

Summary
Spinal sensory ganglia have been shown to contain neuronal subpopulations with different functions and neurotrophin dependencies. Neurotrophins act, in large part, through Trk receptor tyrosine kinases: nerve growth factor (NGF) via TrkA, brain-derived neurotrophic factor (BDNF) and neurotrophin-4/5 (NT-4/5) via TrkB, and neurotrophin-3 (NT-3) via TrkC. In the present paper, we use antibodies to TrkA, TrkB, and TrkC to characterize their expression patterns and to determine which subpopulations of cells are lost in mice lacking individual neurotrophins or Trk receptors. Despite previous reports of Trk receptor mRNAs in neural crest cells, we detect Trk receptor proteins only in neurons and not in neural crest cells or neuronal precursors. Comparisons of neonatal mice deficient in NT-3 or its cognate receptor TrkC have shown that there is a much greater deficiency in spinal sensory neurons in the former, suggesting that NT-3 may activate receptors in addition to TrkC. Using the same antibodies, we show that, during the major period of neurogenesis, NT-3 is required to maintain neurons that express TrkB in addition to those that express TrkC but is not essential for neurons expressing TrkA. Results also indicate that survival of cells expressing both receptors can be maintained by activation of either one alone. NT-3 can thus activate more than one Trk receptor in vivo, which when coexpressed are functionally redundant.
Introduction
Neurotrophic factors have been shown to be important regulators of the development and maintenance of vertebrate nervous systems (reviewed by Reichardt and Fariñas, 1997). In particular, during nervous system development, neuronal populations undergo a process of naturally occurring cell death, which is believed to ensure a balance between neuronal numbers and the sizes of their target territories. Among these, the neurotrophins are a closely related family of trophic factors that have been shown to be secreted in limiting amounts by target tissues, to be internalized into and retrogradely transported within neurons by receptor-mediated processes, and to mediate not only neuronal survival but many aspects of neuronal differentiation and function. In mice, there are four identified neurotrophins: nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and NT-4/5. The actions of these factors are mediated in large part through receptor tyrosine kinases of the Trk family, with NGF interacting with TrkA, BDNF and NT-4 with TrkB, and NT-3 with TrkC. Ligand binding to these receptors has been shown to activate several intracellular signaling pathways, including PI-3 kinase, ras, MAP kinases, and PLC1, some of which promote cell survival and others of which promote differentiation. All of the neurotrophins have also been shown to interact with the low affinity neurotrophin receptor p75NTR, which has been shown also to regulate intracellular signaling pathways, such as sphingomyelin hydrolysis.
Spinal sensory neurons, found in dorsal root ganglia (DRG), include different subpopulations within each ganglion specialized for transfer of different modalities of sensory information (reviewed by Scott, 1992). Distinct subclasses of neurons have been shown to innervate separate types of peripheral sensory organs and specific laminae within the spinal cord. Previous work has shown that these different populations of neurons exhibit different neurotrophin dependencies and that specific subpopulations are absent in mice lacking individual neurotrophins or Trk receptors (reviewed by Reichardt and Fariñas, 1997). Thus, in the DRGs of NGF-or TrkA-deficient mice, ~70% of the normal complement of neurons is missing, and these include essentially all of the neurons that express TrkA postnatally, which are small-diameter neurons with unmyelinated axons that mediate pain perception and express the peptide transmitters calcitonin gene-related peptide (CGRP) and substance P (Crowley et al., 1994; Smeyne et al., 1994; Minichiello et al., 1995). An additional population of small-diameter neurons with C-fibers that mediate non-nociceptive thermal and low threshold mechanoreceptive stimuli are also lost in these mutants (Silos-Santiago et al., 1995). Mice with targeted mutations in either NT-3 or its high affinity receptor TrkC have been shown to lack major populations of proprioceptive neurons, including virtually all group Ia afferents, as assessed by the absence of the Ia projection to the ventral spinal cord and of muscle spindles and Golgi tendon organs in peripheral target fields (Ernfors et al., 1994; Fariñas et al., 1994; Klein et al., 1994; Tessarollo et al., 1994; Tojo et al., 1995). Deficiencies in D-hair cutaneous afferents and Merkel cells, which are induced and maintained by slowly adapting (SA) afferents, are also seen in NT-3–deficient animals postnatally (Airaksinen et al., 1996). While the identities of the ~35% of DRG sensory neurons lost in trkB and BDNF mutant animals have been more elusive, these deficits appear to occur postnatally (Minichiello et al., 1995; Silos-Santiago et al., 1997; I. F. and L. F. R., unpublished data) and may reflect the importance in vivo of an autocrine BDNF–TrkB loop (Acheson et al., 1995).
In most instances, neurotrophin-deficient and cognate Trk receptor–deficient mice have similarities in phenotype in agreement with the biochemical interactions between these molecules characterized in vitro (see Reichardt and Fariñas, 1997, for summary). Surprisingly, mice deficient in NT-3 lose many more neurons than those lacking a functional TrkC receptor (Ernfors et al., 1994; Fariñas et al., 1994, 1996; Klein et al., 1994; Tessarollo et al., 1994, 1997; Minichiello et al., 1995; Tojo et al., 1995; Liebl et al., 1997; Silos-Santiago et al., 1997). In particular, as many as 60%–70% of the neurons are missing in DRGs of neonatal NT-3–deficient mice, whereas only ~20% are lost in a TrkC kinase–deficient mouse strain and ~30% are lost in trkC null mice, suggesting that NT-3 may activate receptors other than TrkC. Consistent with this possibility, it has been shown that NT-3 is capable of activating TrkA and TrkB when expressed in fibroblasts (Ip et al., 1993). However, when these receptors were expressed in PC12 cells, they could only be activated by NT-3 at concentrations 100-fold higher than those of their preferred ligands, NGF and BDNF, respectively. This has suggested that neuronal populations restrict the actions of nonpreferred ligands and that, in vivo, where neurotrophins are present in limiting quantities, NT-3 acts only on its cognate receptor TrkC.
We have previously shown that the lack of NT-3 during embryonic development of DRGs results in increased neuronal apoptosis as early as embryonic day 11 (E11) (Fariñas et al., 1996). It has also been shown that apoptosis is increased in the DRGs of embryos that lack a functional TrkC receptor at E11 (White et al., 1996). This suggests that the absence of NT-3–mediated TrkC signaling causes the loss of proprioceptive neurons, which are missing in both the trkC and the NT-3 mutant mice at birth. In order to understand why so many additional neurons are missing in the NT-3–deficient embryos, we analyzed Trk protein expression on sensory neurons and their precursors in wild-type, NT-3 mutant, and various trk mutant embryos. Using antibodies specific for the extracellular domains of each Trk receptor (Clary et al., 1994; Wilkinson, 1997), we have discovered that, in addition to TrkC-expressing neurons, TrkB-expressing neurons are also lost in NT-3 mutants, providing an explanation for this discrepancy. TrkB neurons are not lost in trkC mutants, implying that NT-3–mediated activation of TrkB is essential for survival of these cells in vivo.
We have also shown previously that the lack of NT-3 during embryonic development of DRGs results in premature differentiation of neuronal precursor cells within the DRG by E12 (Fariñas et al., 1996). As there have been numerous reports of survival and/or mitogenic effects of NT-3 and other neurotrophins on neuronal precursors cultured in vitro (e.g., Sieber-Blum, 1991; Memberg and Hall, 1995), the simplest explanation for the observed premature differentiation of precursors in vivo was a direct mitogenic effect of this neurotrophin on those cells. Using these same antibodies, however, we have discovered that DRG precursors do not express detectable levels of any Trk receptor protein. Together with our recent observations indicating that NGF deficiency also results in neuronal loss followed by premature differentiation of precursors (unpublished data), our results indicate that the actions of neurotrophins on precursor cells in vivo are indirect and the loss of large numbers of neurons reduces interactions that normally inhibit precursor differentiation.
