fear and alters neuronal activity in the fear circuit. Biol Psychiatry. 2007;62:1140–1148. doi: 10.1016/j.biopsych.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Coryell MW, Wunsch AM, Haenfler JM, Allen JE, McBride JL, Davidson BL, Wemmie JA. Restoring acid-sensing ion channel-1a in the amygdala of knock-out mice rescues fear memory but not unconditioned fear responses. J Neurosci. 2008;28:13738–13741. doi: 10.1523/JNEUROSCI.3907-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryan JF, Holmes A. The ascent of mouse: advances in modelling human depression and anxiety. Nat Rev Drug Discov. 2005;4:775–790. doi: 10.1038/nrd1825. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Slattery DA. Animal models of mood disorders: recent developments. Curr Opin Psychiatry. 2007;20:1–7. doi: 10.1097/YCO.0b013e3280117733. [DOI] [PubMed] [Google Scholar]
- David DJ, Renard CE, Jolliet P, Hascoët M, Bourin M. Antidepressant-like effects in various mice strains in the forced swimming test. Psychopharmacology (Berl) 2003;166:373–382. doi: 10.1007/s00213-002-1335-4. [DOI] [PubMed] [Google Scholar]
- Davidson BL, Stein CS, Heth JA, Martins I, Kotin RM, Derksen TA, Zabner J, Ghodsi A, Chiorini JA. Recombinant adeno-associated virus type 2, 4, and 5 vectors: transduction of variant cell types and regions in the mammalian central nervous system. Proc Natl Acad Sci U S A. 2000;97:3428–3432. doi: 10.1073/pnas.050581197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Whalen PJ. The amygdala: vigilance and emotion. Mol Psychiatry. 2001;6:13–34. doi: 10.1038/sj.mp.4000812. [DOI] [PubMed] [Google Scholar]
- Dranovsky A, Hen R. Hippocampal neurogenesis: regulation by stress and antidepressants. Biol Psychiatry. 2006;59:1136–1143. doi: 10.1016/j.biopsych.2006.03.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets WC. Functional anatomical abnormalities in limbic and prefrontal cortical structures in major depression. Prog Brain Res. 2000;126:413–431. doi: 10.1016/S0079-6123(00)26027-5. [DOI] [PubMed] [Google Scholar]
- Drevets WC, Videen TO, Price JL, Preskorn SH, Carmichael ST, Raichle ME. A functional anatomical study of unipolar depression. J Neurosci. 1992;12:3628–3641. doi: 10.1523/JNEUROSCI.12-09-03628.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets WC, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Funct. 2008;213:93–118. doi: 10.1007/s00429-008-0189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubé GR, Lehto SG, Breese NM, Baker SJ, Wang X, Matulenko MA, Honoré P, Stewart AO, Moreland RB, Brioni JD. Electrophysiological and in vivo characterization of A-317567, a novel blocker of acid sensing ion channels. Pain. 2005;117:88–96. doi: 10.1016/j.pain.2005.05.021. [DOI] [PubMed] [Google Scholar]
- Duman RS. Synaptic plasticity and mood disorders. Mol Psychiatry. 2002;7(Suppl 1):S29–S34. doi: 10.1038/sj.mp.4001016. [DOI] [PubMed] [Google Scholar]
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54:597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- Duncan GE, Breese GR, Criswell H, Stumpf WE, Mueller RA, Covey JB. Effects of antidepressant drugs injected into the amygdala on behavioral responses of rats in the forced swim test. J Pharmacol Exp Ther. 1986;238:758–762. [PubMed] [Google Scholar]
- Escoubas P, De Weille JR, Lecoq A, Diochot S, Waldmann R, Champigny G, Moinier D, Ménez A, Lazdunski M. Isolation of a tarantula toxin specific for a class of proton-gated Na+ channels. J Biol Chem. 2000;275:25116–25121. doi: 10.1074/jbc.M003643200. [DOI] [PubMed] [Google Scholar]
- Friese MA, Craner MJ, Etzensperger R, Vergo S, Wemmie JA, Welsh MJ, Vincent A, Fugger L. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat Med. 2007;13:1483–1489. doi: 10.1038/nm1668. [DOI] [PubMed] [Google Scholar]
- Frodl T, Meisenzahl E, Zetzsche T, Bottlender R, Born C, Groll C, Jäger M, Leinsinger G, Hahn K, Möller HJ. Enlargement of the amygdala in patients with a first episode of major depression. Biol Psychiatry. 2002;51:708–714. doi: 10.1016/s0006-3223(01)01359-2. [DOI] [PubMed] [Google Scholar]
