

Abstract
Vascular endothelial growth factor (VEGF) is a potent cytokine involved in the induction of neovascularization. Secreted as a cysteine-linked dimer, it has two binding sites at opposite poles through which it may bind VEGF receptors (VEGFRs), receptor tyrosine kinases found on the surface of endothelial and other cells. The binding of a VEGF molecule to two VEGFR molecules induces transphosphorylation of the intracellular domains of the receptors, leading to signal transduction. The dominant mechanism of receptor dimerization is not clear: the receptors may be present in an inactive pre-dimerized form, VEGF binding first to one of the receptors, the second receptor then ideally located for dimerization; or VEGF may bind receptor monomers on the cell surface, which then diffuse and bind to available unligated receptor monomers to complete the activation. Both processes take place and one or other may dominate on different cell types. We demonstrate the impact of dimerization mechanism on the binding of VEGF to the cell surface and on the formation of active signaling receptor complexes. We describe two methods to determine which process dominates, based on binding and phosphorylation assays. The presence of two VEGF receptor populations, VEGFR1 and VEGFR2, can result in receptor heterodimer formation. Our simulations predict that heterodimers will comprise 10–50% of the active, signaling VEGF receptor complexes, and that heterodimers will form at the expense of homodimers of VEGFR1 when VEGFR2 populations are larger. These results have significant implications for VEGF signal transduction and interpretation of experimental studies. These results may be applicable to other ligand-receptor pairs, in particular PDGF.
Keywords: homodimer, heterodimer, growth factor, receptor tyrosine kinase, mathematical model
INTRODUCTION
Angiogenesis (neovascularization), the growth of new blood vessels from pre-existing vasculature, plays a key role in diseases such as cancer, diabetic retinopathy and arthritis [1–3]. Vascular endothelial growth factor (VEGF) is a critical regulator of angiogenesis [4,5], a cytokine which carries activating, proliferative and chemotactic signals from hypovascularized tissues to endothelial cells of quiescent blood vessels and initiates new vessel sprouting to increase perfusion of the target tissue. VEGF is a member of the cysteine knot family of growth factors and is secreted as a dimer in which the monomers are linked together by two intersubunit disulfide bridges [6,7] (Figure 1A). The receptor binding sites of VEGF are formed by amino acid residues of both monomers which collocate at the poles of the antiparallel dimer [8–10] (Figure 1A). The VEGF Receptor-1 (VEGFR1) and VEGFR2 binding sites are slightly different but overlap, resulting in competition for VEGF binding [10].
Figure 1. Dimerization of VEGF receptors by VEGF.

A, Schematic of dimeric structure of VEGF [60,61], representing the backbone structure of the first 110 amino acids of VEGF. One monomer is represented by a black line, the other by a gray line. The disulfide bonds which hold the monomers together are located approximately as indicated by the dashed lines [7]. The location of binding sites for VEGFR1 and VEGFR2 are indicated by dotted black and gray lines respectively [9,10]. Each site is formed by a combination of residues of both monomers, at both poles of the dimer. The VEGFR1 and VEGFR2 sites overlap, preventing the binding of two VEGF receptors to one pole of VEGF. The binding of one receptor monomer to each pole of VEGF results in the transactivation of the intracellular domains of the two receptor monomers. B, 1:1 binding of VEGF dimers to VEGF receptor dimers, with activation taking place as part of the single binding step. This model has been used in previous descriptions of VEGF interaction with its receptors [18,19,21]. C, Mechanisms of VEGF Receptor dimerization explored: SPD, static pre-dimerization; DPD, dynamic pre-dimerization; LID, ligand-induced dimerization; and PDM, parallel dimerization model, in which both ligand-induced and ligand-independent dimerization can take place. PDM thus includes all the ligand-receptor and receptor-receptor binding steps shown here. Bonds between molecules are shown in blue. For more details, see text.
The VEGF receptors are receptor tyrosine kinases and, like other members of that family, require dimerization to be active. Signaling is initiated when VEGF binds two receptor monomers, resulting in the transactivation of the intracellular domains of the receptors and subsequent signal propagation through SH2-domain containing proteins that are phosphorylated by the active receptors. Prior to the binding of VEGF, the receptor monomers diffuse throughout the cell membrane as singletons or as ligand-independent pre-associated dimers. These dimers are not active, possibly due to the presence of an inhibitory domain in the non-receptor binding part of the extracellular moiety of the receptor [11]. Thus, two distinct pathways exist to dimerization: in the first, the ligand (VEGF) binds an available receptor monomer, and this complex diffuses laterally on the cell surface to bind a second, empty receptor monomer; in the second, the ligand binds to one of the receptors in an available receptor dimer, and then the second pole of the ligand binds to (‘couples’) the second, pre-associated receptor. We will refer to these two mechanisms as ligand-induced dimerization (LID) and dynamic pre-dimerization (DPD), respectively. A more detailed description of the receptor dimerization schemes is contained in the Methods section.
Both of these mechanisms are involved in VEGF receptor dimerization; VEGF can bind to receptor monomers [12–16], and VEGF receptors can associate in the absence of ligand [17], however it is not known to what extent each mechanism actually occurs. One pathway may dominate over the other, and so we examine the complete dimerization model as well as these two pathways alone.
Because signaling is initiated by VEGF-induced receptor dimerization, an understanding of the mechanism of dimerization is essential for correct interpretation of experimental results. The formation of heterodimers of VEGF receptors and the resultant modulation of VEGF signaling, in particular, are difficult to study without a formal description of dimerization. Downstream signal transduction adds further complexity to the study of VEGF, but without a sound footing of knowledge at the cell surface, signal transduction experiments are less informative. In addition, therapeutics targeted to VEGF-VEGFR interactions may be more or less effective than predicted depending on the mechanism of dimerization.
We have previously constructed computational models for in silico simulation of VEGF-VEGFR interactions in in vitro assays [18,19] as well as in vivo [20]. We have also demonstrated using Monte Carlo methods that continuum representation of the receptor-ligand system in terms of concentrations is justified [21]. In those studies, the interactions were approximated as one-to-one reactions, with VEGF binding to and activating pre-dimerized receptors in a single binding step, and being released from those receptors in a single unbinding step. Here, we introduce dimerization reactions and investigate the impact of dimerization on signal transduction by VEGF receptors. In particular, this approach allows us to study the formation of VEGF receptor heterodimers.
Although the principles of LID and DPD have been studied before [22,23], to our knowledge this is the first model of receptor dimerization which incorporates both together. This allows us to explore the signaling of VEGF and its receptors in detail, maintaining the biophysical accuracy of the interactions: a bivalent ligand and monovalent receptors with transient ligand-independent association of receptors. Other ligand-receptor tyrosine kinase pairs have been studied, in which the requirement for signaling is also ligand bound to receptors inducing dimerization, but the interactions are different: EGF is a monovalent ligand – monovalent receptor, with coupling after two ligands bind to two receptors [24–30]; FGF is similar to EGF, though the ligand may also dimerize transiently before binding to its receptors [23,31–33].
The dimerization mechanism proposed here should be similar for other members of the same ligand subfamily, a subset of the cysteine knot family of constitutively dimeric proteins. The other known members of this family, in addition to the protein products of several VEGF genes (VEGF-A,VEGF-B, VEGF-C, VEGF-D) are placental growth factor (PlGF), which also binds to VEGF receptors, and platelet-derived growth factor (PDGF), which binds to PDGF receptors [34,35]. The model presented here would be useful for describing those systems as well. A model of PDGF dimerization has been published [36], which could be applied to the VEGF system. That model considers ligand-induced dimerization, but does not include receptor dimer pre-association. A hypothetical unstable intermediate of two ligand dimers bound to two receptor monomers is introduced to resolve differences between model predictions and experimental data. The model presented here, with receptor pre-association included, is an alternative scheme to describe the dimerization of growth factor receptors. We do not discuss in detail here the observed cooperativity in PDGF binding (not explained by this version of our model), but the addition to this model of a partial decoupling of dimerization and phosphorylation could provide an alternative to the unstable intermediate theory (Mac Gabhann and Popel, unpublished observations).
In this analysis, we will simulate in vitro experiments with cells expressing VEGFR1 and/or VEGFR2. No Neuropilin or extracellular matrix is included, and thus the analysis here is applicable to any isoform of VEGF. We will first examine cells which express a single population of VEGF receptors – e.g. monocytes, which express VEGFR1 but not VEGFR2 (VEGFR1 mediates migratory signals for these cells). Then, we will investigate dimerization of VEGFRs on cells which express two populations of VEGFR – e.g. endothelial cells which typically express both VEGFR1 and VEGFR2, though not necessarily in equal numbers. We will examine the impact of heterodimerization of these receptors on VEGF signaling.
