

Abstract
Ubiquitin is a highly conserved protein that is encoded by a multigene family. It is generally believed that this gene family is subject to concerted evolution, which homogenizes the member genes of the family. However, protein homogeneity can be attained also by strong purifying selection. We therefore studied the proportion (pS) of synonymous nucleotide differences between members of the ubiquitin gene family from 28 species of fungi, plants, and animals. The results have shown that pS is generally very high and is often close to the saturation level, although the protein sequence is virtually identical for all ubiquitins from fungi, plants, and animals. A small proportion of species showed a low level of pS values, but these values appeared to be caused by recent gene duplication. It was also found that the number of repeat copies of the gene family varies considerably with species, and some species harbor pseudogenes. These observations suggest that the members of this gene family evolve almost independently by silent nucleotide substitution and are subjected to birth-and-death evolution at the DNA level.
Ubiquitin is a small protein consisting of 76 amino acids that plays a major role in both cellular stress response and protein degradation in eukaryotes. It is one of the most highly conserved proteins (1), and 72 of the 76 amino acids appear to be invariant among fungi, plants, and animals (2). Ubiquitins are encoded by a small-to-medium-size multigene family, which comprises a monomeric gene class and a polymeric gene class. Monomeric ubiquitin genes consist of 228 nucleotides (76 codons) with an additional C-terminal sequence that encodes a ribosomal protein. By contrast, polymeric genes known as poly-ubiquitins (poly-u) are composed of tandem repeats of a 228-bp gene with no spacer sequence between them (Fig. 1). The number of ubiquitin–gene units in a poly-u locus, the number of poly-u loci, and the number of monomeric genes per genome vary considerably among eukaryotic species (3–5). Yet all ubiquitin genes encode the same amino acid sequence in each species.
Figure 1.

Ubiquitin polymeric (poly-u) loci in humans and C. elegans. A poly-u locus consists of a number of ubiquitin genes that are concatenated with no intervening sequences. A monomeric locus is composed of a ubiquitin gene and a ribosomal protein gene with either 52 or 80 codons. The proteins encoded by polymeric and monomeric genes are identical. Poly-u ψ is a pseudogene locus.
Because ubiquitins are highly conserved and are encoded by a multigene family, this gene family is generally believed to be subject to concerted evolution, which homogenizes the member genes by interlocus recombination or gene conversion (3, 5–7). In concerted evolution, member genes of a family are assumed to evolve as a unit, exchanging genetic information (8–11). However, protein homogeneity can also be attained by strong purifying selection, and in this case there is no need to have concerted evolution. In fact, member genes of a family may evolve independently by silent nucleotide substitution or may be subject to evolution by a birth-and-death process at the DNA level, even if the protein sequence remains unchanged. Birth-and-death evolution is a form of independent evolution and assumes that new genes are created by repeated gene duplication and that some duplicate genes may stay in the genome for a long time, whereas others may be deleted or become nonfunctional (12–14).
Although concerted evolution and birth-and-death evolution are conceptually different, they may not be distinguishable if the rate of concerted evolution is assumed to be very slow. In this paper, however, we define concerted evolution as a rapid process of interlocus recombination or gene conversion so that even closely related species (e.g., two species of the frog genus Xenopus or humans and chimpanzees) have different sets of homogeneous member genes, as was originally proposed with ribosomal RNA genes (8, 10, 15). Under the assumption of a rapid process of interlocus recombination or gene conversion, we would expect that in each species the proportion of synonymous nucleotide differences per synonymous site (pS) between repeat genes is similar to or only slightly higher than the proportion of nonsynonymous differences (pN), whether there is purifying selection or not. However, if birth-and-death evolution with strong purifying selection is the major evolutionary force, pS would be much higher than pN, because the member genes may diverge extensively by silent nucleotide substitution.
The purpose of this paper is to study the major evolutionary force operating in the ubiquitin gene family by using the above criteria. In the case of ubiquitin genes, pN is effectively 0, because there are virtually no amino acid differences even between different species. Therefore, the problem will be whether pS is as small as expected under concerted evolution.
Materials and Methods
Nucleotide Sequences Used.
We have compiled nucleotide sequence data for as many genes as possible from GenBank. The complete set of ubiquitin genes compiled is presented in the supplementary material (Table 7 at www.pnas.org). In the present study, we have decided to examine primarily poly-u genes, because the nucleotide sequences of monomeric genes are known to be very different from those of poly-u genes in most organisms, and the divergence of these two classes of genes is as large as that between the poly-u genes from fungi, plants, and animals (7). We have also excluded protist genes from our analysis, because the number of genes studied is small and the genetic codes used in some protist species were different from the standard genetic code (16).
