

PREFACE
Mitochondria are remarkably dynamic organelles that migrate, divide and fuse. Cycles of mitochondrial fission and fusion ensure metabolite and mitochondrial DNA (mtDNA) mixing and dictate organelle shape, number and bioenergetic functionality. There is mounting evidence that mitochondrial dysfunction is an early and causal event in neurodegeneration. Mutations in mitochondrial fusion GTPases (mitofusin-2 and optic atrophy-1), neurotoxins and oxidative stress all disrupt the cable-like morphology of functional mitochondria. This results in impaired bioenergetics and mitochondrial migration and can trigger neurodegeneration. These findings suggest potential new treatment avenues for neurodegenerative diseases.
INTRODUCTION
The importance of mitochondria in energy production has long been appreciated, but new research on the dynamic nature of mitochondria (that is, their ability to undergo continuous cycles of fission and fusion) has highlighted their role in normal cell physiology and disease. Mitochondria actively communicate and interact with each other and other cellular organelles, such as the endoplasmic reticulum, to satisfy the cell’s changing energetic needs and protect the cell from excessive calcium influx, oxidative damage and mtDNA mutations; events that typically characterize ageing and neurodegenerative processes.
Mitochondria are important organelles in all cell types, but they are particularly important in the nervous system. Mitochondrial function is essential to neuronal processes such as energy production, Ca2+ regulation, maintenance of plasma membrane potential, protein folding by chaperones, axonal and dendritic transport and the release and re-uptake of neurotransmitters at synapses (Figure 1).1-3 Mitochondria help neurons meet the high energy demands that are required for proper neuronal function — unlike other cell types, neurons cannot switch to glycolysis when oxidative phosphorylation becomes limited. Furthermore, mitochondrial transport, together with the dynamic processes of mitochondrial fission and fusion, facilitate the transmission of energy across long distances, which is particularly important in neurons given that axons can extend up to one metre in motor neurons. Whereas mitochondrial fission allows for mitochondrial renewal, redistribution and proliferation into synapses,2, 4 the competing process, mitochondrial fusion, allows mitochondria to interact and communicate with each other, facilitating mitochondrial movement and distribution across long distances and to the synapses.4, 5 As individual mitochondria are subject to injury and dysfunction, it is likely that mitochondrial fusion serves as a protective mechanism, by preventing these deficiencies from damaging the entire neuron while maintaining an adequate level of bioenergetic capacity.5, 6 However, recent studies suggest that mitochondrial fission could also have a protective role: the segregation of damaged and inactive mitochondria that facilitates autophagic clearance.7, 8
Figure 1. Neuronal mitochondria.

Each neuron contains several hundred mitochondria that form cable-like structures along neuronal projections to help them meet their large energy demands. Neurons require energy to transport organelles and cargo along microtubules or actin fibres (motor molecules like dyneins, kinesins and myosin mediate this process) and to maintain ion gradients and the membrane potential by ATP-dependent Ca2+ and Na+/K+ pumps and ion channels. Additionally, neurotransmitter vesicle loading at pre-synaptic terminals and Ca2+-mediated neurotransmitter release into the synaptic cleft are also ATP-dependent events. Glutamate transporters mediate glutamate re-uptake from the synaptic cleft, and at the post-synaptic membrane, glutamate binding to NMDA receptors evokes Ca2+ influx, which in turn can activate Nitric Oxide Synthase (NOS) and stimulate the generation of Nitric Oxide (NO). Both, NO and Ca2+ can directly modulate mitochondrial function by altering the levels of ROS (H2 O2 and O2-) and ATP production.(b) Fluorescence 3D microscope image of mitochondria in a dendritic arbor of a neuron expressing DsRed-Mito, a red fluorescent fusion protein targeted selectively to the mitochondrial matrix (scale bar:5 μm). (c) Slice through an EM tomographic volume showing a mitochondrion in a neuronal process. Mitochondrial length is typically 2-25 μm in neurites with a diameter of 0.5 μm (scale bar: 400 nm). Shown underneath is a view of the surface-rendered volume after segmentation of the same mitochondrion. The outer membrane is a translucent pale blue and individual cristae are shown in different colors.
In the past few years, multiple findings have suggested that disruptions of mitochondrial function and dynamics contribute to neurodegenerative diseases (Box 1). Here, we focus on our current understanding of the mechanisms of mitochondrial fission and fusion, the regulation of these processes, and how they can contribute to neurodegenerative disease.
BOX 1 -Mitochondrial dysfunction during neurodegeneration.
Mitochondrial defects are observed in many common neurodegenerative diseases, as shown in the figure. Oxidative damage is a very early event in human Alzheimer’s disease 137: Amyloid-β (Aβ) peptide inhibits cytochrome c (Cyt c) oxidase activity (complex IV, cIV), thus increasing damaging ROS production in mitochondria.138, 139
Inhibitors of complex I (cI) in mitochondria, such as rotenone and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), cause parkinsonism.140, 141 Moreover, proteins that are mutated in familial forms of Parkinson’s disease (PD), including leucine-rich repeat kinase 2 (LRRK2), alpha-synuclein (α-Syn), parkin, DJ-1 and PTEN-induced putative kinase 1 (PINK1), associate with the mitochondrial outer membrane (OM) and are associated with ROS production or defense. HTRA2/OMI, another protein that is mutated in familial PD, locates to the intermembrane space (IMS) of mitochondria and might be involved in proteolytic processing of mitochondrial proteins.84
Familial amyotrophic lateral sclerosis (FALS) is characterized by defects in Ca2+ loading142, suggesting that the mutant protein in FALS, superoxide dismutase-1 (mtSOD1), might disrupt Ca2+ channels. In addition, mtSOD1 might block protein import at the mitochondrial outer membrane,143 disrupt the respiratory chain and cause aberrant ROS production,144 and by binding to mitochondrial Bcl-2, block its anti-apoptotic actions.145
Mouse models of Huntington’s disease (HD) exhibit early defects in respiration and ATP production.146 Interestingly, 3-nitroproprionic acid (3-NP) is a mitochondrial complex II (cII) inhibitor that produces HD-like symptoms and mutant huntingtin (mtHtt) itself seems to disrupt cII activity.147 MtHtt also disrupts mitochondrial Ca2+ buffering.148
The fact that mutations in the mitochondrial fission and fusion machinery can cause neurodegenerative diseases and that the familial PD disease-specific genes, PINK1 and parkin, seem to play a role in mitochondrial fission 149, 150 underscores the role of these proteins in mitochondrial health and neuronal function. Whether defective mitochondrial fission and fusion contribute to the mitochondrial dysfunction that is characteristic of neurological diseases with unknown aetiology, and whether the mutant proteins that are associated with hereditary neurodegenerative diseases cause mitochondrial dysfunction by affecting mitochondrial dynamics is currently subject of intensive research.

FISSION AND FUSION MECHANISMS
Mitochondrial fission and fusion was first described in yeast.9, 10 Fission and fusion allow mixing of metabolites and mtDNA, proliferation and distribution of mitochondria and cellular adaptation to changing energy demands. A group of large GTPases mediate mitochondrial fission and fusion. Although their precise mechanism of action is unclear, the sequence homology between dynamin, a GTPase involved in the scission of vesicles in endocytosis,11 and these GTPases suggests potential mechanisms for mitochondrial fission and fusion. Furthermore, the conservation of these GTPases across species allows us to correlate the findings from pioneering studies in yeast with the potential mechanisms of mitochondrial fission and fusion in mammalian cells.
Mitochondrial fusion requires both outer and inner mitochondrial membrane components.2 Studies in yeast identified the proteins Fzo1 and Mgm1 as key mitochondrial fusion mediators.12-14 The mammalian orthologues of Fzo1 are mitofusin 1 and mitofusin 2, which derive from two homologous genes (Mfn1 and Mfn2). The mammalian orthologue of Mgm1 is optic atrophy-1 (OPA1).
Outer membrane fusion
Fzo1, Mfn1 and Mfn2 are large GTPases localized to the mitochondrial outer membrane that direct mitochondrial fusion (Figure 2a). The N-terminal region contains the conserved GTPase domain, while the C-terminal region of these proteins consists of a coiled-coil structure (Figure 2). Mutations in the GTPase domain or coiled-coil domains of Fzo1 inhibit mitochondrial fusion in yeast15 and in vitro studies indicated that Fzo1 interacts in trans to align mitochondrial outer membranes for fusion events.16 In addition, Mfn1 and Mfn2 have been found to form homo-oligomeric and hetero-oligomeric complexes in trans.6, 17, 18 It has been proposed that Fzo1, Mfn1 and Mfn2 mediate mitochondrial fusion by tethering outer membranes together via interactions of their coiled-coil domains in trans.18
Figure 2. Mitochondrial fusion.

