

Abstract
Phototropins, major blue-light receptors in plants, are sensitive to blue light through a pair of flavin mononucleotide (FMN)-binding light oxygen and voltage (LOV) domains, LOV1 and LOV2. LOV2 undergoes a photocycle involving light-driven covalent adduct formation between a conserved cysteine and the FMN C(4a) atom. Here, the primary reactions of Avena sativa phototropin 1 LOV2 (AsLOV2) were studied using ultrafast mid-infrared spectroscopy and quantum chemistry. The singlet excited state (S1) evolves into the triplet state (T1) with a lifetime of 1.5 ns at a yield of ∼50%. The infrared signature of S1 is characterized by absorption bands at 1657 cm−1, 1495–1415 cm−1, and 1375 cm−1. The T1 state shows infrared bands at 1657 cm−1, 1645 cm−1, 1491–1438 cm−1, and 1390 cm−1. For both electronic states, these bands are assigned principally to C=O, C=N, C-C, and C-N stretch modes. The overall downshifting of C=O and C=N bond stretch modes is consistent with an overall bond-order decrease of the conjugated isoalloxazine system upon a π-π∗ transition. The configuration interaction singles (CIS) method was used to calculate the vibrational spectra of the S1 and T1 excited ππ∗ states, as well as respective electronic energies, structural parameters, electronic dipole moments, and intrinsic force constants. The harmonic frequencies of S1 and T1, as calculated by the CIS method, are in satisfactory agreement with the evident band positions and intensities. On the other hand, CIS calculations of a T1 cation that was protonated at the N(5) site did not reproduce the experimental FMN T1 spectrum. We conclude that the FMN T1 state remains nonprotonated on a nanosecond timescale, which rules out an ionic mechanism for covalent adduct formation involving cysteine-N(5) proton transfer on this timescale. Finally, we observed a heterogeneous population of singly and doubly H-bonded FMN C(4)=O conformers in the dark state, with stretch frequencies at 1714 cm−1 and 1694 cm−1, respectively.
Introduction
Phototropins (phot) are major blue-light receptors for phototropism, light-directed chloroplast movement, light-induced stomatal opening, rapid inhibition of growth, and gametogenesis, that are sensitive to blue light through a pair of light oxygen and voltage (LOV) domains, LOV1 and LOV2 (1). LOV2 constitutes the main blue-light input sensor of phototropins, whereas LOV1 is hypothesized to play a regulatory role (2). Avena sativa phot 1 LOV2 (AsLOV2) consists of ∼100 amino acids, and noncovalently binds flavin mononucleotide (FMN). The LOV2 domain undergoes a photocycle involving light-driven covalent adduct formation between a conserved cysteine residue and the C(4a) atom of FMN (3–5). Formation of a covalent bond to the FMN triggers protein conformational changes on the surface of the Per-ARNT-Sim (PAS) core, disrupting the interactions of this core with a C-terminal amphiphilic helix, called Jα, packed against its central β-sheet (6). Unfolding of the Jα helix is the critical event that regulates the C-terminal kinase activity of phototropin (7) and downstream signal transduction.
The early steps of the LOV photocycle after photon absorption involve the intersystem crossing of the FMN singlet excited state to the FMN triplet state on a nanosecond timescale (8,9), after which, covalent adduct formation proceeds without apparent intermediates on a microsecond timescale (10–15). In contrast to the apparent consensus on the spectroscopically distinguishable intermediates in the LOV photocycle, the mechanism by which covalent adduct formation occurs in the LOV domains is a matter of considerable debate. Broadly speaking, two reaction mechanisms for covalent adduct formation have been put forward: 1), an ionic mechanism; and 2), a radical-pair mechanism (16). According to the ionic model, which is schematically depicted in Fig. 1 (top), the sharply increased basicity of N5 in the FMN triplet state triggers its protonation. The proton would either originate in the conserved cysteine itself (8,17,18), or from another nearby group (11,19) (the former origin is shown in Fig. 1). This event would change the double-bond of N(5)=C(4a) to a single bond, leaving a very reactive carbocation at the C(4a) position. This carbon atom has sp3 hybridization, which would decrease the distance to the cysteine. Subsequently, a nucleophilic attack by the cysteine thiolate on the C(4a) carbocation would occur, leading to formation of the covalent FMN-C(4a)-thiol adduct. All these events may occur sequentially (8), or in a concerted fashion (17,18). In all cases, the sharp increase in the pKa of N(5) upon triplet formation is the switch that drives the photoreaction, via proton abstraction of a nearby donor. In support of this mechanism, ultrafast ultraviolet-visible (UV-V) experiments indicated a partial protonation of the FMN triplet state in LOV2 on a nanosecond timescale (8).
Figure 1.
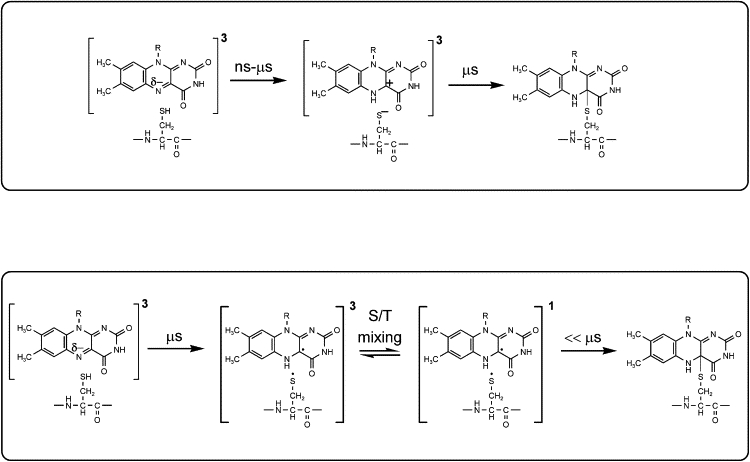
Proposed reaction mechanisms for light-driven covalent flavin-C(4a)-cysteinyl adduct formation in LOV domains. (Top) Ionic mechanism. (Bottom) Radical-pair mechanism. See text for details.
Alternatively, a radical-pair mechanism for covalent adduct formation was put forward, as depicted in Fig. 1 (bottom) (20,21). Upon promotion of the FMN chromophore to the triplet state, a hydrogen atom would be transferred from the conserved cysteine to N5 of the flavin, resulting in an FMNH•-H2C-S• radical pair. In the neutral flavin semiquinone radical, the unpaired electron density resides in the C(4a) atom. Such a radical pair would be formed in a triplet configuration. However, the proximity of the (heavy) sulfur radical to the isoalloxazine ring causes a strong spin-orbit coupling, inducing a rapid triplet-singlet interconversion. After the radical pair obtains an appreciable singlet character, radical-pair recombination between H2C-S• and the unpaired electron at C(4a) may take place, also resulting in the FMN-C(4a)-thiol adduct. In the context of this proposed mechanism, the trigger for adduct formation could either be an electron transfer from the cysteine to the flavin, followed by proton transfer, or a concerted mechanism whereby a hydrogen-atom transfer from the thiol to the flavin occurs (20–23). Ab initio quantum chemical calculations favor the radical-pair over the ionic mechanism, through hydrogen abstraction rather than electron transfer (24–27).
Time-resolved infrared spectroscopy is a powerful tool for assessing the molecular nature of transient intermediates in photoactive biomolecules (28–34). Here, we describe the results of an ultrafast blue-pump, mid-infrared probe spectroscopic study of the LOV2 domain of Avena sativa phototropin 1, to characterize the initial physical/chemical changes of the chromophore and its protein environment on the femtosecond to nanosecond timescale. We obtained the infrared signatures of the FMN singlet and triplet state in AsLOV2. The FMN singlet-excited state mid-infrared spectrum, recorded in a broad spectral window from 1750 cm−1 to 1300 cm−1, provided direct information about the principal stretch modes of the isoalloxazine moiety upon excitation. The primary photoproduct of AsLOV2 corresponds to an FMN triplet state that rises with a lifetime of 1.5 ns, because of the sulfur-induced, enhanced intersystem crossing of FMN by the reactive cysteine in (8,35). We report that the triplet-state spectrum in AsLOV2 is very similar to the nonprotonated triplet states observed in a flavin- model compound in aprotic solution according to time-resolved infrared (IR) spectroscopy (36), and to the photoaccumulated FMN triplet state at low temperature in Adiantum LOV2, as probed by Fourier transform infrared (FTIR) spectroscopy (37). The configuration interaction singles (CIS) method was used to calculate vibrational spectra of the first singlet and triplet excited states. The calculated harmonic frequencies of singlet and triplet excited states are in satisfactory agreement with the evident band positions and intensities. On the other hand, CIS calculations of a T1 cation that was protonated at the N(5) site did not reproduce the experimentally observed FMN triplet spectrum. The implications for the reaction mechanism of covalent adduct formation are discussed.