Results
To characterize the developmental expression patterns and neurotrophin dependencies of cells expressing the different Trk receptors, we prepared antibodies specific for TrkA, TrkB, and TrkC, using the entire extracellular domains of rat TrkA, TrkB, and TrkC purified from either baculovirus-infected Sf29 cell or COS cell supernatants. By antigen blot, antigen immunoprecipitation, and immunocytochemistry, each of these antibodies has been shown to recognize its antigen but not either of the two other Trk receptors or unrelated antigens (e.g., Clary et al., 1994; Wilkinson, 1997). Thus, anti-TrkA recognizes TrkA but not TrkB or TrkC and so on.
Using these specific reagents, we then characterized the expression patterns of each Trk during the major period of neurogenesis in spinal sensory ganglia, which occurs between E10 and E13 (e.g., Fariñas et al., 1996). As illustrated in Figure 1, immunostaining of DRGs with antibodies specific for TrkA, TrkB, or TrkC shows that Trk receptor expression in wild-type embryos is highly dynamic between E11 and E13. Cells with neuronal morphologies expressing each of the Trk receptors are present at E11. Each antibody appears to stain both intracellular and surface membrane compartments containing Trk receptor protein. Prominent staining is detected on axons as well as cell bodies of neurons expressing each of these proteins. We have also observed prominent staining of axons and growth cones in the targets of these neurons at this and later stages of development (data not shown). Even at E11, the average sizes of neurons expressing TrkA appear to be significantly smaller than those of neurons expressing either TrkB or TrkC, as has previously been observed in adult sensory ganglia (e.g., McMahon et al., 1994).
Figure 1. TrkA, TrkB, and TrkC Expression in Early DRGs Is Highly Dynamic.
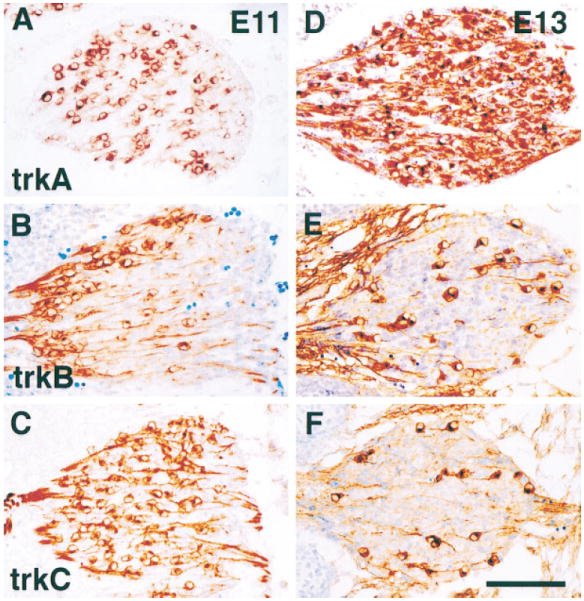
Immunostaining of thoracolumbar DRGs from E11 (A–C) and E13 (D–F) wild-type embryos with antibodies specific to TrkA (A and D), TrkB (B and E), or TrkC (C and F) shows the highly dynamic expression of these receptors over this 2 day period. Scale bar, 100 μm.
In earlier work, we have quantified neurogenesis in both thoracic and lumbar sensory ganglia and have observed that neurogenesis is initiated shortly before E10 and is consummated with a major burst between E12 and E13, after which there is no net neurogenesis within these ganglia (Fariñas et al., 1996). Examination of Trk receptor expression during this interval, as illustrated in Figure 1, shows that the absolute numbers and proportions of cells expressing each Trk receptor vary rapidly over this interval. At E11, TrkC appears to be expressed in a majority of neurons, but comparatively few neurons continue to express this receptor at E13. At E11, a spatially skewed population of neurons expresses TrkB, and this population also appears to be reduced at E13. In contrast, while TrkA-expressing cells are present in reasonable numbers at E11, their numbers increase dramatically over the next 2 days, so that at E13, the vast majority of neurons appear to express this receptor. These data appear generally consistent with previous descriptions of Trk mRNA distributions within DRGs using in situ hybridization techniques (Martín-Zanca et al., 1990; Tessarollo et al., 1993, 1994; Lamballe et al., 1994; White et al., 1996). The numbers of neurons expressing each Trk receptor have been quantified, and the results are presented in Table 1 and Figure 2. Results show that in the L1 DRG ~70% of neurons express TrkC at E11, but <10% continue to express it at E13. The total number of neurons expressing TrkC at E13 is smaller than at E11, as the total number of neurons increases ~2.5-fold during this interval (Fariñas et al., 1996), not sufficient to compensate for the 4-fold decrease in proportion of TrkC-expressing cells. Approximately 40% of the neurons express TrkB at E11, and this fraction is reduced to ~8% at E13. Again, this reflects a decrease in absolute number as well as proportion of neurons expressing this Trk receptor. In contrast, while ~20% of neurons express TrkA at E11, this proportion is increased to almost 80% at E13. This represents an 8-fold increase in the number of cells expressing TrkA and suggests strongly that the vast majority of neurons generated during the burst of neurogenesis that occurs between E12 and E13 express this Trk receptor. While differing in magnitude, the same trends are seen during development of the T1 DRG during this interval (Table 1).
Table 1.
Numbers of Trk-Positive Neurons in DRGs of Wild-Type Embryos
| T1 DRG |
L1 DRG |
|||||
|---|---|---|---|---|---|---|
| E11 | E12 | E13 | E11 | E12 | E13 | |
| TrkA | 718 ± 79 (3) | 1403 ± 260 (2) | 3398 ± 454 (3) | 457 ± 52 (3) | 1611 ± 33 (2) | 3694 ± 461 (3) |
| TrkB | 777 ± 47 (3) | 400 ± 44 (2) | 414 ± 28 (3) | 834 ± 95 (3) | 592 ± 136 (2) | 369 ± 50 (3) |
| TrkC | 1385 ± 100 (4) | 962 ± 70 (2) | 722 ± 22 (2) | 1522 ± 113 (3) | 1310 ± 38 (2) | 426 ± 8 (3) |
Numbers of TrkA-, TrkB-, and TrkC-expressing cells in DRGs of wild-type embryos. Numbers of cells in the T1 and L1 DRGs expressing individual Trk receptors were counted at E11, E12, and E13. The number of embryos analyzed for each determination is indicated in parentheses. The average number of immunopositive cells per ganglion is presented ± SEM (variance for n = 2). These numbers were used in preparing Figures 2A and 2B, together with numbers for neurofilament-expressing cells that were determined earlier (Fariñas et al., 1996).
Figure 2. Development of TrkA-, TrkB-, and TrkC-Expressing Cells in Early DRGs.
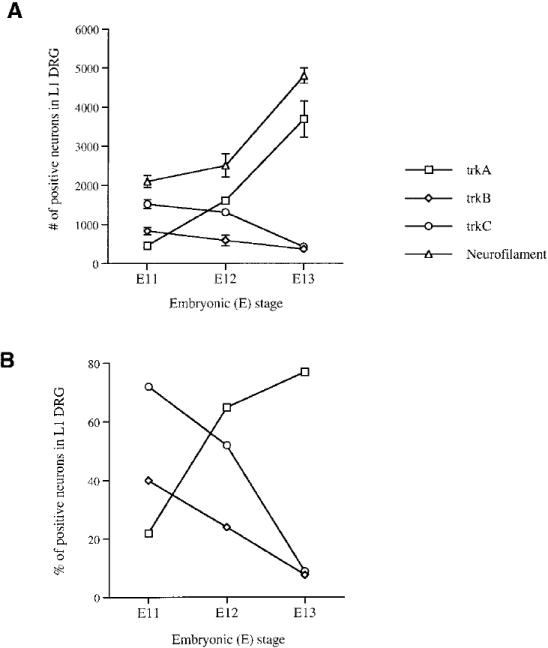
(A) Graph depicts numbers of cells in the L1 DRG expressing individual Trk receptors or neurofilament antigen at E11, E12, and E13. The numbers of cells positive for each Trk were counted in the L1 DRGs of at least two different embryos. Numbers for neurofilament-expressing cells were determined earlier and published in a separate publication (Fariñas et al., 1996).