METHODS
Simulating in vitro experiments
The results presented in this report are for simulations representing in vitro experiments. In these experiments, a confluent monolayer of cells is plated in cell culture chambers (wells) and cell culture medium added, into which growth factors, inhibitors, and other proteins may be added during the course of the experiment, and from which fluid may be drawn to extract data for Scatchard plots and other information. The cells are assumed to have a surface area of 1000 μm2 [18] and the culture medium covers the cells to a height of 1 mm. The results that we obtain from the simulations – in particular, VEGF binding to the cell surface and VEGF receptor activation – are equivalent to, and can be compared to, measurements made in in vitro experiments. The specific experiments required to validate the predictions of the model have not yet been performed, and this study should serve as an impetus to stimulate future experimental work.
Dimerization models
In previous models of VEGF-VEGFR interactions [18–20] the dimerization of VEGF receptors was not included explicitly. Instead, VEGF bound to a pre-dimerized receptor in a single binding and activation step (Fig 1B). In fact there are two parallel pathways by which VEGF may bind to and dimerize VEGF receptors, illustrated in Figure 1C for VEGFR1. First, VEGF may bind to pre-dimerized receptors; these receptors are not activated but are associated together and require the binding of VEGF for activation. The association of the two receptors (Reaction 1) may be direct or indirect (e.g. mediated by heparan sulfate proteoglycans). One pole of VEGF binds to one of the receptors (Reaction 3), and this complex then facilitates the binding of the second pole of VEGF to the associated receptor (Reaction 4). If the receptor dimers are inserted into the membrane and do not dissociate, we refer to it as Static Pre-Dimerization, or SPD. If instead receptor monomers are inserted into the membrane and they can dynamically associate and dissociate from each other, it is known as Dynamic Pre-Dimerization, or DPD. Note that SPD is similar to the 1:1 Langmuir binding shown in Figure 1B except that the binding is a two-step process. Second, VEGF may bind to a VEGF receptor monomer (Reaction 7). This VEGF-VEGFR complex diffuses laterally in the cell membrane, encountering and binding a second unbound receptor monomer (Reaction 6). The receptors transphosphorylate and the signal transduction cascade begins. This pathway is known as Ligand-Induced Dimerization, or LID.
As noted in the introduction, experimental evidence suggests that both of the major dimerization pathways (LID and DPD) are active for the dimerization of VEGF receptors. Including both pathways results in the Parallel Dimerization Model (PDM); within that framework LID or DPD may be balanced or one may dominate; therefore we will study both individually and later propose experiments to determine which mechanism is more important. SPD is a special case of DPD and is therefore implicitly included in the PDM.
We do not deal explicitly with the processes of phosphorylation and dephosphorylation of the VEGF receptors here. We assume that phosphorylation occurs instantly subsequent to VEGF binding two receptor monomers, and that dephosphorylation is instant following VEGF unbinding from one of the receptors. Nonlinear behavior of the phosphorylation and dephosphorylation processes could result in different outcomes than those described here.
Ligand-receptor binding interactions
The dimerization mechanisms (Figure 1C) are converted into reaction-diffusion equations which are similar to those which have been published previously [18,19]. A combination of second-order (e.g. VEGF binding to receptor, or receptor-receptor association) and first-order (e.g. the second pole of receptor-bound VEGF binding to pre-associated receptor) reactions are used to represent the mechanisms.
For the example of VEGFR1 only expressed on the surface, the reactions are:
Here R1 is the VEGFR1 receptor monomer and V is VEGF. The dot (·) connects binding partners, thus V·R1·R1 is VEGF bound to one VEGFR1 monomer of a VEGFR1 dimer pair, while R1·V·R1 is VEGF bound to two VEGFR1 monomers. Δ indicates that each of the three components in that complex is bound to the other two (otherwise links are in the order shown). *denotes that the receptor is activated (phosphorylated). These reactions are used to generate the model equations (below). The kinetic parameters governing each reaction are shown on Figure 1C and described in detail below.
For SPD, reactions 3 and 4 are included; for DPD, reactions 1, 3 and 4; for LID, reactions 6 and 7; and for PDM, reactions 1–7. Reactions 2 and 5 arise only if both ligand-dependent and ligand-independent receptor association are occurring.
For VEGFR2, reactions equivalent to 1–7 and equations for VEGFR2 are included in the model. In addition, VEGFR1-VEGFR2 heterodimers may form, and reactions equivalent to 1 and 5, as well as two reactions each equivalent to reactions 2, 3, 4 and 6 must be included. The total number of reactions in the model is therefore 24 (see Supplemental information for complete list).
Model equations
VEGF is introduced into the cell culture medium and is uniformly distributed at the start of simulations of in vitro experiments. VEGF diffuses in the cell culture medium throughout the simulation:
Here [V] is the concentration of VEGF; DV is the diffusivity of VEGF in the cell culture medium. Only unligated receptors are present on the cell surface at the start of the simulation:
Once VEGF is introduced into the system, binding proceeds and the cell surface receptor and ligand-receptor complexes are determined by:
Here sR1 is the insertion rate of new VEGFR1 monomers into the cell membrane; kint is the internalization rate of receptors and receptor-ligand complexes. Boxed subscripts (1–7) indicate the reaction number from which the term derives. For SPD, terms for reactions 3 and 4 are included; for DPD, reactions 1, 3 and 4; for LID, reactions 6 and 7; and for PDM, reactions 1–7.
For receptor pre-association simulations, there is a lead-in time before VEGF is added to allow the ligand-independent receptor dimerization reactions to come to steady state.
Degradation of VEGF in the cell culture medium is assumed to be negligible over the short times of these simulations. The pathway for removal of VEGF from the medium is binding of VEGF to the cell surface and subsequent internalization. Cell surface binding of VEGF determines the boundary condition for VEGF transport:
At the air-liquid boundary of the cell culture medium, a no-flux boundary condition is used:
This study also includes VEGFR2, for which additional equations are required similar to those presented here and additional terms are included in the above equations. The complete set of equations are included in the online supplement. Placental growth factor (PlGF) is a VEGF family ligand that binds only VEGFR1 (not VEGFR2) and also requires additional equations. These additional equations are not presented here but follow the same form as those above.
Binding kinetics
Binding affinities, receptor numbers and kinetic rates reported previously for the VEGF-VEGFR system are based on an assumption of 1:1 binding [37–44]. For the dimerization schemes shown here, we use kinetics which would result in approximately the same Scatchard and kinetic rate data being observed. The measured affinity and measured 1:1 kinetics are related by:
For SPD and DPD,
And at steady state,
where kon,VR is the rate of the binding of one pole of VEGF to one receptor of the two pre-dimerized receptors, koff,VR is the dissociation rate for that interaction, and kΔ,VR is the rate of binding of the second VEGF pole to the second pre-associated receptor. Thus,
We assume that the value of the on-rate, kon, is half that of the measured (i.e., effective 1:1) rate, |kon,VR| = |kon,meas|/2, as there are two receptor binding sites for VEGF to bind. We take the value of the off-rate, koff,VR, to be proportional to the square root of the value of the measured rate. An off-rate of matches with experimental observations of the off-rate for a single receptor monomer [15]; in those experiments, monomer-specific mutagenesis was used to alter one binding site of the VEGF dimer and make it inactive. By equating the measured binding affinity (Kd = koff,,meas/kon,meas) with the above expression for Kd, the association rate for the second VEGF-VEGFR binding is found to be |kΔ,VR| = 1–2·|koff, VR|. Although koff and kΔ could be adjusted together without significantly affecting the observed kinetics or equilibrium constants, the experimental measurements of single- and double-site off rates are assumed to hold [15]. Note that the units of the measured and single-site kon and koff are the same, but the values must be different for the scheme to give the same observed binding and activation.
For LID,
The number of receptor monomers is divided by two to find the number of receptor dimers (this would be what is measured by the Scatchard plot).
As above, |kon,VR| = |kon,meas|/2 and and the coupling rate for binding to the second VEGFR monomer is |kc,VR| = (1–2 ·|koff,VR|)/|[R1]|. This non-linear result demonstrates why, for LID, there is not a straight line on the Scatchard plot (see Results); it also suggests that where more receptors are present, a lower coupling rate is required to achieve the same observed binding, as we would expect.