(a) Mitochondrial outer membrane fusion occurs by homotypic Mfn2 interaction in trans across two mitochondria. It is possible that Mfn2 mediates outer membrane fusion by binding to and forming clusters at mitochondrial tips - the ends of the tubular organelles. (b) Domain model of Mfn2 showing the conserved GTPase domain and the proposed Neck, Trunk, and Paddle/Tip regions. Ribbon diagram of the bacterial dynamin-like protein (BDLP) homodimer shown with one molecule in gray and the other rainbow color-coded from N (blue) to C (red) terminus. Mfn2 cartoon based on the BDLP structure indicating proposed GTPase (magenta), Neck (green), Trunk (cyan), and Paddle (red) regions. (c) Proposed model of potential buckle and molecular zippering mechanism for Mfn2-mediated mitochondrial fusion. The Mfn2 dimer grabs adjacent mitochondrial outer membranes with its hydrophobic paddle domain and fuses them through a conformational change (induced by GTP hydrolysis) of its trunk and paddle region. Other regulatory proteins are likely to be involved. (d) OPA1-mediated inner membrane fusion. Mitochondrial matrix contents mix after inner membrane fusion. (e) Domain model of OPA1 (Swissprot: O60313) showing location of the mitochondrial targeting sequence (MTS), TM transmembrane region, GTPase domain, helical domain, and putative GED domain.
The crystal structure of the mitofusin homologue from cyanobacteria, bacterial dynamin-like protein (BDLP),19 reveals a compact molecule in which the predicted GTPase and coiled-coil domains do not form discrete entities. Instead, it contains a GTPase domain (residues 68-287) with a long helix-loop-helix amino terminus (1-67), followed by a long four-helix bundle region that forms the neck, trunk and tip of the molecule. Moreover, the two predicted transmembrane helices (residues 572-606) resemble a hydrophobic paddle that could insert into lipid bilayers to promote membrane curvature and fusion (Figure 2b). These findings suggest that the paddle region of Mfn2 is likely to be directly involved in membrane fusion. Based on these structural observations, we propose a new model of Mfn2-mediated mitochondrial fusion (Figure 2c). Whether additional factors assist this process remains uncertain.
Inner membrane fusion
In yeast, inner membrane fusion requires Mgm1, suggesting that the mammalian orthologue OPA1 is also involved in this process (Figure 2d)20, 21. Indeed, OPA1 mutant mice and homozygous OPA1 mutant Drosophila exhibit increased mitochondrial fragmentation.22-24 In addition, in vitro analysis of mitochondrial fusion indicated that mitochondria deficient in Mgm1 can fuse their outer membranes, but not their inner membranes.25 A mitochondrial targeting sequence (MTS) at the N-terminus of OPA1 and Mgm1 mediates mitochondrial import (Figure 2e). Adjacent to the MTS, a transmembrane helix anchors the proteins to the mitochondrial inner membrane (Figure 2e). Proteolytic cleavage in vivo releases both Mgm1 and OPA1 from the membrane, producing functionally distinct isoforms that are necessary for normal fusion activity (for review see REF 2). Studies have identified several proteases that might cleave OPA1, including PARL,26 the i-AAA protease Yme1L 27, 28and the human murine m-AAA protease.29 Interestingly, mutations in paraplegin, a member of the AAA protease family, cause an autosomal recessive form of spastic paraplegia, a peripheral neuropathy.30
Mechanistically, like Fzo1, Mgm1 appears to interact with itself in trans via its GTPase and GED domains to tether mitochondrial inner membranes.25 Because of the sequence similarity between Mgm1 and OPA1, we can speculate that the mechanism of mitochondrial inner membrane fusion in mammalian cells is similar.
Mitochondrial Fission
Dynamin-1 (Dnm1) in yeast and its mammalian homologue dynamin-related protein 1 (Drp1) cluster into large foci at the sites of mitochondrial fission (Figure 3a).13, 31-34 Mitochondrial fission produces spherical mitochondria that have significant ultrastructural differences, including cristae remodeling characterized by fragmentation, occasional cristae dilation or vesiculation, and the disappearance of cristae membrane, when compared with the parent mitochondria (Figure 3 b,c,d,e). Dnm1/Drp1 has three conserved domains, an N-terminal GTPase domain, a central helical domain, and a GED, which contribute to mitochondrial fission (Figure 3f).
Figure 3. Mitochondrial fission.

(a) Drp1 is found in the cytoplasm, but cycles on and off mitochondria, possibly by indirect interaction with the outer-membrane associated hFis1. Once bound to the mitochondrial outer membrane, Drp1 forms large clusters or foci, which mediate membrane fission. OM indicates the outer membrane and IM indicates the inner membrane. (b) Domain model of Drp1 (Swissprot: O00429) showing the conserved GTPase domain, helical domain, and GTP effector domain (GED). (c) Fluorescent 3D microscopic image of a fused, elongated mitochondrion (red) in a healthy neuron and round, fissioned mitochondria in a neuron exposed to nitrosative stress. The mitochondrial labeling results from DsRed-Mito expression.(d) Slice through an EM tomographic volume showing four fragmented mitochondria (indicated by arrows) in a neuronal process after exposure to an NO donor, which triggers mitochondrial fission. Mitochondria are recognizable because of their cristae structure, even with some cristae membrane degradation. (e) Top view of the surface-rendered volume after segmentation of the same four mitochondria as shown in (d). The outer membrane is shown in pale blue and the cristae in various colors. Cristae fragmentation is evident from the smaller and regionally confined cristae. (f) Side view of the surface-rendered volume. The outer membrane is made transparent to better visualize the cristae. Fission induces a profound remodeling of the inner membrane with cristae vesiculation.
In yeast, Fis1, a small molecule localized to the mitochondrial outer membrane, targets cytosolic Dnm1 to the mitochondrial membrane through an indirect interaction.35-38 The role of the mammalian homologue, hFis1, in recruitment of Drp1 to the mitochondria is less clear because Drp1 cycling on and off of mitochondria does not require hFis1; 39, 40 however, hFis1 silencing inhibited fission, suggesting that hFis1 acts downstream of Drp1 recruitment.40
Studies in yeast favour a model in which Dnm1 self-assembles into spirals and localizes to mitochondrial membrane constriction sites, recognized under electron microscopy by accumulation of Dnm1 oligomers as large foci or belts around mitochondria and reduction of mitochondrial diameter (∼0.5 μm - 0.1 μm), through an indirect interaction, via Mdv1 and Caf4, with Fis1. When bound to analogues of GTP that cannot be hydrolyzed to GDP, Dnm1 assembles into ring and spiral-like structures.41 Mutations that impair GTP binding prevent spiral formation and mitochondrial fission.33, 34 Interestingly, the addition of hydrolyzable GTP causes the disassembly of the highly ordered rings and spirals.41 Thus, GTP hydrolysis by Dnm1, which is required for fission but not the assembly of oligomeric complexes, may cause a conformational change in the protein that helps complete the fission event.41 In mammals, the mechanism is likely to be similar, with hFis1, and potentially other proteins such as ganglioside-induced differentiation associated protein-1 (GDAP1),42 interacting with Drp1 to mediate conversion of Drp1 into an active, fission-promoting conformation.2
REGULATION OF FISSION AND FUSION
Cells continually adjust the rate of mitochondrial fission and fusion in response to changing energy demands and to facilitate the distribution of mitochondria.43 Recent studies have identified three post-translational modifications that seem to regulate mitochondrial fission and fusion proteins: phosphorylation, sumoylation and ubiquitination.
Protein Kinase A (PKA, cAMP-dependent protein kinase) phosphorylates Drp1 at Ser637 in the variable domain of humans and Ser656 in the conserved GED in rats.44, 45 PKA phosphorylation caused a significant decrease in GTPase activity 44 and inhibited mitochondrial fission.44, 45 Interestingly, another study found that mitosis-promoting factor (MPF, Cdk1/cyclin B) phosphorylates Drp1 at Ser585 in rats during mitosis.46 Unlike phosphorylation by PKA, Cdk1/cyclin B phosphorylation seems to stimulate mitochondrial fission during mitosis.46 Thus, it appears that phosphorylation of Drp1 by different kinases at different amino acids causes opposite effects. Finally, cyclin-dependent kinase 5 (Cdk5) is another key regulator of mitochondrial fission.47 Whether Cdk5 phosphorylates Drp1 or other proteins involved in mitochondrial fission, such as components of the cytoskeleton, remains unclear. Additionally, whether kinases implicated in Parkinson’s disease pathogenesis, such as LRRK2 and PINK1, regulate Drp1 or related factors remains a question for future investigation.
Sumoylation is another means of Drp1 regulation. SUMO1 interacts with Drp1 and associates with mitochondria at fission sites before and after fission.48 Time-lapse microscopy showed that Drp1 and SUMO1 initially localize to the middle of the mitochondrion, and after fission, Drp1 and SUMO1 remain localized on the tip of one of the divided mitochondria. 48 Overexpression of SUMO1 leads to mitochondrial fragmentation and apoptosis, probably because SUMO1 protects Drp1 from degradation, stabilizes the Drp1 pool and enhances Drp1 binding to mitochondria.40, 48
Ubiquitination has also been associated with mitochondrial dynamics. Several studies have identified a mitochondrial outer membrane protein, membrane-associated RING-CHV (MARCH-V or MARCH5), that ubiquitinates Drp1, but the effect of MARCH5 on mitochondrial dynamics remains unclear.49-51 Cells expressing MARCH5 mutants and MARCH5 RNAi exhibit cable-like mitochondria with varying degrees of thickness.49 These findings suggest MARCH5 might ubiquitinate Drp1 to promote fission or ubiquitinate and deactivate an unknown repressor of fission.49 These findings contradict two earlier studies, which found that MARCH5 ubiquitination of Drp1 produced abnormal, elongated mitochondria.50, 51 These differences could be a result of using different imaging techniques and/or criteria for evaluating mitochondrial morphology.