(B) Graph showing the percentage of neurons expressing each Trk in DRGs of different embryonic stages as quantified by division by the total number of neurons (neurofilament-expressing cells) present in these ganglia at each stage (Fariñas et al., 1996). Each point in the graph represents the percentage of Trk-positive neurons in the L1 ganglia.
In studies on peripheral sensory neuron precursors in vitro, several neurotrophins have been reported to promote survival proliferation or differentiation (e.g., Sieber-Blum, 1991; Kalcheim et al., 1992; Henion et al., 1995; Memberg and Hall, 1995). Thus, we were surprised to observe that, at high magnification in vivo, every cell expressing TrkA, TrkB, or TrkC in the DRGs had a clearly neuronal profile (Figures 3A–3C). Even at E11, when >60% of the DRG cells are precursors (Fariñas et al., 1996), those immunoreactive for Trk receptors present a clear neuronal morphology with the bipolar shape characteristic of young sensory neurons. To examine this issue more thoroughly, we injected wild-type females bearing E11 embryos with 2′-bromo-5′-deoxyuridine (BrdU) 2 hr before sacrifice and determined whether proliferating precursors express any of the Trk receptors. In spite of the large numbers of progenitor cells present at that age, none of the Trk-positive cells had incorporated the deoxyribonucleotide (Figures 3D–3F). For quantitation, we counted a total of 636 Trk-expressing cells (including 162 positive for TrkA, 150 for TrkB, and 324 for TrkC) and 735 BrdU-positive cells in single sections through lumbar DRGs of three different embryos and did not find any BrdU-positive cells that coexpressed a Trk receptor. Consistent with this, in a separate set of colabeling experiments, virtually all cells expressing a Trk receptor were also labeled by antibodies to the 150 kDa neurofilament subunit (data not shown). Altogether, these data indicate that only neurons express Trk receptors in the developing DRGs. Results are consistent with our observations that initial formation of DRG sensory ganglia occurs normally in NT-3– and NGF-deficient mice (Fariñas et al., 1996, and unpublished data). Possible explanations to reconcile the in vivo and in vitro observations will be presented in the Discussion.
Figure 3. Trk Proteins Are Found in Developing Sensory Neurons but Not in Precursors.
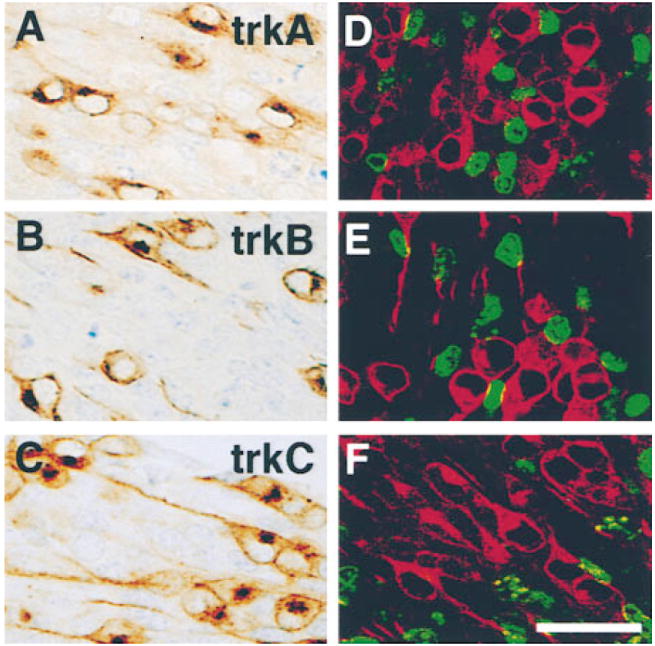
Immunostaining of lumbar DRGs of E11 embryos with antibodies specific to TrkA (A and D), TrkB (B and E), and TrkC (C and F) illustrate the neuronal morphology of cells expressing each of the Trk receptors. Sections (D–F) through lumbar DRGs of E11 embryos stained with each anti-Trk (red) and with anti-BrdU (green) show a complete lack of colocalization between the nucleotide and each Trk receptor. Scale bar, 20 μm.
Neural crest cells, precursors of sensory neurons, have been reported to express mRNA for TrkB and TrkC but not TrkA (Klein et al., 1990; Tessarollo et al., 1993). As we failed to detect Trk receptor protein in the derivatives of these cells, the proliferating precursors within sensory ganglia, we extended our analysis to earlier stages of development when neural crest cells are migrating to sensory and autonomic ganglia. As illustrated in Figure 4, staining of transverse sections through the trunks of E9 embryos, when neural crest cells are actively migrating, indicates that these cells do not express detectable levels of TrkC (Figure 4B) or TrkB (data not shown) proteins but do express the low affinity neurotrophin receptor p75 (Figure 4A). Previous in situ analyses have not detected TrkA mRNA in neural crest (e.g., Martín-Zanca et al., 1990). The data thus indicate that Trk receptors are not present in migrating murine neural crest cells in vivo. We also determined when TrkC protein expression first appears in developing spinal ganglia by staining spinal ganglia at different rostral–caudal levels at E10. Ganglion coalescence and the initiation of neurogenesis occurs at E10 and follows a rostrocaudal gradient. As illustrated in Figures 4C–4E, coinciding with the spatial gradient of neurogenesis, the number of TrkC-immunoreactive cells at E10 is low in caudal DRGs (Figure 4C) and increases progressively in more anterior DRGs (Figures 4D–4E). Even at the time when the first TrkC-positive cell is seen in a DRG (Figure 4C), it shows a clear bipolar neuronal morphology. Similar results were seen using the antibodies to the other Trk receptors (data not shown). Each of these antibodies is a polyclonal prepared using the entire extracellular domain of a Trk receptor as an immunogen and is able to detect Trk protein in intracellular compartments as well as on the cell surface (see Figures 1 and 3). Thus, these data indicate that, in murine DRG sensory ganglia, Trk expression is restricted to postmitotic neurons. When compared to previous in situ analyses (Klein et al., 1990; Tessarollo et al., 1994), our results suggest that translational control mechanisms may help regulate neurotrophin responsiveness. This possibility will be explored more extensively in the Discussion.
Figure 4. Absence of Trk Expression in Neural Crest and Appearance with Neurogenesis in DRGs.
