

Abstract
hERG1 is a member of the cyclic nucleotide binding domain family of K+ channels. Alignment of cyclic nucleotide binding domain channels revealed an evolutionary conserved sequence HwX(A/G)C in the S5 domain. We reasoned that histidine 562 in hERG1 could play an important structure-function role. To explore this role, we created in silica models of the hERG1 pore domain based on the KvAP crystal structure with Rosetta-membrane modeling and molecular-dynamics simulations. Simulations indicate that the H562 residue in the S5 helix spans the gap between the S5 helix and the pore helix, stabilizing the pore domain, and that mutation at the H562 residue leads to a disruption of the hydrogen bonding to T618 and S621, resulting in distortion of the selectivity filter. Analysis of the simulated point mutations at positions 562/618/621 showed that the reciprocal double mutations H562W/T618I would partially restore the orientation of the 562 residue. Matching hydrophobic interactions between mutated W562 residue and I618 partially compensate for the disrupted hydrogen bonding. Complementary in vitro electrophysiological studies confirmed the results of the molecular-dynamics simulations on single mutations at positions 562, 618, and 621. Experimentally, mutations of the H562 to tryptophan produced a functional channel, but with slowed deactivation and shifted V1/2 of activation. Furthermore, the double mutation T618I/H562W rescued the defects seen in activation, deactivation, and potassium selectivity seen with the H562W mutation. In conclusion, interactions between H562 in the S5 helix and amino acids in the pore helix are important determinants of hERG1 potassium channel function, as confirmed by theory and experiment.
Introduction
hERG1 is a member of the ether-a-go-go (EAG) family of genes that encode voltage-gated potassium channels (1–3). More broadly, hERG1 is a member of the cyclic nucleotide binding domain (CNBD) family of channels, which includes hyperpolarization activated channels, cyclic nucleotide gated channels, and numerous plant and bacterial channels (4). Human mutations in the hERG1 gene lead to long-QT syndrome, which can lead to sudden cardiac death (3). Promiscuous binding of drugs within the inner vestibule of the hERG1 channel leads to drug-induced long-QT syndrome (5–8). This is a major problem for the pharmaceutical industry in terms of drug development, resulting in a need to further understand the structure-function relationship of hERG1.
Several potassium channel structures have been solved by x-ray crystallography (9–20). These known structures have been used as a basis to model the hERG1 channel (21–25). An issue for any model is how closely it resembles the true structure. Arguably, the best approach in the absence of a high-resolution crystal structure is to create validated homology models that can be prospectively tested in vitro (26). Reciprocal mutagenesis was used in previous studies of the plant KAT1 channel and is a plausible experimental strategy to test and guide theoretical modeling of potassium channels with unknown structure (27,28). Alignment of CNBD channels from bacteria, plants, and animals revealed a conserved sequence HwX(A/G)C in the S5 transmembrane domain. The evolutionary conservation of this region suggests that it could play an important, but as yet unknown role in the functional activity of the hERG1 channel. To explore the role of this conserved element of the pore domain (PD), we created an in silica model of hERG1 based on the KvAP crystal structure using the Rosetta-membrane program and extensive molecular-dynamics (MD) simulations. The S5-pore helix interactions predicted by the modeling were then explored by means of reciprocal mutagenesis and electrophysiology.
Materials and Methods
Theoretical protocol
Homology modeling
Homology modeling is unavoidable when the actual crystal structure of the ion channel of interest is unknown; however, high-resolution structures of homologous crystallized proteins are available. A critical step in any homology modeling is careful alignment of the amino acid sequences. We used Clustalw (29) to make alignments within the known K channel subfamilies, and then, for the more difficult to align S5 domain, attempted to achieve a consensus alignment based on charged residues conserved within and between the subfamilies (Fig. 1).
Figure 1.
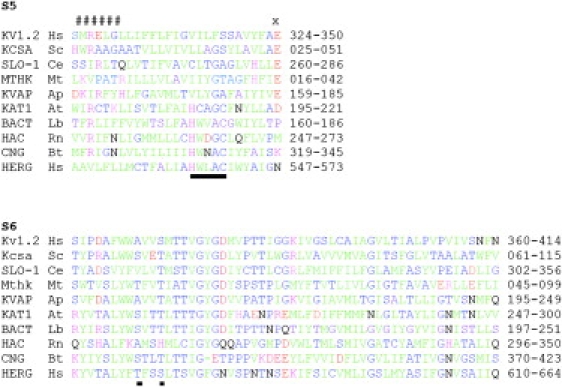
Amino acid sequence alignments of the S5 sequence alignment of HERG with KVAP. (A and B) For S5, CNBD channels were first aligned based on a conserved consensus HwX(A/G)C. This is underlined. All other channels were aligned using the conserved COOH-terminal glutamic acid (X). The two groups were aligned by trying to minimize the incursion of NH2-terminal basic residues (#####) into the membrane. The channels aligned are Homo sapiens Kv1.2 NP_004965, Streptomyces coelicolor Kcsa NP_631700, Caenorhabditis elegans SLOwpoke-1 NP_001024260, Methanothermobacter thermautotrophicus Mthk NP_276634, Aeropyrum pernix KVAP NP_147625, Arabidopsis thaliana KAT1 NP_199436, Leptospira biflexa bacterial CNBD channel ABZ94327, Rattus norvegicus hyperpolarization activated channel NP_446137, Bos taurus cyclic nucleotide gated channel NP_776704, and Homo sapiens HERG1 ABF71886. (C) Pore-S6 alignment for the above ion channels. Green, large nonpolar residues I, L, M, V, F, Y, and W. Blue, mainly small (except T) mainly polar (except A) residues A, C, G, S, and T. Black, large polar residues Q and N. Red, acidic residues D and E. Purple, basic residues K, R, and H.
De novo modeling with Rosetta-membrane
The Rosetta program package was used to model missed elements in the S5-pore helix linker (30). A novel sampling strategy implemented in Rosetta-membrane block (31) enables generation of the membrane-embedded and solvent-exposed parts of the PD at moderate-to-high resolution; ∼10,000 different models in two different trials were generated for the PD of the hERG1 channel followed by clustering. The root mean-square deviation of the cluster centers relative to the native structure of the conserved motif (S5, pore-S6 helix of KvAP) was used to constrain sampled conformational space, in addition to global distance testing. The average structure representing the cluster with minimum energy was chosen for further evaluation with MD simulations.