In the present study, we used sequence data for 8 poly-u loci from 8 fungal species, 18 poly-u loci from 9 plant species, and 16 poly-u loci from 11 animal species. When there are several poly-u loci in a species, they are denoted by A, B, C, etc. or by UbA, UbB, UbC, etc., which are identifiable by GenBank accession numbers (Table 7 at www.pnas.org). Note that these notations are not necessarily the same as those of previous authors (3, 8). To examine the extent of sequence divergence, we computed the number of synonymous differences per synonymous sites (pS) for all pairs of ubiquitin genes within and between poly-u loci in all species. For some groups of species, we also computed the pS value for interspecific comparison of poly-u genes. In the computation of pS, we used the modified Nei–Gojobori method (17). There was no problem in sequence alignment of the coding regions, because the amino acid sequences are highly conserved.
Results
Sequence Divergence of Ubiquitin Genes Within Poly-u Loci.
As mentioned above, it would be relatively easy to distinguish between concerted and birth-and-death evolution if there were ubiquitin gene sequences from many pairs of closely related species. Unfortunately, the ubiquitin gene sequences available now come mostly from distantly related species. However, intraspecific comparison of repeat genes would also reject the importance of concerted evolution if the pS values are very large, because the frequent occurrence of interlocus recombination or gene conversion is expected to give small pS values within each species.
We therefore computed the pS values for all poly-u genes compiled. The results for the repeat genes within poly-u loci from representative fungal, plant, and animal species are presented in Table 1. In most species, the pS value is generally greater than 0.20 and is often 0.4–0.6. The latter values are near or at the saturation level (0.4–0.74) of pS, as will be shown later. In some species (e.g., Gibberella pulicaris), there are gene pairs that show a pS value of about 0.06. However, even these pS values are still quite high compared with those for other genes from closely related species. For example, the average pS value between the human and chimpanzee globin genes is about 0.016 (18). This suggests that the ubiquitin gene repeats are not homogenized as often as the rRNA gene repeats (10), and that they are consistent with the model of birth-and-death evolution. Note that because birth-and-death evolution allows for the occurrence of gene duplication, pairs of genes that are duplicated recently are expected to be closely related or even identical. Our study has shown that this conclusion applies to almost all fungal and plant ubiquitin genes so far studied (see below for the exceptional cases of green algae and fission yeast).
Table 1.
Synonymous differences per synonymous site (pS × 100) within poly-u loci
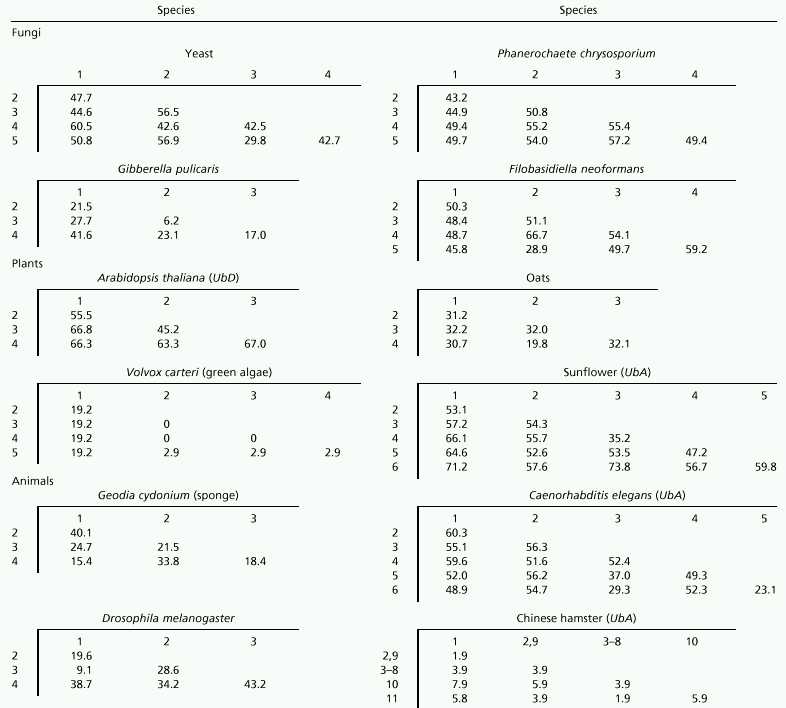 |
The pattern of intraspecific variation of ubiquitin genes in animals is also generally similar to that of fungi and plants. This can be seen from the pS values for sponge, Caenorhabditis elegans, and Drosophila (Table 1). The pS values in these species are generally very high. This was also the case with humans, chicken, and Xenopus.