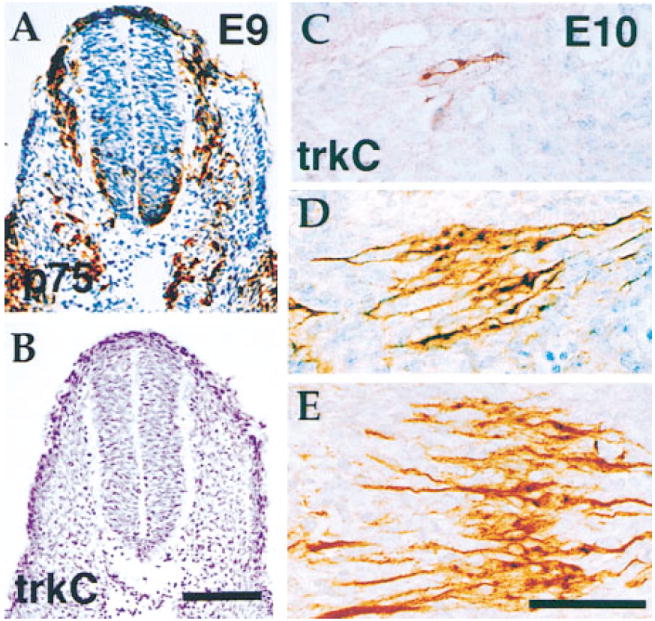
Consecutive transverse sections through the body of an E9 embryo stained for p75NTR to reveal the migrating neural crest (A) and for TrkC (B) demonstrate that neural crest cells do not express the TrkC protein. Neural crest cells were also immunonegative for TrkB and TrkA (data not shown). DRGs in an E10 embryo at successive levels in the caudorostral direction were stained with anti-TrkC antibodies (C–E). Coincident with the spatiotemporal progress of the neurogenic process, the number of cells positive for TrkC increases in the caudal-to-rostral direction. Even in the most caudal DRGs, where as few as one positive cell can be observed, the morphology of the labeled cells is clearly neuronal (C). Scale bars, 190 μm (A and B); 70 μm (C–E).
In earlier work, we have shown that neurons, not precursors, die in the DRGs of NT-3 mutant embryos at E11 (Fariñas et al., 1996). Comparisons of neuronal losses in NT-3–deficient and TrkC-deficient mice also have shown that many more spinal sensory neurons are lost in the former (e.g., Ernfors et al., 1994; Fariñas et al., 1994; Klein et al., 1994; Tessarollo et al., 1997). Among many possibilities, a simple explanation for this discrepancy would be an essential role for NT-3 in maintaining survival of neurons expressing TrkA or TrkB. To explain this discrepancy, it is clearly necessary to identify the populations lost in the absence of NT-3. With this objective, we compared the morphological profiles and quantitated the numbers of neurons expressing each Trk receptor at E11 in wild-type and mutant DRGs (Figure 5 and Table 2). When cells expressing TrkC were examined, we observed, not unexpectedly, a dramatic disruption in staining in NT-3–deficient embryos (Figure 5A). Positive neuronal numbers were drastically reduced and immunoreactivity was, in most instances, associated with pyknotic profiles. We also used TUNEL (terminal deoxynucleotide transferase [TdT] -mediated dUTP nick end labeling) staining, which visualizes nuclei with fragmented DNA, to demonstrate apoptosis of TrkC-positive neurons (Figure 5B). Many but not all TUNEL-positive cells were also stained with the TrkC antibodies. When total numbers of cells expressing TrkC were quantitated in the T1 and L1 DRGs, a dramatic reduction in the numbers of TrkC-positive neurons was observed in the mutant at E11 (Table 2). Thus, almost all TrkC-positive neurons have either died or are dying in the absence of NT-3. When examined a day later at E12, TrkC-expressing cells are completely absent (data not shown), consistent with a previous report that no TrkC mRNA–expressing neurons remain in these ganglia at E13.5 (Tessarollo et al., 1994). At E15.5 and later stages, we have detected reappearance of TrkC expression in a subset of neurons within the DRGs in NT-3 mutants, consistent with the reported presence of TrkC mRNA within a subset of these DRG neurons at E17 and P0 (Tojo et al., 1995; Liebl et al., 1997).
Figure 5. TrkB- and TrkC- but Not TrkA-Positive Neurons Die in Lumbar DRGs of E11 NT-3–Deficient Embryos.
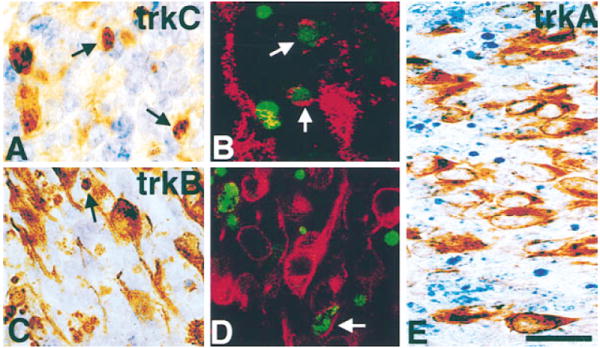
Pyknotic figures (arrows) can be observed that are positive for TrkC (A) and TrkB (C) but not for TrkA (E). Double labeling for either TrkC ([B], red) or TrkB ([D], red) and TUNEL (green) in lumbar DRGs of E11 NT-3–deficient embryos reveals cells positive for both a Trk and TUNEL (arrows). Scale bar, 20 μm.
Table 2.
Numbers of Trk-Positive Neurons in DRGs of Wild-Type and Mutant E11 Embryos
| T1 DRG |
L1 DRG |
|||||
|---|---|---|---|---|---|---|
| Wild-Type | NT-3−/− | % Reduction | Wild-Type | NT-3−/− | % Reduction | |
| TrkA | 718 ± 79 (3) | 770 ± 10 (3) | NS | 457 ± 52 (3) | 390 ± 34 (2) | NS |
| TrkB | 777 ± 47 (3) | 494 ± 98 (2) | 37* | 834 ± 95 (3) | 540 ± 12 (2) | 35* |
| TrkC | 1385 ± 100 (4) | 270 ± 70 (2) | 80** | 1522 ± 113 (3) | 494 ± 254 (2) | 68** |
| Wild-Type | trkC−/− | % Reduction | Wild-Type | trkC−/− | % Reduction | |
| TrkB | 777 ± 47 (3) | 640 ± 36 (2) | NS | 834 ± 95 (3) | 844 ± 24 (2) | NS |
| Wild-Type | trkB−/− | % Reduction | Wild-Type | trkB−/− | % Reduction | |
| TrkC | 1385 ± 100 (4) | 1606 ± 133 (3) | NS | 1522 ± 113 (3) | 1140 ± 168 (2) | NS |
Quantification of Trk-expressing neurons present in NT-3-, TrkB-, and TrkC-deficient embryos at E11. The numbers of cells positive for each of the Trks were counted in the T1 and L1 DRGs (number of embryos in parentheses). The percentages of reduction in mutants compared to wild-type controls are shown. Statistical significance was tested with a one-tailed Student’s t test: NS, not significant;
p < 0.05;
p < 0.001.
The numbers of TrkB- and TrkC- but not TrkA-positive cells are reduced in the absence of NT-3. No significant deficits in the numbers of TrkB- or TrkC-positive cells are seen in the trkC or trkB mutant embryos, respectively.
As NT-3 is the only known ligand for TrkC, it was not surprising to observe such a drastic effect in the NT-3 mutant on survival of TrkC-expressing neurons. As this result seemed insufficient to explain the much more severe deficiency in neuronal numbers observed in NT-3– compared to TrkC-deficient neonatal animals, we also determined whether absence of NT-3 affected the morphological differentiation or survival of subpopulations of DRG neurons expressing either of the other two Trk receptors. As illustrated in Figures 5C–5D, we also observed apoptosis of TrkB-positive neurons in the absence of NT-3 as assessed by the presence of many TrkB-positive apoptotic profiles and by colabelling of TUNEL-positive profiles with anti-TrkB. While TrkB-expressing cells are clearly undergoing apoptosis in the mutant ganglia, it is worth noting that, in constrast to TrkC-expressing cells, numerous neurons expressing TrkB appear morphologically normal. Quantification of numbers of TrkB-expressing cells, presented in Table 2, shows that NT-3 deficiency results in a loss of ~40% of the TrkB-expressing neurons at this stage, a significantly less severe phenotype than the reduction of TrkC-expressing cells. In contrast to observations with TrkC-expressing cells, significant numbers of TrkB-expressing cells remain in these ganglia in subsequent days (E12: T1, 318 ± 78 [2]; L1, 395 ± 49 [3]; E13: T1, 208 ± 24 [2]; L1, 252 ± 20 [2]). The effect of NT-3 deficiency is specific at this stage for TrkC and TrkB-expressing neurons. When we examined the ganglia of mutant animals for possible effects on TrkA-expressing cells, these neurons appeared to be completely normal. At this age, TrkA immunoreactivity was never associated with pyknotic profiles (Figure 5E). When quantified, the total number of TrkA neurons was not reduced in the mutant embryos (Table 2).