In summary, although phosphorylation, sumoylation and ubiquitination appear to be important processes in the regulation of mitochondrial fission and fusion, the pathways and proteins involved are far from clear. Further research on the modulation of mitochondrial dynamics in human disease is necessary.
MITOCHONDRIAL FISSION AND CELL DEATH
Programmed cell death is an important process in both health and disease.39, 52Although mitochondrial fission is an early event in cell death, the precise role of fission in cell death remains unclear. Expression of the dominant-negative Drp1 mutant, Drp1K38A, in cell lines decreases mitochondrial fragmentation and blocks cell death in response to staurosporine, gamma radiation and etoposide.52, 53 In addition, downregulation of Fis1 inhibited HeLa cell death,39 inhibition of Drp1 partially protected C. elegans cells against cell death,54 inhibition of Drp1 reduced and delayed mitochondrial fragmentation and cell death in embryonic Drosophila cells 55, 56 and inactivation of Drp1 by phosphorylation protected PC12 cells from various apoptotic insults.45 Finally, a fungal ageing model indicates that deletion of the Dnm1 gene extends lifespan and delays apoptosis.57
The discovery that Bax and Bak, two pro-apoptotic regulators, interact with mitochondrial fission and fusion GTPases further supports the functional link between mitochondrial dynamics and apoptosis. Bax forms large foci with Drp1 and Mfn2 on mitochondria during apoptosis53 and hyperglycemic injury of dorsal root ganglion cells causes Drp1 and Bax to associate.58 Furthermore, neurons challenged with toxic levels of nitric oxide exhibit Bax foci on mitochondria as they undergo mitochondrial fission. Inhibiting Drp1 function delays mitochondrial fission, Bax foci formation and neuronal loss.7, 59 Bak also appears to have a crucial function in mitochondrial dynamics during apoptosis as it complexes with Mfn1 and Mfn2.60, 61
Despite the strong correlation between mitochondrial fission and cell death, some studies have questioned the importance of mitochondrial fission and fragmentation in apoptosis. For example, inhibition of Fis1 and Drp1 prevents mitochondrial fragmentation, but does not block apoptosis.62-64 Conversely, Fis1 and Drp1 might be able to promote apoptosis without causing fission.40, 65 Thus, the pro-apoptotic and fission promoting functions of Fis1 and Drp1 might be distinct and mitochondrial fragmentation per se might not cause apoptosis. Supporting this belief, cells deficient in both Fis1 and OPA1 exhibited fragmented mitochondria, similar to OPA1 RNAi cells, but were very resistant to apoptosis, similar to Fis1 RNAi cells.39
Attempting to correlate studies in cell lines to neurodegeneration is difficult. First, it is reasonable to assume that neurons, which are post-mitotic, behave differently from actively dividing cells. Second, the classical caspase-dependent apoptosis does not adequately account for the slow onset and progression of neurodegenerative diseases. It is possible that mitochondrial fission contributes to chronic neurodegeneration through other non-apoptotic cell-death pathways such as type II and type III PCD, autophagic or ‘necrosis-like’ pathways that are caspase independent.7, 66, 67 Alternatively, it is possible that persistent mitochondrial fission could lead to cell dysfunction (including synaptic damage 68 and bioenergetic failure 7) and subsequent neurodegeneration rather than cell death.
MITOCHONDRIAL DYNAMICS AND BIOENERGETICS
Because mitochondria provide most of the energy that is necessary to maintain neuronal function, it is important to consider the possible link between changes in mitochondrial dynamics and bioenergetic failure. Nitrosative stress-induced mitochondrial fission in cultured neurons results in bioenergetic defects, neuronal dysfunction and cell death.7 Similarly, blocking Drp1 activity with RNAi in HeLa cells prevented mitochondrial fission and decreased respiratory activity and ATP production.69 Interestingly, a lack of mitochondrial fusion per se causes bioenergetic defects in cultured non-neuronal cells, 20 as cells lacking both Mfn1 and Mfn2, in which mitochondria are unable to fuse, have significant bioenergetic defects.20In addition, cells lacking either Mfn1 or Mfn2 or overexpressing OPA1 exhibit fragmented mitochondria, but are not bioenergetically defective.20 Thus, it appears that a lack of fusion factors might cause bioenergetic defects independently of changes in mitochondrial morphology. Intriguingly, there seems to be a bi-directional relationship between mitochondrial fragmentation and bioenergetics as a decrease in ATP can also stimulate fragmentation.69 Thus, although there is convincing evidence that disruption of mitochondrial dynamics and bioenergetic defects are closely linked, the precise relationship remains unclear. Further supporting the idea of multiple roles for mitochondrial fission and fusion proteins in bioenergetics, another study found that the translocation of Drp1 to mitochondria can cause a decrease in respiration in a GTP-independent process in normal and leukemic B lymphocytes.66This suggests that Drp1 may also function in two distinct ways. First, as a mediator of mitochondrial fission, via a GTP-dependent mechanism. Second, as an inhibitor of mitochondrial function and possible mediator of Type III programmed cell death, via a GTP-independent mechanism.
In summary, although it is becoming clear that proteins directing mitochondrial fission and fusion can contribute to bioenergetic failure under certain conditions, the precise role of mitochondrial dynamics requires further investigation.
FISSION AND FUSION PROTEIN MUTATIONS AND NEUROLOGICAL DISORDERS
Loss-of-function mutations in the genes that encode mitochondrial fusion GTPases cause neurodegenerative disease.70 Mutations in Mfn2 cause Charcot-Marie-Tooth subtype 2A (CMT2A), a peripheral neuropathy characterized by muscle weakness and axonal degradation of sensory and motor neurons,71, 72 and hereditary motor and sensory neuropathy type VI (HMSN VI), which is clinically similar to CMT with the addition of optic atrophy and visual impairment.73 Mutations in OPA1 cause the most common form of optic atrophy, autosomal dominant optic atrophy (ADOA). Patients with ADOA exhibit progressive loss of vision and degeneration of the optic nerve and retinal ganglion cells.74 In addition, some mutations in the OPA1 GTPase domain cause ‘ADOA-plus’ phenotypes that are also characterized by deafness, sensory-motor neuropathy and muscle movement disorders.75, 76 The similarity of the symptoms caused by Mfn2 and OPA1 mutations support the idea that these proteins are functionally similar.
A recent study characterized nine Mfn2 mutants associated with CMT2A 77, five of which cannot induce mitochondrial fusion. The ability of the other four Mfn2 mutants to fuse mitochondria suggests that Mfn2 has other roles, not related to fusion, in disease pathology. One study suggests that Mfn2 might have a role in mitochondrial trafficking, and disruption of this function could lead to peripheral axon degeneration.4 Additionally, the non-functional Mfn2 mutants promoted mitochondrial fusion in Mfn2-null cells, but not in Mfn-1 null cells77, suggesting that Mfn1 complementation, possibly resulting from heteromeric associations,6 can rescue mitochondrial fusion. These results might also explain why mutations in Mfn2, despite being present in all cells, only affect specific neurons in CMT2A as cells with limited Mfn1 activity, and therefore less able to compensate for the Mfn2 mutations, might be more susceptible.77 Whether the levels of Mfn1 are indeed reduced in degenerating neurons in patients with CMT2A is an important question for future study.
The current mitochondrial fusion hypothesis states that Mfn2 and OPA1 are mechano-enzymes that use GTP hydrolysis to switch between distinct conformations that facilitate membrane fusion.78, 79 Intriguingly, the majority of missense mutations found in Mfn2 in patients with Charcot-Marie-Tooth disease and in OPA1 in patients with ADOA reside in the highly conserved GTPase domains (Figure 4) and thus, could interfere with nucleotide binding and hydrolysis. However, a number of missense mutations are also located outside the GTPase domain (Mfn2 W740S, Figure 4a, OPA1 L939P, Figure 4c) pointing to critical residues that will be valuable starting points to further dissect the mechanisms by which these molecules recognize and fuse mitochondrial membranes.
Figure 4. Mutations in Mfn2 and OPA1 present in human patients.

(a) Domain model of Mfn2 (Swissprot: O95140) showing location of the GTPase domain and the putative Neck, Trunk, and Paddle domains. Red arrows indicate locations of missense mutations found in CMT-2A patients (OMIM database: 608507). (b) Homology model of human Mfn2 residues 24-757 shown in ribbon presentation, indicating the GTPase, Neck, Tip, Paddle, and Trunk region based on the crystal structure of bacterial dynamin-like protein (PDB-ID 2j68 (2), FFAS (1) score - 67.0, sequence identity 11%). Red spheres indicate mutations found in CMT2A. (c) Domain model of OPA1 with locations of missense mutations found in ADOA patients (top) (OMIM database: 165500) indicated by red arrows and ‘OPA1-plus’ disorders (bottom) indicated by black arrows. (d) Homology model of the OPA1 GTPase domain (residues 244-575) based on the dynamin GTPase domain (1jw1.pdb) shown in ribbon representation with a GDP molecule bound in the putative active site shown in ball and stick. Red spheres indicate residues mutated in ADOA patients (eOPA1)5. A close up view of the active site in OPA-1 shows native (gray) and mutant (yellow) side-chains for comparison. The model clearly shows that several mutations directly interfere with GTP binding and most probably impair the catalytic function. Models were prepared with PyMOL (DeLano Scientific).