- Hansson AC, Sommer WH, Metsis M, Strömberg I, Agnati LF, Fuxe K. Corticosterone actions on the hippocampal brain-derived neurotrophic factor expression are mediated by exon IV promoter. J Neuroendocrinol. 2006;18:104–114. doi: 10.1111/j.1365-2826.2005.01390.x. [DOI] [PubMed] [Google Scholar]
- Heinrichs S, Koob G. Applications of experimental stressors in laboratory rodents. In: Crawley J, Gerfen C, McKay R, Rogawski M, Sibley D, Skolnick P, editors. Current protocols in neuroscience. New York: Wiley; 2005. pp. 5–6. Chap 8. [DOI] [PubMed] [Google Scholar]
- Hettema JM, An SS, Neale MC, van den Oord EJ, Kendler KS, Chen X. Lack of association between the amiloride-sensitive cation channel 2 (ACCN2) gene and anxiety spectrum disorders. Psychiatr Genet. 2008;18:73–79. doi: 10.1097/YPG.0b013e3282f08a2a. [DOI] [PubMed] [Google Scholar]
- Heurteaux C, Lucas G, Guy N, El Yacoubi M, Thümmler S, Peng XD, Noble F, Blondeau N, Widmann C, Borsotto M, Gobbi G, Vaugeois JM, Debonnel G, Lazdunski M. Deletion of the background potassium channel TREK-1 results in a depression-resistant phenotype. Nat Neurosci. 2006;9:1134–1141. doi: 10.1038/nn1749. [DOI] [PubMed] [Google Scholar]
- Holtzheimer PE, 3rd, Nemeroff CB. Emerging treatments for depression. Expert Opin Pharmacother. 2006;7:2323–2339. doi: 10.1517/14656566.7.17.2323. [DOI] [PubMed] [Google Scholar]
- Kent JM, Rauch SL. Neurocircuitry of anxiety disorders. Curr Psychiatry Rep. 2003;5:266–273. doi: 10.1007/s11920-003-0055-8. [DOI] [PubMed] [Google Scholar]
- Kessler RC. The effects of stressful life events on depression. Annu Rev Psychol. 1997;48:191–214. doi: 10.1146/annurev.psych.48.1.191. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Stang PE, Wittchen HU, Ustun TB, Roy-Burne PP, Walters EE. Lifetime panic-depression comorbidity in the National Comorbidity Survey. Arch Gen Psychiatry. 1998;55:801–808. doi: 10.1001/archpsyc.55.9.801. [DOI] [PubMed] [Google Scholar]
- Lange C, Irle E. Enlarged amygdala volume and reduced hippocampal volume in young women with major depression. Psychol Med. 2004;34:1059–1064. doi: 10.1017/s0033291703001806. [DOI] [PubMed] [Google Scholar]
- Lubin FD, Roth TL, Sweatt JD. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci. 2008;28:10576–10586. doi: 10.1523/JNEUROSCI.1786-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Manji H, Lu B. New insights into BDNF function in depression and anxiety. Nat Neurosci. 2007;10:1089–1093. doi: 10.1038/nn1971. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. 2002;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2001. [Google Scholar]
- Ploski JE, Newton SS, Duman RS. Electroconvulsive seizure-induced gene expression profile of the hippocampus dentate gyrus granule cell layer. J Neurochem. 2006;99:1122–1132. doi: 10.1111/j.1471-4159.2006.04156.x. [DOI] [PubMed] [Google Scholar]
- Porsolt RD, Bertin A, Jalfre M. Behavioral despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther. 1977;229:327–336. [PubMed] [Google Scholar]
- Pothion S, Bizot JC, Trovero F, Belzung C. Strain differences in sucrose preference and in the consequences of unpredictable chronic mild stress. Behav Brain Res. 2004;155:135–146. doi: 10.1016/j.bbr.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Rauch SL, Shin LM, Wright CI. Neuroimaging studies of amygdala function in anxiety disorders. Ann N Y Acad Sci. 2003;985:389–410. doi: 10.1111/j.1749-6632.2003.tb07096.x. [DOI] [PubMed] [Google Scholar]
- Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, Niederehe G, Thase ME, Lavori PW, Lebowitz BD, McGrath PJ, Rosenbaum JF, Sackeim HA, Kupfer DJ, Luther J, Fava M. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163:1905–1917. doi: 10.1176/ajp.2006.163.11.1905. [DOI] [PubMed] [Google Scholar]
- Sahay A, Drew MR, Hen R. Dentate gyrus neurogenesis and depression. Prog Brain Res. 2007;163:697–722. doi: 10.1016/S0079-6123(07)63038-6. [DOI] [PubMed] [Google Scholar]