MD simulations
All-atom MD simulations were carried out with the use of the CHARMM program package (32). The protein was first minimized in the presence of harmonic constraints in the implicit bilayer and then subjected to a short MD run (∼1000 ps) in the implicit bilayer/solvent to remove steric clashes. The equilibrated PD of the hERG1 channel (wild-type (WT) and mutant) was embedded in a lipid membrane with explicit solvent. The system contains one hERG1, three bound ions (3 K+) in the S0:S2:S4 binding sites in the selectivity filter, and 116 dimyristoyl-phosphatidylcholine (DMPC), solvated by a 100 mM KCl aqueous salt solution. The simulation systems, comprising a total of ∼47,000 atoms, are represented in Fig. 2. All of the computations were carried out using the CHARMM program version c33b2 (32,33). The simulation methodology is similar to that used previously to study ion selectivity in the KcsA K+ channel (34). Briefly, the simulation system was constructed using a membrane-building protocol (35). The NPaT ensemble was used to maintain pressure of 1 atm, e.g., the hexagonal periodic boundary was used with the dimensions 88.1 × 88.1 × 70.1 Å. The system was allowed to vary only in the z-dimension via an Andersen constant pressure barostat (36). A Nose-Hoover thermostat was used to maintain an average temperature of 315 K. The system was electroneutralized, accounting for a net protein charge, and electrostatic interactions were treated using a particle mesh Ewald (PME) algorithm (37). The protocol chosen is known to produce stable bilayers for simulations. The bilayer was stabilized in the presence of constrained protein (hERG1) for over 4 ns before production simulations were run. Several properties were monitored to ensure proper preparation of the bilayer, e.g., density and area per lipid. The area per lipid was found to reach a plateau region after ∼3 ns of simulation with an average area ∼64 Å2 in good accord with the experimentally reported value of 60.6 Å2. The average root mean-square values for the most stable model (Cα) were on the order of 2.2–2.5 Å (values reported at T = 315 K). The most mobile element of the structure is the S5P linker. The initial ion load for the filter was chosen according to recent studies of filter stability in modeled hERG channels (38,39). All simulations started from ions being present in sites S0:S2:S4. No restraints were used to prevent ion hopping from site to site.
Figure 2.
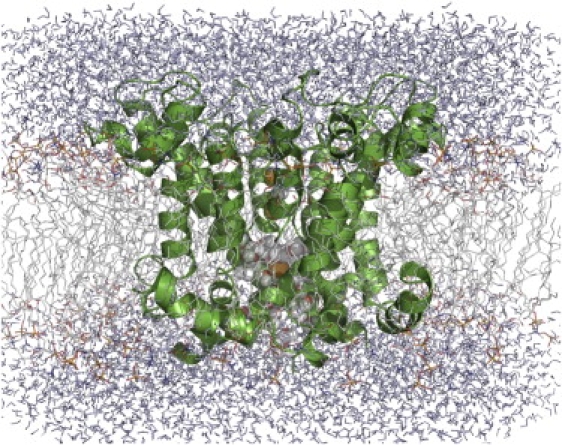
Snapshot of the full system of hERG1 embedded into a lipid membrane surrounded by water. Ions and water inside the IC cavity are shown in spheres. The lipid bilayer (DMPC) is represented in sticks.
The de novo protein design with Rosetta-membrane (40) led to identification of the two stable amphipathic helices located in the linker structure (stable for over 20 ns of simulation), in good accord with the reported NMR structure for the S5P linker (41) and biophysical measurements (42). The total simulation time was >40 ns. Mutations of interest were introduced using SCWRL (43) and then reequilibrated for 10 ns to analyze stability and dynamics in the proximity of the selectivity filter.
Hydrogen-bonding patterns analysis
To analyze the hydrogen-bonding patterns in the vicinity of the selectivity filter, we used geometry criteria from a previous study (44). The donor–acceptor distance for hydrogen bonding was set to be <2.7 Å and the heavy–heavy atom distance <3.5 Å. The “occupancy” criteria for the hydrogen bond (H-bond) used in the text below represents the probability of a certain H bond in the trajectory.
Experimental protocol
Heterologous expression
Site-directed mutagenesis was carried out by overlap extension using polymerase chain reaction according to Ho et al. (45) hERG1 was cloned into pIRES-hr GFP-1a (Stratagene) for coexpression with humanized Renilla reniformis green fluorescent protein. Transfected HEK cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% horse serum (GIBCO, Invitrogen, Carlsbad, CA). HEK cells were selected because their background potassium currents are tiny, and, more importantly, because no tail current is ever observed using the voltage clamp protocol in untransfected cells (see Fig. 4). Thus, the tail currents are specific to hERG1 overexpression.
Figure 4.
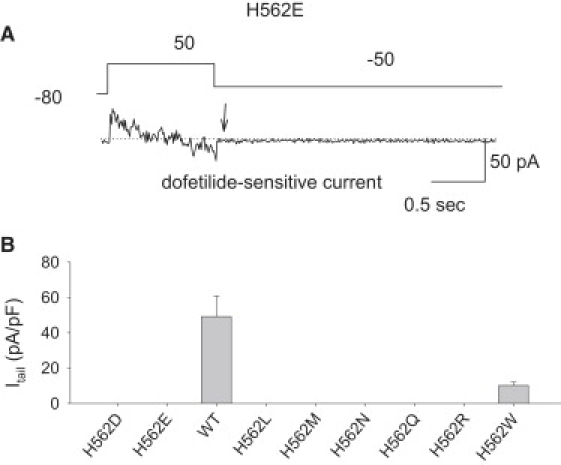
Mutations created at the 562 site. (A) A representative example of a cell that does not manifest a dofetilide-sensitive tail current. The arrowhead shows the absence of a tail current. (B) H562W was the only mutation that created a channel with functional expression. The dofetilide tail current expression is substantial in this mutation.
Electrophysiology
The cells were placed on glass coverslips in a chamber mounted on a modified stage of an inverted microscope. The chamber was superfused at a rate of 2 mL/min with normal external solution. IKr was recorded using whole-cell standard patch-clamp methods and an AXOPATCH 200B amplifier (Axon Instruments). Micropipettes were pulled from borosilicate glass capillary tubes on a programmable horizontal puller (Sutter Instruments, Novato, CA). When the pipettes were filled with the intracellular solution, the pipette tip resistance was 2–3 MΩ. The pipette solution contained (in mM) KCl 10, K-aspartate 110, MgCl2 5, Na2 ATP 5, EGTA 10, HEPES 5, CaCl2 1, corrected to pH 7.4 with KOH. The extracellular solution contained (in mM) NaCl 140, KCl 5.4, CaCl2 1.8, MgCl2 1, HEPES 10, glucose 5.5, pH 7.4, with NaOH. Cells were superfused at 2 mL/min at 36°C. For IKr measurements the whole cell configuration of the patch clamp method was used. The series resistance was <7 MΩ. Data were sampled at 1 kHz. The patch clamp protocol is shown in the top of Figure 4, A. In H562W, a −120 mV, 3 s prepulse was applied before each test pulse to completely deactivate the channel. To study the voltage dependence of activation, the amplitudes of the tail currents were recorded during repolarization to −50 mV for all constructs, except for the H562W, S621I, and H562W/S621I constructs. All tail currents reported in this work represent dofetilide-sensitive currents. In these constructs the tail currents were recorded at repolarization to −120 mV. The peak amplitudes of the tail currents were normalized by the maximum tail current.