A recent case study reported a dominant-negative mutation in the helical domain of Drp1 in a human patient.80 The patient exhibited elongated and tangled mitochondria that were concentrated around the nucleus, characteristic of impaired mitochondrial fission.80 Unfortunately, the patient died 37 days after birth, displaying some symptoms that were similar to ADOA and CMT2A. Obviously, the Drp1-related disease had a much earlier onset and the impact of Drp1 mutations was much more severe than the fusion mutation diseases. Finally, mutations in GDAP1, which localizes to the outer membrane and appears to participate in Drp1-dependent mitochondrial fission, 42 cause CMT4A, another subtype of Charcot-Marie-Tooth syndrome.81-83 CMT4A combines demyelination with axonal loss and the homozygous mutation causes early onset and more severe progression.83
MITOCHONDRIAL FISSION AND SPORADIC NEURODEGENERATIVE DISEASE
Mitochondrial dysfunction is a characteristic of many neurodegenerative disorders, but is mitochondrial fission activated in- or causally related to sporadic neurodegenerative disease? If so, what might activate this pathological mitochondrial fission? Several triggers might contribute to this cell-specific shift in the balance between fission/fusion including oxidative stress and altered regulation by cell cycle kinases.
Oxidative stress
Because mitochondria are the primary producers of reactive oxygen species (ROS), oxidative damage of mitochondrial proteins or DNA is likely to contribute to the mitochondrial dysfunction that is characteristic of many neurodegenerative diseases.84 However, another potential deleterious effect of increased ROS levels is chronic mitochondrial fission. Nitrosative stress causes profound mitochondrial fission in neurons prior to the onset of neuronal loss in an animal model of stroke7 and treatment with antioxidants has been shown to reduce mitochondrial fission. Expression of Mfn or dominant-negative Drp1 partially prevented mitochondrial fission and neuronal cell death by nitric oxide, which is known to be largely caspase independent. Another study found that oxidative stress promoted mitochondrial fission in cerebellar granule neurons and that Mfn2 expression was protective.85 Whether oxidative stress directly regulates the mitochondrial fission and fusion GTPases is currently unclear.
Cell cycle kinases in neurodegeneration
A growing body of research suggests that cyclin-dependent kinases (Cdks), many of which are important players in cell cycle regulation, have an important role in neuronal death.86 Multiple in vitro models of neuronal death indicate that activation of Cdks is a required step in the cell death cascade.86-88 In addition, studies have found increased levels of Cdk4 in ischemic brain tissue and mouse models of ALS,89, 90 increased levels of Cdk2 and Cdk4 in postmortem brain samples from AD patients,91 and increased Cdk5 activity in MPTP models of PD.92 Furthermore, genetic studies have linked certain polymorphisms of Cdk1 with increased susceptibility to Alzheimer’s disease.93, 94 As mentioned above, Cdk1 can phosphorylate Drp1, increase its activity and stimulate mitochondrial fragmentation.46 Although this process is likely to serve a functional role in cell division, aberrant activation of Cdk1 in post-mitotic neurons could cause deleterious mitochondrial fragmentation, sensitizing the neurons to dysfunction and cell death. It is also known that neuronal mitochondria favor different morphologies at different time points in their life cycle. Immature neurons have smaller, less-connected mitochondria whereas mature, highly functional neurons have elongated, connected mitochondria.95 Keeping this in mind, it is conceivable that factors that induce mitochondrial fission in mature neurons could cause energy depletion, neuronal dysfunction and eventual neuronal demise. Such a mechanism of neuronal dysfunction would help explain the late onset and progressive nature of many neurodegenerative diseases, such as AD.
MITOCHONDRIAL DYNAMICS AND ONSET OF NEURODEGENERATIVE DISEASE
Mitochondrial diseases caused by hereditary mtDNA mutations and diseases associated with mitochondrial dysfunction have unique characteristics that are hard to explain: they only affect specific cell types and, despite mutations being present from birth, they show delayed disease onset. The emerging roles of mitochondrial fission and fusion, including synaptic maintenance, bioenergetics and gene drift of mtDNA subpopulations, along with an increased appreciation of differences in the mitochondrial proteome in different cell types, might shed light on these properties.
Synaptic maintenance
Synaptic function and maintenance are crucial for neuronal survival and efficient cell-to-cell communication. Mitochondria are highly relevant to synaptic maintenance because synapses have high energy requirements.96 In addition, the synaptic micro-environment is highly dynamic, awash with ions and neurotransmitters moving in and out of the synaptic cleft. Mitochondria buffer Ca2+ ions, which modulate action potential firing and the release of neurotransmitter-containing vesicles (Figure 1a).
A decline in mitochondrial activity at synapses might be among the earliest events in neurodegenerative diseases, initiating clinical symptoms such as memory loss and cognitive decline.97 Studies also indicate that mitochondrial fission and fusion are necessary for synaptic maintenance. First, Drp1 overexpression in hippocampal neurons from rat embryos promoted synaptogenesis whereas OPA1 and mutant Drp1 overexpression had the opposite effect, likely because of differences in mitochondrial distribution.68 Both Drp1 and OPA1 overexpression cause mitochondrial fragmentation, yet they have the opposite effect on synaptogenesis and function. What could account for this apparent contradiction? OPA1 exists in multiple proteolytically-processed isoforms and both the short and long isoforms are necessary for mitochondrial fusion. Thus, OPA1 overexpression could overwhelm the system with too much of either the long or short isoform, irreversibly blocking fusion. On the other hand, Drp1 overexpression stimulates fission, but fusion can still occur. This creates the perfect situation for synaptic maintenance as mitochondria can fuse, which helps them reach the synapse, 4 yet they actively divide, which is critical for proliferation into the synapse.68, 98 Further supporting the role of mitochondrial fission in synaptic health, the cell survival protein Bcl-xL increases synapse number and function by stimulating Drp1’s GTPase activity (and thus Drp1-mediated fission) in cultured hippocampal neurons99 and in Drosophila, a Drp1 mutation prevented the mobilization of the neurotransmitter reserve pool.100
These findings suggest a mechanism that might explain why mutations in fission/fusion proteins only affect specific types of cells in hereditary neurodegenerative diseases (e.g. ADOA and CMT2A) and, potentially, why specific types of neurons selectively degenerate in sporadic neurodegenerative diseases.
Selection and accumulation of mtDNA mutations
The mtDNA genome is small (16.5 kbp in mammals) and codes for only 13 proteins in humans,101 but its integrity is important for proper mitochondrial function and mutations or loss of mtDNA can damage cells.102, 103 Mutations in mtDNA can cause aging,104-107 which is the leading risk factor for neurodegenerative disease.
The potential role of mitochondrial fusion in the expansion of mtDNA deletion mutants is controversial. A modeling study of mitochondrial fusion and the proliferation of mtDNA mutations, in which fusion was assumed to occur regularly, creating a shared compartment of mtDNA and structural components, found that the size of mtDNA deletions increases with age.108 The authors of this study suggest that, whereas the wild-type mtDNA maintain mitochondrial function and keep the mitochondria viable, smaller mutated mtDNAs (i.e. those with larger deletions) might have a survival advantage because of more rapid replication.109, 110 Strikingly, the model produced a deletion mutant profile that was remarkably similar to an experimental rat profile.108 However, the relevance of this model to the physiological situation in post-mitotic neurons is unclear because it relies on the proposed selective advantage of faster replication of small mtDNA mutants, which might be of relevance only to actively dividing cells. In addition, other models have proposed that random drift can account for the clonal expansion of mtDNA mutants with age.111
Another study of Purkinje cells with impaired mitochondrial fusion suggests that a lack of fusion might cause a loss of mtDNA.5 Mfn2-deficient Purkinje cells appeared to exhibit a greater number of mitochondria completely lacking mtDNA compared with wild-type cells, although this study lacked quantitative analysis and requires further confirmation.5 As is the case with mtDNA deletion mutants, a lack of fusion might cause the proliferation of mtDNA-deficient mitochondria by preventing the mixing and exchange of mitochondrial components. A loss of mitochondrial fusion, over time, could allow a build up of mitochondria lacking mtDNA. Although this would not be initially harmful, at some point the cell would become defective because there would not be enough viable mitochondria for efficient respiration and energy production.
Two new studies indicate that OPA1 has an important role in mtDNA stability and maintenance.75, 76 Mutations in the OPA1 GTPase domain, known as ‘OPA1-plus’ mutants, result in increased breakage or truncation of mtDNA molecules. These new findings suggest an mtDNA stabilizing function for OPA1 in addition to its established role in mitochondrial fusion. Because both OPA1 and nucleoids (the structural units of mtDNA consisting of 5-7 copies of mtDNA and multiple mitochondrial proteins) associate closely with the mitochondrial inner membrane, it is conceivable that OPA1 mutations could disrupt the nucleoids and promote mtDNA mutations.23, 112-114
Although mitochondrial fusion probably protects cells from mtDNA loss and point mutations, it might also promote the accumulation of mtDNA mutants with larger deletions. Thus, the precise relationship between mitochondrial fusion and mtDNA mutations is complicated and requires further investigation. Upregulation of mitochondrial fusion could have both positive and negative effects on overall health. In addition, activity of fusion proteins such as OPA1, as opposed to mitochondrial fusion per se, might be important for mtDNA maintenance because these proteins probably have multiple functions.