Materials and Methods
LOV2 expression, purification, and preparation
The LOV2 domain of Avena sativa (As, oat) phototropin 1 was expressed and purified as previously described (6). AsLOV2 was expressed from a construct spanning residues 404–560 (construct generously provided by K. Gardner of the Southwestern Medical Center, University of Texas, Dallas, TX). The samples in H2O and D2O buffers (20 mM Tris/HCl, pH/pD 8.0, and 50 mM NaCl) were concentrated (Amicon YM10, Millipore, Billerica,MA) and placed between two 2-mm-thick CaF2 plates separated by a 6-μm and 20-μm Teflon spacer, respectively. The samples had an absorbance of ∼0.2 at 447 nm and 1.2 and 0.8 at 1650 cm−1 for H2O and D2O samples. A more concentrated H2O sample, with an absorbance of ∼0.4 at 447 nm, was prepared to resolve the carbonyl region better between 1730 cm−1 and 1670 cm−1. During the experiments, the sample cell was continuously translated with a Lissajous scanner, which ensured sample refreshment after each laser shot and a time interval of 1 min between successive exposures to laser beams. In H2O, the dark recovery rate amounts to (40 s)−1. With a fraction of <20% of the LOV domains entering the photocycle with each shot and the given Lissajous return and dark recovery times, a fraction of <10% of the AsLOV2 sample is continuously accumulated in the covalent adduct state. Because the FMN-cysteinyl covalent adduct does not absorb at an excitation wavelength of 475 nm, the latter state will not be photo-excited, and hence will not contribute to the observed dynamics. The dark recovery rate of AsLOV2 is slowed down about three times in D2O (10) to (120 s)−1. We introduced 1 mM imidazole in the D2O samples, to catalyze the dark recovery to a rate of about (12 s)−1 (38), which ensured that a major fraction of AsLOV2 decays between successive laser shots (1 min).
Time-resolved mid-infrared spectroscopy
The experimental setup was a homebuilt spectrometer, based on a 1-kHz amplified Ti:sapphire laser system operating at 1 kHz (Spectra Physics Hurricane, Mountain View,CA) that allows for a visible-pump/mid-infrared probe in a time window from 180 fs to 3 ns, as previously described (31,39). The blue excitation pulse was generated by means of a noncollinear optical parametric amplifier and centered around 475 nm, at an excitation energy of 500 nJ. The infrared probe had a spectral width of 200 cm−1, was spectrally dispersed after the sample, and was detected with a 32-element array detector, leading to a spectral resolution of 6 cm−1. Vibrational spectra between 1720 cm−1 and 1300 cm−1 were taken in intervals and simultaneously analyzed. Spectra were recorded at 74 time-delay points between −15 ps and 3 ns.
Global analysis
Time-resolved spectra were analyzed with a global analysis program (40), using a kinetic model consisting of sequentially interconverting species, i.e., 1 → 2 → …, in which the arrows indicate successive monoexponential decays of increasing time constants. Associated with each species were a lifetime and a difference spectrum, denoted the evolution-associated difference spectrum (EADS). The ultrafast IR signals before and around zero time delay were affected by perturbed free induction decay (FID) and by cross-phase modulation artifacts. To avoid inclusion of the perturbed FID and cross-phase modulation signals in the global analysis, the signals from −10 ps to 200 fs were given a low weight in the fitting process.
Differential FTIR spectroscopy
Infrared light-minus-dark spectra were recorded using an FTIR spectrometer (IFS 66s, Bruker, Ettlingen, Germany) equipped with a nitrogen-cooled photovoltaic mercury cadmium telluride detector (20 MHz, KV 100, Kolmar Technologies Newburyport, MA). Background and sample interferograms are the average of 500 and 2000 interferograms, recorded at 4-cm−1 resolution, respectively, and were repeated three times. After Fourier transform, the obtained absorption spectrum represents the light-minus-dark FTIR difference spectrum. A blue LED emitting at 470 nm (5-mW output) was used for photo-conversion at saturating intensity.
Quantum-chemical calculations
Harmonic vibrations of a lumiflavin molecule in the electronically excited singlet and triplet states were computed with a quantum-chemical program PC GAMESS/Firefly (from Alex A. Granovsky, PC GAMESS/Firefly version 7.1.C, www http://classic.chem.msu.su/gran/gamess/index.html). We used the CIS/6–31(d)G method to calculate and compare the excited-state equilibrium geometries, Löowdin atomic charges, and harmonic vibrational spectra and normal modes. We also compared the energies of the N(1)-H and N(5)-H protonated lumiflavin cations in the first triplet state at their equilibrium geometries. The harmonic vibrations of the triplet N(5)-H cation were computed and compared with the neutral triplet lumiflavin.
Results and Discussion
Ultrafast mid-infrared spectroscopy of AsLOV2
Time-resolved mid-infrared (mid-IR) spectra of AsLOV2 in H2O were collected at frequencies between 1730 cm−1 and 1300 cm−1 and globally analyzed. Two components were required for an adequate description of the time-resolved data, with a lifetime of 1.5 ns and a component that did not decay on the timescale of the experiment (3 ns). The resulting EADS of AsLOV2 in H2O are shown in Fig. 2, with the 1.5-ns component represented by a black line and the nondecaying component by a gray line. Kinetic traces at representative vibrational frequencies are shown in Fig. 3. The EADS represent the mid-IR absorbance difference spectra of the molecular species in question with respect to those of the ground state, and show a bleaching of the FMN ground-state bands (negative signals) and induced absorption bands of the groups of the isoalloxazine ring of FMN and/or the protein that undergo photophysical/chemical transformation (positive signals). In this sense, the EADS can be interpreted in the same way as conventional differential FTIR spectra.
Figure 2.
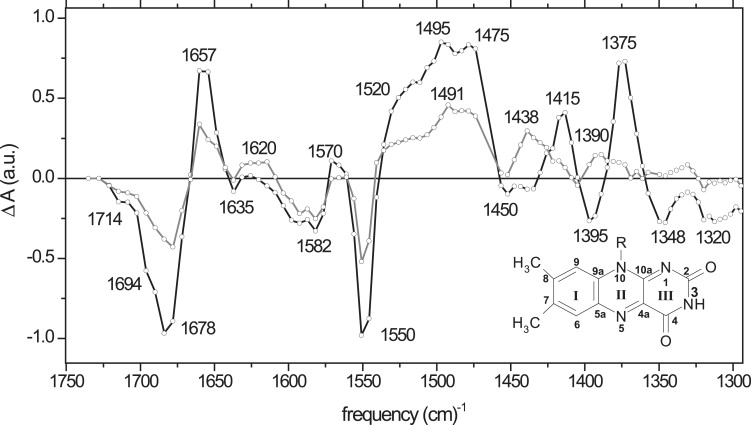
EADS that follow from a global analysis of ultrafast IR transient absorption experiments on A. sativa phot1 LOV2 (AsLOV2) domain in H2O. The excitation wavelength was 475 nm. The first EADS (black line) evolves in 1.5 ns to the second EADS (gray line), which does not decay on the timescale of the experiment.
Figure 3.