Electrophysiologic analysis
Clampfit (Axon Instruments) was used to assess the double exponential fits to the deactivation time course. Previous studies showed that hERG1 deactivation is best fit by such a double exponential (7). In this study, the deactivation time course was fit to a double exponential decay. A single exponential model created obvious errors, whereas the decay process was well fit to a biexponential model. The fit was extrapolated to the beginning of the repolarizing pulse.
To assess the g-V relationship of the various constructs, tail current amplitudes were normalized to the values at +50 mV and the data were normalized to a Boltzmann function establishing the following parameters: V1/2, and k the slope factor of the conductance-voltage (g-V) relationship where G was established as the tail current value minus the reversal potential (Vt − Erev). Since one of the channels (e.g., H562W) failed to measurably deactivate at potentials substantially positive to EK, the deactivation time course was established during repolarizing pulses to −120 mV.
Statistical analysis
Statsview (Abacus Concepts, Berkeley, CA) was used to analyze the data. Data are presented as mean ± standard error. An unpaired Student's t-test was used to compare data, with a two-tailed p-value of 0.05 designated as significant.
Results
To make a model of hERG1 based on K channels of known structure, we first aligned the amino acid sequences. The pore/S6 transmembrane domains of most K channels can be simply aligned by leaving no gaps between the start of the pore helix and the end of S6 (Fig. 1). For many K channels, a strong alignment of the S5 transmembrane domains can also be obtained by anchoring the sequence with a COOH-terminal glutamic acid. These include several channels of known structure, such as KVAP, KCSA, and Kv1.2. The diverse family of CNBD channels including hERG1 does not possess this conserved COOH-terminal glutamic acid in S5, which makes alignment to other families difficult. There is, however, a conserved sequence motif, HwXG(A/G)C (underlined in Fig. 1), that makes alignment within the CNBD channel family possible. When CNBD channels are compared as a group to other families, the NH2-terminal arginine residues found in some CNBD members can be closely aligned to those found in other families (Fig. 1).
Modeling of the hERG1 potassium channel was initiated by building missing elements of the secondary structure using the Rosetta-membrane de novo modeling system complemented by extensive MD simulations in an explicit lipid bilayer for >30 ns. The average root mean-square values for heavy atoms were used to monitor the stability of the WT hERG1 channel and all mutations. A full account of the developed algorithm and model validation is summarized in a separate work (J. Subbotina, V. Yarov-Yarovoy, J. Lees-Miller, H.J. Duff, and S.Y. Noskov, unpublished).
S5-pore helix interactions from MD simulations
The 3D structures of the S5, selectivity filter, and pore helix averaged from the last 20 ns of simulation are shown in Fig. 3. The protocol for the H-bond statistics from MD-generated trajectories was based on an evaluation of average distances and angles, as in our previous studies (44). In Fig. 3 A, the MD simulations highlight the existence of the three bifurcating H-bonds formed between the imidazole ring of H562 in S5 and the hydroxyl groups of both T618 (two bonds) and S621 (one bond) in the pore helix of the WT channel. The average interaction energy due to these H-bonds is −1.5 kcal/mol per H-bond or −4.5 kcal/mol per network of bonds (averaged over the last 10 ns of simulation). To assess the impact of the loss of these H-bonds, a series of substitutions were created at the H562 site using MD simulations. To illustrate an extensive network of bonds involving residues from the S5 and pore helix, we performed a statistical analysis of the hydrogen-bonding patterns collected in Table 1. In good accord with previous models and experiments, the backbone atoms of H562 are involved in the formation of stable H-bonds with residues forming S5, whereas side-chain donor and acceptor sites interact extensively with hydroxyl groups provided by S621 and T618 side chains. Of interest, the H-bond between Nɛ2 and hydroxyl groups of S621 and T618 appears to be bifurcating.
Figure 3.
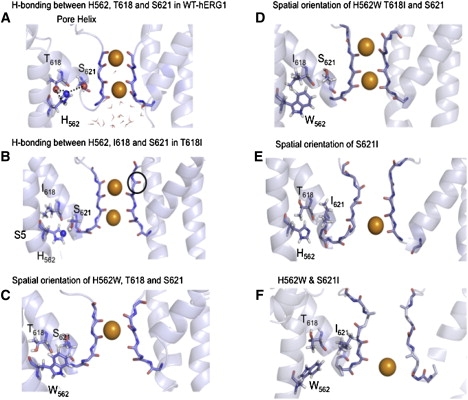
(A) Proximity and bifurcating hydrogen bonding between H562, T618, and S621. (B) Disrupted hydrogen bonding between H562, I618, and S621. (C) Disrupted orientation of W562 in the presence of T618 and S621. (D) The reestablished connectivity between W562, I618, and S621. (E) The disrupted H562 orientation with altered selectivity filter in S621. (F) Persistently disrupted selectivity filter in S621 in the presence of W562.
Table 1.
Hydrogen-bonding statistics for residues S621, T618, and H562 in the WT hERG1 channel
| Protein | Donor | Acceptor | Occupancy∗ |
|---|---|---|---|
| WT | HN S621 | O P617 | 0.49 |
| HN S621 | O H618 | 0.51 | |
| HG1 S621 | NE2 H562 | 0.45 | |
| HG1 S624 | O S621 | 0.98 | |
| HG1 T618 | NE2 H562 | 0.55 | |
| NH C566 | H562 O | 0.88 | |
| HN H562 | A558 O | 0.89 |
Occupancy refers to the average lifetime of the particular H-bond in the MD simulations.
In silica mutagenesis
To qualitatively assess the impact of loss of hydrogen bonding due to substitutions of hydrophobic amino acids for the hydrophilic T618 and S621 residues, in silica mutagenesis was performed. The decrease in the hydrogen-bonding connectivity between the mutated T618I residue and the native H562 residue was manifest. The T618I mutation contained only a single long-lived H-bond between the imidazole group of the H562 and the hydroxyl oxygen of S621. In contrast, three stable H-bonds were found for the WT-hERG1 channel (Fig. 3 B). Introduction of a single mutation at position 618 leads to a change in the orientation of the H562 side chain (see Fig. 3 D), which modestly destabilized the selectivity filter.
The in silica replacement of the serine at the position 621 to isoleucine produced a more dramatic effect on the stability of the selectivity filter, with a sharp increase in the filter fluctuations from 1.7–1.9 Å for the WT compared to 2.9–3.5 Å in the S621I mutant. The average filter structure resulting from our simulation is reminiscent of the low-K structure of KcsA (47), which is thought to be representative of the inactivated/nonconductive state. It should be stressed that these mutations preserved extensive hydrogen bonding involving main-chain atoms, but bifurcating bonds due to side chains were diminished.
One way to compensate for the loss of hydrogen bonding between H562W and the Thr/Ser-rich pore helix is to create reciprocal mutations. In this scenario one has to account for the relative packing of the side chains and carefully balance all interactions. Molecular simulations offer arguably the best theoretical route to address the effects of single-point mutations on interaction energies. We hypothesized that substituting T618 with a hydrophobic residue could recover some of the interaction energy with H562W, as the tryptophan is closely packed against the pore helix. To test this hypothesis, we created the double mutation H562W/T618I in silica.