Cell type-specific mitochondrial fission
One of the more mysterious characteristics of neurodegenerative mitochondrial disease is the cell-type specific pathogenesis. Compared with other cell types, neurons seem to be particularly vulnerable to changes in mitochondrial morphology and connectivity, probably due to their great energy requirements and unique energy transmission demands. But why can some neurons survive while others cannot? Proteomic studies indicate that mitochondrial gene expression profiles are tissue specific.94, 115 These findings suggest that neuronal mitochondria in different areas of the brain have different properties and may respond to stressors such as ROS, increased mitochondrial fission and genetic mutations more or less efficiently.
ARE CHANGES IN MITOCHONDRIAL DYNAMICS REVERSIBLE?
If mitochondrial fission and fusion contribute to neurodegeneration and aging, it is necessary to consider whether they offer new opportunities for therapy. Three avenues that warrant further examination are transcriptional regulation of mitochondrial biogenesis, exercise and ROS management.
What can compensate for too much fission?
Because too much mitochondrial fission negatively affects mitochondrial function, stimulating mitochondrial biogenesis could compensate for the deleterious effects. Peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) is a transcriptional co-activator that is a master regulator of mitochondrial biogenesis and respiration.116, 117 PGC-1α regulates the expression of many mitochondrial genes, including those associated with mitochondrial biogenesis and antioxidant defense.116 Thus, increased expression of PGC-1α in degenerating neurons suffering from excessive mitochondrial fission might be neuroprotective and a potential therapeutic option. There is considerable evidence that PGC-1α has neuroprotective function.117-119
Exercise and mtDNA mutations
Exercise is a well-established tool in the fight against aging and age-related diseases such as neurodegenerative disease. For example, voluntary exercise in mice increases neurogenesis in the hippocampus120, 121 and increases metabolic capacity in the motor cortex.122 Does exercise improve mitochondrial function? A large body of research indicates that it does. First, exercise improves energy metabolism by stimulating mitochondrial biogenesis 123and increasing the expression of PGC-1α.124, 125 Mitochondrial biogenesis is important because it increases the amount of wild-type mtDNA and the loss of wild-type mtDNA in mutant cells is a major cause of respiratory defects.126 In addition, resistance exercise reverses many aspects of the aging transcriptome in skeletal muscle, increasing expression of many proteins related to mitochondrial function.127 Finally, expression of Mfn1 and Mfn2 increases 24 hours after endurance exercise 128 and increased levels of Mfn2 in developing muscle tissue correlates with more efficient mitochondrial function and energy transmission.129 Thus, exercise has a significant impact on mitochondrial function. The relationship between exercise, nutrition, metabolism, and mitochondrial fission and fusion is less clear, but is a subject of increasing interest.
Another area of interest is the relationship between exercise and mtDNA mutations. Accumulation of mutations in mtDNA with age is one mechanism of mitochondrial dysfunction. An early case study showed that resistance exercise training produced an increase in wild-type, non-mutant mtDNA in a mitochondrial myopathy patient carrying a mutation in more than 90% of his skeletal muscle mtDNA.130 The decrease in mutant mtDNA probably results from gene shifting — the fusion of mitochondria and transfer of mtDNA from wild-type satellite cells, without the mutation, to mitochondria from mutant cells (Figure 5). Does resistance exercise induce gene shifting and a decrease in mtDNA mutations in healthy older adults? A study of healthy older adults found no decrease in mtDNA mutations following resistance exercise.131 However, exercise could provide functional benefits by decreasing the mutant mtDNA load in key areas below a pathological threshold, through fusion-mediated redistribution of mtDNA mutations within cells or tissues, while increasing or failing to decrease the total occurrence of mtDNA mutations.132, 133 Thus, the potential of exercise to revert mutations in mtDNA through a fusion-related pathway is certainly of great interest and deserves further research. The applicability of these lines of research in muscle cells to mitochondrial dysfunction in neurodegenerative disease is also an open question.
Figure 5. Gene shift and exercise.
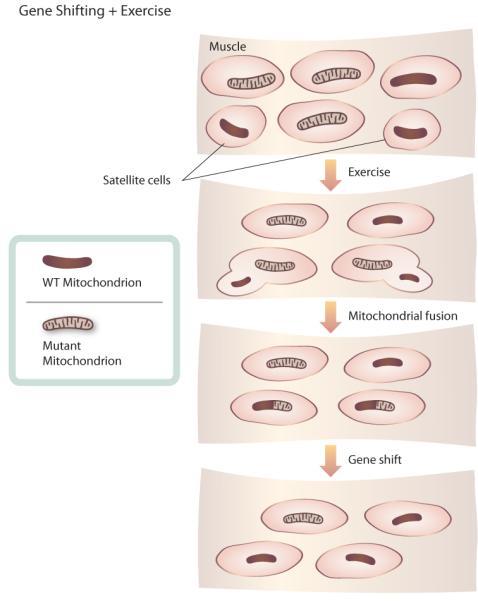
Model of how exercise might reduce the prevalence of mtDNA point mutations in muscle cells through gene shift. Satellite cells, undifferentiated cells found in mature muscle, possess wild-type mtDNA. Exercise stimulates differentiation and growth of satellite cells. When wild-type satellite cells fuse with muscle cells carrying mtDNA point mutations, mitochondria mix and fuse. Fusion of mitochondria allows wild-type mtDNA from satellite cells to compensate for mutant mtDNA, decreasing the prevalence of mtDNA mutations.
ROS management
Because ROS are an important cause and effect of increased mitochondrial fission, effective ROS management might be crucial in the fight against fission damage. Nitrosative stress induces mitochondrial fission that is often asymmetric, with one fissioned mitochondrion retaining normal ultrastructure and the other exhibiting significant damage.7 This stress-induced fission in immature neurons and neurons expressing survival molecules such as Bcl-xL was frequently reversible (our unpublished observation). Thus, transitory fission induced by oxidative stress could increase cell survival by allowing excision and degradation of damaged mitochondria by an autophagic process. However, if the oxidative stress persists, resulting in chronic and persistent fission, a shift to cell death might occur. It is likely that differential signal transduction pathways determine whether mitochondrial fission-mediated cell survival or cell death predominates.
In addition to their role in ROS production, mitochondria also contain an extensive ROS-defense system, which neutralizes ROS and includes coenzyme Q10, cytochrome c, glutathione, manganese superoxide dismutase, catalase and glutathione peroxidase.84 ROS induces PGC-1α activity, which increases the expression of the ROS-detoxifying enzymes 134, 135 and protects neuronal cells from oxidative stress.117
While ROS-directed therapies for neurodegenerative disease are conceptually appealing, their therapeutic viability is questionable. Clinical antioxidant trials have been largely ineffective in treating neurodegenerative disease. This comes as no surprise. The ROS system is highly complex, consisting of different types of compounds with different properties and functions. In addition, it stands to reason that beginning antioxidant treatment after disease onset probably cannot undo the damage done by years of oxidative stress. At best, it could slow progression which is a positive outcome, but far from a cure. Finally, we must also consider the negative effects of antioxidant therapy, which could include disruption of the beneficial roles of ROS (e.g. cell signaling and induction of mitochondrial fission-mediated cell survival) and desensitization of the native ROS-defense mechanisms.84
PGC-1α treatments could hold greater promise because of the potential for more widespread effects. PGC-1α can enhance the native ROS-defense system and stimulate mitochondrial biogenesis. However, overexpression of PGC-1α in mice causes cardiac abnormalities,136 presenting a potential problem for the use of PGC-1α in humans. In addition, the same basic flaw that applies to antioxidant therapies also applies to potential PGC-1α therapies. Such strategies are strictly responsive and do not get at the underlying cause of the problem. Even if oxidative stress causes mitochondrial dysfunction in neurodegenerative diseases, we must improve our understanding of what happens earlier in the disease process to cause increased ROS production. Only then will we be able to intervene early enough in the pathogenic cascade to provide anything more than temporary relief.
OUTLOOK
Because of their important role in energy production, mitochondria are vital to health maintenance throughout the aging process. It is becoming increasingly clear that mitochondrial dysfunction leads to neurodegeneration and aging. The dynamic nature of mitochondria, characterized by tightly controlled fission and fusion, is an important part of mitochondrial health and function. In addition, there is a growing appreciation of the range of effects that mitochondrial fission and fusion events have in cells and of the proteins involved in these processes. Fission and fusion might also have an important role in processes such as the modulation of bioenergetics and complementation of mtDNA mutations. Furthermore, fission and fusion proteins, beyond maintaining mitochondrial morphology, probably contribute to other cellular processes such as cell death and development. We are just beginning to understand the importance of mitochondrial fission and fusion to aging and neurodegeneration, and continued research into mitochondrial dynamics, focusing on both the big picture and the individual players, will enhance our understanding of mitochondrial health and will hopefully lead to breakthroughs in the diagnosis, treatment and prevention of a wide variety of neurodegenerative diseases.