The number of receptors and their binding affinities for VEGF have been measured for several cell types. Monocytes express VEGFR1; endothelial cells VEGFR2 and possibly VEGFR1. There is a wide range of receptor expression levels depending on the type of cell in question; cells from different vessels, or from the same vessel in different species, express different numbers of each receptor type; for cells with a single receptor population, we assume 20,000 receptor dimers (40,000 monomers) with affinity of 150 pM, consistent with both endothelial VEGFR2 and monocyte VEGFR1 [37–44]. For cells with two receptor populations, these studies note that VEGFR1 has a 4–10 fold higher affinity for VEGF than does VEGFR2. We assume 20,000 total VEGF receptor dimers, divided in 1:10 or 1:1 ratio between VEGFR1 and VEGFR2. We assume affinities of 30 pM for VEGFR1 and 150 pM for VEGFR2, again within the range of the same experimental studies.
Cell-free systems can be used to measure kinetics of binding of VEGF to immobilized receptors. The cell-free measured on-rates of VEGF binding to VEGFR2 range from 6.6·104–5.23·106 M−1s−1; the associated off-rates were 1.27·10−5 – 4.1·10−4 s−1 [12–15]. For VEGFR1, the cell-free kinetics were measured to be 4·106 M−1s−1 and 3·10−5 s−1 [16]. However, in these cell-free assays, the co-receptors (e.g. heparan sulfate proteoglycans) which affect binding on the cell surface, are missing, and cell-based assays reveal faster kinetics for binding of VEGF to cell surface receptors: application of our model to experimental data of cell-surface binding of VEGF over time to monocytes [39] reveals an on-rate of 8.4·106 M−1s−1and an off-rate of 1.3·10−3 s−1. For endothelial cells which express both VEGFR1 and VEGFR2, we assume the same off rate, but use a different on rate to match the binding affinity.
Ligand-receptor coupling rates
Above it was noted that the coupling rate for a ligand-receptor complex to bind a second empty receptor should be inversely proportional to the number of free receptors in order to maintain a constant effective Kd, but here we use a constant coupling rate; we will note that the resulting Scatchard plot is close to linear under typical experimental conditions. We have shown that the ligand-induced coupling rate for VEGF binding two different receptor types [19] agreed with estimates of the theoretical diffusion-limited rate [33,45], and with experimental estimates of coupling rates from other experimental systems [46,47]. This rate is almost constant over a wide range of receptor densities (varies by a factor of 3 for 1–106 receptors/cell), and so we choose for each simulation a constant value of the coupling rate, but one which is based on the total number of receptors present, to ensure adequate agreement between the experimental and simulated Scatchard plots. For cells expressing two receptors, this coupling rate is adjusted to reflect the disparity in the on-rates between the two receptors, thus satisfying the requirements of detailed balance. This can be interpreted as follows: if one receptor binds VEGF more tightly, then it would also couple to a VEGF-receptor complex more often.
The dissociation of a ligand-induced receptor dimer involves the release of an empty receptor monomer and we assume that this occurs with the same VEGF-VEGFR off rate as the first reaction.
Receptor-receptor coupling rates
Receptors may also dimerize in the absence of ligands. As noted above, a theoretical coupling rate, kc,RR, of 1–3 1014 cm2 mol−1 s−1 was predicted for VEGF receptors, more specifically for coupling between VEGFR2 and Neuropilin-1 [19]; we use 2.125 1014 cm2 mol−1 s−1 as the value throughout this paper; though the sensitivity of the results to variation in this parameter are noted at various points. The rate of association for receptors which are coupled together by a ligand but which are not themselves pre-associated (this is analogous to the reaction in which the second pole of VEGF binds to the second pre-dimerized receptor) is required by detailed balance to be kΔ,RR= kΔ,VR(kc,RR/kc,VR,. The rate of dissociation of two ligated or unligated receptors is taken to be 10−2 s−1; the balance of coupling and dissociation results in the steady-state balance between monomers and dimers.
Internalization
Internalization is slow or nonexistent at 4 °C [48]. At higher temperatures, the internalization is faster for complexes which are phosphorylated [48]. The values assumed here are zero internalization at the lower temperature; 10−5 s−1 and 2.8·10−4 s−1 for inactive and activated receptors, respectively [18].
Ligand diffusivity
An estimated diffusivity is used based on the molecular weight of each species, and adjusted for the temperature of the experiment. The diffusivity at 23°C was estimated by Berk et al [49] as:
We adjust this to other temperatures using the Stokes-Einstein equation,
where η is the viscosity of the solvent. In the range 0°C–37°C, the latter function is described well by:
The molecular weight of VEGF is approximately 45 kDa and that of PlGF 40 kDa.
Scatchard Plots
We generate Scatchard plots that would be observed experimentally if certain dimerization mechanisms were present. To do this, both free VEGF and bound VEGF are measured in the simulation in a similar manner to the experimentally-derived plots. We use a sample of fluid away from the cells – not the concentration at the cell surface – to determine the free ligand concentration; this leads to a slight discrepancy between the measured and actual Kd, but experimentally the concentration cannot be measured at the cell surface, and the difference is less than 5%. The value for bound VEGF used is the total number of VEGF bound to the surface, not the total number of receptors bound, as typically the ligand is radiolabeled for the generation of these graphs. This is an important distinction in comparing the different dimerization pathways.
RESULTS
Single Receptor: VEGFR1 or VEGFR2
Comparison of dimerization models
Not all endothelial cells express both VEGFR1 and VEGFR2. Monocytes also express only one receptor for VEGF, VEGFR1, and it is implicated in signal transduction, including migration [50]. Thus cells with single receptor populations are a useful starting point for our study. Each of the dimerization models was used to estimate the VEGF bound and activated VEGF receptors on cells expressing 20,000 VEGF receptors, with a binding affinity of 150 pM and a measured off-rate of 1.32·10−3 s−1. Each of the dimerization mechanisms (using the kinetics as derived in the Methods section) would generate similar Scatchard plots at steady state (Figure 2A).
Figure 2. Dimerization mechanisms on cells expressing one receptor population.
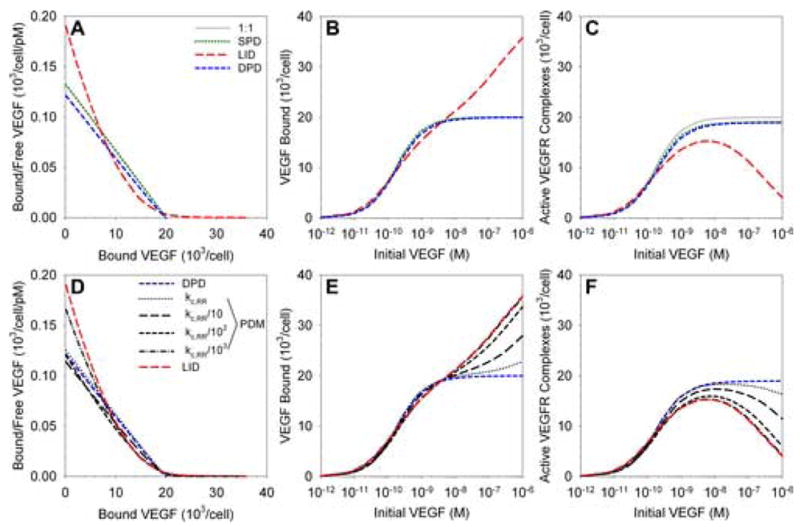
Results of in silico simulations of in vitro experiments for a monolayer of cells expressing various densities of VEGFR. A–C, Comparison of SPD, DPD and LID dimerization models as described in Figure 1 and in the text; scatchard plot (A), total VEGF bound to the cell surface (B) and active (signaling) VEGF-VEGFR complexes (C) are shown for increasing concentrations of VEGF. D–F, The parallel dimerization model (PDM) balances ligand–dependent and –independent dimerization. PDM includes all reactions shown in Figure 1C. By decreasing the rate of association between receptors, the balance of dimerization mechanisms shifts from DPD to LID. Graphs as in A–C.
For VEGF concentrations below 1 nM, binding to the cell surface is predicted to be similar for each of the mechanisms (Figure 2B). Above this threshold, for LID, increasing VEGF results in the formation of complexes of a single VEGF receptor monomer bound to VEGF. We assume that these complexes cannot bind to each other due to the steric hindrance of VEGF, and thus cannot form functional active VEGF receptor dimers. This has two effects: first, twice as many VEGF molecules can bind to the cell, as each receptor monomer is occupied, as opposed to one molecule per receptor dimer in other mechanisms; second, there is an optimal VEGF concentration for the formation of active VEGF receptors, above which the formation of nonsignaling receptor monomer complexes begins to dominate (Figure 2C). This phenomenon (high-dose inhibition) is characteristic of LID and although it has not been explicitly noted at the level of VEGFR phosphorylation, it is not unusual for such experiments not to be continued beyond the VEGF concentration considered to result in peak activation. In addition, there are many instances of high-dose inhibition at the level of cellular outcome – including DNA synthesis [9,41] and proliferation [51,52]. Those observations may not be the result of high-dose inhibition at the level of VEGF binding and VEGFR phosphorylation at the surface, but could instead be the result of intracellular signaling. Nonetheless, we suggest that more study is required to investigate this mechanism.