However, there are several exceptional poly-u loci. For example, the UbA locus of Chinese hamster contains 11 repeat genes, and genes 2 and 9 show the identical nucleotide sequence. Genes 3 to 8 also show the same sequence, although it is not the same as that for genes 2 and 9 (Table 1). Furthermore, the pS values for all other gene pairs are 0.079 or smaller. This suggests that this gene family has recently arisen by repeated gene duplication or has been subject to gene conversion. The UbB locus of Chinese hamster also showed rather low pS values (0–0.074), but the average pS (0.050) was considerably higher than that of UbA (0.017) (data not shown). Another poly-u locus that showed small pS values is the rat UbA. This locus includes a number of genes with the identical nucleotide sequence, and the average pS was 0.026. In the rat UbB locus, all sequences were different, yet the average pS was 0.047 (Table 2). The mouse UbB locus also showed relatively low pS values (average pS being 0.098), but all four sequences were different.
Table 2.
pS values (×100) for intraspecific and interspecific comparisons of poly-u B loci from humans (H), mice (M), and rats (R)
| Gene* | H1 | H2 | H3 | M1 | M2 | M3 | M4 | R1 | R2 | R3 |
|---|---|---|---|---|---|---|---|---|---|---|
| H1 | ||||||||||
| H2 | 12 | |||||||||
| H3 | 19 | 7 | ||||||||
| M1 | 28 | 24 | 25 | |||||||
| M2 | 30 | 24 | 25 | 7 | ||||||
| M3 | 31 | 25 | 26 | 4 | 9 | |||||
| M4 | 27 | 22 | 24 | 15 | 7 | 16 | ||||
| R1 | 31 | 27 | 25 | 10 | 9 | 15 | 16 | |||
| R2 | 33 | 27 | 25 | 10 | 6 | 12 | 13 | 6 | ||
| R3 | 30 | 25 | 24 | 6 | 7 | 10 | 15 | 4 | 5 | |
| R4 | 30 | 24 | 22 | 7 | 3 | 9 | 10 | 6 | 3 | 5 |
Gene symbols are abbreviated.
In addition to these rodent species, one fungal (Schizosaccharomyces pombe, fission yeast) and one plant (Volvox carteri, green algae) species showed a low level of sequence divergence among the poly-u repeat genes. In V. carteri, three of the five genes showed the identical sequence, and pS varied from 0 to 0.192, the average pS being 0.081 (Table 1). S. pombe has a poly-u locus consisting of eight genes, and pS varied from 0 to 0.247, the average being 0.066 (data not shown). There were two pairs of genes that had the identical nucleotide sequence. In these two species, however, there were some genes that were substantially different from the others, unlike the rodent genes.
Divergence of Repeat Genes Within and Between Poly-u Loci from the Same Species.
In rRNA genes, it has been shown that the homogenization of member genes occurs even between genes located on different chromosomes (10). Comparing poly-u genes from different loci, Tan et al. (5) concluded that ubiquitin genes on different chromosomes are also homogenized by gene conversion. This conclusion is based on the observation that in some species (e.g., sunflower), a gene from a poly-u locus is very similar to a gene from another poly-u locus. We therefore computed the pS values for intralocus and interlocus comparisons of ubiquitin repeat genes for maize, sunflower, humans, and other species, where several poly-u loci exist. In maize, there are at least three poly-u loci (A, B, and C), and all member genes of the three loci are completely sequenced. In this species, pS is generally very large and is nearly the same for both within-locus and between-locus comparisons, although there are some exceptions (Table 3). This suggests that the times of divergence of within-locus repeats are generally as old as those of divergence of between-locus repeats. In fact, this extent of divergence is close to the saturation level (0.4–0.74). One obvious exception is the relatively small value (0.05) of pS between genes C1 and C3. This pair of genes must have been produced by a recent gene duplication event, yet the duplication appears to have occurred about 15 million years (MY) ago if the rate of synonymous nucleotide substitution is 1.7 × 10−9 per site per year, as will be discussed later. All other within-locus gene pairs show a much higher pS value. Similar results were obtained in many other species such as humans, chicken, and rice.
Table 3.