The spatial orientations of key residues in the H562W mutation are shown in Fig. 3 D. The simulations indicate that the double H562W/T618I mutant could potentially be capable of “almost” normal function. The hydrophobic interactions between side chains of W562 and I618 were capable of recovering at least 70% of the interaction losses due to the disruption of the H-bond network. The energy of interaction between these two amino acids was ∼−2.5 kcal/mol, averaged over the last 20 ns of the trajectory. In this double mutation, H562W/T618I, the interaction between H562W and S621 is virtually abolished (−0.12 kcal/mol). Simulations indicated that the reciprocal H562W/S621I mutation would have a smaller effect to recover function because the H562 has on average two H-bonds with T618 and only one with S621. Moreover, the single mutation S621I caused a more substantial distortion of the selectivity filter. This distortion was not corrected in the double mutation H562W/S621I. These are important outcomes; however, validation of these results by the simulation alone is challenging. Mutation effects were rendered out from a single trajectories based on a homology model. Nevertheless, these MD simulations performed on a homology model of hERG1 provide fresh impetus for experimental testing of the hypothesis formulated above.
In vitro mutagenesis
Single-point mutagenesis
Fig. 4 reviews the in vitro experimental results of these substitutions at the H562 site. Only one mutation, H562W, produced a robust functional channel. A representative example of the absence of a dofetilide-tail current for H562E is shown Fig. 4 A. In contrast, H562W creates a functional channel with a substantial dofetilide sensitive tail-current.
H562W. Fig. 5 illustrates representative examples of the family of currents induced in WT channels and the mutations of hERG1 explored in this study. Although H562W is the only mutation that creates a functional channel, it manifests a grossly altered electrophysiology: a substantial slowing of deactivation of the channel such that its deactivation properties can only be examined at hyperpolarized potentials (Fig. 6). The fast component of deactivation that slowed from 47 ± 5 ms in WT to 394 ± 44 ms in H562W modestly shifts activation to the left (from −16 ± 3 in WT to −25 ± 5; p < 0.05; Table 2). The current-voltage relationships of activation are shown in Fig. 7. The H562W mutation produces a significant shift to the left (more hyperpolarized potentials) by 11 mV from −16 ± 3 mV in WT to −25 ± 5 mV in H562W (Fig. 7). Examples of measurements of the reversal potentials are shown in Fig. 8. The reversal potential of H562W was −73 ± 3 mV, a value significantly different from that seen with WT (−77 ± 3 mV; p = 0.048). Although this change is statistically significant, it is modest in magnitude.
Figure 5.
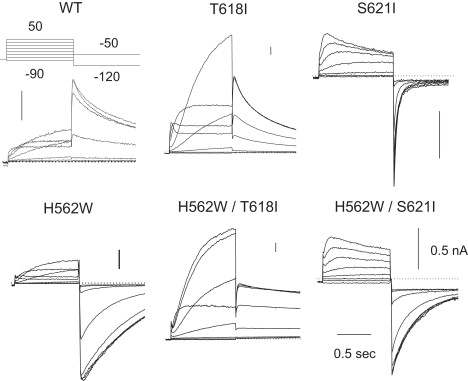
Representative examples of the family of currents elicited by the current-voltage protocol shown in the inset. The representative current traces of WT-HergA and the mutations of T618I, S621I, H562W, H562W/T618I, and H562W/S621I elicited by the patch clamp protocol are shown at the top of the WT panel. Because of the slow deactivation in H562W, a −120 mV, 3 s prepulse was given to complete deactivation before the next depolarizing test pulse. The vertical bar in the upper right of each panel represents the amplitude of 500 pA. In T618I, the test pulses were depolarized to as high as +90 mV to cover its entire activation range.
Figure 6.
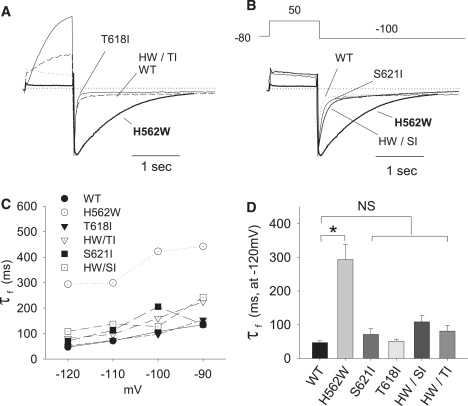
Overlay of the deactivation time course comparing the various constructs. The time courses of deactivation of the tail currents of WT-Herg, H562W, T618I, and H562W/T618I (A) as well as WT-Herg, H562W, S621I, and H562W/S621I (B) are shown after repolarization to −120 mV. The currents were normalized to have the same magnitude of peak tail currents. (C) The mean fast time constants of the deactivations for every mutation plotted against the depolarization potential. (D) The statistical analysis of the average fast time constant of the deactivations of these mutations. The time constants of the fast and slow components of deactivation are enumerated in Table 2.
Table 2.
Mean electrophysiological measurements
| Activation | V1/2 ± SE | S ± SE | n |
|---|---|---|---|
| WT | −16 ± 3 | 6 ± 2 | 14 |
| H562W | −25 ± 5∗ | 10 ± 1 | 6 |
| S621I | −28 ± 3 | 3 ± 5 | 3 |
| T618I | +16 ± 2∗ | 19 ± 3 | 5 |
| H562W/S621I | −35 ± 4∗ | 8 ± 1 | 5 |
| H562W/T618I | −12 ± 4 | 11 ± 1 | 13 |
| Deactivation | τ-f | τ-s | n |
| WT | 47 ± 5 | 649 ± 39 | 5 |
| H562W | 394 ± 44∗ | 730 ± 94 | 8 |
| S621I | 71 ± 17 | 565 ± 162 | 3 |
| T618I | 51 ± 6 | 402 ± 64 | 4 |
| H562W/S621I | 135 ± 11 | 993 ± 268 | 7 |
| H562W/T618I | 81 ± 16 | 484 ± 104 | 4 |
| Reversal potential | SE | n | |
| WT | −77 ± 3 | 4 | |
| H562W | −73 ± 3∗ | 6 | |
| T618I | −75 ± 7 | 3 | |
| H562W/T618I | −77 ± 3 | 3 | |
p < 0.05, t-test, compared to WT.
Figure 7.