Using the Parallel Dimerization Model (PDM), i.e. both DPD and LID mechanisms operational, we note that for the theoretical estimate of the receptor coupling parameter, the DPD model appears to dominate (Figure2D–F, dotted line, kc,RR = 1014 cm2/mol/s). Decreasing the pre-association rate of receptors (and thus also the rate of association of receptors coupled by VEGF), we see that the VEGF binding and VEGFR activation results move from similarity to DPD to that of LID. Thus, with this single parameter we can vary the predominance of the DPD and LID models. The reason for this is shown in Figure S1A (online supplement). Decrease in the coupling rate of receptors results in the formation of fewer dimers. As the DPD model relies on pre-formed dimers to bind VEGF, more VEGF binds to receptor monomers and thus LID dominates. Note that if only the DPD mechanism is functional, a decrease in receptor pre-dimerization rate does not significantly affect the VEGF binding and active VEGFR complex formation (Figure S1B; active VEGFR complex formation and VEGF binding are the same for DPD); but when the LID pathway is available, then a decrease in receptor pre-dimerization rate will cause that mechanism to dominate (Figure 2E,F). Increased receptor density also results in increased pre-dimerization of receptors, and thus to the results of the complete model being closer to the DPD results (Figure S1A).
Single-site binding variants: predictions and comparison to experimental data
As each binding site for the VEGF receptors is located across one pole of the VEGF dimer, formed by residues from both monomer units, a constitutively monomeric VEGF (i.e. with the cysteines responsible for intersubunit binding mutated [7]) does not contain one entire binding site, does not compete for binding to receptors and thus is not useful for the study of the ligand-receptor kinetics [9]. Instead, to study single sites, the goal is to affect just one of the binding sites on the dimer and this has been accomplished by experimental investigators in two different ways. First, by creating two sets of VEGF monomers with each missing one of the cysteines involved in intersubunit binding, these monomers can only form VEGF heterodimers and not homodimers [6]. Using such a construct, each monomer can be mutated separately to affect the two parts of the binding pocket at only one end of the dimer [9,53]. Second, by creating a single-chain version of the VEGF dimer which is expressed as one continuous peptide chain, mutations can be introduced that affect only one binding pole [15]. In either case, the result is a VEGF dimer with normal binding activity at one pole and impaired binding at the other pole. Computational simulation results for the binding of such a variant of VEGF to cell surface receptors are shown in Figure 3.
Figure 3. Dimerization inhibited by single-site VEGF binding variants.
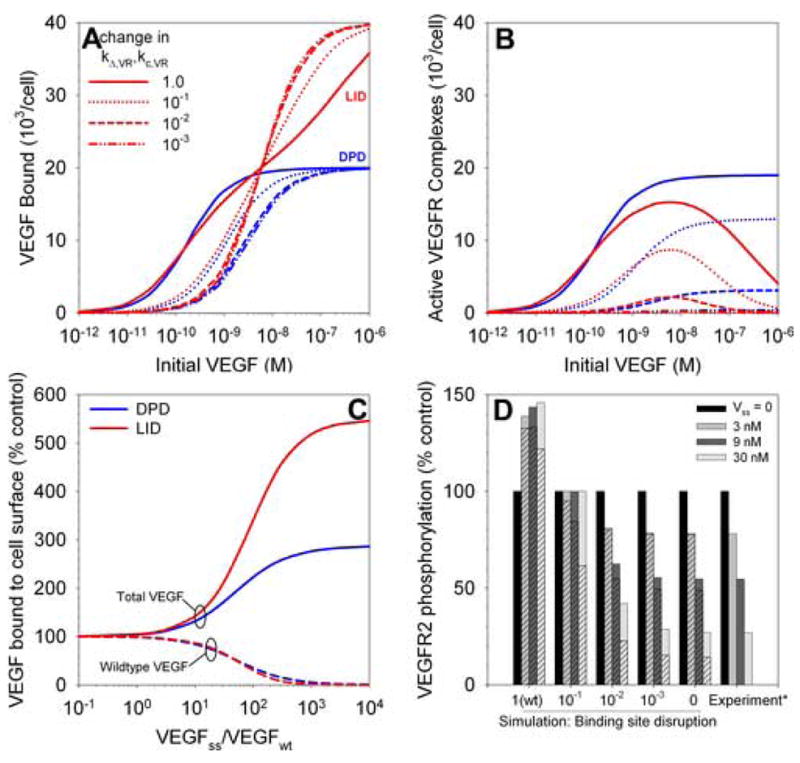
A and B, Binding of single-site variants which have one impaired VEGFR binding site on one pole of the VEGF dimer, using LID or DPD models. Decreasing the activity of one pole of VEGF decreases activation (B) more than binding (A). C, Inhibition of wildtype VEGF (VEGFwt) signaling by addition of competing single-site binding variant (VEGFss). Total VEGF bound increases while VEGFwt binding decreases. D, Single-site variants inhibit VEGF-induced VEGFR2 phosphorylation. *Experimental data from [53].
We represent the decrease in binding affinity of the second pole by decreasing the ligand-receptor coupling rate (kc,VR, affects LID) as well as the association rate of a ligand to a second pre-associated receptor (kΔ,RR, affects DPD). It was noted that a single-site variant with greater than 100-fold lower affinity for receptor dimers lost most of its ability to induce DNA synthesis in HUVECs [9], and this is in agreement with our results (Figure 3B, dashed lines) for either DPD or LID.
Interestingly, in a study which introduced gradually more severe mutations into one binding site, high-dose inhibition of proliferation was noted under the action of the VEGF variants [15]. As noted before, this inhibition does not occur (at the level of VEGF-induced receptor dimerization) for DPD models (Figure 3B) and is only marginally present for parallel dimerization models (PDM) (not shown), in the absence of any reduction in the rate of formation of VEGF receptor pre-associated dimers (kc,RR). The proliferation results (though significantly downstream from receptor activation, and therefore possibly due to other signal processing effects) suggest that the LID model, or a PDM model with a reduced rate of receptor preassociation may be dominant in those cells.
The single-site VEGF variant can be used to antagonize wildtype VEGF signaling and this is illustrated in Figure 3C. Experiments show that excess quantities of the variant (VEGFss) can completely inhibit VEGF-induced DNA synthesis [9,53] and proliferation [15], in agreement with our results. Only the wildtype VEGF is generating significant receptor activation, and thus the decline in binding of that form of VEGF as the variant is added (Figure 3C) results in a similar decrease in signaling. The single-site variant is slightly more effective in inhibiting VEGF binding through the LID mechanism (Figure 3C).
The inhibition of VEGFR phosphorylation by the addition of increasing amounts of a single-site variant has been observed [53] and this is compared to the results of simulations for addition of wildtype VEGF (wt) and VEGF with a second binding site of decreasing affinity (Figure 3D). The simulation results are in close agreement (assuming a correlation between VEGF-induced VEGFR dimerization and VEGFR phosphorylation) with the experimental results [53] for the complete loss of binding to one pole of VEGF, for either DPD (solid bars) or LID (hatched bars). A 100-fold decrease in the binding affinity of one site results in almost complete loss of that binding pole.
Sequential addition of competing ligands
Two ligands competing for binding to the receptor result in dynamic changes in the binding depending on the kinetics of the binding of each ligand, and also on the kinetics and mechanisms of dimerization. Since it is difficult to infer the dimerization mechanism from the binding of a single ligand to its receptor (Figure 2), we simulated the sequential addition of two VEGFR1 ligands to investigate the possibility of further distinguishing the LID and DPD mechanisms. First, VEGF was added and binding was allowed to proceed to equilibrium. Next, PlGF was added and the resulting changes in PlGF and VEGF binding and in VEGFR1 activation were observed over 60 minutes. PlGF and VEGF compete for the same binding site on VEGFR1 [54]. Displacement of VEGF by PlGF from VEGFR1 requires the prior dissociation of VEGF, and thus the results are different for the two dimerization mechanisms: in LID, the dissociation of one pole of VEGF from its receptor allows that receptor to now bind another ligand (potentially a PlGF molecule); in DPD, dissociation of the receptor from a single pole of VEGF does not permit the binding of another ligand, as the receptor remains associated with the bound receptor. Thus, to become available for binding, the VEGF molecule must dissociate completely from both receptor monomers. This should take place more slowly than the release from a single receptor, and this is confirmed by the results of our simulations (Figure 4, Figure S2).