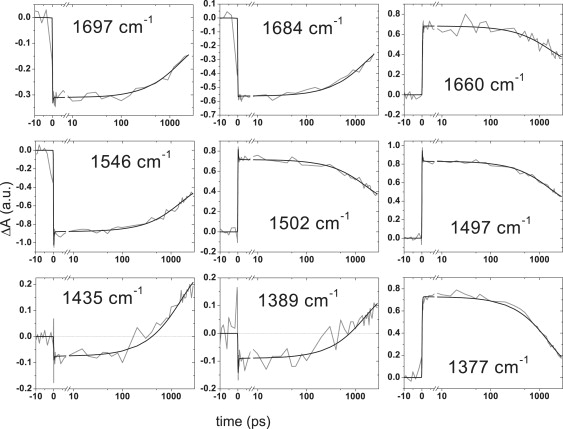
Kinetic traces at indicated mid-IR vibrational frequencies recorded in the AsLOV2 domain (gray line). The excitation wavelength was 475 nm. The result of global analysis is shown as a black line. Time axis is linear from −10 ps to 7 ps, and logarithmic thereafter.
The first EADS (Fig. 2, black line), with a 1.5-ns lifetime, is assigned to a population of the lowest singlet excited state of LOV2-bound FMN. It features negative signals at 1714 cm−1, 1694 cm−1, 1678 cm−1, 1637 cm−1, 1592 cm−1, 1582 cm−1, 1550 cm−1, 1395 cm−1, 1348 cm−1, and 1320 cm−1. Positive bands were found at 1657 cm−1, 1645 cm−1 (shoulder), 1570 cm−1, 1415 cm−1, and 1375 cm−1. Notably, around 1500 cm−1, a multiple-band structure is evident, with peaks at 1475 cm−1 and 1495 cm−1, and a pronounced shoulder at 1520 cm−1. The frequencies of the bleaching bands agree with those recorded with FTIR, Raman, and fluorescence line-narrowing spectroscopy on AsLOV2 and several other LOV domains upon covalent adduct formation (5,35,41,42). The observation of a single-exponential decay of the FMN singlet excited state in AsLOV2 agrees with that observed for essentially all LOV domains (8,9,43,44), in contrast with the multiexponential excited-state decays found in the flavin adenine dinucleotide (FAD)-binding blue-light sensing using FAD (BLUF) domains (34,45–47). The 1.5-ns time constant that was found here fairly agrees with that obtained previously with ultrafast visible spectroscopy (2.0 ns (8)).
The second EADS (Fig. 2, gray line) did not decay on the timescale of the experiment, had overall decreased amplitude, and was formed in 1.5 ns from the FMN singlet excited state. In accordance with previous ultrafast UV-V experiments, it is assigned to a population of the FMN triplet state. The second EADS exhibits essentially the same ground-state bleaching features as the first EADS, except at frequencies below 1425 cm−1, where the bleach pattern appears to be different. Relatively minor changes of induced absorption bands occur, with the appearance of a shoulder at 1645 cm−1, absorption near 1620 cm−1, a broadening and partial separation of bands at 1491 cm−1 and 1520 cm−1, and the appearance of absorption at 1438 cm−1 and 1390 cm−1.
Notably, the overall amplitude of the second EADS diminished with respect to the first EADS: the amplitude of the dominant 1678-cm−1 and 1550-cm−1 bleach signals decreased by ∼50%. Although compensation effects of partly overlapping bleach and induced absorption bands render a quantitative estimate uncertain, this observation indicates that the triplet yield in LOV2 amounts to ∼50%, which reasonably agrees with a previous estimate from ultrafast visible spectroscopy (∼60% (8)).
Infrared signature of AsLOV2 FMN singlet excited state
The first EADS of Fig. 2 shows a pattern of negative and positive absorption features that is characteristic of a population of the FMN singlet excited state in LOV2. The 1714-cm−1, 1694-cm−1, and 1678-cm−1 bleaches can be assigned to the C(4)=O and C(2)=O stretch frequencies of FMN in the ground state (5,35,41,42,48). The occurrence of three rather than two C=O frequencies will be discussed below. Upon promotion of FMN to the singlet excited state, the FMN C=O frequencies downshift, to result in a single resolved positive absorption at 1657 cm−1. A small bleach signal at 1637 cm−1 can be assigned to the ring I vibration of FMN (5,35,41,42). The bleach bands at 1582 cm−1 and 1550 cm−1 can be assigned to in-phase and out-of-phase C=N stretch modes of FMN, respectively (5,35,41,49). These bands correlate with the large, positive, multiple-band feature at 1475 cm−1, 1495 cm−1, and 1520 cm−1. Thus, the C=N stretch frequencies of FMN downshift in the singlet excited state. The small positive feature at 1565 cm−1 may also follow from a shifted C=N stretch frequency. The overall downshifting of the C=O and C=N (double) bond stretch modes in the singlet excited state is consistent with an overall bond-order decrease of the conjugated isoalloxazine system upon a π-π∗ transition. The bleach bands at 1395 cm−1, 1348 cm−1, and 1320 cm−1 contain the single-bond character of FMN, such as the C(4)-N(3), C(4)-C(4a), C(4a)-C(10a), and C(10a)-N(10) stretches (48,49).
The IR spectrum of the LOV2-bound FMN singlet excited state may be compared with those obtained of flavins in other systems. Wolf et al. reported on the IR spectrum of the singlet excited state of riboflavin in organic solvent (48). The observed bands of singlet-excited riboflavin were similar to those observed here, with slightly different frequencies and varying band intensities. The band assignments for the FMN singlet excited state in AsLOV2, as described above, are in line with those determined by CIS calculations for riboflavin by Wolf et al. (48).
Stelling et al. (50) and Kondo et al. (51) performed an ultrafast IR study on FAD in aqueous solution and bound to the AppA photoreceptor. In those studies, the ground-state FAD C=O bands in D2O solution were positioned at ∼1700 cm−1 and 1650 cm−1, i.e., significantly lower frequencies than observed here, which presumably resulted from extensive hydrogen bonding with the (deuterated) solvent water molecules. No positive absorption features of downshifted C=O frequencies were evident in the FAD singlet-excited state. The pattern of downshifted C=N stretches of FAD in D2O was similar to that observed here. In FAD bound to the AppA photoreceptor, the FAD C=O bleach maxima were similar to those observed in our study. Positive absorption at 1666 cm−1 (which appeared within the instrument response time) was assigned by the authors to an ultrafast keto-enol tautomerization of a glutamine side chain in proximity to FAD, resulting in a positive C=N absorption band. Strikingly, that band is very similar to the one observed in our study for the LOV2 domain (1657 cm−1). In LOV2, a conserved glutamine side chain is located in proximity to FMN, hydrogen-bonded to its C(4)=O group (17,52). However, we consider it unlikely that a keto-enol tautomerization of this glutamine occurs in LOV2 upon formation of the FMN singlet excited state. Rather, we think that the induced absorption at 1657 cm−1 results from a downshift of FMN C=O frequencies in the singlet excited state, because: 1), a downshift of C=O stretch frequencies is expected for a π-π∗ transition; 2) the 1657-cm−1 band appears within the instrument response function of 200 fs; and 3), the conserved glutamine has no role in LOV2 photochemistry (53). In our opinion, the 1666-cm−1 induced absorption observed in the AppA BLUF domain may have the same origin. Accordingly, a similar band that we observed in the Synechocystis Slr-1694 BLUF domain was interpreted as a downshifted C=O mode in the singlet excited state (34). In riboflavin in organic solvent, positive bands were observed at 1652 cm−1 and 1642 cm−1, which could be assigned to downshifted C(4)=O and C(2)=O stretches by means of CIS calculations (48).
In previous experiments on riboflavin in solution, AsLOV2, and FAD-binding BLUF domains, a vibrational cooling process occurring in ∼2 ps was observed in the singlet excited state after excitation at 400 nm (16,45,47,48). Here, the FMN cofactor is excited at 475 nm, in the 0-0 vibronic transition, and thus no excess vibrational energy is deposited in the system. Hence, no effects of vibrational cooling are observed in the transient IR spectra.
Infrared signature of FMN triplet state in AsLOV2
The nondecaying EADS of Fig. 2 was assigned to a population of the FMN triplet state in LOV2. The bleaches remain at the same frequencies as those of the singlet excited state, except at frequencies below 1425 cm−1, where the bleach pattern appears different. Strikingly, the induced absorption features of the C=O and C=N stretches are overall similar to those in the singlet excited state. The differences are relatively minor, with the appearance of a shoulder at 1645 cm−1, absorption near 1620 cm−1, and a broadening and partial separation of the bands at 1491 cm−1 and 1520 cm−1. Moreover, the pronounced absorption bands at 1415 cm−1 and 1375 cm−1 in the singlet excited state disappear, and are replaced by absorptions at 1438 cm−1 and 1390 cm−1 with lower amplitude.