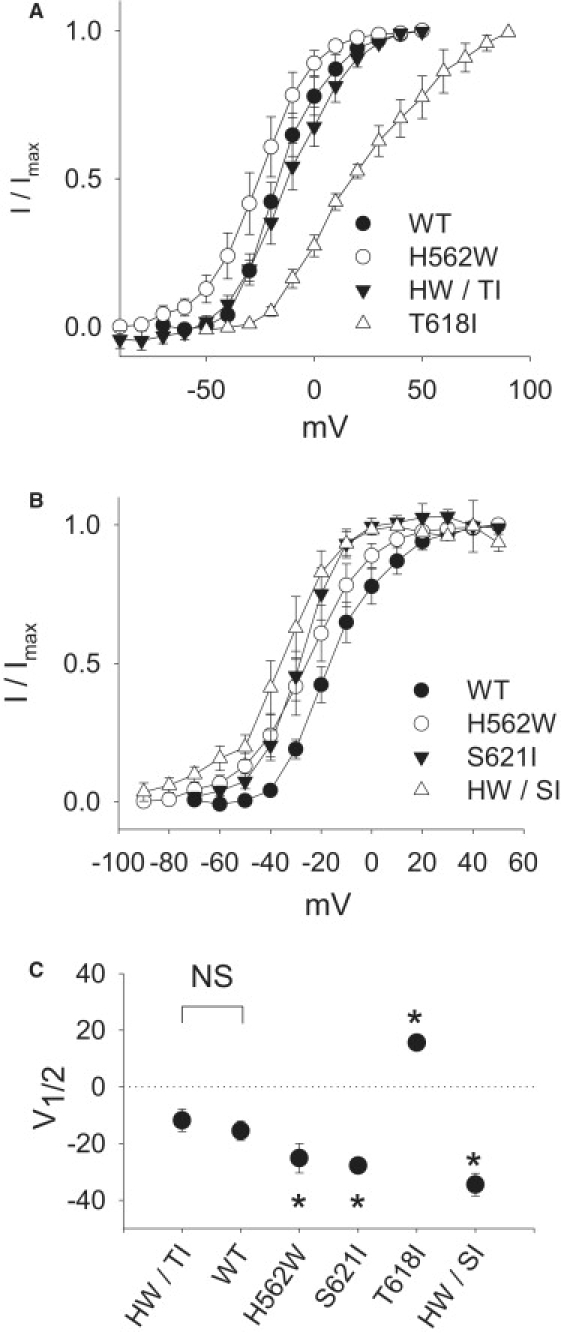
(A) Dofetilide-sensitive tail current-voltage relationships. Activation curves of WT-Herg, H562W, S621I, and H562W/S621I (HW/SI) (B), as well as WT-Herg, H562W, T618I, and H562W/T618I (HW/TI) (C) are shown. The amplitudes of the tail currents are obtained during reversal to −50 mV (for most constructs) except for H562W, S621I, and H562W/S621I, where the tail currents were obtained during repolarization to −120 mV.
Figure 8.
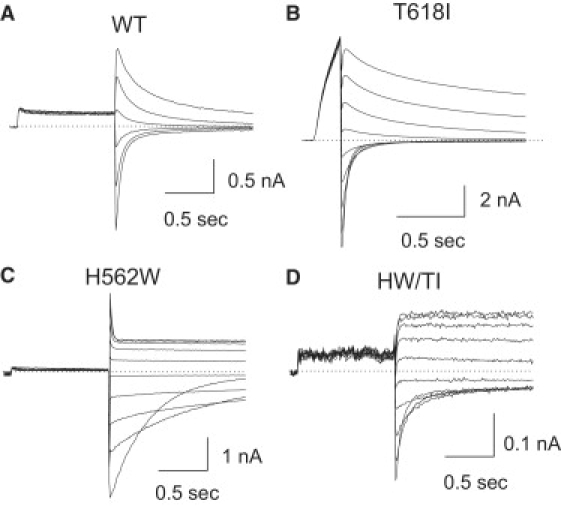
Examples of the reversal potential protocol used with WT, H562W, T618I, and the double mutation H562W/T618I. Dofetilide-sensitive currents are shown.
T618I. The T618I mutation also shifted the V1/2 of activation to +16 ± 2 mV from the WT value of −16 ± 3 mV; Table 2 and Fig. 7). Experimentally the T618I mutation produced a modest, nonsignificant shift in the reversal potential from −77 ± 1 mV in WT hERG1 to −75 ± 2 mV in T618I (NS) at an extracellular K+ of 5.4 mMol/L.
S621I. The in silica simulations suggested that the S621I mutation would more substantially distort the selectivity filter. We observed that although S621I only modestly shifts the voltage dependence of activation and slows deactivation, it substantially alters selectivity filter function. At an extracellular K+ of 5.4 mMol/L, S621I still did not show positive outward tail currents, and thus we were not able to measure its reversal potential. One possible explanation for the lack outward tail currents is a hyperpolarized shift in the voltage dependence of inactivation. Accordingly, to record an outward tail current out of the range of voltages potentially showing inactivation, the external K+ concentration was reduced to 1 mM/L (from 5.4 mM/L). Fig. 9 shows that in WT the reversal potential negatively shifted from −81 mV to −115 mV, as predicted by the Nernst equation for a highly selective potassium channel (the predicted EK for a channel with virtually 100% potassium selectivity would be −125 mV under these conditions). In S621I (n = 5), however, the polarity of the tail current did not change at all; the currents continued to show clear inward tails at potentials more depolarized than −90 mV. These data indicate that the S621I mutation has substantially altered K+ selectivity compared to WT.
Figure 9.
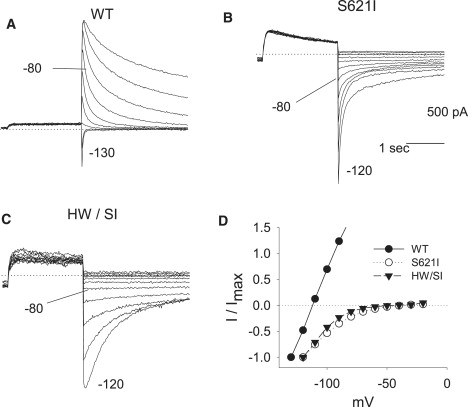
Reversal potentials comparing WT with that seen in mutated channels S621I and H562W/S621I. Representative tail current traces of WT (A), S621I (B), and H562W/S621I (C) at different potentials in 1 mMol/L extracellular K+ solution. (D) The amplitudes of tail currents were plotted against the repolarizing membrane potentials. At an extracellular K+ of 5.4 mMol/L, the S621I did not show positive outward deactivation tails; therefore, it was impossible to measure the reverse potential of S621I and H562W/S621I. To record the outward tail current out of the range of voltages showing inactivation, the external K+ concentration was reduced to 1 mM. In WT the reverse potential negatively shifted from −81 mV to −115 mV, as predicted by the Nernst equation for a highly selective potassium channel (the predicted EK for a channel with virtually 100% potassium selectivity would be −125 mV under these conditions). However, in S621I (n = 3) and H562W/S621I, the polarity of the tail current did not change at all. They still showed clear inward tails at potentials as high as −80 mV. Accordingly, for S621I and H562W/S621I K+, the selectivity filter characteristics are substantially altered compared to WT. The same results were obtained in the five S621I and seven H562W/S621I experiments.