The simplest interpretation of the experiments described above is that, in vivo, NT-3 promotes spinal sensory neuron survival by activation of either TrkB or TrkC. However, it seemed possible that apoptosis in the TrkB-positive population of neurons only reflects a requirement for activation of TrkC, not TrkB, within neurons coexpressing both receptors. To pursue this possibility, we determined whether significant numbers of neurons coexpress both receptors at E11. As documented in Figures 6A–6C, we did observe neurons coexpressing both TrkB and TrkC (especially in medial and posterior parts of the ganglia) as well as neurons expressing only TrkB (especially in anterior parts of the ganglia, where TrkB is very prominent) or TrkC. To determine whether a deficiency in TrkC activation accounted for the loss of TrkB-expressing neurons, we determined whether the survival of TrkB-positive neurons was impaired in a trkC mutant (Klein et al., 1993). As illustrated in Figure 6D, despite extensive apoptosis at this stage in the sensory ganglia derived from the trkC mutant, TrkB staining was not associated with the pyknotic figures. When quantified, we also observed normal numbers of TrkB-positive neurons within these ganglia at this stage (Table 2). Conversely, in trkB mutant DRGs at E11, many fewer pyknotic profiles (Figure 6E) or TUNEL-positive nuclei (data not shown) were observed, but these pyknotic figures were not immunoreactive for TrkC, and the number of TrkC-positive neurons was also normal (Table 2). Thus, absence of TrkC does not result in apoptosis of TrkB-expressing neurons and vice versa, even though loss of NT-3 results in losses in both populations of cells. Altogether, our data indicate that activation by NT-3 of either TrkB or TrkC alone is sufficient to maintain the survival of those neurons that coexpress both receptors. The recent report that the loss of DRG neurons in a double trkB/trkC mutant homozygote appears to be larger than the sum of the deficits in the two single trk mutants is also consistent with this conclusion (Silos-Santiago et al., 1997).
Figure 6. TrkB- and TrkC-Positive Cells Do Not Die in TrkC- and TrkB-Deficient DRGs, Respectively.
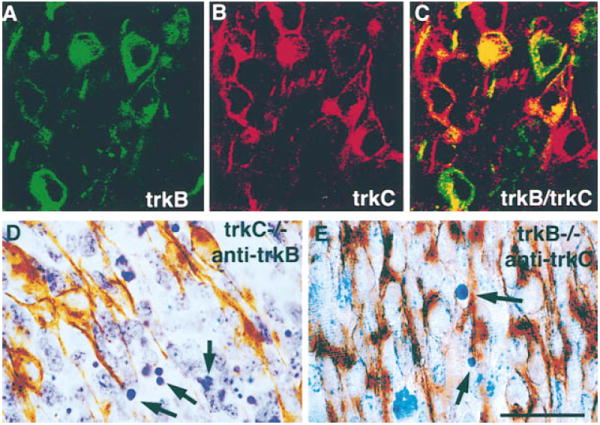
(A–C) Confocal microscopy of lumbar DRGs (middle portion of the ganglion) in a wild-type E11 embryo stained with anti-TrkB ([A], green) and anti-TrkC ([B], red) shows in a combined image that some cells express both receptors (C). Section through a lumbar DRG of a TrkC-deficient E11 embryo stained with antibodies to TrkB (D). Notice that, in spite of the presence of numerous pyknotic figures (arrows), none are positive for TrkB. Section through a lumbar DRG of a TrkB-deficient E11 embryo was stained with antibodies to TrkC (E). Only a few pyknotic profiles are observed (arrows), none of which are positive for TrkC. Scale bar, 20 μm.
Discussion
Results presented in the present paper support several major conclusions. First, expression of Trk receptors appears to be exclusively neuronal within developing spinal sensory ganglia. Secondly, differences in Trk expression between neuronal subpopulations appears at the time of initial neurogenesis of these cells, indicating that they have been previously directed to assume specific fates. Thirdly, NT-3 appears essential to maintain the survival of all or virtually all of the TrkC-expressing neurons. In addition, NT-3 is essential for viability of a major fraction of the TrkB-expressing neurons and acts by direct activation of TrkB. Finally, the apparent absence of apoptosis among the TrkB-expressing cells in a trkC mutant and vice versa indicates that those neurons that coexpress both receptors at this stage can be sustained by activation of either Trk receptor.
Our observation that Trk receptor proteins can be detected exclusively within neurons and not in migrating neural crest or intraganglionic precursor cells is consistent with the apparently normal migration of trunk neural crest cells and the normal initial formation of spinal ganglia we have observed in NT-3 mutants (Fariñas et al., 1996). The antibodies used in these experiments are polyclonal sera that have been shown to detect Trk receptor proteins by immunoblot, immunoprecipitation, and immunocytochemistry. By blot, these antibodies clearly recognize precursors of mature Trk proteins as determined by size in SDS-PAGE. By immunocytochemistry, they clearly recognize Trk proteins in intracellular, almost certainly ER and Golgi, as well as plasmalemmal compartments. We therefore believe that absence of detectable antigen in these experiments must reflect absence of significant levels of the Trk proteins from neural crest and sensory ganglia precursor cells. There is a report of TrkC receptor mRNA expression in murine neural crest cells in vivo using radioactive in situ analysis (Tessarollo et al., 1993). Another report, however, failed to observe such expression using the same methodology (Lamballe et al., 1994). Thus, although the first of these in situ reports raises the interesting possibility that there is accumulation of Trk receptor mRNAs whose translation is repressed within neural crest cells and sensory precursors, it is also possible that major transcription of these genes is initiated at the onset of neurogenesis. In zebrafish, TrkC mRNA expression does not appear to precede neurogenesis (Martin et al., 1998). In the chick, in contrast, stronger evidence for the presence of TrkC within a subset of migrating neural crest cells with neurogenic potential has been obtained using in situ techniques at cellular resolution (Henion et al., 1995). TrkC protein has been observed within a small subset of these cells using a polyclonal antibody (Lefcort et al., 1996).