Acknowledgments
We apologize to all colleagues whose articles we were unable to cite due to space limitations. We wish to acknowledge the support of NIH grants RO1 EY016164 (EBW), RO1 NS047456 (EBW), RO1 NS055193 (EBW), P41 RR004050 (GP)(PI: Mark Ellisman), and P42 ES010337 (GP)(PI: Robert Tukey), as well as EU grants MCEXT-033534 and MCIRG-046536 (RS). We thank Adam Wilson for help with illustrations.
GLOSSARY TERMS
- Synapses
Specialized junctions through where neurons communicate with each other and other cell types (e.g. muscles) via exchange of chemical messengers.
- Oxidative phosphorylation
Pathway that converts nutrients into adenosine triphosphate (ATP) by transferring electrons from donors to acceptors, such as molecular oxygen. The enzymes mediating the process, the electron transport chain, reside in the mitochondrial inner membrane.
- Mitochondrial fission
Separation of a long mitochondrion (2-25 μm) into two or more round (0.5 μm) mitochondria.
- Mitochondrial fusion
Combination of two mitochondria into a single organelle. Occurs on the tips or sides of the mitochondrial filament.
- GTPases
A large family of enzymes that bind and hydrolyze high-energy guanosine triphosphate (GTP) into guanosine diphosphate (GDP), releasing energy to drive changes in protein conformation.
- Cristae
Mitochondrial inner membrane folds that protrude into the mitochondrial matrix. Contain electron transport chain enzymes and ATP synthase.
- Phosphorylation
The addition of a phosphate group to a protein by a kinase, a specific class of enzyme. Phosphorylation is a common post-translational modification that switches proteins on and off.
- Sumoylation
The addition of a small ubiquitin-related modifier (SUMO) to a protein. A post-translational modification that generally stabilizes and extends the lifespan of a protein.
- Ubiquitination
The addition of a small ubiquitin protein to another protein. Marks proteins for degradation by the proteasome.
- Staurosporine
A non-selective protein kinase inhibitor that induces apoptosis.
- Etoposide
Inhibitor of topoisomerase II that induces apoptosis and is an anti-cancer drug.
- Oxidative Stress
Increased reactive oxygen species, free radicals, and peroxides that damage lipids, proteins and DNA.
- ROS
Reactive Oxygen Species. Include oxygen ions, free radicals and peroxides that form as byproducts of oxygen metabolism. Mitochondria are a major source of ROS.
- Purkinje cells
Large, GABAergic neurons found in the cerebellar cortex that may play a role in motor coordination.
Footnotes
TOC Blurb:
There is mounting evidence that mitochondrial dysfunction is an early and causal event in neurodegeneration. Here, Bossy-Wetzel and colleagues discuss how aberrant mitochondrial fission and fusion can contribute to neurodegenerative disease.
AT A GLANCE
- Mitochondria are dynamic organelles that undergo continuous cycles of fission and fusion. These dynamic processes allow mitochondria to communicate, migrate and adapt to changing energy demands and cellular conditions.
- A group of large GTPases mediate mitochondrial fission and fusion. Important players in mammalian cells include mitofusins 1 and 2 (Mfn1, 2), optic atrophy-1 (OPA1) and dynamin-related protein 1 (Drp1).
- The crystal structure of the mitofusin homologue from cyanobacteria, bacterial dynamin-like protein (BDLP), suggests a new model of Mfn2-mediated mitochondrial fusion. Two predicted transmembrane helices in Mfn2 may form a hydrophobic paddle that could insert into lipid bilayers and promote mitochondrial outer membrane curvature and fusion.
- Cells continually adjust the rate of mitochondrial fission and fusion in response to changing energy demands and to facilitate the distribution of mitochondria. Three post-translational modifications that seem to regulate mitochondrial fission and fusion proteins are: phosphorylation, sumoylation and ubiquitination.
- While there is substantial evidence linking mitochondrial fragmentation to cell death, the precise role of mitochondrial fission in neuronal death remains uncertain. Because classical, caspase-dependent apoptosis cannot account for the slow onset and progression of neurodegenerative diseases, it is likely that other cell death mechanisms play important roles.
- Mutations in the mitochondrial fission and fusion GTPases Mfn2, OPA1 and Drp1 cause neurodegenerative diseases. Mfn2 mutations cause Charcot-Marie-Tooth subtype 2A (CMT2A) and sensory neuropathy type VI (HMSN VI), OPA1 mutations cause autosomal dominant optic atrophy (ADOA) and a Drp1 mutation caused a lethal defect in a human infant.
- Several triggers, including oxidative stress and altered regulation by cell cycle kinases, contribute to the cell-specific shift in the balance between fission/fusion characteristic of sporadic neurodegenerative diseases.
- The emerging roles of mitochondrial fission and fusion, including synaptic maintenance, bioenergetics and gene drift of mtDNA subpopulations, along with an increased appreciation of differences in the mitochondrial proteome in different cell types, might help explain some of the unique characteristics of sporadic neurodegenerative diseases, such as late onset and slow progression.
- Transcriptional regulation of mitochondrial biogenesis, exercise and reactive oxygen species (ROS) management are three potential pathways of reversing deleterious changes in mitochondrial dynamics.
References
- 1.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–52. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 2.Hoppins S, Lackner L, Nunnari J. The machines that divide and fuse mitochondria. Annu Rev Biochem. 2007;76:751–80. doi: 10.1146/annurev.biochem.76.071905.090048. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, Chan DC. New insights into mitochondrial fusion. FEBS Lett. 2007;581:2168–73. doi: 10.1016/j.febslet.2007.01.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baloh RH, Schmidt RE, Pestronk A, Milbrandt J. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J Neurosci. 2007;27:422–30. doi: 10.1523/JNEUROSCI.4798-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen H, McCaffery JM, Chan DC. Mitochondrial Fusion Protects against Neurodegeneration in the Cerebellum. Cell. 2007;130:548–62. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 6.Chen H, et al. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barsoum MJ, et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. Embo J. 2006;25:3900–11. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Twig G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. Embo J. 2008;27:433–46. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bereiter-Hahn J, Voth M. Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microsc Res Tech. 1994;27:198–219. doi: 10.1002/jemt.1070270303. [DOI] [PubMed] [Google Scholar]
- 10.Nunnari J, et al. Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol Biol Cell. 1997;8:1233–42. doi: 10.1091/mbc.8.7.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Urrutia R, Henley JR, Cook T, McNiven MA. The dynamins: redundant or distinct functions for an expanding family of related GTPases? Proc Natl Acad Sci U S A. 1997;94:377–84. doi: 10.1073/pnas.94.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hermann GJ, et al. Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J Cell Biol. 1998;143:359–73. doi: 10.1083/jcb.143.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sesaki H, Jensen RE. Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J Cell Biol. 1999;147:699–706. doi: 10.1083/jcb.147.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong ED, et al. The intramitochondrial dynamin-related GTPase, Mgm1p, is a component of a protein complex that mediates mitochondrial fusion. J Cell Biol. 2003;160:303–11. doi: 10.1083/jcb.200209015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griffin EE, Chan DC. Domain interactions within Fzo1 oligomers are essential for mitochondrial fusion. J Biol Chem. 2006;281:16599–606. doi: 10.1074/jbc.M601847200. [DOI] [PubMed] [Google Scholar]
- 16.Meeusen S, McCaffery JM, Nunnari J. Mitochondrial fusion intermediates revealed in vitro. Science. 2004;305:1747–52. doi: 10.1126/science.1100612. [DOI] [PubMed] [Google Scholar]
- 17.Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci. 2004;117:6535–46. doi: 10.1242/jcs.01565. [DOI] [PubMed] [Google Scholar]
- 18.Koshiba T, et al. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–62. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- 19.Low HH, Lowe J. A bacterial dynamin-like protein. Nature. 2006;444:766–9. doi: 10.1038/nature05312. [DOI] [PubMed] [Google Scholar]
- Provided the first structure of a full length dynamin-like protein.