The IR spectrum of the LOV2 FMN triplet state may be compared with the mid-IR triplet and radical spectra observed in a riboflavin tetra-acetate (RBTA) model compound in organic solvent (36). The triplet state of RBTA exhibited major bleach signals at 1716 cm−1, 1684 cm−1, 1588 cm−1, 1548 cm−1, and 1348 cm−1, and induced absorption maxima at 1652 cm−1, with a shoulder around 1645 cm−1, 1484 cm−1, 1436 cm−1, and 1380 cm−1, and thus showed a large similarity with the LOV2 triplet spectrum. On the other hand, the mid-IR spectra of the RBTA anion radical (RBTA•−) and the neutral semiquinone radical (RBTAH•) showed C=N-induced absorptions at 1524 cm−1 and 1500 cm−1 (anion radical) and 1532 cm−1 (neutral semiquinone) (36), and thus did not contribute to the LOV2 triplet spectrum in a major fashion. The LOV2 triplet spectrum is also very similar to the photo-accumulated FMN triplet spectrum of the Adiantum phys3 LOV2 domain observed with FTIR at low temperature (37).
On the basis of the UV-V signature of the FMN triplet in LOV2, it was previously proposed that protonation of the FMN triplet state would take place on a nanosecond timescale. The triplet spectra of free FMN in solution were found to be pH-dependent. At neutral pH, the triplet spectrum of free FMN recorded after several nanoseconds exhibited two distinct absorption bands near 650 nm and 710 nm. At pH 2, only one band was observed at 660 nm, which had previously been assigned to protonation of the FMN triplet at the N(5) site (54). The triplet spectra of LOV2-bound FMN of Adiantum phy3 and Avena phot1 could be well-approximated by a superposition of these two species, and may be interpreted as a partial protonation of the flavin triplet at N(5), taking place on a nanosecond timescale. Proton transfer would occur from the conserved cysteine's thiol to the FMN N(5), triggered by the increased basicity in the triplet state of the latter (8). However, this scenario is not supported by our results, because the FMN triplet IR signature in LOV2 is essentially identical to that of the model compound RBTA (36). The latter was dissolved in aprotic solvent (deutero-acetonitrile), implying that RBTA remained nonprotonated. As will be shown by the use of quantum chemical calculations, a putative protonation event at N(5) would immediately become apparent in the IR spectrum of the FMN triplet, through the appearance of distinct IR bands at other frequencies. Moreover, ultrafast IR experiments on a highly concentrated AsLOV2 sample, which probed the S-H stretch region around 2760 cm−1, did not reveal the bleach that would be expected from deprotonation of the conserved cysteine's thiol (results not shown). Thus, our data support the notion that LOV2-bound FMN in the triplet state remains nonprotonated during the first nanoseconds.
Sato et al. (37) recorded the FTIR spectrum of the photoaccumulated FMN triplet state of Adiantum phy3 LOV2 at low temperature, which featured C=N stretch downshifts from 1590–1546(−) cm−1 to 1493(+) cm−1 and 1439(+) cm−1, essentially identical to the shifts observed in this study. In addition, they observed a protonated cysteine thiol (37). We conclude that the low-temperature photo-accumulated FMN triplet in LOV2 and the one observed in real time, using ultrafast IR spectroscopy, have similar molecular properties, although the former corresponds to a nonreactive fraction of LOV2 domains that does not form the covalent adduct at low temperature.
Quantum chemical calculations on lumiflavin
For analysis of the lumiflavin vibrations in the singlet and triplet excited electronic states, we performed CIS calculations. To examine how the molecular properties of lumiflavin change upon electronic excitation, the results of the CIS calculation for the excited state were compared with the results of Hartree-Fock (HF) calculations for the ground state. In Fig. S1 of the Supporting Material, the optimized structures, atomic charges, and dipole moments of lumiflavin in the ground state, singlet excited state, and triplet state are shown. In Table S1, the vibrational frequencies of lumiflavin in the ground state, as they follow from the HF calculations, are summarized and compared with the observed modes. Table S2 and Table S3 show the vibrational frequencies of lumiflavin in the singlet and triplet excited states that resulted from CIS calculations, together with the experimentally observed vibrational modes. Fig. 4 shows the calculated frequencies in a bar plot, with the lumiflavin ground-state frequencies indicated negatively, and the singlet (black) and triplet (gray) frequencies as positive. For the top horizontal axis, a correction factor of 0.88 was applied, allowing a convenient comparison with the experimentally observed IR spectra of the LOV2-bound FMN singlet and triplet states depicted in Fig. 2. The results for the S1 state are essentially identical to those obtained for riboflavin (48).
Figure 4.
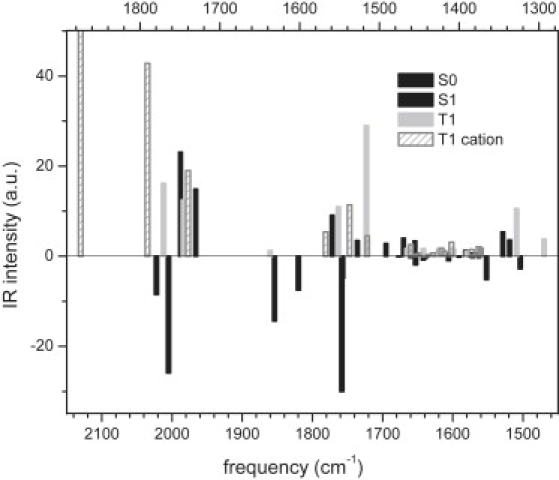
Calculated vibrational frequencies of lumiflavin, calculated by HF and CIS methods, for electronic ground state (negative-going solid bars), singlet excited state S1 (positive-going solid bars), triplet excited state T1 (gray bars), and triplet T1 cation protonated at N(5) (dashed bars). Bottom axis indicates frequencies as they follow directly from calculations, whereas for top axis, a correction factor of 0.88 was applied.
In the S1 and T1 equilibrium geometries, the C(4a)-N(5) and C(5a)-C(9a) distance, but not the C(10a)-N(1) distance, increases, and the N(5) atom becomes more electronegative compared with the ground electronic state (Fig. S1). The calculated result, that N(5) is more basic than N(1) in the flavin triplet state (11,17,36,55,56), indicates that in LOV domains, the increased basicity of N(5) likely constitutes a significant factor in the advancement of the adduct-formation reaction.
Although the S1 and T1 excited states originate from the same highest occupied molecular orbital-lowest unoccupied molecular orbitals ππ∗ electronic excitation, there are pronounced differences in their equilibrium structures, charge distribution, and harmonic frequencies. The C(4a)-C(10a) distance decreases more at S1 than at T1 equilibrium geometry. Lumiflavin is more polar, and the negative charge on the N(5) atom is larger in the S1 state than in the T1 state. The geometry of the benzene ring undergoes more pronounced changes in the S1 state than in the T1 state.
The fairly good correspondence between the computed and experimental frequencies (Fig. 4 and Fig. S2) allowed us to assign all observed positive bands (Table S1 and Table S3) except for the weak absorption around 1625 cm−1 in the excited singlet state, which has no corresponding frequency in the S1 CIS vibrational spectrum. As expected, the C=O double-bond stretching frequencies are more greatly overestimated, compared with other observed frequencies, because the model does not account for the hydrogen bonding of flavin O(4) and O(2) to the protein (52). We conclude that the CIS method provides a satisfactory description, accounting for the different molecular and electronic structures of the isoalloxazine ring in the S1 and T1 state. Importantly, the CIS method predicts the triplet isoalloxazine biradical with the spin density on C(4a), N(5), and the benzene ring (Fig. 5). A similar structure can be obtained from configuration interaction singles and doubles and complete active space self-consistent field calculations (results not shown) (26). However, this electronic structure is different from the one derived from the B3LYP calculations by Martin et al. (36), with unpaired spin density at N(5) and N(1). Next, we describe band assignments to the CIS harmonic normal modes.