Reciprocal double mutations
Deactivation properties
Fig. 6 compares the deactivation time courses of the WT, H562W, T618I, and T618I/H562W (A), and S621I and H562W/ S621I (B). As shown in the examples, although deactivation is markedly slowed in H562W, this phenotype is restored toward WT by both of the double mutations. Fig. 6 C shows the mean voltage-deactivation τ relationships, and Fig. 6 D shows the mean fast τ for the various constructs. Table 2 shows both the slow and fast τ. In review, the reciprocal mutations in large measure rescue the consequences of H562W on deactivation.
Voltage dependence of activation
Fig. 7 shows the mean activation current-voltage relationships for the tail currents of the WT compared with the individual and double mutations (Table 2). The V1/2 for the double mutation H562W/T618I mutation is −11 ± 4 mV, a value similar to WT (−16 ± 3 mV). In contrast, the V1/2 for the double mutation H562W/S621I is shifted even more leftward to −35 ± 4 mV (p < 0.05). These data indicate that the double H562W/T618I shifts the voltage dependence of activation toward that seen with WT.
Reversal potential
To address potential changes in selectivity filter function, reversal potentials of the individual channels were evaluated. At an extracellular potassium of 5.4 mMol/L, the mean reversal potentials of WT, H562W, T618I, and H562/T618I were −77, −73, −75, and −77 mV, respectively (Table 2). Of interest, whereas the single mutation H562W modestly but significantly shifted the reversal potential to more positive potentials (−73 ± 3 mV), the double mutation H562W/T618I restored selectivity filter function back to values equivalent to WT (−77 ± 3 mV).
Measurement of the reversal potential of S621/T621I is more complex. As can be seen in Fig. 8 B, at an extracellular K+ of 5.4, the H562W/ S621I did not show any positive outward deactivation dofetilide-sensitive tails. Without outward tail currents, it is not feasible to quantify the reversal potential. Accordingly, reversal potentials were measured at a [K+]o of 1 mM. As shown in Fig. 8, even under these conditions, the polarity of tail current did not change and still showed clear inward tails at potentials as depolarized as −80 mV. These data indicate that the double mutation S621I/H562W has markedly altered selectivity filter characteristics, like those observed with the single mutation S621I.
Together, these data indicate that the double H562W/T618I mutation has channel activity similar to that of WT, whereas the double mutation H562W/S621I restores deactivation characteristics but retains abnormal activation and selectivity filter characteristics.
Controls
To provide further evidence that the normalization of function by the double mutation H562W/T618I was specific, we assessed whether the T618I mutation would normalize two other mutations with remarkably slowed deactivation: R528D (in the mid S4 domain) and M651T (in the distal S6 domain). The double mutations, R528D/T618I and M651T/T618I, did not correct the slow deactivation profiles. For example, the mean fast and slow deactivation τ for M651T were 114 ± 24 and 1035 ± 71 ms respectively, whereas for M651T/T618I the fast and slow τ were 80 ± 14 and 1027 ± 194 ms, respectively (n = 8; NS). For R528D and R528D/T618, the deactivation time course was so slow that it was not well fit by either single or double exponentials. The deactivation time course is estimated as the ratio of the current at 1 s of deactivation relative to the peak tail current (I1s/Ipeak). The values for R528D were 0.96 ± 0.03 vs. 0.89 ± 0.05 for R528D/T618 (n = 7, NS).
Discussion
For the first time, to our knowledge, this study provides:
-
1.
Evidence for an interaction (hydrogen bonding) between an amino acid in S5, H562 of hERG1, and the amino acids T618 and S621 in the pore helix. This interaction is functionally important for deactivation, V1/2 of activation, and, to a lesser extent, ion selectivity. Thus, histidine in position 562 plays a critical role in stabilizing the structure and function of the S5-pore helix.
-
2.
Experimental validation for a model of hERG1 based on the crystallographic structure of KvAP.
Below we discuss the main findings and implications of our current understanding of hERG1 structure-function.
H562 plays an important structural role in stabilizing the S5-pore-S6 complex
The sequence HwX(A/G)C in the middle of the S5 of hERG1 is evolutionarily conserved in virtually all CNBD channels, including those found in bacteria, plants, and animals. This strict evolutionary conservation suggests that the orientation and interactions of H562 with other domains of the channel may have functional importance. Our study confirms this. Of the seven mutations at position 562, only H562W generates measurable currents, albeit with altered properties: deactivation is slowed and activation is modestly shifted to hyperpolarized potentials.
The second site mutation T618I rescues the phenotype of H562W
In simulations, the doubly mutated H562W/T618I of the hERG1 channel is predicted to have almost normal function. The hydrophobic interaction between side chains of tryptophan (562) and isoleucine (618) are capable of recovering at least 70% of the interaction losses due to disruption of the H-bond network. The results of these simulations were confirmed in vitro. The main consequence of the H562W mutation is slowed deactivation. Double mutations of H562W/T618I and H562W/S621I restore deactivation to near-normal values. It is important to note that the T618I and S621I mutations have fast deactivation τ, similar to that of WT, and therefore do not compensate for the greatly slowed H562W fast τ in a simple subtractive manner as might be expected for a mutation with a faster than normal τ. There also does not appear to be any relationship between the activation threshold and the ability to normalize the deactivation τ, as S621I and T618I result in opposite shifts in the activation threshold. These studies demonstrate that intermolecular interactions between H562 in the S5 and amino acids in the pore helix are necessary for normal or near-normal function. Appropriate interaction(s) between an amino acid at the 562 site and the T618 and S621 sites may consist of hydrogen bonding or hydrophobic interactions. As long as the intermolecular connectivity has the requisite strength, structural stability will be fostered.
Further support for the specificity of the H562-pore helix interaction comes from the finding that the T618I and S621I mutations do not correct the slow deactivation phenotypes of mutations in S4 or in the activation gate (R528D (48) and M651T (6)). The finding that H562, an S5 residue close to the extracellular face of the channel, affects closing kinetics, may appear to be at odds with the cytoplasmic location of the activation gate. However, it is in agreement with experiments demonstrating that the equivalent residue, H210, of KAT1 (Fig. 1) plays a role in the interaction between S5 and the voltage sensor (S4) (27,28). This interaction was demonstrated by identifying specific S4 mutations that compensate for H210E after expressing this plant CNBD channel as a substitute potassium conduit in yeast. In our model we do not envisage a direct interaction between the S5 histidine side chain and S4. Rather, we suggest that S4 interacts with the S5 hydrophobic side chains and or backbone in the vicinity of the histidine. Histidine mutations then alter the local conformation of the S5 helix, resulting in reduced contact with S4.
This model is in agreement with our present understanding that there are no highly specific interactions between the S4 and S5 helices of Kv1.2 (15,16,49). Crystallographic studies have indicated modest interactions between S4 and S5 (16), and the Shaker voltage sensor was found to function with the nonvoltage-gated KcsA channel (49). It is not surprising that mutations in the pore helix can change the function of the selectivity filter and alter inactivation, but the finding that mutations in the pore helix can have substantial effects on activation and deactivation may be unexpected. However, our finding is in keeping with a number of previous studies that confirmed that mutations within the pore helix of hERG1 alter the V1/2 of activation and slow the time constant of deactivation (50,51).