- 20.Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–92. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 21.Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A. 2004;101:15927–32. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davies VJ, et al. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum Mol Genet. 2007;16:1307–18. doi: 10.1093/hmg/ddm079. [DOI] [PubMed] [Google Scholar]
- 23.Olichon A, et al. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 2002;523:171–6. doi: 10.1016/s0014-5793(02)02985-x. [DOI] [PubMed] [Google Scholar]
- 24.Yarosh W, et al. The molecular mechanisms of OPA1-mediated optic atrophy in Drosophila model and prospects for antioxidant treatment. PLoS Genet. 2008;4:e6. doi: 10.1371/journal.pgen.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meeusen S, et al. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell. 2006;127:383–95. doi: 10.1016/j.cell.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 26.Frezza C, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–89. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 27.Griparic L, Kanazawa T, van der Bliek AM. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol. 2007;178:757–64. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–55. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duvezin-Caubet S, et al. OPA1 processing reconstituted in yeast depends on the subunit composition of the m-AAA protease in mitochondria. Mol Biol Cell. 2007;18:3582–90. doi: 10.1091/mbc.E07-02-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Casari G, et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–83. doi: 10.1016/s0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
- 31.Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM. C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell. 1999;4:815–26. doi: 10.1016/s1097-2765(00)80391-3. [DOI] [PubMed] [Google Scholar]
- 32.Legesse-Miller A, Massol RH, Kirchhausen T. Constriction and Dnm1p recruitment are distinct processes in mitochondrial fission. Mol Biol Cell. 2003;14:1953–63. doi: 10.1091/mbc.E02-10-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naylor K, et al. Mdv1 interacts with assembled dnm1 to promote mitochondrial division. J Biol Chem. 2006;281:2177–83. doi: 10.1074/jbc.M507943200. [DOI] [PubMed] [Google Scholar]
- 34.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–56. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cerveny KL, Jensen RE. The WD-repeats of Net2p interact with Dnm1p and Fis1p to regulate division of mitochondria. Mol Biol Cell. 2003;14:4126–39. doi: 10.1091/mbc.E03-02-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karren MA, Coonrod EM, Anderson TK, Shaw JM. The role of Fis1p-Mdv1p interactions in mitochondrial fission complex assembly. J Cell Biol. 2005;171:291–301. doi: 10.1083/jcb.200506158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, Chan DC. Structural basis for recruitment of mitochondrial fission complexes by Fis1. Proc Natl Acad Sci U S A. 2007;104:18526–30. doi: 10.1073/pnas.0706441104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23:5409–20. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–11. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. 2007;177:439–50. doi: 10.1083/jcb.200610042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ingerman E, et al. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol. 2005;170:1021–7. doi: 10.1083/jcb.200506078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Niemann A, Ruegg M, La Padula V, Schenone A, Suter U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot-Marie-Tooth disease. J Cell Biol. 2005;170:1067–78. doi: 10.1083/jcb.200507087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–9. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- 44.Chang CR, Blackstone C. Cyclic AMP-dependent Protein Kinase Phosphorylation of Drp1 Regulates Its GTPase Activity and Mitochondrial Morphology. J Biol Chem. 2007;282:21583–7. doi: 10.1074/jbc.C700083200. [DOI] [PubMed] [Google Scholar]
- 45.Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939–44. doi: 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–9. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 47.Meuer K, et al. Cyclin-dependent kinase 5 is an upstream regulator of mitochondrial fission during neuronal apoptosis. Cell Death Differ. 2007;14:651–61. doi: 10.1038/sj.cdd.4402087. [DOI] [PubMed] [Google Scholar]
- 48.Harder Z, Zunino R, McBride H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol. 2004;14:340–5. doi: 10.1016/j.cub.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 49.Karbowski M, Neutzner A, Youle RJ. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol. 2007;178:71–84. doi: 10.1083/jcb.200611064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakamura N, Kimura Y, Tokuda M, Honda S, Hirose S. MARCH-V is a novel mitofusin 2-and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 2006;7:1019–22. doi: 10.1038/sj.embor.7400790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yonashiro R, et al. A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. Embo J. 2006;25:3618–26. doi: 10.1038/sj.emboj.7601249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frank S, et al. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–25. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 53.Karbowski M, et al. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159:931–8. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- References 52 and 53 were two of the first studies to describe the relationship between mitochondrial fission and cell death.
- 54.Jagasia R, Grote P, Westermann B, Conradt B. DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature. 2005;433:754–60. doi: 10.1038/nature03316. [DOI] [PubMed] [Google Scholar]
- 55.Abdelwahid E, et al. Mitochondrial disruption in Drosophila apoptosis. Dev Cell. 2007;12:793–806. doi: 10.1016/j.devcel.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 56.Goyal G, Fell B, Sarin A, Youle RJ, Sriram V. Role of mitochondrial remodeling in programmed cell death in Drosophila melanogaster. Dev Cell. 2007;12:807–16. doi: 10.1016/j.devcel.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scheckhuber CQ, et al. Reducing mitochondrial fission results in increased life span and fitness of two fungal ageing models. Nat Cell Biol. 2007;9:99–105. doi: 10.1038/ncb1524. [DOI] [PubMed] [Google Scholar]
- 58.Leinninger GM, et al. Mitochondria in DRG neurons undergo hyperglycemic mediated injury through Bim, Bax and the fission protein Drp1. Neurobiol Dis. 2006;23:11–22. doi: 10.1016/j.nbd.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 59.Yuan H, et al. Mitochondrial fission is an upstream and required event for bax foci formation in response to nitric oxide in cortical neurons. Cell Death Differ. 2007;14:462–71. doi: 10.1038/sj.cdd.4402046. [DOI] [PubMed] [Google Scholar]
- 60.Brooks C, et al. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci U S A. 2007;104:11649–54. doi: 10.1073/pnas.0703976104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–62. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
- 62.Estaquier J, Arnoult D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 2007;14:1086–94. doi: 10.1038/sj.cdd.4402107. [DOI] [PubMed] [Google Scholar]
- 63.James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003;278:36373–9. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- 64.Parone PA, et al. Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak-dependent apoptosis. Mol Cell Biol. 2006;26:7397–408. doi: 10.1128/MCB.02282-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alirol E, et al. The mitochondrial fission protein hFis1 requires the endoplasmic reticulum gateway to induce apoptosis. Mol Biol Cell. 2006;17:4593–605. doi: 10.1091/mbc.E06-05-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bras M, et al. Drp1 mediates caspase-independent type III cell death in normal and leukemic cells. Mol Cell Biol. 2007;27:7073–88. doi: 10.1128/MCB.02116-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–87. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Underscores the importance of mitochondrial fission and fusion in synaptic development.
- 69.Benard G, et al. Mitochondrial bioenergetics and structural network organization. J Cell Sci. 2007;120:838–48. doi: 10.1242/jcs.03381. [DOI] [PubMed] [Google Scholar]
- 70.Santel A. Get the balance right: mitofusins roles in health and disease. Biochim Biophys Acta. 2006;1763:490–9. doi: 10.1016/j.bbamcr.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 71.Kijima K, et al. Mitochondrial GTPase mitofusin 2 mutation in Charcot-Marie-Tooth neuropathy type 2A. Hum Genet. 2005;116:23–7. doi: 10.1007/s00439-004-1199-2. [DOI] [PubMed] [Google Scholar]
- 72.Zuchner S, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–51. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- References 71 and 72 were the first to show that mutations in Mfn2 cause CMT2A.
- 73.Zuchner S, et al. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol. 2006;59:276–81. doi: 10.1002/ana.20797. [DOI] [PubMed] [Google Scholar]
- 74.Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res. 2004;23:53–89. doi: 10.1016/j.preteyeres.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 75.Amati-Bonneau P, et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain. 2008;131:338–51. doi: 10.1093/brain/awm298. [DOI] [PubMed] [Google Scholar]
- 76.Hudson G, et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain. 2008;131:329–37. doi: 10.1093/brain/awm272. [DOI] [PubMed] [Google Scholar]
- References 75 and 76 describe new optic atrophy ‘plus’ phenotypes caused by mutations in OPA1.
- 77.Detmer SA, Chan DC. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J Cell Biol. 2007;176:405–14. doi: 10.1083/jcb.200611080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- 79.Griffin EE, Detmer SA, Chan DC. Molecular mechanism of mitochondrial membrane fusion. Biochim Biophys Acta. 2006;1763:482–9. doi: 10.1016/j.bbamcr.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 80.Waterham HR, et al. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. 2007;356:1736–41. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- First report of a Drp1 mutation in a human patient.