Figure 5.
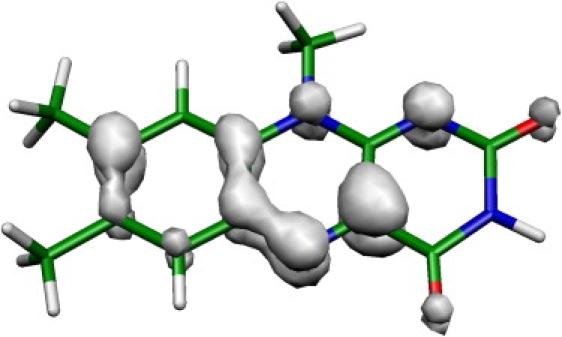
Calculated spin density in lumiflavin T1 electronic state.
Our calculations indicate different frequencies for the C(4)=O and C(2)=O vibrations in the ground, S1, and T1 states (Table S1 and Table S3). In LOV2, only one carbonyl frequency is resolved experimentally in the S1 state at 1657 cm−1, which likely indicates that the C(4)=O and C(2)=O frequencies have merged. In the T1 state, two frequencies are evident at 1660 cm−1 and 1645 cm−1. However, given the hydrogen bonding to the carbonyls, it is difficult to decide which frequency belongs to C(4)=O and C(2)=O. The weak 1620-cm−1 band in the experimentally observed triplet spectrum can be associated with the calculated 1860-cm−1 mode (Table S3), and is in part assigned to C=N double-bond stretching in the triplet state. In the singlet excited state, the first C=C and C=N double-bond stretch frequency appears at 1570 cm−1 (1772 cm−1 in the calculations). These two bands seem to be characteristic, and reveal the different structure of the benzene ring in the singlet and triplet excited states. The observed double-bond structure at 1520 cm−1 and 1491 cm−1, which is slightly different in the two excited states, can be assigned to the downshifted stretching vibrations of the C=N and C=C double bonds. In the T1 state, the corresponding computed modes at 1763 cm−1 and 1721 cm−1 contain a contribution from N(5) atom displacements. In the S1 state, the N(5) displacements contribute to an observed band near 1480 cm−1 (computed modes at 1670 cm−1 and 1654 cm−1). In the S1 state, the shortened C(4a)-C(10a) bond contributes to the observed 1495-cm−1 band (calculated at 1695 cm−1). In the triplet state, the C(4a)-C(10a) distance is somewhat larger, and its stretching vibration can be assigned to the 1438-cm−1 band (calculated frequency at 1642 cm−1), in agreement with the previous assignment. The band at 1390 cm−1 in the T1 state and at 1415 cm−1 in the S1 state are assigned to the ring I and II single-bond stretching vibrations (calculated frequencies at 1599 cm−1 and 1620 cm−1, respectively). The band at 1375 cm−1 in the singlet state is attributable to the stretching vibrations of uracil ring III (calculated at 1582 cm−1). According to our computations, there should be a similar vibration with a close frequency in the triplet state that can be assigned to the shoulder at 1375 cm−1.
We conclude that CIS calculations on lumiflavin reproduce the vibrational spectra of the LOV2-bound FMN singlet and triplet excited states with reasonable accuracy. This good agreement supports the notion that in LOV2, the FMN triplet state remains nonprotonated on the nanosecond timescale. To investigate this point further, we calculated both N(5)-H and N(1)-H protonated lumiflavin cations in the first triplet excited state. At the CIS/6–31(d)G level of theory, the former is characterized by a 16.5-kcal/mol lower energy. Hence, in agreement with previous data, the N(5) site is more basic compared with the N(1) site. In the triplet N(5)-H isoalloxazine cation, electronic density is redistributed such that conjugation between rings is disrupted, and some “double” bonds acquire even more double-bond character than in the ground-state neutral isoalloxazine. Computed geometry (Fig. S3) and vibrations (Table S4) indicate these bonds to be C(6)=C(7), C(8)=C(9), and C(2)=O. Calculations predict an upshift of one of the C=O stretching vibrations, and a significant upshift of the first C=C and C=N double-bond stretching frequency. Therefore, upon N(5) protonation, we expect the 1620-cm−1 triplet-state absorption to move up in the C=O stretching region, as indicated in Fig. 4 (dashed bars). None of these predicted frequency upshifts were observed experimentally, which strengthens our conclusion (see above) that the FMN triplet state does not become protonated in the triplet excited state, at least not on a nanosecond timescale.
Implications of LOV2 covalent adduct formation reaction mechanism
Our results of ultrafast IR spectroscopy on AsLOV2 indicate that the FMN triplet state is formed from the FMN singlet excited state, at a considerable yield of ∼50%. Furthermore, we demonstrated that the FMN triplet state in LOV2 remains nonprotonated on the nanosecond timescale. Therefore, the previously hypothesized model that protonation of the FMN triplet on a nanosecond timescale by cysteine takes place, which in turn would lead to adduct formation through a nucleophilic attack on a microsecond timescale (8), is probably not correct. In the context of an ionic model, this may imply that excited-state proton transfer from cysteine to FMN occurs in concert with a nucleophilic attack (Fig. 1, top) (11,17,18), and in fact constitutes the rate-limiting step. The latter possibility is supported by the observation that hydrogen/deuterium (H/D) exchange in LOV2 slows down covalent adduct formation from 2 μs to 10 μs (10).
Kay et al. (20) and Schleicher et al. (21) discussed the improbability of an ionic mechanism for adduct formation by noting that in such a case, the covalent adduct has to be formed in its triplet state, which would be energetically highly unfavorable. A direct formation of the adduct singlet ground state from the FMN triplet state would be a spin-forbidden process, and if preceded by a triplet-to-singlet transition to the FMN ground state, the FMN would deprotonate rapidly before the adduct could be formed. Instead, by observing FMNH• neutral semiquinone radicals in cysteine mutants of AsLOV2, Kay et al. (20) and Schleicher et al. (21) proposed that adduct formation proceeds via a radical-pair mechanism (Fig. 1, bottom). This mechanism was supported by experiments on a site-directed mutant of Chlamydomonas LOV1, where cysteine was replaced by methionine (22,23). In addition, quantum-chemical calculations by a number of research groups invariably favored the radical-pair over the ionic mechanism, through energetic considerations of the transition state (24–27). On the basis of cryotrapping experiments, it was argued that electron transfer from the conserved cysteine to FMN constitutes the rate-limiting reaction, after which, proton transfer leads to FMNH• formation (21). However, the observation that H/D exchange in LOV2 slows down the time constant of covalent adduct formation fivefold (10) supports hydrogen transfer as the rate-limiting step for FMNH• formation.
No direct spectroscopic observation of the occurrence of flavin radicals was reported in the photocycle of LOV domains (8,10–13). However, by their nature, such radicals would be a short-lived intermediate between the flavin triplet and covalent adduct states, and thus would easily escape detection. We conclude that so far, the question remains unsettled of whether an ionic or radical-pair mechanism is in operation in LOV domains.
Heterogeneity in FMN C=O vibrational frequencies
Fig. 6 C reproduces the EADS of Fig. 2, zooming in on the spectral region of the FMN carbonyl signals (1750–1600 cm−1). Close inspection of the carbonyl bleach signals at 1714–1678 cm−1 shows a fine structure, with bleach minima at 1678 cm−1 and 1714 cm−1, and a shoulder at 1694 cm−1. Fig. 6 D shows the EADS of a highly concentrated AsLOV2 sample in H2O, where the carbonyl bleach bands are better resolved, clearly showing three distinct bleach minima. The three bleach bands rise within the instrument response of the apparatus (200 fs), indicating that these modes can be assigned to the LOV2-bound FMN chromophore. Thus, three carbonyl vibrational modes are associated with FMN in dark-state AsLOV2, with frequencies at 1678 cm−1, 1694 cm−1, and 1714 cm−1. This is a rather striking observation, because only two carbonyl modes are expected for flavins, i.e., the C(2)=O and C(4)=O modes (49). Previously, the 1714-cm−1 mode was assigned to LOV-bound FMN C(4)=O, whereas the 1678-cm−1 mode was assigned to C(2)=O (5,41,42). The 1694-cm−1 mode was evident in several LOV FTIR spectra, but not assigned. Here, we unambiguously demonstrate that the latter is associated with the FMN chromophore.