In this study we found that the simulated mutation at the H562 residue to a tryptophan acid, H562W, altered the interaction with its partner, T618, and disrupted hydrogen bonding, which destabilized the pore helix and the selectivity filter. The orientation of the carbonyl moieties in the backbone of the selectivity filter was significantly altered by the H562W mutation. This is relevant because these carbonyls coordinate the potassium ions within the selectivity filter and are essential for efficient ion permeation. The prediction of altered selectivity filter characteristics by simulation for H562W was confirmed by in vitro electrophysiology. The reversal potential was significantly shifted from −77 ± 3 mV in WT to −73 ± 3 mV in the H562W mutated channel. Of importance, the reciprocal double mutation, H562W/T618I, which was predicted by simulation to stabilize the pore helix and the selectivity filter, restored the reversal potential back to −77 ± 3 mV, consistent with normalization of the selectivity filter function. Although statistically significant changes in selectivity filter function were observed with H562W, the magnitude of the change was relatively modest.
The simulations predicted more dramatic changes in the configuration in the S621I mutation that would not be corrected in the double mutation, H562W/S621I. This in silica finding was consistent with our in vitro results.
Conclusions
In this work, ab initio structure modeling was combined with subsequent MD simulations to refine the structure of the PD for the hERG1 channel. The combination of these methods with molecular mutagenesis led to the identification of a previously unknown interaction between H562 in the S5 transmembrane domain and the pore helix, which is an important determinant of hERG1 function.
Acknowledgments
We thank Drs. B. Roux, J. Faraldo-Gomez, J. Robertson, and G. N. Tseng for helpful comments and discussions.
This work was supported by grants from the National Institute of Mental Health Career Development Research Program (K01 MH67625 to V.Y.-Y.), the Canadian Institutes of Health Research (MOP-186232 to S.N. and H.J.D.), and the Heart and Stroke Foundation of Alberta (to H.J.D.). S.N. is an Alberta Heritage Foundation for Medical Research Scholar, and H.J.D. is an Alberta Heritage Foundation for Medical Research Medical Scientist. The computational support for this work was provided by the West-Grid Canada through a resource allocation award to S.N.
Footnotes
James P. Lees-Miller and Julia O. Subbotina contributed equally to this work.
Contributor Information
Sergei Y. Noskov, Email: snoskov@ucalgary.ca.
Henry J. Duff, Email: hduff@ucalgary.ca.
References
- 1.Warmke J.W., Ganetzky B. A family of potassium channel genes related to Eag in Drosophila and mammals. Proc. Natl. Acad. Sci. USA. 1994;91:3438–3442. doi: 10.1073/pnas.91.8.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Curran M.E., Splawski I., Timothy K.W., Vincent G.M., Green E.D. A molecular basis for cardiac-arrhythmia-Herg mutations cause long Qt syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 3.Splawski I., Shen J.X., Timothy K.W., Lehmann M.H., Priori S. Spectrum of mutations in long-QT syndrome genes KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 4.Yu F.H., Yarov-Yarovoy V., Gutman G.A., Catterall W.A. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol. Rev. 2005;57:387–395. doi: 10.1124/pr.57.4.13. [DOI] [PubMed] [Google Scholar]
- 5.Lees-Miller J.P., Duan Y.J., Teng G.Q., Thorstad K., Duff H.J. Novel gain-of-function mechanism in K+ channel-related long-QT syndrome: altered gating and selectivity in the HERG1 N629D mutant. Circ. Res. 2000;86:507–513. doi: 10.1161/01.res.86.5.507. [DOI] [PubMed] [Google Scholar]
- 6.Lees-Miller J.P., Duan Y.J., Teng G.Q., Duff H.J. Molecular determinant of high-affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: involvement of S6 sites. Mol. Pharmacol. 2000;57:367–374. [PubMed] [Google Scholar]
- 7.Sanguinetti M.C., Mitcheson J.S. Predicting drug-hERG channel interactions that cause acquired long QT syndrome. Trends Pharmacol.Sci. 2005;26:119–124. doi: 10.1016/j.tips.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Vandenberg J.I., Walker B.D., Campbell T.J. HERG K+ channels: friend and foe. Trends Pharmacol Sci. 2001;22:240–246. doi: 10.1016/s0165-6147(00)01662-x. [DOI] [PubMed] [Google Scholar]
- 9.Jiang Y., Lee A., Chen J., Cadene M., Chait B.T. The open pore conformation of potassium channels. Nature. 2002;417:523–526. doi: 10.1038/417523a. [DOI] [PubMed] [Google Scholar]
- 10.Jiang Y., Lee A., Chen J., Cadene M., Chait B.T. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 2002;417:515–522. doi: 10.1038/417515a. [DOI] [PubMed] [Google Scholar]
- 11.Jiang Y., Ruta V., Chen J., Lee A., MacKinnon R. The principle of gating charge movement in a voltage-dependent K+ channel. Nature. 2003;423:42–48. doi: 10.1038/nature01581. [DOI] [PubMed] [Google Scholar]
- 12.Jiang Y., Lee A., Chen J., Ruta V., Cadene M. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 13.Jiang Q., Wang D., MacKinnon R. Electron microscopic analysis of KvAP voltage-dependent K+ channels in an open conformation. Nature. 2004;430:806–810. doi: 10.1038/nature02735. [DOI] [PubMed] [Google Scholar]
- 14.Long S.B., Campbell E.B., MacKinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- 15.Long S.B., Campbell E.B., MacKinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 16.Long S.B., Tao X., Campbell E.B., MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 17.Doyle D.A., Morais Cabral J., Pfuetzner R.A., Kuo A., Gulbis J.M. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 18.Kuo A., Gulbis J.M., Antcliff J.F., Rahman T., Lowe E.D. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 2003;300:1922–1926. doi: 10.1126/science.1085028. [DOI] [PubMed] [Google Scholar]
- 19.Morais-Cabral J.H., Zhou Y., MacKinnon R. Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature. 2001;414:37–42. doi: 10.1038/35102000. [DOI] [PubMed] [Google Scholar]
- 20.Zhou M., Morais-Cabral J.H., Mann S., MacKinnon R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 2001;411:657–661. doi: 10.1038/35079500. [DOI] [PubMed] [Google Scholar]
- 21.Osterberg F., Aqvist J. Exploring blocker binding to a homology model of the open hERG K+ channel using docking and molecular dynamics methods. FEBS Lett. 2005;579:2939–2944. doi: 10.1016/j.febslet.2005.04.039. [DOI] [PubMed] [Google Scholar]