- Salomé N, Stemmelin J, Cohen C, Griebel G. Differential roles of amygdaloid nuclei in the anxiolytic- and antidepressant-like effects of the V1b receptor antagonist, SSR149415, in rats. Psychopharmacology (Berl) 2006;187:237–244. doi: 10.1007/s00213-006-0424-1. [DOI] [PubMed] [Google Scholar]
- Schildkraut JJ. The catecholamine hypothesis of affective disorders: a review of supporting evidence. 1965. J Neuropsychiatry Clin Neurosci. 1995;7:524–533. doi: 10.1176/jnp.7.4.524. discussion 523–524. [DOI] [PubMed] [Google Scholar]
- Selye H. Stress and disease. Science. 1955;122:625–631. doi: 10.1126/science.122.3171.625. [DOI] [PubMed] [Google Scholar]
- Smith MA, Makino S, Kvetnansky R, Post RM. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci. 1995;15:1768–1777. doi: 10.1523/JNEUROSCI.15-03-01768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares JC, Mann JJ. The functional neuroanatomy of mood disorders. J Psychiatr Res. 1997;31:393–432. doi: 10.1016/s0022-3956(97)00016-2. [DOI] [PubMed] [Google Scholar]
- Solich J, Pałach P, Budziszewska B, Dziedzicka-Wasylewska M. Effect of two behavioral tests on corticosterone level in plasma of mice lacking the noradrenaline transporter. Pharmacol Rep. 2008;60:1008–1013. [PubMed] [Google Scholar]
- Stokes PE. The potential role of excessive cortisol induced by HPA hyperfunction in the pathogenesis of depression. Eur Neuropsychopharmacol. 1995;5(Suppl):77–82. doi: 10.1016/0924-977x(95)00039-r. [DOI] [PubMed] [Google Scholar]
- Sulser F, Watts J, Brodie BB. On the mechanism of antidepressant action of imipramine-like drugs. Ann N Y Acad Sci. 1962;96:279–288. doi: 10.1111/j.1749-6632.1962.tb50122.x. [DOI] [PubMed] [Google Scholar]
- Tabuchi A. Synaptic plasticity-regulated gene expression: a key event in the long-lasting changes of neuronal function. Biol Pharm Bull. 2008;31:327–335. doi: 10.1248/bpb.31.327. [DOI] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid-sensing. Nature. 1997;386:173–177. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- Welsh MJ, Price MP, Xie J. Biochemical basis of touch perception: mechanosensory function of degenerin/epithelial Na+ channels. J Biol Chem. 2002;277:2369–2372. doi: 10.1074/jbc.R100060200. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Chen J, Askwith CC, Hruska-Hageman AM, Price MP, Nolan BC, Yoder PG, Lamani E, Hoshi T, Freeman JH, Jr, Welsh MJ. The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron. 2002;34:463–477. doi: 10.1016/s0896-6273(02)00661-x. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Askwith CC, Lamani E, Cassell MD, Freeman JH, Jr, Welsh MJ. Acid-sensing ion channel 1 is localized in brain regions with high synaptic density and contributes to fear conditioning. J Neurosci. 2003;23:5496–5502. doi: 10.1523/JNEUROSCI.23-13-05496.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wemmie JA, Coryell MW, Askwith CC, Lamani E, Leonard AS, Sigmund CD, Welsh MJ. Overexpression of acid-sensing ion channel 1a in transgenic mice increases fear-related behavior. Proc Natl Acad Sci U S A. 2004;101:3621–3626. doi: 10.1073/pnas.0308753101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wemmie JA, Price MP, Welsh MJ. Acid-sensing ion channels: advances, questions and therapeutic opportunities. Trends Neurosci. 2006;29:578–586. doi: 10.1016/j.tins.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Zha X, Welsh MJ. Acid-sensing ion channels (ASICs) and pH in synapse physiology. In: Hell JW, Ehlers MD, editors. Structural and functional organization of the synapse. New York: Springer; 2008. [Google Scholar]
- Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Young SN, Smith SE, Pihl RO, Ervin FR. Tryptophan depletion causes a rapid lowering of mood in normal males. Psychopharmacology (Berl) 1985;87:173–177. doi: 10.1007/BF00431803. [DOI] [PubMed] [Google Scholar]
- Zha XM, Wemmie JA, Green SH, Welsh MJ. ASIC1a is a postsynaptic proton receptor that influences the density of dendritic spines. Proc Natl Acad Sci U S A. 2006;103:16556–16561. doi: 10.1073/pnas.0608018103. [DOI] [PMC free article] [PubMed] [Google Scholar]