Figure 6.
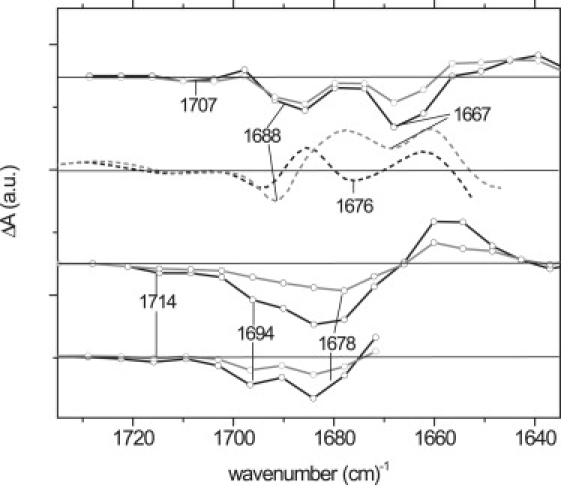
EADS that follow from a global analysis of ultrafast IR transient absorption experiments on A. sativa AsLOV2 domain in carbonyl region in D2O (A) and H2O (C). Black lines denote first EADS, with a lifetime of 1.5 ns, and gray lines denote nondecaying EADS. (D) EADS of a highly concentrated AsLOV2 sample in H2O, where three FMN carbonyls bands are clearly resolved. (B) AsLOV2 light-minus-dark FTIR spectra in H2O (dotted black line) and D2O (dotted gray line).
To characterize further the FMN carbonyl vibrational pattern in LOV2, we performed ultrafast IR spectroscopy on AsLOV2 dissolved in D2O. The time-resolved data were globally analyzed, using a sequential kinetic scheme. As with AsLOV2 in H2O, one lifetime of 1.5 ns and a component that did not decay on the timescale of the experiment were found. The resulting EADS are shown in Fig. 6 A. The first EADS (black line) is assigned to the FMN singlet excited state. Compared with AsLOV2 in H2O, significant shifts were observed between 1730 cm−1 and 1650 cm−1, with bleach minima at 1707 cm−1, 1688 cm−1, and 1667 cm−1, and an induced absorption at 1642 cm−1. Thus, the band previously assigned to C(4)=O at 1714 cm−1 in H2O (42) downshifts to 1707 cm−1 in D2O. The large carbonyl bleach envelope in H2O, containing the 1694-cm−1 shoulder and 1678-cm−1 minimum, downshifts and splits into two distinct bands at 1688 cm−1 and 1667 cm−1 in D2O. The downshift of carbonyl frequencies is mainly attributable to deuteration of the FMN N(3)-H bond, because both of these modes also involve a substantial contribution from the N(3)-H(D) bend (57). For comparison, the light-minus-dark FTIR spectra of AsLOV2 in H2O and D2O are shown in Fig. 6 B (dashed black and gray lines, respectively). Similar downshifts of FMN upon H/D exchange are evident in these spectra.
The extent of downshift upon H/D exchange indicates that the 1694(−)-cm−1 band belongs to FMN C(4)=O. We observe that the 1694(−)-cm−1 band downshifts by 6 cm−1, to 1688(−) cm−1. This is comparable to the shift of the C(4)=O band at 1714 cm−1, which downshifts by 7 cm−1 to 1707 cm−1. In contrast, the C(2)=O band at 1678(−) cm−1 undergoes a significantly larger downshift by 11 cm−1 to 1667 cm−1. Thus, the 1714-cm−1 and 1694-cm−1 bands belong to FMN C(4)=O, and the 1678-cm−1 band to FMN C(2)=O. Comparable FMN carbonyl downshifts were evident in the Slr-1694 BLUF domain from Synechocystis upon H/D exchange (58).
The presence of two distinct FMN C(4)=O vibrational frequencies at 1714 cm−1 and 1694 cm−1 in AsLOV2 is interpreted in terms of conformational heterogeneity in the LOV2 domain involving two different C(4)=O conformer populations that are singly and doubly H-bonded with the protein environment, respectively, and that coexist in the dark state. Such conformers display slightly shifted absorption spectra, and likely cause a splitting of the 475-nm band in the UV-V spectra of LOV domains at low temperature (Alexandre et al. in the accompanying article).
Conclusions
We report on an ultrafast blue-pump, mid-IR probe spectroscopic study of the LOV2 domain of Avena sativa phototropin 1 (AsLOV2). Two components were required for an adequate description of the time-resolved data, with a lifetime of 1.5 ns and a component that did not decay on the timescale of the experiment. These two kinetic components were assigned to the singlet excited (S1) and the triplet excited (T1) states of LOV2-bound FMN, respectively. The T1 state is formed from the S1 state, with a yield of ∼50%. As expected for a π-π∗ electronic transition, the C=O and C=N bonds of the isoalloxazine moiety acquire a single-bond character in the S1 state, characterized by bandshifts of their main normal modes at ∼1700(−)/1657(+) cm−1 and 1582;1550(−)/1520;1495;1475(+) cm−1, respectively. Upon triplet formation, the IR bands associated with C=O and C=N stretches are overall similar to the singlet excited state, but show some significant shifts. Pronounced absorption bands at 1415 cm−1 and 1375 cm−1 in the S1 state disappear, and are replaced by absorptions at 1438 cm−1 and 1390 cm−1 in the T1 state. The harmonic frequencies of the S1 and T1 excited states, as calculated by the CIS method, are in satisfactory agreement with the observed band positions. On the other hand, CIS calculations of a T1 cation protonated at the N(5) site did not reproduce the experimentally observed FMN T1 spectrum in LOV2. This study indicates that the FMN triplet state of AsLOV2 remains nonprotonated on the nanosecond timescale, which rules out a previously hypothesized ionic mechanism for covalent adduct formation involving cysteine-N(5) proton transfer on this timescale (8). In addition, we report that the LOV2-bound FMN chromophore in the dark exhibits three carbonyl stretch modes at 1678 cm−1, 1714 cm−1, and 1694 cm−1, of which the former is assigned to C(2)=O and the latter two to C(4)=O. The dual C(4)=O frequencies are explained by a heterogeneous population of FMN C(4)=O conformers with single and double H-bonds with the protein environment.
Supporting Material
Three figures and four tables are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)00804-2.
Supporting Material
Acknowledgments
The A. sativa phototropin 1 LOV2 construct was generously provided by Kevin Gardner (Southwestern Medical Center, University of Texas, Dallas, TX).
J.T.M.K. was supported by the Earth and Life Sciences Council of the Netherlands Foundation for Scientific Research via a VIDI grant. M.T.A.A. was supported by the Netherlands Foundation for Scientific Research through the Molecule to Cell Program and the Human Frontier Science Program. T.D. was supported by the Deutsche Forschungsgemeischaft through Research Group FOR526, Sensory Blue Light Receptors.
Footnotes
Luuk J. G. W. van Wilderen's present address is the Division of Molecular Biosciences, Faculty of Natural Sciences, South Kensington Campus, Imperial College London, London SW7 2AZ, United Kingdom.