The data also argue that reported effects in vivo of NT-3 deficiency on murine sensory neuron precursors are likely to be indirect, as these cells do not express Trk receptors (e.g., Fariñas et al., 1996). While NT-3 might conceivably act through p75NTR, in analyses of proliferation rate and survival of sensory precursors in vivo, we failed to detect any effects of NT-3 deficiency on these cells in vivo (Fariñas et al., 1996). Premature differentiation of precursors into neurons, though, did result in depletion of the precursor pool by E12, suggesting that many of the sensory neurons not present at birth in the NT-3 mutant were not born during the major period of neurogenesis between E12 and E13 because precursors were not present in normal numbers. Interestingly, premature differentiation of precursor cells is also seen in the DRGs of NGF-deficient embryos along with increased apoptosis of TrkA-positive neurons (I. F., unpublished data). Thus, the inhibition of either of two distinct ligand–tyrosine kinase pathways, both of which result in extensive apoptosis of largely or completely distinct neuronal populations, causes premature precursor differentiation. Consequently, these results suggest that large reductions in sensory neurons of either type create an environment in which precursor differentiation is enhanced, probably by removal of inhibitors of neurogenesis (e.g., Haddon et al., 1998). As predicted by this model, despite the presence in the NT-3 mutant of normal numbers of TrkA-expressing DRG neurons at E11, the deficiency in precursor cell numbers results in fewer TrkA-expressing neurons within DRGs at E13 and E15 (I. F., unpublished data). It is worth noting that NT-3 has been shown to induce survival, differentiation, and/or proliferation of cultured neural crest cells in vitro. While the majority of these studies have used cultured avian neural crest cells (e.g., Sieber-Blum, 1991; Kalcheim et al., 1992; Henion et al., 1995), a few have examined cells derived from rodent embryos (e.g., Memberg and Hall, 1995; ElShamy and Ernfors, 1996). Extrapolating from in vitro data, direct effects of NT-3 on precursor populations in vivo have been proposed to explain effects on precursor populations observed after inhibition of NT-3 action in murine or avian embryos (e.g., Gaese et al., 1994; ElShamy and Ernfors, 1996). Our data, however, provide independent lines of evidence arguing that effects in vivo are instead indirect, at least in murine embryos. We suggest that the in vivo and in vitro studies are not directly comparable, because the loss of cell–cell interactions or another aspect of cell culture results in expression of the TrkC protein and NT-3 responsiveness in these cells in vitro.
Trk protein expression does not appear to precede neurogenesis, because no expression of any receptor was seen in proliferating cells as identified with BrdU incorporation. At the same time, the initial steps in sensory neurogenesis appear to include induction of Trk receptor expression, as Trk receptor–expressing cells were observed at essentially the same time as neurofilament-expressing cells in these ganglia. Our observations indicate that neurons are generated expressing each of the Trk receptors at early stages in neurogenesis within DRGs. As development precedes, the bias in neurogenesis shifts from generation of predominantly TrkC-or TrkB-expressing neurons at early stages to generation of predominantly TrkA-expressing cells between E12 and E13. In addition, examination of the percentages in Figure 2B suggests that a sizable fraction of the neurons present at E11 coexpress more than one Trk receptor, but very few do so 2 days later. Thus, neuronal phenotype appears to be further refined with time after neurogenesis. In the future, it will clearly be interesting to characterize the intrinsic and extrinsic factors that regulate the generation and further development of these different sets of neurons (e.g., Alexiades and Cepko, 1997).
The striking discrepancy between the ~20% loss of DRG neurons in neonatal TrkC-deficient mice and 60% loss in NT-3–deficient mice has suggested that NT-3 deficiency affects populations of neurons that do not require signaling through TrkC for survival (Fariñas et al., 1994). Results in the present paper show that NT-3 has the physiologically essential role of activating TrkB in addition to TrkC within cells during sensory ganglia neurogenesis, providing a plausible explanation for this discrepancy. At E11 in either thoracic or lumbar DRGs, NT-3 deficiency resulted in loss of virtually all TrkC neurons with normal morphologies. The reduced numbers of profiles remaining appeared to be associated with degenerating or apoptotic cells. It thus seems likely that, as neurons expressing TrkC are generated, they become almost immediately dependent upon this neurotrophin and undergo apoptosis in its absence. Our work is consistent with previous reports of the very early loss of TrkC-expressing cells from the sensory ganglia of this mutant (e.g., Tessarollo et al., 1994). In previous work, we have documented the extensive expression of NT-3 within mesenchymal cells adjacent to these ganglia between E10 and E12, so the mesenchyme appears capable of providing trophic support to these neurons before their axons have reached their final targets (Fariñas et al., 1996). At E11, NT-3 deficiency also resulted in severe depletion of TrkB-expressing neurons and appearance of numerous degenerating profiles of cells expressing this receptor. Notably, there was no deficiency in these TrkB-expressing cells in TrkC-deficient embryos, indicating that NT-3 is not supporting these cells via activation of TrkC coexpressed within the same neurons. Consequently, the results indicate that TrkB-expressing neurons also become dependent upon local sources of NT-3 almost immediately after neurogenesis, which sustains them until their axons innervate targets that express other neurotrophins capable of activating this receptor, such as BDNF and NT-4/5. Expression of BDNF mRNA has been detected in dermal mesenchyme of targets of sensory neurons but not within mesenchyme immediately adjacent to DRGs at E11.5 in rat and murine embryos (Schecterson and Bothwell, 1992; A. P., unpublished data). Taken together, previous in situ analyses and our data indicate that BDNF and NT-4/5 are not accessible to DRG sensory neurons immediately after neurogenesis.
While NT-3 is clearly required to support a significant fraction of the TrkB-expressing population of sensory neurons, many TrkB-expressing neurons remained and many of these had normal morphologies in the NT-3 mutant. As our data strongly argues that other ligands able to activate this receptor are not available to these neurons immediately after neurogenesis, we think these cells very likely survive because they coexpress TrkA. Since both sera were prepared in rabbits, it was technically difficult to examine coexpression of TrkA and TrkB, and we have not performed these studies. The percentages in Figure 2B, though, suggest that significant numbers of cells coexpress more than one Trk receptor at E11. TrkA and TrkB have been definitively shown to be coexpressed in DRG neurons of adult rats (McMahon et al., 1994). In analyses of mutant mice, double trkB/trkA homozygous mutants have been reported to have essentially the same number of neurons as trkA homozygotes (Minichiello et al., 1995), consistent with the possibility that a significant fraction of TrkB-expressing neurons are supported by NGF at the times of our examinations.
At E11, although significant numbers of TrkA-expressing cells have been generated, evidence in the present paper indicates that they are not affected by the absence of NT-3. Elevated levels of apoptosis of TrkA-expressing neurons were not observed within NT-3 mutants, and the neurons present had normal morphological profiles. While not addressed in this paper, it seems most likely that these neurons also acquire neurotrophin dependence immediately after neurogenesis, but are sustained in the NT-3 mutant by another neurotrophin, almost certainly NGF. NGF mRNA has been detected at E11.5 with strongest expression in epidermal cells in murine and rat embryos (e.g., Davies et al., 1987; Schecterson and Bothwell, 1992). In other results (I. F., unpublished data), we have observed losses of TrkA-expressing cells in an NGF mutant at this stage, which is consistent with this model and also suggests that the ability of NT-3 to compensate for NGF absence within these ganglia is limited at this stage in development. High levels of NT-3 have been shown to be capable of activating TrkA in cultured PC12 cells or sympathetic neurons in vitro (e.g., Clary et al., 1994; Bamji et al., 1998). Both NT-3 and NGF have been shown to collaborate in promoting survival of normal numbers of sympathetic neurons in vivo, but NT-3 clearly cannot sustain these neurons in the absence of NGF (Davies et al., 1987). In the case of sympathetic neurons, evidence has implicated the low affinity receptor p75 as a negative regulator of NT-3 activation of survival signals through TrkA (Bamji et al., 1998). It will be interesting to determine why NT-3 is not more effective in supporting the TrkA-expressing population of sensory neurons in vivo.
In summary, the present study has provided direct in vivo evidence that interactions of NT-3 with TrkB are important for the regulation of neuronal survival in DRG sensory populations and explains the large discrepancy between the neuronal losses observed in the NT-3 and trkC mutants. Elegant in vitro experiments have shown that neurons isolated from cranial ganglia of TrkC-deficient embryos indeed respond to high doses of NT-3 at certain stages of development (Davies et al., 1995). However, it is difficult to know whether such high concentrations of neurotrophin are attained in vivo. Here, we show that interactions of NT-3 with TrkB, at least, do occur in a physiological context. Furthermore, the finding that Trk proteins are not expressed in precursors and neural crest cells raises further questions on regulation of Trk expression, as well as mechanisms of indirect effects of neurons on precursor population differentiation.