- 22.Rajamani R., Tounge B.A., Li J., Reynolds C.H. A two-state homology model of the hERG K+ channel: application to ligand binding. Bioorg. Med. Chem. Lett. 2005;15:1737–1741. doi: 10.1016/j.bmcl.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 23.Witchel H.J., Dempsey C.E., Sessions R.B., Perry M., Milnes J.T. The low-potency, voltage-dependent HERG blocker propafenone—molecular determinants and drug trapping. Mol. Pharmacol. 2004;66:1201–1212. doi: 10.1124/mol.104.001743. [DOI] [PubMed] [Google Scholar]
- 24.Pearlstein R.A., Vaz R.J., Kang J.S., Chen X.L., Preobrazhenskaya M. Characterization of HERG potassium channel inhibition using CoMSiA 3D QSAR and homology modeling approaches. Bioorg. Med. Chem. Lett. 2003;13:1829–1835. doi: 10.1016/s0960-894x(03)00196-3. [DOI] [PubMed] [Google Scholar]
- 25.Stansfeld P.J., Gedeck P., Gosling M., Cox B., Mitcheson J.S. Drug block of the hERG potassium channel: insight from modeling. Proteins. 2007;68:568–580. doi: 10.1002/prot.21400. [DOI] [PubMed] [Google Scholar]
- 26.Kaback R., Sahin-Toth M., Weinglass A.B. The kamikaze approach to membrane transport. Nat. Rev. Mol. Cell Biol. 2001;2:610–620. doi: 10.1038/35085077. [DOI] [PubMed] [Google Scholar]
- 27.Grabe M., Lai H.C., Jain M., Jan Y.N., Jan L.Y. Structure prediction for the down state of a potassium channel voltage sensor. Nature. 2007;445:550–557. doi: 10.1038/nature05494. [DOI] [PubMed] [Google Scholar]
- 28.Lai H.C., Grabe M., Jan Y.N., Jan L.Y. The S4 voltage sensor packs against the pore domain in the KAT1 voltage-gated potassium channel. Neuron. 2005;47:395–406. doi: 10.1016/j.neuron.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 29.Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 30.Yarov-Yarovoy V., Schonbrun J., Baker D. Multipass membrane protein structure prediction using Rosetta. Proteins. 2006;62:1010–1025. doi: 10.1002/prot.20817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pathak M., Yarov-Yarovoy V., Agarwal G., Roux B., Barth P. Closing in on the resting state of the shaker K+ channel. Neuron. 2007;56:124–140. doi: 10.1016/j.neuron.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 32.Brooks B.R., Bruccoleri R.E., Olafson B.D., States D.J., Swaminathan S. CHARMM: a program for macromolecular energy minimization and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 33.MacKerell A.D., Brooks C.L., Nilsson L., Roux B., Won Y. CHARMM: the energy function and its parameterization with an overview of the program. In: Schleyer P., editor. The Encyclopedia of Computational Chemistry. John Wiley & Sons; Chichester, UK: 1998. pp. 271–277. [Google Scholar]
- 34.Noskov S.Y., Berneche S., Roux B. Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature. 2004;431:830–834. doi: 10.1038/nature02943. [DOI] [PubMed] [Google Scholar]
- 35.Woolf T.B., Roux B. Molecular dynamics simulation of the gramicidin channel in a phospholipid bilayer. Proc. Natl. Acad. Sci. USA. 1994;91:11631–11635. doi: 10.1073/pnas.91.24.11631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feller S., Zhang Y., Pastor R., Brooks B. Constant pressure molecular dynamics simulation—the Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 37.Essman U., Perera M., Berkowitz M., Darden T., Lee H. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 38.Stansfeld P.J., Grottesi A., Sands Z.A., Sansom M.S.P., Gedeck P. Insight into the mechanism of inactivation and pH sensitivity in potassium channels from molecular dynamics simulations. Biochemistry. 2008;47:7414–7422. doi: 10.1021/bi800475j. [DOI] [PubMed] [Google Scholar]
- 39.Kutteh R., Vandenberg J.I., Kuyucak S. Molecular dynamics and continuum electrostatics studies of inactivation in the HERG potassium channel. J. Phys. Chem. B. 2007;111:1090–1098. doi: 10.1021/jp066294d. [DOI] [PubMed] [Google Scholar]
- 40.Yarov-Yarovoy V., Baker D., Catterall W.A. Voltage sensor conformations in the open and closed states in ROSETTA structural models of K+ channels. Proc. Natl. Acad. Sci. USA. 2006;103:7292–7297. doi: 10.1073/pnas.0602350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Torres A.M., Bansal P., Alewood P.F., Bursill J.A., Kuchel P.W. Solution structure of CnErg1 (Ergtoxin), a HERG specific scorpion toxin. FEBS Lett. 2003;539:138–142. doi: 10.1016/s0014-5793(03)00216-3. [DOI] [PubMed] [Google Scholar]
- 42.Tseng G.N., Sonawane K.D., Korolkova Y.V., Zhang M., Liu J. Probing the outer mouth structure of the hERG channel with peptide toxin footprinting and molecular modeling. Biophys. J. 2007;92:3524–3540. doi: 10.1529/biophysj.106.097360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dunbrack R.L., Karplus M. Backbone-dependent rotamer library for proteins. Application to side-chain prediction. J. Mol. Biol. 1993;230:543–574. doi: 10.1006/jmbi.1993.1170. [DOI] [PubMed] [Google Scholar]
- 44.Noskov S.Y., Roux B. Control of Na+ selectivity in LeuT: two binding sites, two mechanisms. J. Mol. Biol. 2008;377:804–817. doi: 10.1016/j.jmb.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ho S.N., Hunt H.D., Horton R.M., Pullen J.K., Pease L.R. Site-directed mutagenesis by overlap extension using the polymerase chain-reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 46.Reference deleted in proof.
- 47.Lockless S.W., Zhou M., MacKinnon R. Structural and thermodynamic properties of selective ion binding in a K+ channel. PLoS Biol. 2007;5:e21. doi: 10.1371/journal.pbio.0050121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang M., Liu J., Jiang M., Wu D.M., Sonawane K. Interactions between charged residues in the transmembrane segments of the voltage-sensing domain in the hERG channel. J. Membr. Biol. 2005;207:169–181. doi: 10.1007/s00232-005-0812-1. [DOI] [PubMed] [Google Scholar]
- 49.Lu Z., Klem A.M., Ramu Y. Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J. Gen. Physiol. 2002;120:663–676. doi: 10.1085/jgp.20028696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perry M., Stansfeld P.J., Leaney J., Wood C., de Groot M.J. Drug binding interactions in the inner cavity of hERG channels: molecular insights from structure-activity relationships of clofilium and ibutilide analogs. Mol. Pharmacol. 2006;69:509–519. doi: 10.1124/mol.105.016741. [DOI] [PubMed] [Google Scholar]
- 51.Siebrands C.C., Friederich P. Structural requirements of human ether-a-go-go-related gene channels for block by bupivacaine. Anesthesiology. 2007;106:523–531. doi: 10.1097/00000542-200703000-00017. [DOI] [PubMed] [Google Scholar]