Figure 4. Competition for VEGFR1.

VEGF binding reaches steady state and then PlGF (or unlabeled VEGF) is added. The progression to a new steady state depends on the dimerization method. A, Decrease in VEGF binding and increase in PlGF binding 5 minutes after PlGF addition as a percentage of the change after 60 minutes for LID or DPD as the prevailing dimerization method. B, The difference in the speed of response of LID and DPD (measured as the ratio of the LID and DPD lines in part A) depends on the kinetics of PlGF and VEGF binding. Results shown for VEGF binding decrease after addition of 10 nM PlGF.
The total VEGF bound is decreased by addition of increasing amounts of PlGF (Figure S2A); however this process goes to completion in approximately 15 minutes for LID, and to 50–95% completion in 5 minutes (Figure 4A). For DPD, in contrast, the process takes at least 60 minutes, and is only 20–30% complete after 5 minutes (Figure 4A). Similar numbers are calculated for the increase in PlGF binding (Figure 4A, Figure S2B). This suggests that we can distinguish between the dimerization mechanisms on the basis of the dynamics of competitive ligand binding. Instead of VEGF and PlGF, it would be equally possible to use labeled and unlabeled VEGF, allowing for straightforward determination of the time-course of ligand binding and dissociation. It is timely to reinforce the point that the outcome of the Parallel Dimerization Model (PDM), with both DPD and LID pathways available, lies between the two extremes of the DPD and LID models; therefore this result suggests that the relative dominance of one over the other can be measured by the time course of binding. As before (Figure2D–F), changes in the receptor pre-dimerization rate result in moving from a DPD-characteristic curve to a LID-characteristic curve.
The difference in timing between LID and DPD depends primarily on the kinetics of VEGF release from the receptors (Figure 4B). If the measured off rate were 10-fold faster, then there would be little difference between LID and DPD. Note that the 10-fold increase would be a 3.2-fold increase in the single-site VEGF off rate (see Methods). In contrast, the difference in PlGF-VEGF competition between LID and DPD is only a weak function of the PlGF kinetics (Figure 4B).
Note that there are, as before, differences in receptor activation between the DPD and LID models (Figure S2C–D), however binding studies are more common and straightforward than phosphorylation/activation measurements, and thus a binding-based method to distinguish the dimerization mechanism is advantageous.
Two Receptor Populations: VEGFR1 and VEGFR2
Homodimerization
On most endothelial cells, both VEGFR1 and VEGFR2 are present, though in different ratios for each subtype of endothelial cell [37–40,42–44]. Here, we model endothelial cells as containing 20,000 VEGF receptors, which are present in the ratios 1:10, 1:1, or 10:1 VEGFR1:VEGFR2, to investigate the sensitivity of the results to this ratio.
For homodimerization, (that is, not allowing the formation of VEGFR1-VEGFR2 heterodimers), the presence of two receptors has an effect on the overall binding of VEGF to activation of VEGF receptors (Figure S3A,B), but this is a result of the differing affinities of each of the two receptors. The percentage of active dimers that are VEGFR1-VEGFR1 is dependent on the number of VEGFR1 present but only slightly on the VEGF concentration (data not shown).
Heterodimerization
There is significant recent evidence that VEGFR1-VEGFR2 heterodimers do exist on the cell surface and are significant conduits for VEGF signaling [17,55,56], and thus we consider their formation – either ligand-induced or as pre-dimerizing receptors – in the model. For pre-dimerizing receptors (DPD or PDM models), the formation of dimers before the addition of ligand depends on the coupling rate of empty receptors (Figure S3C). The overall prevalence of dimers is close to uniform across the ratios of VEGFR1:VEGFR2. Of these dimers, homodimers are most prevalent only when one receptor is in significant excess over the other; where the receptors are present in equivalent quantities, preformed heterodimers represent up to a third of the preformed dimers present (Figure S3D). This is independent of the coupling rate of the receptors, though the relative quantities could be affected by a mechanism which preferentially increases or inhibits the formation of homo- vs. heterodimers.
The formation of signaling receptor heterodimers is significant upon addition of VEGF (Figure S3E–F). The total number of active dimers formed in response to VEGF is dependent on the relative affinities of VEGFR1 and VEGFR2 as well as the ratios of receptor expression (Figure S3F). The ratio of expression also plays an important role in the specific signal transduction (Figure 5). We note that the formation of active heterodimers is significant, 10–50% of all dimers across a range of VEGFR1:VEGFR2 ratios and VEGF concentrations. The homodimer formation is split between VEGFR1 and VEGFR2 depending on their ratios and relative kinetics and affinities; the less-expressed receptor does not form significant numbers of homodimers.
Figure 5. Two receptor populations with heterodimerization: VEGF binding.
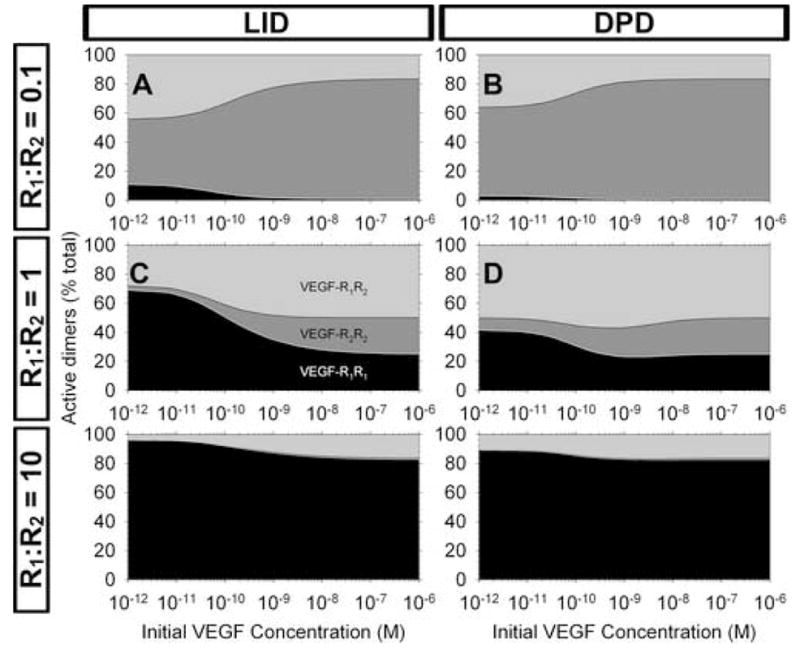
A–F, Signaling profiles: characterization of the active receptors for different ratios of receptors.
While the formation of these three separate signaling units (activated homo- and heterodimers) increases the complexity needed to understand experimental results on phosphorylation of receptors and subsequent signaling, we can see that the problem can be simplified by studying cell types which have more of one receptor than another (Figure5A–B, 5E–F), resulting in negligible formation of one signaling homodimer, and reducing the study to the heterodimer and the other homodimer.
As before (Figure 4, Figure S2), the signaling profile for LID is similar after 5 minutes and after 60 minutes, whereas for DPD there are time-dependent differences in the VEGF-dependent formation of signaling complexes (data not shown).
Competing ligands reveal timing differences in dimerization mechanisms
As noted above, studying cell types with unequal receptor populations can simplify the problem of the variety of signaling complexes on the surface. Another method is to use ligands which cannot take part in receptor heterodimer complexes, such as PlGF, which binds only VEGFR1, or VEGF-C, which binds VEGFR2 but not VEGFR1. Addition of PlGF results in the formation of PlGF-VEGFR1-VEGFR1 complexes, and a decrease in the formation of heterodimers. This is true for both LID and DPD (Figure 6, Figure S4), though we note again a difference in the timing of the changes in binding and activation, receptors using LID reacting faster than DPD (Figure6AS4C–D). Note, however, that it requires a significant quantity of PlGF to prevent the formation of heterodimers altogether (Figure 6B); this is in agreement with an experimental study which noted that addition of VEGF-C compared to VEGF decreased but did not eliminate VEGFR1-VEGFR2 heterodimers [17].
Figure 6. Competition for one of two receptors: VEGF with increasing amounts of PlGF added to cells expressing VEGFR1 and VEGFR2.