Experimental Procedures
Preparation of Antibodies
The rabbit and goat polyclonal antibodies specific for TrkA, TrkB, TrkC, and p75NTR (low affinity neurotrophin receptor) used in this study have been previously characterized (Weskamp and Reichardt, 1991; Clary et al., 1994; Wilkinson, 1997). In brief, the TrkA and p75NTR antisera were prepared with the entire extracellular domains of rat TrkA and rat p75NTR expressed in Sf9 cells using a baculovirus expression vector. After purification of each antigen to homogeneity from the conditioned media of these cells, each antigen was used to immunize rabbits. The purified sera have been shown to be specific for their respective antigens and to inhibit strongly interactions of NGF with the receptor used as an immunogen (Weskamp and Reichardt, 1991; Clary et al., 1994). The TrkB and TrkC antisera were prepared with the entire extracellular domains of rat TrkB and rat TrkC expressed in COS cells. After purification of each antigen from the conditioned media of these cells, anti-TrkB was prepared by immunizing rabbits and anti-TrkC by immunizing a goat. To improve specificity, it was necessary to affinity purify the antibodies in each serum using affinity columns in which each antigen was coupled to Sepharose. The affinity purified Ig preparations were shown to be specific for their respective antigens as assessed using antigen blots, immunoprecipitations, and immunocytochemistry (Wilkinson, 1997). It has not been determined whether they inhibit neurotrophin interactions with these receptors.
Animal Husbandry and Histological Procedures
Procedures for the generation, genotyping, and processing of the embryos used in this analysis have been described elsewhere (Fariñas et al., 1994, 1996).
Immunocytochemistry
For single labelings, primary antibodies were used at the following concentrations in blocking solution (10 mM Tris HCl [pH 7.5], 150 mM NaCl, containing 0.4% Triton X-100, 3% bovine serum albumin, and 10% normal serum from the host species of the secondary antibody to be used): rabbit anti-TrkA IgG (5 μg/ml; Clary et al., 1994), affinity-purified rabbit anti-TrkB (5 μg/ml; Wilkinson, 1997), affinity-purified goat anti-TrkC (5 μg/ml; Wilkinson, 1997), and rabbit anti-p75NTR IgG (5 μg/ml; Weskamp and Reichardt, 1991). Biotinylated goat anti-rabbit IgG or biotinylated rabbit anti-goat IgG and the ABC complex from the Vectastain-peroxidase detection kit (Vector Laboratories, Burlingame, CA) were used following the manufacturer’s instructions. Methods for BrdU injection and labeling and for TUNEL staining have been described previously (Fariñas et al., 1996). For double-labeling experiments, TrkC immunoreactivity was detected with Cy3-coupled donkey anti-goat antibody, and TrkA and TrkB immunoreactivity was detected using Cy3-coupled donkey anti-rabbit IgG or FITC-coupled donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA) at 1:200.
Acknowledgments
We thank Drs. M. Barbacid and I. Silos-Santiago for providing the TrkB and TrkC kinase–deficient mouse strains. This work has been supported by research grants from the United States Public Health Service (National Institutes of Health grant MH48200) and the How-ard Hughes Medical Institute; I. F. was the recipient of a long-term fellowship from the Human Frontier Science Program Organization; A. P. is a fellow of the Damon Runyon–Walter Winchell Cancer Research Foundation; and L. F. R. is an Investigator of the Howard Hughes Medical Institute.
References
- Acheson A, Conover JC, Fandl JP, DeChiara TM, Russell M, Thadani A, Squinto SP, Yancopoulous GD, Lindsay RM. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature. 1995;374:450–453. doi: 10.1038/374450a0. [DOI] [PubMed] [Google Scholar]
- Airaksinen MS, Koltzenburg M, Lewin GR, Masu Y, Helbig C, Wolf E, Brem G, Toyka KV, Thoenen H, Meyer M. Specific subtypes of cutaneous mechanoreceptors require neurotrophin-3 following peripheral target innervation. Neuron. 1996;16:287–295. doi: 10.1016/s0896-6273(00)80047-1. [DOI] [PubMed] [Google Scholar]
- Alexiades MR, Cepko CL. Subsets of retinal progenitors display temporally regulated and distinct biases in the fates of their progeny. Development. 1997;124:1119–1131. doi: 10.1242/dev.124.6.1119. [DOI] [PubMed] [Google Scholar]
- Bamji SX, Madjan M, Pozniak CD, Belliveau DJ, Alyz R, Kohn J, Causing CG, Miller FD. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol. 1998;140:911–927. doi: 10.1083/jcb.140.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clary DO, Weskamp G, Austin LAR, Reichardt LF. TrkA cross-linking mimics neuronal responses to nerve growth factor. Mol Biol Cell. 1994;5:549–563. doi: 10.1091/mbc.5.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley C, Spencer SD, Nishimura MC, Chen KS, Pitts-Meek S, Armanini MP, Ling GH, McMahon SB, Shelton DL, Levinson AD, Phillips HS. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell. 1994;76:1001–1011. doi: 10.1016/0092-8674(94)90378-6. [DOI] [PubMed] [Google Scholar]
- Davies AM, Bandtlow CE, Heumann R, Korsching S, Rohrer H, Thoenen H. Timing and site of nerve growth factor synthesis in developing skin in relation to innervation and expression of the receptor. Nature. 1987;326:353–357. doi: 10.1038/326353a0. [DOI] [PubMed] [Google Scholar]
- Davies AM, Minichiello L, Klein R. Developmental changes in NT3 signaling via TrkA and TrkB in embryonic neurons. EMBO J. 1995;14:4482–4489. doi: 10.1002/j.1460-2075.1995.tb00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElShamy WM, Ernfors P. A local action of neurotrophin-3 prevents the death of proliferating sensory neuron precursor cells. Neuron. 1996;16:963–972. doi: 10.1016/s0896-6273(00)80119-1. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Lee KF, Kucera J, Jaenisch R. Lack of neurotrophin-3 leads to deficiencies in the peripheral nervous system and loss of limb proprioceptive afferents. Cell. 1994;77:503–512. doi: 10.1016/0092-8674(94)90213-5. [DOI] [PubMed] [Google Scholar]
- Fariñas I, Jones KR, Backus C, Wang X-Y, Reichardt LF. Severe sensory and sympathetic deficits in mice lacking neurotrophin-3. Nature. 1994;369:658–661. doi: 10.1038/369658a0. [DOI] [PubMed] [Google Scholar]
- Fariñas I, Yoshida CK, Backus C, Reichardt LF. Lack of neurotrophin-3 results in death of spinal sensory neurons and premature differentiation of their precursors. Neuron. 1996;17:1065–1078. doi: 10.1016/s0896-6273(00)80240-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaese F, Kolbeck R, Barde YA. Sensory ganglia require neurotrophin-3 in early development. Development. 1994;120:1613–1619. doi: 10.1242/dev.120.6.1613. [DOI] [PubMed] [Google Scholar]
- Haddon C, Smithers L, Schneider-Maunoury S, Coche T, Henrique D, Lewis J. Multiple delta genes and lateral inhibition in zebrafish primary neurogenesis. Development. 1998;125:359–370. doi: 10.1242/dev.125.3.359. [DOI] [PubMed] [Google Scholar]
- Henion PD, Garner AS, Large TH, Weston JA. TrkC-mediated NT-3 signaling is required for the early development of a subpopulation of neurogenic neural crest cells. Dev Biol. 1995;172:602–613. doi: 10.1006/dbio.1995.8054. [DOI] [PubMed] [Google Scholar]