References
- 1.Christie J.M., Briggs W.R. Blue-light sensing and signaling by the phototropins. In: Briggs W.R., Spudich J.L., editors. Handbook of Photosensory Receptors. Wiley-VCH Verlag GmbH & Co.; Weinheim, Germany: 2005. pp. 277–304. [Google Scholar]
- 2.Christie J.M. Phototropin blue-light receptors. Annu. Rev. Plant Biol. 2007;58:21–45. doi: 10.1146/annurev.arplant.58.032806.103951. [DOI] [PubMed] [Google Scholar]
- 3.Salomon M., Eisenreich W., Durr H., Schleicher E., Knieb E. An optomechanical transducer in the blue light receptor phototropin from. Avena sativa. Proc. Natl. Acad. Sci. USA. 2001;98:12357–12361. doi: 10.1073/pnas.221455298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crosson S., Moffat K. Photoexcited structure of a plant photoreceptor domain reveals a light-driven molecular switch. Plant Cell. 2002;14:1067–1075. doi: 10.1105/tpc.010475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swartz T.E., Wenzel P.J., Corchnoy S.B., Briggs W.R., Bogomolni R.A. Vibration spectroscopy reveals light-induced chromophore and protein structural changes in the LOV2 domain of the plant blue-light receptor phototropin 1. Biochemistry. 2002;41:7183–7189. doi: 10.1021/bi025861u. [DOI] [PubMed] [Google Scholar]
- 6.Harper S.M., Neil L.C., Gardner K.H. Structural basis of a phototropin light switch. Science. 2003;301:1541–1544. doi: 10.1126/science.1086810. [DOI] [PubMed] [Google Scholar]
- 7.Harper S.M., Christie J.M., Gardner K.H. Disruption of the LOV-J alpha helix interaction activates phototropin kinase activity. Biochemistry. 2004;43:16184–16192. doi: 10.1021/bi048092i. [DOI] [PubMed] [Google Scholar]
- 8.Kennis J.T.M., Crosson S., Gauden M., van Stokkum I.H.M., Moffat K. Primary reactions of the LOV2 domain of phototropin, a plant blue-light photoreceptor. Biochemistry. 2003;42:3385–3392. doi: 10.1021/bi034022k. [DOI] [PubMed] [Google Scholar]
- 9.Holzer W., Penzkofer A., Fuhrmann M., Hegemann P. Spectroscopic characterization of flavin mononucleotide bound to the LOV1 domain of Phot1 from. Chlamydomonas reinhardtii. Photochem. Photobiol. 2002;75:479–487. doi: 10.1562/0031-8655(2002)075<0479:scofmb>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 10.Corchnoy S.B., Swartz T.E., Lewis J.W., Szundi I., Briggs W.R. Intramolecular proton transfers and structural changes during the photocycle of the LOV2 domain of phototropin 1. J. Biol. Chem. 2003;278:724–731. doi: 10.1074/jbc.M209119200. [DOI] [PubMed] [Google Scholar]
- 11.Swartz T.E., Corchnoy S.B., Christie J.M., Lewis J.W., Szundi I. The photocycle of a flavin-binding domain of the blue light photoreceptor phototropin. J. Biol. Chem. 2001;276:36493–36500. doi: 10.1074/jbc.M103114200. [DOI] [PubMed] [Google Scholar]
- 12.Kottke T., Heberle J., Hehn D., Dick B., Hegemann P. Phot-LOV1: photocycle of a blue-light receptor domain from the green alga Chlamydomonas reinhardtii. Biophys. J. 2003;84:1192–1201. doi: 10.1016/S0006-3495(03)74933-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo H.M., Kottke T., Hegemann P., Dick B. The Phot LOV2 domain and its interaction with LOV1. Biophys. J. 2005;89:402–412. doi: 10.1529/biophysj.104.058230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Losi A., Quest B., Gartner W. Listening to the blue: the time-resolved thermodynamics of the bacterial blue-light receptor YtvA and its isolated LOV domain. Photochem. Photobiol. Sci. 2003;2:759–766. doi: 10.1039/b301782f. [DOI] [PubMed] [Google Scholar]
- 15.Losi A., Kottke T., Hegemann P. Recording of blue light-induced energy and volume changes within the wild-type and mutated Phot-LOV1 domain from Chlamydomonas reinhardtii. Biophys. J. 2004;86:1051–1060. doi: 10.1016/S0006-3495(04)74180-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kennis J.T.M., Alexandre M.T.A. Mechanisms of light activation in flavin-binding photoreceptors. In: Silva E., Edwards A.M., editors. Flavins: Photochemistry and Photobiology. Royal Society for Chemistry Publishing; Cambridge, UK: 2006. pp. 287–319. [Google Scholar]
- 17.Crosson S., Moffat K. Structure of a flavin-binding plant photoreceptor domain: insights into light-mediated signal transduction. Proc. Natl. Acad. Sci. USA. 2001;98:2995–3000. doi: 10.1073/pnas.051520298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fedorov R., Schlichting I., Hartmann E., Domratcheva T., Fuhrmann M. Crystal structures and molecular mechanism of a light-induced signaling switch: the Phot-LOV1 domain from Chlamydomonas reinhardtii. Biophys. J. 2003;84:2474–2482. doi: 10.1016/S0006-3495(03)75052-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swartz T.E., Bogomolni R.A. LOV-domain photochemistry. In: Briggs W.R., Spudich J.L., editors. Handbook of Photosensory Receptors. Wiley-VCH Verlag GmbH & Co.; Weinheim, Germany: 2005. pp. 305–323. [Google Scholar]
- 20.Kay C.W.M., Schleicher E., Kuppig A., Hofner H., Rudiger W. Blue light perception in plants—detection and characterization of a light-induced neutral flavin radical in a C450A mutant of phototropin. J. Biol. Chem. 2003;278:10973–10982. doi: 10.1074/jbc.M205509200. [DOI] [PubMed] [Google Scholar]
- 21.Schleicher E., Kowalczyk R.M., Kay C.W.M., Hegemann P., Bacher A. On the reaction mechanism of adduct formation in LOV domains of the plant blue-light receptor phototropin. J. Am. Chem. Soc. 2004;126:11067–11076. doi: 10.1021/ja049553q. [DOI] [PubMed] [Google Scholar]
- 22.Kottke T., Dick B., Fedorov R., Schlichting I., Deutzmann R. Irreversible photoreduction of flavin in a mutated Phot-LOV1 domain. Biochemistry. 2003;42:9854–9862. doi: 10.1021/bi034863r. [DOI] [PubMed] [Google Scholar]
- 23.Bittl R., Kay C.W.M., Weber S., Hegemann P. Characterization of a flavin radical product in a C57M mutant of a LOV1 domain by electron paramagnetic resonance. Biochemistry. 2003;42:8506–8512. doi: 10.1021/bi034123i. [DOI] [PubMed] [Google Scholar]
- 24.Neiss C., Saalfrank P. Ab initio quantum chemical investigation of the first steps of the photocycle of phototropin: a model study. Photochem. Photobiol. 2003;77:101–109. doi: 10.1562/0031-8655(2003)077<0101:aiqcio>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 25.Dittrich M., Freddolino P.L., Schulten K. When light falls in LOV: a quantum mechanical/molecular mechanical study of photoexcitation in Phot-LOV1 of Chlamydomonas reinhardtii. J. Phys. Chem. B. 2005;109:13006–13013. doi: 10.1021/jp050943o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Domratcheva T., Fedorov R., Schlichting I. Analysis of the primary photocycle reactions occurring in the light, oxygen, and voltage blue-light receptor by multiconfigurational quantum-chemical methods. J. Chem. Theory Comput. 2006;2:1565–1574. doi: 10.1021/ct0600114. [DOI] [PubMed] [Google Scholar]
- 27.Zenichowski K., Gothe M., Saalfrank P. Exciting flavins: absorption spectra and spin-orbit coupling in light-oxygen-voltage (LOV) domains. J. Photochem. Photobiol. Chem. 2007;190:290–300. [Google Scholar]
- 28.Herbst J., Heyne K., Diller R. Femtosecond infrared spectroscopy of bacteriorhodopsin chromophore isomerization. Science. 2002;297:822–825. doi: 10.1126/science.1072144. [DOI] [PubMed] [Google Scholar]
- 29.Heyne K., Mohammed O.F., Usman A., Dreyer J., Nibbering E.T.J. Structural evolution of the chromophore in the primary stages of trans/cis isomerization in photoactive yellow protein. J. Am. Chem. Soc. 2005;127:18100–18106. doi: 10.1021/ja051210k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Thor J.J., Ronayne K.L., Towrie M. Formation of the early photoproduct Lumi-R of cyanobacterial phytochrome Cph1 observed by ultrafast mid-infrared spectroscopy. J. Am. Chem. Soc. 2007;129:126–132. doi: 10.1021/ja0660709. [DOI] [PubMed] [Google Scholar]