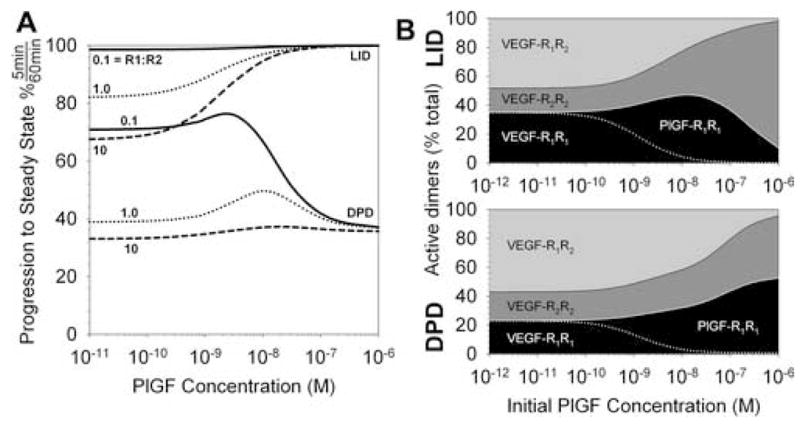
A, Increase in PlGF binding 5 minutes after PlGF addition as a percentage of the increase after 60 minutes for LID or DPD as the prevailing dimerization method. Three different ratios of VEGFR1:VEGFR2 expression are noted. B, Content of active receptor dimers for VEGFR1=VEGFR2. Formation of active (signaling) homo- and hetero-dimers for LID (top) and DPD (bottom).
As before, the presence of VEGFR1 in excess almost eliminates VEGFR2 homodimer formation (data not shown).
Differences in heterodimer and homodimer signaling
It is important to note that two active VEGFR1-VEGFR2 heterodimers should not, and do not, result in the same signal transduction as one VEGFR1-VEGFR1 plus one VEGFR2-VEGFR2. Although the same number of receptor monomers of each type is involved and they are all activated, the mechanism of activation means that they will not transduce the same signals. Each receptor has kinase activity in the intracellular domain. For each receptor, this kinase domain is different, and thus the substrate on which each will act is different. The result is that a VEGFR2 kinase domain will phosphorylate different VEGFR1 tyrosine sites, with different kinetics, than will a VEGFR1 kinase domain (and vice versa). This has been observed experimentally [57]. It was also observed for the interaction of VEGFR2 with VEGFR3, a lymphatic endothelial cell surface receptor [55]. Thus the heterodimers can produce entirely novel signaling which may not have been seen on cells which express one receptor type alone [56]. The result is summarized in Figure 7A: four independent signal transduction molecules can be formed: VEGFR1*1 (“VEGFR1 activated by VEGFR1”), VEGFR1*2, VEGFR2*1 and VEGFR2*2, each of which can result in independent, unique signals, which may interact further downstream or lead to feedback on one or all of the other active receptors [57]. VEGFR signaling schema should be further analyzed to improve our understanding of how VEGF transduces its signals on cells with multiple VEGF receptors.
Figure 7. PlGF synergy with VEGF: receptor phosphorylation by VEGF and PlGF.
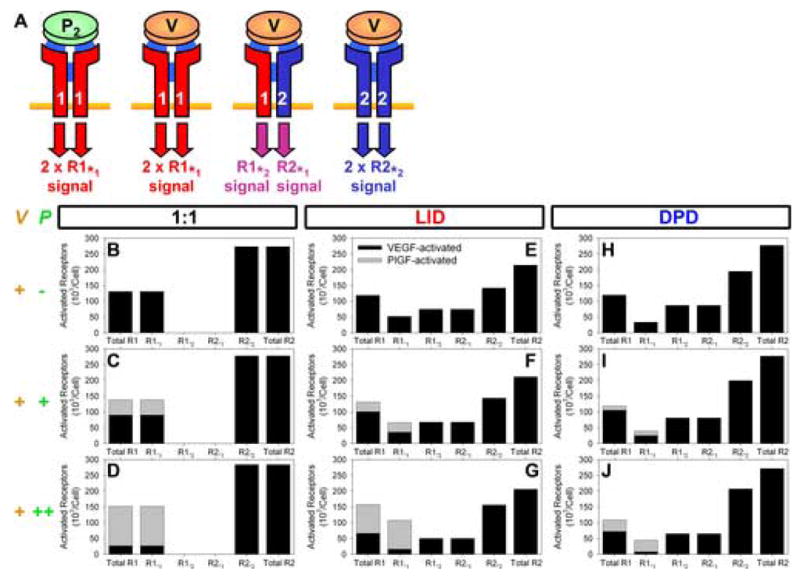
A closer look at the signaling complexes. A, VEGF and PlGF binding results in the activation of both receptor monomers. Each receptor monomer is the substrate for the kinase domain of the other. Changing kinase-substrate pairs can result in distinct signaling. B–J, Phosphorylation of VEGFR1 and VEGFR2 by VEGF, VEGF and PlGF in equal quantities (+) and VEGF with PlGF in 10-fold excess (++). R1*1V, VEGFR1 activated by VEGF-VEGFR1; R1*1P, VEGFR1 activated by PlGF-VEGFR1; R1*2, VEGFR1 activated by VEGF-VEGFR2; R2*2, VEGFR2 activated by VEGF-VEGFR2; R2*1, VEGFR2 activated by VEGF-VEGFR1.
PlGF-VEGF synergy revisited
We noted in previous work [18] that the experimentally observed synergy between VEGF and PlGF in inducing proliferation, survival and migration [58] and VEGFR2 phosphorylation [57] in cells in vitro was unlikely to be the result of PlGF ‘displacing’ VEGF from VEGFR1 to VEGFR2. We revisit those simulations here by using our dimerization models to illuminate the formation of homo- and heterodimeric signaling complexes. Three cases are simulated: VEGF added; equal amounts of VEGF and PlGF added; and VEGF added along with an excess of PlGF. The first two cases were the subject of experimental work previously published [58], and computer simulations based on that work [18]; the third is a hypothetical case included here for comparison. The resulting peak formation of each subtype of activated receptor is shown in Figure 7B–J, with the active VEGFR1 separate from active VEGFR2. The peak total number of active VEGFR2 is almost unchanged by the addition of PlGF for the 1:1 ligand:receptor binding case explored in the previous paper [18], and for both LID and DPD. However, as PlGF is added, the shift from heterodimers to homodimers results in a shift from VEGFR2*1 (VEGFR2 activated by VEGFR1) to VEGFR2*2 (compare Figure 7F,G to Figure 7E; Figure 7I,J to Figure 7H). In contrast, the change in the total number of VEGFR1 activated is significant, especially when dimerization is included explicitly. Looking at the VEGFR1 activation in more detail, we note two changes in the makeup of the activation profile: first, there is a change from VEGF-activated VEGFR1 to PlGF-activated VEGFR1; second, where dimerization is explicitly included, there is also an observable shift from VEGFR1*2 to homodimerically activated VEGFR1*1. It has been noted that PlGF and VEGF induce differential phosphorylation of VEGFR1 [57], but to what extent the observed synergy is a result of the alternate ligand versus the accompanying change from heterodimeric to homodimeric activation remains to be studied. Thus, the presence of VEGFR heterodimers contributes to the complexity of the VEGF signal transduction system, and should be studied further.
DISCUSSION
One of the first questions to ask about the dimerization of VEGF receptors is, what is the mechanism? Experimental evidence exists supporting both ligand-independent and ligand-dependent dimerization of receptors, with activation occurring only when both receptor monomers are bound to one pole of the VEGF molecule. Our simulation results suggest that, depending on the receptor density and other factors, one of the mechanisms may dominate, and confirmation of this would be an important validation of the model presented here. For receptor pre-association rates similar to those predicted theoretically, the pre-dimerization mechanism is predicted to dominate. However, this depends on the receptor density, and the pre-association rate has yet to be measured experimentally.
Measurement of the pre-association rate would be difficult. One possibility is a fluorescence resonance energy transfer (FRET)-based assay to measure the proximity of receptors to each other in the presence and absence of ligand. Unfortunately, such a system could interfere with the receptor autotransactivation. Another method would be to interfere with the pre-association, the change in behavior giving an indication of the absolute value of the rate constant. It would, however, be difficult to change the receptor association behavior without affecting ligand-receptor binding directly, especially without knowledge of the mechanism of pre-dimerization e.g. intracellular scaffolding or mutual association with proteoglycans. One possibility is the cellular expression of a fused receptor dimer; signaling bell-curves that appear in downstream signaling could not therefore be used as evidence of LID.