- Ip NY, Stitt TN, Tapley P, Klein R, Glass DJ, Fandl J, Greene LA, Barbacid M, Yancopoulos GD. Similarities and differences in the way neurotrophins interact with the Trk receptors in neuronal and nonneuronal cells. Neuron. 1993;10:137–149. doi: 10.1016/0896-6273(93)90306-c. [DOI] [PubMed] [Google Scholar]
- Kalcheim C, Carmeli C, Rosenthal A. Neurotrophin-3 is a mitogen for cultured neural crest cells. Proc Natl Acad Sci USA. 1992;89:1661–1665. doi: 10.1073/pnas.89.5.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Martín-Zanca D, Barbacid M, Parada LF. Expression of the tyrosine kinase receptor gene trkB is confined to the murine embryonic and adult nervous system. Development. 1990;109:845–850. doi: 10.1242/dev.109.4.845. [DOI] [PubMed] [Google Scholar]
- Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL, Barbacid M. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell. 1993;75:113–122. [PubMed] [Google Scholar]
- Klein R, Silos-Santiago I, Smeyne RJ, Lira SA, Brambilla R, Bryant S, Zhang L, Snider WD, Barbacid M. Disruption of the neurotrophin-3 receptor gene trkC eliminates Ia muscle afferents and results in abnormal movements. Nature. 1994;368:249–251. doi: 10.1038/368249a0. [DOI] [PubMed] [Google Scholar]
- Lamballe F, Smeyne R, Barbacid M. Developmental expression of TrkC, the neurotrophin-3 receptor, in the mammalian nervous system. J Neurosci. 1994;14:14–28. doi: 10.1523/JNEUROSCI.14-01-00014.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefcort F, Clary DO, Rusoff AC, Reichardt LF. Inhibition of the NT-3 receptor TrkC, early in chick embryogenesis, results in severe reductions in multiple neuronal subpopulations in the dorsal root ganglia. J Neurosci. 1996;16:3704–3713. doi: 10.1523/JNEUROSCI.16-11-03704.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebl DJ, Tessarollo L, Palko ME, Parada LF. Absence of sensory neurons before target innervation in brain-derived neurotrophic factor-, neurotrophin-3-, and TrkC-deficient embryonic mice. J Neurosci. 1997;17:9113–9121. doi: 10.1523/JNEUROSCI.17-23-09113.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SC, Sandell JH, Heinrich G. Zebrafish TrkC1 and TrkC2 receptors define two different cell populations in the nervous system during the period of axonogenesis. Dev Biol. 1998;195:114–130. doi: 10.1006/dbio.1997.8839. [DOI] [PubMed] [Google Scholar]
- Martín-Zanca D, Barbacid M, Parada LF. Expression of the trk protooncogene is restricted to the sensory cranial and spinal ganglia of neural crest origin in mouse development. Genes Dev. 1990;4:683–694. doi: 10.1101/gad.4.5.683. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Armanini MP, Ling LH, Phillips HS. Expression and coexpression of trk receptors in subpopulations of adult primary sensory neurons projecting to identified peripheral targets. Neuron. 1994;12:1161–1171. doi: 10.1016/0896-6273(94)90323-9. [DOI] [PubMed] [Google Scholar]
- Memberg SP, Hall AK. Proliferation, differentiation, and survival of rat sensory neuron precursors in vitro require specific trophic factors. Mol Cell Neurosci. 1995;6:323–335. doi: 10.1006/mcne.1995.1025. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Piehl F, Vazquez E, Schimmang T, Hokfelt T, Represa J, Klein R. Differential effects of combined Trk receptor mutations on dorsal root ganglion and inner ear sensory neurons. Development. 1995;121:4067–4075. doi: 10.1242/dev.121.12.4067. [DOI] [PubMed] [Google Scholar]
- Reichardt LF, Fariñas I. Neurotrophic factors and their receptors: roles in neuronal development and function. In: Cowan MW, Jessell TM, Zipurski L, editors. Molecular Approaches to Neural Development. New York: Oxford University Press; 1997. pp. 220–263. [Google Scholar]
- Schecterson LC, Bothwell M. Novel roles for neurotrophins are suggested by BDNF and NT-3 mRNA expression in developing neurons. Neuron. 1992;9:449–463. doi: 10.1016/0896-6273(92)90183-e. [DOI] [PubMed] [Google Scholar]
- Scott SA. Sensory Neurons: Diversity, Development, and Plasticity. New York: Oxford University Press; 1992. [Google Scholar]
- Sieber-Blum M. Role of the neurotrophic factors BDNF and NGF in the commitment of pluripotent neural crest cells. Neuron. 1991;6:949–955. doi: 10.1016/0896-6273(91)90235-r. [DOI] [PubMed] [Google Scholar]
- Silos-Santiago I, Molliver DC, Ozaki S, Smeyne RJ, Fagan AM, Barbacid M, Snider WD. Non-TrkA-expressing small DRG neurons are lost in TrkA-deficient mice. J Neurosci. 1995;15:5929–5942. doi: 10.1523/JNEUROSCI.15-09-05929.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silos-Santiago I, Fagan AM, Garber M, Fritzsch B, Barbacid M. Severe sensory deficits but normal CNS development in newborn mice lacking TrkB and TrkC tyrosine protein kinase receptors. Eur J Neurosci. 1997;9:2045–2056. doi: 10.1111/j.1460-9568.1997.tb01372.x. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA, Barbacid M. Severe sensory and sympathetic neuropathies in mice carrying a disrupted trk/NGF receptor gene. Nature. 1994;368:246–248. doi: 10.1038/368246a0. [DOI] [PubMed] [Google Scholar]
- Tessarollo L, Tsoulfas P, Martín-Zanca D, Gilbert DJ, Jenkins NA, Copeland NG, Parada LF. TrkC, a receptor for neurotrophin-3, is widely expressed in the developing nervous system and in nonneuronal tissues. Development. 1993;118:463–475. doi: 10.1242/dev.118.2.463. [DOI] [PubMed] [Google Scholar]
- Tessarollo L, Vogel KS, Palko ME, Reid SW, Parada LF. Targeted mutation in the neurotrophin-3 gene results in loss of muscle sensory neurons. Proc Natl Acad Sci USA. 1994;91:11844–11848. doi: 10.1073/pnas.91.25.11844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessarollo L, Tsoulfas P, Donovan MJ, Palko ME, Blair-Flynn J, Hempstead BL, Parada LF. Targeted deletion of all isoforms of the trkC gene suggests the use of alternate receptors by its ligand neurotrophin-3 in neuronal development and implicates TrkC in normal cardiogenesis. Proc Natl Acad Sci USA. 1997;94:14776–14781. doi: 10.1073/pnas.94.26.14776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tojo H, Kaisho Y, Nakata M, Matsuoka K, Kitagawa M, Abe T, Takami K, Yamamoto M, Shino A, Igarashi K, et al. Targeted disruption of the neurotrophin-3 gene with lacZ induces loss of TrkC-positive neurons in sensory ganglia but not in spinal cords. Brain Res. 1995;669:163–175. doi: 10.1016/0006-8993(94)01219-8. [DOI] [PubMed] [Google Scholar]
- Weskamp G, Reichardt LF. Evidence that biological activity of NGF is mediated through a novel subclass of high affinity receptors. Neuron. 1991;6:649–663. doi: 10.1016/0896-6273(91)90067-a. [DOI] [PubMed] [Google Scholar]
- White FA, Silos-Santiago I, Molliver DC, Nishimura M, Phillips H, Barbacid M, Snider WD. Synchronous onset of NGF and TrkA survival dependence in developing dorsal root ganglia. J Neurosci. 1996;16:4662–4672. doi: 10.1523/JNEUROSCI.16-15-04662.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson GA. PhD thesis. University of California; San Francisco, CA: 1997. Neurotrophin-3 and Trk receptors in the developing mouse trigeminal ganglion; pp. 1–163. [Google Scholar]