Intralocus and interlocus synonymous differences per site (pS × 100) for poly-u loci A, B, and C of maize
| Gene | A1 | A2 | A3 | A4 | A5 | B1 | B2 | B3 | B4 | B5 | C1 | C2 | C3 | C4 | C5 | C6 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A1 | ||||||||||||||||
| A2 | 26 | |||||||||||||||
| A3 | 33 | 27 | ||||||||||||||
| A4 | 41 | 40 | 44 | |||||||||||||
| A5 | 52 | 48 | 48 | 36 | ||||||||||||
| B1 | 34 | 38 | 32 | 50 | 47 | |||||||||||
| B2 | 45 | 44 | 35 | 39 | 39 | 33 | ||||||||||
| B3 | 48 | 46 | 34 | 41 | 41 | 35 | 35 | |||||||||
| B4 | 42 | 49 | 37 | 50 | 41 | 24 | 40 | 38 | ||||||||
| B5 | 48 | 46 | 50 | 39 | 20 | 44 | 38 | 46 | 46 | |||||||
| C1 | 28 | 34 | 31 | 49 | 44 | 31 | 39 | 40 | 41 | 42 | ||||||
| C2 | 30 | 29 | 32 | 44 | 44 | 27 | 38 | 37 | 42 | 44 | 20 | |||||
| C3 | 27 | 32 | 29 | 47 | 45 | 29 | 40 | 45 | 40 | 44 | 5 | 22 | ||||
| C4 | 42 | 39 | 38 | 33 | 36 | 35 | 34 | 26 | 47 | 36 | 32 | 31 | 34 | |||
| C5 | 42 | 45 | 37 | 30 | 38 | 38 | 37 | 29 | 43 | 46 | 43 | 44 | 45 | 27 | ||
| C6 | 40 | 49 | 47 | 30 | 32 | 47 | 41 | 41 | 45 | 36 | 39 | 43 | 41 | 38 | 37 | |
| C7 | 49 | 50 | 50 | 40 | 24 | 51 | 43 | 47 | 52 | 27 | 43 | 55 | 45 | 35 | 43 | 38 |
The pS values for poly-u loci A and B of sunflower are of special interest (Table 4). Here the intralocus pS values and most of the interlocus pS values are very high and are at the saturation level. However, several interlocus gene comparisons show a relatively small pS value. For example, the pS's for gene pairs A1-B1, A2-B2, and A3-B4 are 0.186, 0.218, and 0.138, respectively. This observation led Tan et al. (5) to suggest that these sequence similarities were generated by gene conversion. If this hypothesis is correct, the gene conversion events must have occurred 40–64 MY ago. This is not a rapid homogenization process, if we follow the definition of concerted evolution given by Smith (15) and Arnheim (10). Actually, a more plausible explanation of the sequence similarity of the above three pairs of genes is that poly-u loci A and B were generated by duplication of an ancestral poly-u locus (O) and subsequent deletion and inversion of some genes, as shown in Fig. 2. Locus duplication can be generated easily when chromosome duplication occurs.
Table 4.
Synonymous differences per site (pS × 100) within and between poly-u loci A and B of sunflower
| Gene | A1 | A2 | A3 | A4 | A5 | A6 | B1 | B2 | B3 |
|---|---|---|---|---|---|---|---|---|---|
| A1 | |||||||||
| A2 | 53 | ||||||||
| A3 | 57 | 54 | |||||||
| A4 | 66 | 56 | 35 | ||||||
| A5 | 65 | 53 | 54 | 47 | |||||
| A6 | 71 | 58 | 74 | 57 | 60 | ||||
| B1 | 19 | 45 | 60 | 65 | 63 | 70 | |||
| B2 | 45 | 22 | 55 | 60 | 55 | 62 | 42 | ||
| B3 | 63 | 45 | 60 | 61 | 52 | 74 | 52 | 58 | |
| B4 | 54 | 59 | 14 | 40 | 61 | 75 | 57 | 63 | 54 |
Figure 2.

A scenario of the evolution history of poly-u loci A and B in sunflower. O represents the common ancestral poly-u locus of UbA and UbB, whereas A′, B′, etc., stand for more recent ancestors. a, b, c, etc., represent ancestral ubiquitin genes. The X mark indicates gene deletion. In the ancestral poly-u locus, B′, genes c and e are inverted. The present-day ubiquitin genes are indicated by numbers.
A more interesting case of generation of duplicate poly-u loci is provided by the sequence data for Arabidopsis. Arabidopsis has five poly-u loci, and the pS values for interlocus and intralocus comparisons of ubiquitin genes for loci A, B, and C are presented in Table 5. Here again, interlocus pS values are generally as large as intralocus pS values. However, in each comparison of different poly-u loci, some pS values (printed in bold face) on the diagonal or close to it are considerably smaller than those off the diagonal. This suggests that loci A, B, and C are generated by locus duplication and subsequent repeat gene deletion and duplication within poly-u loci, as shown in Fig. 3. In fact, intralocus and interlocus comparisons of repeat genes for all of the five loci, A–E, suggest that loci D and E are also products of locus duplication (Table 8 at www.pnas.org). Therefore, it seems that the relatively high sequence similarities for some gene pairs are caused by locus duplication and subsequent repeat gene deletion and duplication, and there is no need to invoke gene conversion.