- 31.Groot M.L., van Wilderen L.J.G.W., Di Donato M. Femtosecond time-resolved and dispersed infrared spectroscopy on proteins. Photochem. Photobiol. Sci. 2007;6:501–507. doi: 10.1039/b613023b. [DOI] [PubMed] [Google Scholar]
- 32.van Wilderen L.J.G.W., van der Horst M.A., van Stokkum I.H.M., Hellingwerf K.J., van Grondelle R. Ultrafast infrared spectroscopy reveals a key step for successful entry into the photocycle for photoactive yellow protein. Proc. Natl. Acad. Sci. USA. 2006;103:15050–15055. doi: 10.1073/pnas.0603476103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kennis J.T.M., Groot M.L. Ultrafast spectroscopy of biological photoreceptors. Curr. Opin. Struct. Biol. 2007;17:623–630. doi: 10.1016/j.sbi.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 34.Bonetti C., Mathes T., van Stokkum I.H.M., Mullen K.M., Groot M.L. Hydrogen bond switching among flavin and amino acid side chains in the BLUF photoreceptor observed by ultrafast infrared spectroscopy. Biophys. J. 2008;95:4790–4802. doi: 10.1529/biophysj.108.139246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alexandre M.T.A., van Grondelle R., Hellingwerf K.J., Robert B., Kennis J.T.M. Perturbation of the ground-state electronic structure of FMN by the conserved cysteine in phototropin LOV2 domains. Phys. Chem. Chem. Phys. 2008;10:6693–6702. doi: 10.1039/b810040c. [DOI] [PubMed] [Google Scholar]
- 36.Martin C.B., Tsao M.L., Hadad C.M., Platz M.S. The reaction of triplet flavin with indole. A study of the cascade of reactive intermediates using density functional theory and time resolved infrared spectroscopy. J. Am. Chem. Soc. 2002;124:7226–7234. doi: 10.1021/ja0123711. [DOI] [PubMed] [Google Scholar]
- 37.Sato Y., Iwata T., Tokutomi S., Kandori H. Reactive cysteine is protonated in the triplet excited state of the LOV2 domain in Adiantum phytochrome3. J. Am. Chem. Soc. 2005;127:1088–1089. doi: 10.1021/ja0436897. [DOI] [PubMed] [Google Scholar]
- 38.Alexandre M.T.A., Arents J.C., van Grondelle R., Hellingwerf K.J., Kennis J.T.M. A base-catalyzed mechanism for dark state recovery in the Avena sativa phototropin-1 LOV2 domain. Biochemistry. 2007;46:3129–3137. doi: 10.1021/bi062074e. [DOI] [PubMed] [Google Scholar]
- 39.Groot M.L., van Wilderen L., Larsen D.S., van der Horst M.A., van Stokkum I.H.M. Initial steps of signal generation in photoactive yellow protein revealed with femtosecond mid-infrared spectroscopy. Biochemistry. 2003;42:10054–10059. doi: 10.1021/bi034878p. [DOI] [PubMed] [Google Scholar]
- 40.van Stokkum I.H.M., Larsen D.S., van Grondelle R. Global and target analysis of time-resolved spectra. Biochim. Biophys. Acta (Bioenerg.) 2004;1657:82–104. doi: 10.1016/j.bbabio.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 41.Ataka K., Hegemann P., Heberle J. Vibrational spectroscopy of an algal Phot-LOV1 domain probes the molecular changes associated with blue-light reception. Biophys. J. 2003;84:466–474. doi: 10.1016/S0006-3495(03)74866-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iwata T., Nozaki D., Sato Y., Sato K., Nishina Y. Identification of the C=O stretching vibrations of FMN and peptide backbone by 13C-labeling of the LOV2 domain of Adiantum phytochrome3. Biochemistry. 2006;45:15384–15391. doi: 10.1021/bi061837v. [DOI] [PubMed] [Google Scholar]
- 43.Holzer W., Penzkofer A., Hegemann P. Absorption and emission spectroscopic characterisation of the LOV2-His domain of phot from Chlamydomonas reinhardtii. Chem. Phys. 2005;308:79–91. doi: 10.1016/j.jphotobiol.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 44.Schuttrigkeit T.A., Kompa C.K., Salomon M., Rudiger W., Michel-Beyerle M.E. Primary photophysics of the FMN binding LOV2 domain of the plant blue light receptor phototropin of Avena sativa. Chem. Phys. 2003;294:501–508. [Google Scholar]
- 45.Gauden M., Yeremenko S., Laan W., van Stokkum I.H.M., Ihalainen J.A. Photocycle of the flavin-binding photoreceptor AppA, a bacterial transcriptional antirepressor of photosynthesis genes. Biochemistry. 2005;44:3653–3662. doi: 10.1021/bi047359a. [DOI] [PubMed] [Google Scholar]
- 46.Gauden M., van Stokkum I.H., Key J.M., Luhrs D.C., van Grondelle R. Hydrogen-bond switching through a radical pair mechanism in a flavin-binding photoreceptor. Proc. Natl. Acad. Sci. USA. 2006;103:10895–10900. doi: 10.1073/pnas.0600720103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gauden M., Grinstead J.S., Laan W., van Stokkum I.H.M., Avila-Perez M. On the role of aromatic side chains in the photoactivation of BLUF domains. Biochemistry. 2007;46:7405–7415. doi: 10.1021/bi7006433. [DOI] [PubMed] [Google Scholar]
- 48.Wolf M.M.N., Schumann C., Gross R., Domratcheva T., Diller R. Ultrafast infrared spectroscopy of riboflavin: dynamics, electronic structure, and vibrational mode analysis. J. Phys. Chem. B. 2008;112:13424–13432. doi: 10.1021/jp804231c. [DOI] [PubMed] [Google Scholar]
- 49.Abe M., Kyogoku Y. Vibrational analysis of flavin derivatives—normal coordinate treatments of lumiflavin. Spectrochim. Acta [A] Mol. Biomol. Spectrosc. 1987;43:1027–1037. [Google Scholar]
- 50.Stelling A.L., Ronayne K.L., Nappa J., Tonge P.J., Meech S.R. Ultrafast structural dynamics in BLUF domains: transient infrared spectroscopy of AppA and its mutants. J. Am. Chem. Soc. 2007;129:15556–15564. doi: 10.1021/ja074074n. [DOI] [PubMed] [Google Scholar]
- 51.Kondo M., Nappa J., Ronayne K.L., Stelling A.L., Tonge P.J. Ultrafast vibrational spectroscopy of the flavin chromophore. J. Phys. Chem. B. 2006;110:20107–20110. doi: 10.1021/jp0650735. [DOI] [PubMed] [Google Scholar]
- 52.Halavaty A.S., Moffat K. N- and C-terminal flanking regions modulate light-induced signal transduction in the LOV2 domain of the blue light sensor phototropin 1 from Avena sativa. Biochemistry. 2007;46:14001–14009. doi: 10.1021/bi701543e. [DOI] [PubMed] [Google Scholar]
- 53.Nozaki D., Iwata T., Ishikawa T., Todo T., Tokutomi S. Role of Gln1029 in the photoactivation processes of the LOV2 domain in Adiantum phytochrome3. Biochemistry. 2004;43:8373–8379. doi: 10.1021/bi0494727. [DOI] [PubMed] [Google Scholar]
- 54.Sakai M., Takahashi H. One-electron photoreduction of flavin mononucleotide: time-resolved resonance Raman and absorption study. J. Mol. Struct. 1996;379:9–18. [Google Scholar]
- 55.Song P.S. On basicity of excited state of flavins. Photochem. Photobiol. 1968;7:311–313. doi: 10.1111/j.1751-1097.1968.tb08018.x. [DOI] [PubMed] [Google Scholar]
- 56.Song P.S. Electronic structure and photochemistry of flavins. 4. Sigma-electronic structure and lowest triplet configuration of a flavin. J. Phys. Chem. 1968;72:536–542. [Google Scholar]
- 57.Kim M., Carey P.R. Observation of a carbonyl feature for riboflavin bound to riboflavin-binding protein in the red-excited Raman spectrum. J. Am. Chem. Soc. 1993;115:7015–7016. [Google Scholar]
- 58.Masuda S., Hasegawa K., Ishii A., Ono T. Light-induced structural changes in a putative blue-light receptor with a novel FAD binding fold sensor of blue-light using FAD (BLUF): Slr1694 of Synechocystis sp PCC6803. Biochemistry. 2004;43:5304–5313. doi: 10.1021/bi049836v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.