Our results suggest that while receptor phosphorylation data could provide a clear indication of which mechanism dominated, VEGF binding data may also be used to distinguish the relative strength or prevalence of the different dimerization mechanisms – through the dynamics of competition experiments (Figures4, 6,, S2, S4). However, no time-dependent data for binding or phosphorylation was found in literature. Inclusion of explicit phosphorylation and dephosphorylation steps in the model would aid comparison, but again no data is available for these processes.
Typically, reported quantitative results are at one of two points in the pathway: binding of ligand to the cell; and outcome of the signaling at the cellular level, e.g. survival, migration or DNA synthesis and proliferation. This latter point is so far downstream of the focus of our study (ligand-receptor binding and receptor activation) that the VEGF-response profile of those outcomes may instead be a result of other events along the signal transduction pathways. Experimental investigation of the phosphorylation of VEGF receptors has increased in recent years and a combination of experimental results and models such as this one may lead to insight into the activation of the VEGF signaling pathways, and how the pathway might be up- or down-regulated for therapeutic purposes.
Ligand-induced dimerization exhibits high-dose inhibition of receptor phosphorylation, which has been neither directly reported nor ruled out definitively for VEGF receptors. PDGF receptors, which are related and are also involved in a bivalent ligand-monovalent receptor binding and activation scheme, were reported not to demonstrate high-dose inhibition of receptor activation. To explain the dimerization scheme of those receptors, an unstable intermediate complex of two PDGF ligands and two PDGF receptor monomers was introduced [22]. Here we have presented an alternative scheme, including receptor dimer pre-association, that agrees with some of the experimental observations, including the lack of high-dose inhibition for typical values of receptor pre-association. Our model does not reproduce the positive cooperativity observed in the PDGFR system. While the VEGFR and PDGFR system may behave differently, the decoupling of dephosphorylation from the unbinding of the ligand can be used to produce similar cooperativity (Mac Gabhann and Popel, unpublished observations).
The formation of heterodimers of VEGFR1 and VEGFR2 has been observed [17,56]. It has been shown that the presence of both receptor types results in a calcium release profile in response to VEGF that was significantly different (earlier and biphasic) to profiles on cells expressing either receptor alone [56]. Prostacyclin synthesis has been reported to be under the control of VEGFR heterodimers [17]. These results suggest that the signaling of heterodimers is unique and significant. However, no quantification of the relative abundance of heterodimers and homodimers has been reported. Our simulation suggests that the level of heterodimer formation is significant, at all concentrations of VEGF and for a wide range of VEGFR1 and VEGFR2 expression levels. In fact, for unbalanced expression of VEGFR1 and VEGFR2, it is predicted that homodimer formation by the less-abundant receptor is suppressed. VEGFR1 is typically present in lower quantities than VEGFR2, and the results of VEGFR1 binding VEGF (in which case VEGFR1-VEGFR2 heterodimers would be formed) or PlGF (only VEGFR1 homodimers would be formed) are different: different tyrosine sites are phosphorylated and different signaling occurs downstream, including impact on the activation of VEGFR2 [57]. Thus our simulations may explain the observations that the strength of VEGFR1 phosphorylation in response to VEGF is often measured to be quite low, as the VEGFR2 in the heterodimers may activate VEGFR1 less well than VEGFR1 in homodimers.
It is not clear at this point whether the differences between the VEGF and PlGF-induced VEGFR1 activation is a direct result of the different ligands (e.g. due to conformational changes), or due to the change from VEGFR2- to VEGFR1-mediated activation of VEGFR1. In order to experimentally measure the phosphorylation of heterodimers, it may be possible to generate a VEGFR heterodimer-specific VEGF ligand, for example, combining the strategies mentioned previously to generate single-site binding variants of VEGF [15,53] and the method used to generate receptor-specific variants of VEGF [59]. The resulting VEGF variant would bind only VEGFR1 at one pole and VEGFR2 at the other. Note that this is distinct from the endogeneously produced VEGF-PlGF heterodimers, which should be capable only of binding VEGFR1 homodimers.
We have represented both the activation and deactivation of receptors here as being an instantaneous process, occurring at the moment the second receptor monomer binds to or unbinds from VEGF. We have also represented activation as being a single process, that all the phosphorylation sites on the receptor are activated or deactivated simultaneously. However, since the activation is a result of phosphate addition by a receptor kinase, and since each intracellular site is different, we would expect each site to have different and finite kinetics. Similarly, each site is dephosphorylated by phosphatases (not necessarily the same phosphatase for each site). However, in the absence of more detailed information on the kinetics of each site, our current simulations are useful in understanding the effects of dimerization on activation of a representative site. Inclusion of phosphorylation and dephosphorylation of VEGF-bound VEGFR dimers may result in a non-linear relationship between active receptors and dimerized receptors. However, the relationship should be monotonic and direct, even if non-linear, thus affecting the quantitative results but not necessarily the qualitative shape of the response.
Supplementary Material
Table 1. Parameters for simulations.
The numbers marked * below are within the range of values reported for receptor densities and binding affinities [37–44]. The remaining parameters are calculated or estimated and this is detailed in the text.
| Receptor density | 20,000 receptors/cell | * |
|---|---|---|
| 1:1 Binding affinity (Kd,meas) | 150 pM, 30 pM | * |
| 1:1 On-rate for VEGF (kon,meas) | 8.8 106 M−1 s−1, 4.4 107 M−1 s−1 | * |
| 1:1 Off-rate for VEGF (koff,meas) | 1.32 10−3 s−1 | * |
| On-rate for one pole of VEGF (kon) | 4.4 106 M−1 s−1, 2.2 107 M−1 s−1 | See Text |
| Off-rate for one pole of VEGF (koff) | 2.6 10− 2 s−1 | See Text |
| Coupling rate of bound receptors (kc,VR) (LID) | 2.1 1014 cm2 mol−1 s− 1 | See Text |
| Coupling rate of empty receptors (kc,RR) (DPD) | 1014 cm2 mol− 1 s−1 | See Text |
| Rate of VEGF binding second receptor (kΔ,VR) | 0.949 s−1 | See Text |
| Rate of receptor binding second receptor (kΔ,RR) | 0.446 s−1 | See Text |
| Dissociation rate of empty receptors (kd,RR) | 10− 2 s−1 | See Text |
Acknowledgments
This work was supported by NHLBI RO1 HL079653 and R33 HL87351.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Folkman J. Antiangiogenesis in cancer therapy--endostatin and its mechanisms of action. Exp Cell Res. 2006;312:594–607. doi: 10.1016/j.yexcr.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 2.Maruotti N, Cantatore FP, Crivellato E, Vacca A, Ribatti D. Angiogenesis in rheumatoid arthritis. Histol Histopathol. 2006;21:557–566. doi: 10.14670/HH-21.557. [DOI] [PubMed] [Google Scholar]
- 3.Wilkinson-Berka JL. Vasoactive factors and diabetic retinopathy: vascular endothelial growth factor, cycoloxygenase-2 and nitric oxide. Curr Pharm Des. 2004;10:3331–3348. doi: 10.2174/1381612043383142. [DOI] [PubMed] [Google Scholar]
- 4.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 5.Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res. 2006;312:549–560. doi: 10.1016/j.yexcr.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 6.Potgens AJ, Lubsen NH, van Altena MC, Vermeulen R, Bakker A, Schoenmakers JG, Ruiter DJ, de Waal RM. Covalent dimerization of vascular permeability factor/vascular endothelial growth factor is essential for its biological activity. Evidence from Cys to Ser mutations. J Biol Chem. 1994;269:32879–32885. [PubMed] [Google Scholar]
- 7.Walsh TP, Grant GH. Computer modelling of the receptor-binding domains of VEGF and PIGF. Protein Engineering. 1997;10:389–398. doi: 10.1093/protein/10.4.389. [DOI] [PubMed] [Google Scholar]