Table 5.
Synonymous differences per site (pS × 100) within and between poly-u loci A, B, and C of Arabidopsis
| Gene | A1 | A2 | A3 | A4 | B1 | B2 | B3 | B4 | B5 | C1 | C2 | C3 | C4 | C5 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A2 | 53 | |||||||||||||
| A3 | 58 | 49 | ||||||||||||
| A4 | 52 | 45 | 51 | |||||||||||
| B1 | 12 | 45 | 60 | 51 | ||||||||||
| B2 | 50 | 14 | 53 | 47 | 45 | |||||||||
| B3 | 69 | 51 | 30 | 56 | 67 | 54 | ||||||||
| B4 | 46 | 49 | 39 | 38 | 40 | 49 | 44 | |||||||
| B5 | 49 | 46 | 52 | 23 | 48 | 49 | 58 | 38 | ||||||
| C1 | 9 | 54 | 63 | 56 | 19 | 51 | 69 | 50 | 48 | |||||
| C2 | 49 | 12 | 55 | 49 | 42 | 14 | 55 | 53 | 52 | 51 | ||||
| C3 | 61 | 48 | 30 | 55 | 58 | 52 | 21 | 45 | 52 | 64 | 50 | |||
| C4 | 60 | 48 | 26 | 49 | 55 | 52 | 18 | 33 | 44 | 58 | 50 | 23 | ||
| C5 | 44 | 54 | 41 | 38 | 40 | 51 | 47 | 18 | 43 | 52 | 56 | 45 | 35 | |
| C6 | 48 | 45 | 49 | 28 | 43 | 50 | 56 | 33 | 11 | 46 | 54 | 52 | 43 | 44 |
Figure 3.

A scenario of the evolutionary history of poly-u loci A, B, C, D, and E in Arabidopsis. The notations O, A′, B′, etc., and a, b, c, etc., are the same as those in Fig. 2. The present-day ubiquitin genes are indicated by numbers. The pS values for the five poly-u loci are presented in Table 8 (see supplementary material at www.pnas.org).
Interkingdom Sequence Divergence of Ubiquitin Genes.
A number of authors (3, 5, 7) have claimed that concerted evolution occurs whenever the extent of intergenic sequence divergence within species is lower than that between species. For example, Tan et al. (5) computed the average number of nucleotide differences (d̄W) between 11 repeat genes of the C. elegans poly-u locus and the average number of nucleotide differences (d̄B) between the C. elegans genes and other poly-u genes from other species, including animals, plants, fungi, and protists. They obtained d̄W = 32.2 and d̄B = 49.0, respectively. Because the former was significantly smaller than the latter, they concluded that the C. elegans genes have been homogenized by concerted evolution. However, the definition of concerted evolution used here is very different from the original one mentioned earlier. If we accept Tan et al.'s definition (d̄B > d̄W), it would be inferred that almost all multigene families, including retroposon families, have experienced concerted evolution, because gene duplication has occurred many times in higher organisms (19, 20). For this reason, it is not very meaningful to compare ubiquitin genes from distantly related organisms for the purpose of studying concerted evolution.
However, comparison of ubiquitin genes between different kingdoms would give information about the upper limit of pS when there is no or virtually no amino acid substitution. When there is no selection and the Jukes–Cantor model of nucleotide substitution applies, the expected upper limit of pS is approximately 0.75. In ubiquitin genes, however, this assumption does not apply, because virtually no amino acid substitution has occurred, and there is some extent of codon usage bias in ubiquitin genes (4). Therefore, the simplest way to infer the upper limit of pS is to evaluate the pS values between different eukaryotic kingdoms, which apparently diverged over one billion years ago (21). For this reason, we computed the pS values between poly-u genes from animals (C. elegans), fungi (Neurospora), and plants (Arabidopsis) (Table 6). The pS value for the interkingdom comparison varies from 0.398 to 0.738, the average being 0.605. Variation in pS is probably due to sampling errors and a small degree of codon usage differences. The number of potentially synonymous sites (S) (16) was nearly the same for all genes, and the average was about 65 in the present case. Therefore, even if the sampling error alone is considered, the SE becomes (0.605 × 0.395/65)1/2 = 0.061. This indicates that the lower and the upper two standard-error limits from the mean are 0.48 and 0.73, respectively, and thus a large portion of the variation in pS can be explained by chance alone.
Table 6.