- 81.Baxter RV, et al. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat Genet. 2002;30:21–2. doi: 10.1038/ng796. [DOI] [PubMed] [Google Scholar]
- 82.Cuesta A, et al. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat Genet. 2002;30:22–5. doi: 10.1038/ng798. [DOI] [PubMed] [Google Scholar]
- 83.Kabzinska D, et al. Early onset Charcot-Marie-Tooth disease caused by a homozygous Leu239Phe mutation in the GDAP1 gene. Acta Myol. 2006;25:34–7. [PubMed] [Google Scholar]
- 84.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–95. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 85.Jahani-Asl A, et al. Mitofusin 2 protects cerebellar granule neurons against injury-induced cell death. J Biol Chem. 2007;282:23788–98. doi: 10.1074/jbc.M703812200. [DOI] [PubMed] [Google Scholar]
- 86.Smith PD, O’Hare MJ, Park DS. CDKs: taking on a role as mediators of dopaminergic loss in Parkinson’s disease. Trends Mol Med. 2004;10:445–51. doi: 10.1016/j.molmed.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 87.Park DS, Levine B, Ferrari G, Greene LA. Cyclin dependent kinase inhibitors and dominant negative cyclin dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J Neurosci. 1997;17:8975–83. doi: 10.1523/JNEUROSCI.17-23-08975.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Park DS, et al. Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J Cell Biol. 1998;143:457–67. doi: 10.1083/jcb.143.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nguyen MD, et al. Cell cycle regulators in the neuronal death pathway of amyotrophic lateral sclerosis caused by mutant superoxide dismutase 1. J Neurosci. 2003;23:2131–40. doi: 10.1523/JNEUROSCI.23-06-02131.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Osuga H, et al. Cyclin-dependent kinases as a therapeutic target for stroke. Proc Natl Acad Sci U S A. 2000;97:10254–9. doi: 10.1073/pnas.170144197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Busser J, Geldmacher DS, Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer’s disease brain. J Neurosci. 1998;18:2801–7. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Smith PD, et al. Calpain-regulated p35/cdk5 plays a central role in dopaminergic neuron death through modulation of the transcription factor myocyte enhancer factor 2. J Neurosci. 2006;26:440–7. doi: 10.1523/JNEUROSCI.2875-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Johansson A, et al. Genetic association of CDC2 with cerebrospinal fluid tau in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2005;20:367–74. doi: 10.1159/000088634. [DOI] [PubMed] [Google Scholar]
- 94.Johnson DT, Harris RA, Blair PV, Balaban RS. Functional consequences of mitochondrial proteome heterogeneity. Am J Physiol Cell Physiol. 2007;292:C698–707. doi: 10.1152/ajpcell.00109.2006. [DOI] [PubMed] [Google Scholar]
- 95.Chang DT, Reynolds IJ. Differences in mitochondrial movement and morphology in young and mature primary cortical neurons in culture. Neuroscience. 2006;141:727–36. doi: 10.1016/j.neuroscience.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 96.Jonas E. BCL-xL regulates synaptic plasticity. Mol Interv. 2006;6:208–22. doi: 10.1124/mi.6.4.7. [DOI] [PubMed] [Google Scholar]
- 97.Reddy PH, Beal MF. Are mitochondria critical in the pathogenesis of Alzheimer’s disease? Brain Res Brain Res Rev. 2005;49:618–32. doi: 10.1016/j.brainresrev.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 98.Rikhy R, Kamat S, Ramagiri S, Sriram V, Krishnan KS. Mutations in dynamin-related protein result in gross changes in mitochondrial morphology and affect synaptic vesicle recycling at the Drosophila neuromuscular junction. Genes Brain Behav. 2007;6:42–53. doi: 10.1111/j.1601-183X.2006.00218.x. [DOI] [PubMed] [Google Scholar]
- 99.Li H, et al. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 2008;105:2169–74. doi: 10.1073/pnas.0711647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Verstreken P, et al. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–78. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 101.Anderson S, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–65. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 102.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wallace DC. Why do we still have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu Rev Biochem. 2007;76:781–821. doi: 10.1146/annurev.biochem.76.081205.150955. [DOI] [PubMed] [Google Scholar]
- 104.Cao Z, Wanagat J, McKiernan SH, Aiken JM. Mitochondrial DNA deletion mutations are concomitant with ragged red regions of individual, aged muscle fibers: analysis by laser-capture microdissection. Nucleic Acids Res. 2001;29:4502–8. doi: 10.1093/nar/29.21.4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gokey NG, et al. Molecular analyses of mtDNA deletion mutations in microdissected skeletal muscle fibers from aged rhesus monkeys. Aging Cell. 2004;3:319–26. doi: 10.1111/j.1474-9728.2004.00122.x. [DOI] [PubMed] [Google Scholar]
- 106.Kujoth GC, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–4. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 107.Trifunovic A, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–23. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 108.Kowald A, Jendrach M, Pohl S, Bereiter-Hahn J, Hammerstein P. On the relevance of mitochondrial fusions for the accumulation of mitochondrial deletion mutants: a modelling study. Aging Cell. 2005;4:273–83. doi: 10.1111/j.1474-9726.2005.00169.x. [DOI] [PubMed] [Google Scholar]
- 109.Diaz F, et al. Human mitochondrial DNA with large deletions repopulates organelles faster than full-length genomes under relaxed copy number control. Nucleic Acids Res. 2002;30:4626–33. doi: 10.1093/nar/gkf602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Moraes CT, Kenyon L, Hao H. Mechanisms of human mitochondrial DNA maintenance: the determining role of primary sequence and length over function. Mol Biol Cell. 1999;10:3345–56. doi: 10.1091/mbc.10.10.3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Elson JL, Samuels DC, Turnbull DM, Chinnery PF. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am J Hum Genet. 2001;68:802–6. doi: 10.1086/318801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bogenhagen DF, Rousseau D, Burke S. The layered structure of human mitochondrial DNA nucleoids. J Biol Chem. 2008;283:3665–75. doi: 10.1074/jbc.M708444200. [DOI] [PubMed] [Google Scholar]
- 113.Garrido N, et al. Composition and dynamics of human mitochondrial nucleoids. Mol Biol Cell. 2003;14:1583–96. doi: 10.1091/mbc.E02-07-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jacobs HT, Lehtinen SK, Spelbrink JN. No sex please, we’re mitochondria: a hypothesis on the somatic unit of inheritance of mammalian mtDNA. Bioessays. 2000;22:564–72. doi: 10.1002/(SICI)1521-1878(200006)22:6<564::AID-BIES9>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 115.Johnson DT, et al. Tissue heterogeneity of the mammalian mitochondrial proteome. Am J Physiol Cell Physiol. 2007;292:C689–97. doi: 10.1152/ajpcell.00108.2006. [DOI] [PubMed] [Google Scholar]
- 116.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–68. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 117.St-Pierre J, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 118.Cui L, et al. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- References 117 and 118 describe a potential role for PGC-1α and mitochondrial biogenesis in neurodegenerative disease.
- 119.Handschin C, et al. PGC-1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev. 2007;21:770–83. doi: 10.1101/gad.1525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.van Praag H, Christie BR, Sejnowski TJ, Gage FH. Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad Sci U S A. 1999;96:13427–31. doi: 10.1073/pnas.96.23.13427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.van Praag H, Kempermann G, Gage FH. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci. 1999;2:266–70. doi: 10.1038/6368. [DOI] [PubMed] [Google Scholar]
- 122.McCloskey DP, Adamo DS, Anderson BJ. Exercise increases metabolic capacity in the motor cortex and striatum, but not in the hippocampus. Brain Res. 2001;891:168–75. doi: 10.1016/s0006-8993(00)03200-5. [DOI] [PubMed] [Google Scholar]
- 123.Irrcher I, Adhihetty PJ, Joseph AM, Ljubicic V, Hood DA. Regulation of mitochondrial biogenesis in muscle by endurance exercise. Sports Med. 2003;33:783–93. doi: 10.2165/00007256-200333110-00001. [DOI] [PubMed] [Google Scholar]
- 124.Pilegaard H, Saltin B, Neufer PD. Exercise induces transient transcriptional activation of the PGC-1alpha gene in human skeletal muscle. J Physiol. 2003;546:851–8. doi: 10.1113/jphysiol.2002.034850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Short KR, et al. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005;102:5618–23. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Krishnan KJ, Greaves LC, Reeve AK, Turnbull DM. Mitochondrial DNA mutations and aging. Ann N Y Acad Sci. 2007;1100:227–40. doi: 10.1196/annals.1395.024. [DOI] [PubMed] [Google Scholar]
- 127.Melov S, Tarnopolsky MA, Beckman K, Felkey K, Hubbard A. Resistance exercise reverses aging in human skeletal muscle. PLoS ONE. 2007;2:e465. doi: 10.1371/journal.pone.0000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cartoni R, et al. Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise. J Physiol. 2005;567:349–58. doi: 10.1113/jphysiol.2005.092031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bach D, et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem. 2003;278:17190–7. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- 130.Taivassalo T, et al. Gene shifting: a novel therapy for mitochondrial myopathy. Hum Mol Genet. 1999;8:1047–52. doi: 10.1093/hmg/8.6.1047. [DOI] [PubMed] [Google Scholar]
- 131.Parise G, Brose AN, Tarnopolsky MA. Resistance exercise training decreases oxidative damage to DNA and increases cytochrome oxidase activity in older adults. Exp Gerontol. 2005;40:173–80. doi: 10.1016/j.exger.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 132.DiMauro S, Hirano M, Schon EA. Approaches to the treatment of mitochondrial diseases. Muscle Nerve. 2006;34:265–83. doi: 10.1002/mus.20598. [DOI] [PubMed] [Google Scholar]
- 133.Taivassalo T, Haller RG. Exercise and training in mitochondrial myopathies. Med Sci Sports Exerc. 2005;37:2094–101. doi: 10.1249/01.mss.0000177446.97671.2a. [DOI] [PubMed] [Google Scholar]
- 134.Kukidome D, et al. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes. 2006;55:120–7. [PubMed] [Google Scholar]
- 135.St-Pierre J, et al. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. J Biol Chem. 2003;278:26597–603. doi: 10.1074/jbc.M301850200. [DOI] [PubMed] [Google Scholar]
- 136.Lehman JJ, et al. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–56. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nunomura A, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–67. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 138.Crouch PJ, et al. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42. J Neurosci. 2005;25:672–9. doi: 10.1523/JNEUROSCI.4276-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Manczak M, et al. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–49. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 140.Betarbet R, et al. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301–6. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 141.Fornai F, et al. Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc Natl Acad Sci U S A. 2005;102:3413–8. doi: 10.1073/pnas.0409713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Damiano M, et al. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J Neurochem. 2006;96:1349–61. doi: 10.1111/j.1471-4159.2006.03619.x. [DOI] [PubMed] [Google Scholar]
- 143.Liu J, et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 144.Mattiazzi M, et al. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–33. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- 145.Pasinelli P, et al. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43:19–30. doi: 10.1016/j.neuron.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 146.Milakovic T, Johnson GV. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J Biol Chem. 2005;280:30773–82. doi: 10.1074/jbc.M504749200. [DOI] [PubMed] [Google Scholar]
- 147.Benchoua A, et al. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell. 2006;17:1652–63. doi: 10.1091/mbc.E05-07-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Panov AV, et al. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–6. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 149.Poole AC, et al. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105:1638–43. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yang Y, et al. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A. 2008;105:7070–5. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]