Synonymous differences per site (pS × 100) within and between animal (C, C. elegans), fungal (N, Neurospora crassa), and plant (A, A. thaliana) poly-u genes
| Gene | C7 | C8 | C9 | C10 | C11 | N1 | N2 | N3 | N4 | A1 | A2 | A3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C8 | 41 | |||||||||||
| C9 | 44 | 11 | ||||||||||
| C10 | 46 | 34 | 40 | |||||||||
| C11 | 52 | 57 | 58 | 57 | ||||||||
| N1 | 62 | 64 | 60 | 55 | 60 | |||||||
| N2 | 65 | 59 | 55 | 54 | 64 | 48 | ||||||
| N3 | 64 | 64 | 66 | 55 | 62 | 54 | 53 | |||||
| N4 | 57 | 71 | 72 | 66 | 67 | 56 | 75 | 49 | ||||
| A1 | 54 | 66 | 66 | 51 | 74 | 61 | 57 | 58 | 77 | |||
| A2 | 66 | 65 | 63 | 54 | 62 | 54 | 54 | 49 | 53 | 53 | ||
| A3 | 55 | 52 | 50 | 55 | 63 | 67 | 60 | 58 | 67 | 58 | 49 | |
| A4 | 61 | 55 | 55 | 57 | 67 | 60 | 57 | 43 | 54 | 53 | 45 | 51 |
In C. elegans, genes 1–6 were not used to save space (see Table 2 for these genes).
In Tables 1 and 3, we have seen that many intralocus gene comparisons show a pS value of about 0.30 or higher. The above computation suggests that they are either at or close to the saturation level. Many interlocus gene comparisons in Arabidopsis (Table 5) also show high pS values. Apparently, they are also at the saturation level.
Rates of Synonymous Substitution in Ubiquitin Genes.
To convert the number of synonymous substitutions per site (dS) into the time of divergence between the two sequences, a number of authors (3, 5) used the average rate of synonymous substitution for many mammalian genes, which is 4.61 × 10−9 per site per year. However, because there are virtually no amino acid substitutions between ubiquitin genes, this rate is probably an overestimate for ubiquitin genes. The synonymous rate is also known to vary substantially from gene to gene. We therefore reexamined this problem by using the average number of synonymous substitutions between humans and rodents and between mice and rats (Table 2). For this purpose, we used the UbB locus, which is known to be orthologous because of the high sequence similarity of the 3′ untranslated region (ref. 7; unpublished results). The time of divergence (T) between humans and rodents is not well established, but we used a molecular estimate of 100 MY (22, 23). Similarly, we used 40 MY as a rough estimate for the divergence time between mice and rats (23).
The average pS value (p̄S) between the human UbB repeat genes and their orthologous counterparts from mice and rats was 0.263. Inserting this p̄S into the Jukes–Cantor formula, we obtained dS = 0.324 as a rough estimate of the number of synonymous substitutions per site between humans and mice. The rate of synonymous substitution (r) is therefore obtained by r = dS/(2T) = 0.324/(200 × 106) = 1.6 × 10−9. Similarly, we obtain p̄S = 0.133 and dS = 0.146 for mice and rats. Therefore, if we use T = 40 MY, the rate of synonymous substitution becomes r = 1.8 × 10−9. We use the average (1.7 × 10−9) of these estimates in this paper.
Divergence Times for Closely Related Sequences.
Previously we have seen that in some species there are repeat gene pairs that show a small pS value. Let us estimate the time of divergence between these genes. In the poly-u locus of Gibberella, the pS for genes 2 and 3 is 0.062, and this can be converted into a Jukes–Cantor distance of dS = 0.065. Therefore, the time of divergence between these two genes is estimated to be T = dS/(2r) = 18 MY. The next closest pair of genes in this species is genes 3 and 4, which show pS = 0.170. This is translated into dS = 0.193 and T = 57 MY. These time estimates are much higher than the time of divergence (about 5 MY) between humans and chimpanzees. In our view, therefore, it is inappropriate to claim that this locus has been subject to concerted evolution.
In many other species, pS is close to its upper limit, and therefore it is difficult to estimate divergence times. However, it is clear that many repeat genes have diverged by silent mutation probably for hundreds of millions of years. As was mentioned earlier, there are exceptional poly-u loci, in which repeat genes show identical or very closely related sequences. In our view, however, the repeat genes of these loci were generated by recent gene duplication (see below).
Discussion
We have seen that in the ubiquitin gene family, the pS values between repeat genes are generally very high, and this supports the model of birth-and-death evolution under the influence of strong purifying selection. The homogeneity of the proteins produced is apparently caused by the functional constraints of the ubiquitin protein rather than by concerted evolution. Despite the extremely high degree of evolutionary conservation of the protein, the nucleotide sequences have diverged extensively by silent mutations.
In some species, such as Chinese hamster, we have seen a high degree of sequence similarity between repeat genes of a poly-u locus. This result is consistent with the model of concerted evolution, but it is also consistent with birth-and-death evolution if gene duplication is assumed to have occurred recently. Only if DNA sequence homogeneity is attained by gene conversion does this observation reject the model of birth-and-death evolution. However, Nenoi et al. (7) have shown that the sequence similarity in Chinese hamster was actually caused by recent gene duplication rather than by gene conversion. In fact, a number of authors have found rare variations of the poly-u A locus with respect to copy number in Chinese hamster and humans (7, 24, 25).
Nenoi et al.'s (7) study suggests that the increase or decrease of copy number in poly-u loci is caused by interlocus recombination. In the case of ubiquitin genes, however, the number of poly-u loci also occasionally increases or decreases in the evolutionary process, as is clear from the comparison of the number of poly-u loci among different species (Table 7 of supplemental materials at www.pnas.org). It appears that the increase of poly-u loci is caused by genome duplication or some other large-scale DNA duplication. It is therefore reasonable that higher organisms with larger genome sizes (e.g., maize and humans) have a larger number of poly-u loci. By contrast, the decrease of the number of poly-u loci apparently occurs by inactivation of redundant poly-u loci.
Inactivation of a poly-u locus is unique, because a locus contains contiguous copies of ubiquitin genes without intervening nucleotides. Therefore, a single nonsense mutation or an insertion or deletion of a single or a few nucleotides in the first gene from the 5′ side of the locus may inactivate the entire set of genes in the locus. This has occurred in the poly-u pseudogenes of humans and Arabidopsis (Fig. 4). However, if a nonsense or frameshift mutation occurs in the last gene (3′ side) of the locus, only this gene will be inactivated. This event has happened with the Chinese hamster poly-u A locus (26).
Figure 4.

Poly-u pseudogenes from humans, Arabidopsis, and Chinese hamster.
For the last two decades, it has been commonly believed that most multigene families are subject to concerted evolution (9, 11, 27). This idea comes primarily from studies of ribosomal and small nuclear RNA genes and globin genes (27). In recent years, however, a number of authors have shown that multigene families associated with immune response or disease resistance are generally subject to birth-and-death evolution, and this mode of evolution is important in generating a set of diversified member genes to cope with different pathogens (12–14, 28–32). We have now shown that even the ubiquitin gene family encoding a highly conserved protein is subject to birth-and-death evolution at the DNA level, and the homogeneity of ubiquitin sequences is attained by strong purifying selection. This suggests that the occurrence of birth-and-death evolution may be more widespread than previously thought. In fact, we have obtained evidence that the multigene family encoding histone, another highly conserved protein, is also subject to birth-and-death evolution at the DNA level (A. P. Rooney, H.P., and M.N., unpublished work). It now seems to be time to reevaluate the modes of evolution of various multigene families.
Supplementary Material
Acknowledgments
We thank Claude dePamphilis, Walter Fitch, Mitsuru Nenoi, Alex Rooney, and Shozo Yokoyama for their comments on an earlier version of this paper. This work was supported by grants from the National Institutes of Health (GM20293) and the National Aeronautics and Space Administration (NCC2–1057) to M.N.
Abbreviations
- poly-u
poly-ubiquitin
- MY
million years
- pS
proportion of synonymous nucleotide differences per synonymous site
References
- 1.Jentsch S, Seufert W, Hauser H P. Biochim Biophys Acta. 1991;1089:127–139. doi: 10.1016/0167-4781(91)90001-3. [DOI] [PubMed] [Google Scholar]
- 2.Graham R W, Jones D, Candido E P. Mol Cell Biol. 1989;9:268–277. doi: 10.1128/mcb.9.1.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharp P M, Li W H. J Mol Evol. 1987;25:58–64. doi: 10.1007/BF02100041. [DOI] [PubMed] [Google Scholar]
- 4.Mita K, Ichimura S, Nenoi M. J Mol Evol. 1991;33:216–225. doi: 10.1007/BF02100672. [DOI] [PubMed] [Google Scholar]
- 5.Tan Y, Bishoff S T, Riley M A. Mol Phylogenet Evol. 1993;2:351–360. doi: 10.1006/mpev.1993.1035. [DOI] [PubMed] [Google Scholar]
- 6.Vrana P B, Wheeler W C. Mol Phylogenet Evol. 1996;6:259–269. doi: 10.1006/mpev.1996.0075. [DOI] [PubMed] [Google Scholar]
- 7.Nenoi M, Mita K, Ichimura S, Kawano A. Genetics. 1998;148:867–876. doi: 10.1093/genetics/148.2.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dover G. Nature (London) 1982;299:111–117. doi: 10.1038/299111a0. [DOI] [PubMed] [Google Scholar]
- 9.Ohta T. Theor Popul Biol. 1983;23:216–240. doi: 10.1016/0040-5809(83)90015-1. [DOI] [PubMed] [Google Scholar]
- 10.Arnheim N. In: Evolution of Genes and Proteins. Nei M, Koehn R K, editors. Sunderland, MA: Sinauer; 1983. pp. 38–61. [Google Scholar]
- 11.Li W-H. Molecular Evolution. Sunderland, MA: Sinauer; 1997. , chap. 11. [Google Scholar]
- 12.Nei M, Hughes A L. In: 11th Histocompatibility Workshop and Conference. Tsuji K, Aizawa M, Sasazuki T, editors. Vol. 2. Oxford: Oxford Univ. Press; 1992. pp. 27–38. [Google Scholar]
- 13.Ota T, Nei M. Mol Biol Evol. 1994;11:469–482. doi: 10.1093/oxfordjournals.molbev.a040127. [DOI] [PubMed] [Google Scholar]
- 14.Nei M, Gu X, Sitnikova T. Proc Natl Acad Sci USA. 1997;94:7799–7806. doi: 10.1073/pnas.94.15.7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith G P. Cold Spring Harbor Symp Quant Biol. 1974;38:507–513. doi: 10.1101/sqb.1974.038.01.055. [DOI] [PubMed] [Google Scholar]
- 16.Nei M, Kumar S. Molecular Evolution and Phylogenetics. Oxford: Oxford Univ. Press; 2000. [Google Scholar]
- 17.Zhang J, Rosenberg H F, Nei M. Proc Natl Acad Sci USA. 1998;95:3708–3713. doi: 10.1073/pnas.95.7.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li W-H, Tanimura M. Nature (London) 1987;326:93–96. doi: 10.1038/326093a0. [DOI] [PubMed] [Google Scholar]
- 19.Nei M. Nature (London) 1969;221:40–42. doi: 10.1038/221040a0. [DOI] [PubMed] [Google Scholar]
- 20.Ohno S. Evolution by Gene Duplication. Berlin: Springer; 1970. [Google Scholar]
- 21.Dickerson R E. J Mol Evol. 1971;1:26–45. doi: 10.1007/BF01659392. [DOI] [PubMed] [Google Scholar]
- 22.Li W-H, Gouy M, Sharp P M, O'hUigin C, Yang Y-W. Proc Natl Acad Sci USA. 1990;87:6703–6707. doi: 10.1073/pnas.87.17.6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar S, Hedges B. Nature (London) 1998;392:917–919. doi: 10.1038/31927. [DOI] [PubMed] [Google Scholar]
- 24.Baker R T, Board P G. Am J Hum Genet. 1989;44:534–542. [PMC free article] [PubMed] [Google Scholar]
- 25.Nenoi M, Mita K, Ichimura S, Cartwright I L, Takahashi E, Yamauchi M, Tsuji H. Gene. 1996;175:179–185. doi: 10.1016/0378-1119(96)00145-x. [DOI] [PubMed] [Google Scholar]
- 26.Nenoi M, Mita K, Ichimura S, Cartwright I L. Biochim Biophys Acta. 1994;1204:271–278. doi: 10.1016/0167-4838(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 27.Liao D. Am J Hum Genet. 1999;64:24–30. doi: 10.1086/302221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Date A, Satta Y, Takahata N, Chigusa S I. Immunogenetics. 1998;47:417–429. doi: 10.1007/s002510050379. [DOI] [PubMed] [Google Scholar]
- 29.Michelmore R W, Meyers B C. Genome Res. 1998;8:1113–1130. doi: 10.1101/gr.8.11.1113. [DOI] [PubMed] [Google Scholar]
- 30.Sitnikova T, Nei M. Mol Biol Evol. 1998;15:50–60. doi: 10.1093/oxfordjournals.molbev.a025846. [DOI] [PubMed] [Google Scholar]
- 31.Gu X, Nei M. Mol Biol Evol. 1999;16:147–156. doi: 10.1093/oxfordjournals.molbev.a026097. [DOI] [PubMed] [Google Scholar]
- 32.Zhang J, Dyer K D, Rosenberg H F. Proc Natl Acad Sci USA. 2000;97:4701–4706. doi: 10.1073/pnas.080071397. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.