- 8.Wiesmann C, Fuh G, Christinger HW, Eigenbrot C, Wells JA, de Vos AM. Crystal structure at 1.7 A resolution of VEGF in complex with domain 2 of the Flt-1 receptor. Cell. 1997;91:695–704. doi: 10.1016/s0092-8674(00)80456-0. [DOI] [PubMed] [Google Scholar]
- 9.Fuh G, Li B, Crowley C, Cunningham B, Wells JA. Requirements for binding and signaling of the kinase domain receptor for vascular endothelial growth factor. J Biol Chem. 1998;273:11197–11204. doi: 10.1074/jbc.273.18.11197. [DOI] [PubMed] [Google Scholar]
- 10.Keyt BA, Nguyen HV, Berleau LT, Duarte CM, Park J, Chen H, Ferrara N. Identification of vascular endothelial growth factor determinants for binding KDR and FLT-1 receptors. Generation of receptor-selective VEGF variants by site-directed mutagenesis. J Biol Chem. 1996;271:5638–5646. doi: 10.1074/jbc.271.10.5638. [DOI] [PubMed] [Google Scholar]
- 11.Tao Q, Backer MV, Backer JM, Terman BI. Kinase insert domain receptor (KDR) extracellular immunoglobulin-like domains 4–7 contain structural features that block receptor dimerization and vascular endothelial growth factor-induced signaling. J Biol Chem. 2001;276:21916–21923. doi: 10.1074/jbc.M100763200. [DOI] [PubMed] [Google Scholar]
- 12.Cunningham SA, Tran TM, Arrate MP, Brock TA. Characterization of vascular endothelial cell growth factor interactions with the kinase insert domain-containing receptor tyrosine kinase. A real time kinetic study. J Biol Chem. 1999;274:18421–18427. doi: 10.1074/jbc.274.26.18421. [DOI] [PubMed] [Google Scholar]
- 13.Shinkai A, Ito M, Anazawa H, Yamaguchi S, Shitara K, Shibuya M. Mapping of the sites involved in ligand association and dissociation at the extracellular domain of the kinase insert domain-containing receptor for vascular endothelial growth factor. J Biol Chem. 1998;273:31283–31288. doi: 10.1074/jbc.273.47.31283. [DOI] [PubMed] [Google Scholar]
- 14.Huang X, Gottstein C, Brekken RA, Thorpe PE. Expression of soluble VEGF receptor 2 and characterization of its binding by surface plasmon resonance. Biochem Biophys Res Commun. 1998;252:643–648. doi: 10.1006/bbrc.1998.9717. [DOI] [PubMed] [Google Scholar]
- 15.Boesen TP, Soni B, Schwartz TW, Halkier T. Single-chain vascular endothelial growth factor variant with antagonist activity. J Biol Chem. 2002;277:40335–40341. doi: 10.1074/jbc.M204107200. [DOI] [PubMed] [Google Scholar]
- 16.von Tiedemann B, Bilitewski U. Characterization of the vascular endothelial growth factor-receptor interaction and determination of the recombinant protein by an optical receptor sensor. Biosens Bioelectron. 2002;17:983–991. doi: 10.1016/s0956-5663(02)00090-8. [DOI] [PubMed] [Google Scholar]
- 17.Neagoe PE, Lemieux C, Sirois MG. Vascular endothelial growth factor (VEGF)-A165-induced prostacyclin synthesis requires the activation of VEGF receptor-1 and -2 heterodimer. J Biol Chem. 2005;280:9904–9912. doi: 10.1074/jbc.M412017200. [DOI] [PubMed] [Google Scholar]
- 18.Mac Gabhann F, Popel AS. Model of competitive binding of vascular endothelial growth factor and placental growth factor to VEGF receptors on endothelial cells. Am J Physiol Heart Circ Physiol. 2004;286:H153–164. doi: 10.1152/ajpheart.00254.2003. [DOI] [PubMed] [Google Scholar]
- 19.Mac Gabhann F, Popel AS. Differential binding of VEGF isoforms to VEGF receptor 2 in the presence of neuropilin-1: a computational model. Am J Physiol Heart Circ Physiol. 2005;288:H2851–2860. doi: 10.1152/ajpheart.01218.2004. [DOI] [PubMed] [Google Scholar]
- 20.Mac Gabhann F, Ji JW, Popel AS. Computational Model of VEGF Spatial Distribution in Muscle and Pro-Angiogenic Cell Therapy. PLoS Comp Biol. 2006 doi: 10.1371/journal.pcbi.0020127. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mac Gabhann F, Yang MT, Popel AS. Monte Carlo simulations of VEGF binding to cell surface receptors in vitro. Biochim Biophys Acta. 2005;1746:95–107. doi: 10.1016/j.bbamcr.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 22.Haugh JM. Mathematical model of human growth hormone (hGH)-stimulated cell proliferation explains the efficacy of hGH variants as receptor agonists or antagonists. Biotechnol Prog. 2004;20:1337–1344. doi: 10.1021/bp0499101. [DOI] [PubMed] [Google Scholar]
- 23.Gopalakrishnan M, Forsten-Williams K, Tauber UC. Ligand-induced coupling versus receptor pre-association: cellular automaton simulations of FGF-2 binding. J Theor Biol. 2004;227:239–251. doi: 10.1016/j.jtbi.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 24.Schlessinger J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell. 2002;110:669–672. doi: 10.1016/s0092-8674(02)00966-2. [DOI] [PubMed] [Google Scholar]
- 25.Klein P, Mattoon D, Lemmon MA, Schlessinger J. A structure-based model for ligand binding and dimerization of EGF receptors. Proc Natl Acad Sci U S A. 2004;101:929–934. doi: 10.1073/pnas.0307285101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schlessinger J. Common and distinct elements in cellular signaling via EGF and FGF receptors. Science. 2004;306:1506–1507. doi: 10.1126/science.1105396. [DOI] [PubMed] [Google Scholar]
- 27.Hendriks BS, Opresko LK, Wiley HS, Lauffenburger D. Quantitative analysis of HER2-mediated effects on HER2 and epidermal growth factor receptor endocytosis: distribution of homo- and heterodimers depends on relative HER2 levels. J Biol Chem. 2003;278:23343–23351. doi: 10.1074/jbc.M300477200. [DOI] [PubMed] [Google Scholar]
- 28.Mayawala K, Vlachos DG, Edwards JS. Computational modeling reveals molecular details of epidermal growth factor binding. BMC Cell Biol. 2005;6:41. doi: 10.1186/1471-2121-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayawala K, Vlachos DG, Edwards JS. Spatial modeling of dimerization reaction dynamics in the plasma membrane: Monte Carlo vs. continuum differential equations. Biophys Chem. 2006;121:194–208. doi: 10.1016/j.bpc.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 30.Blinov ML, Faeder JR, Goldstein B, Hlavacek WS. A network model of early events in epidermal growth factor receptor signaling that accounts for combinatorial complexity. Biosystems. 2006;83:136–151. doi: 10.1016/j.biosystems.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 31.Forsten-Williams K, Chua CC, Nugent MA. The kinetics of FGF-2 binding to heparan sulfate proteoglycans and MAP kinase signaling. J Theor Biol. 2005;233:483–499. doi: 10.1016/j.jtbi.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 32.Chua CC, Rahimi N, Forsten-Williams K, Nugent MA. Heparan sulfate proteoglycans function as receptors for fibroblast growth factor-2 activation of extracellular signal-regulated kinases 1 and 2. Circ Res. 2004;94:316–323. doi: 10.1161/01.RES.0000112965.70691.AC. [DOI] [PubMed] [Google Scholar]
- 33.Filion RJ, Popel AS. A reaction-diffusion model of basic fibroblast growth factor interactions with cell surface receptors. Ann Biomed Eng. 2004;32:645–663. doi: 10.1023/b:abme.0000030231.88326.78. [DOI] [PubMed] [Google Scholar]
- 34.Fredriksson L, Li H, Eriksson U. The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004;15:197–204. doi: 10.1016/j.cytogfr.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 35.Roy H, Bhardwaj S, Yla-Herttuala S. Biology of vascular endothelial growth factors. FEBS Lett. 2006;580:2879–2887. doi: 10.1016/j.febslet.2006.03.087. [DOI] [PubMed] [Google Scholar]
- 36.Park CS, Schneider IC, Haugh JM. Kinetic analysis of platelet-derived growth factor receptor/phosphoinositide 3-kinase/Akt signaling in fibroblasts. J Biol Chem. 2003;278:37064–37072. doi: 10.1074/jbc.M304968200. [DOI] [PubMed] [Google Scholar]
- 37.Bikfalvi A, Sauzeau C, Moukadiri H, Maclouf J, Busso N, Bryckaert M, Plouet J, Tobelem G. Interaction of vasculotropin/vascular endothelial cell growth factor with human umbilical vein endothelial cells: binding, internalization, degradation, and biological effects. J Cell Physiol. 1991;149:50–59. doi: 10.1002/jcp.1041490108. [DOI] [PubMed] [Google Scholar]
- 38.Brogi E, Schatteman G, Wu T, Kim EA, Varticovski L, Keyt B, Isner JM. Hypoxia-induced paracrine regulation of vascular endothelial growth factor receptor expression. J Clin Invest. 1996;97:469–476. doi: 10.1172/JCI118437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li S, Peck-Radosavljevic M, Koller E, Koller F, Kaserer K, Kreil A, Kapiotis S, Hamwi A, Weich HA, Valent P, Angelberger P, Dudczak R, Virgolini I. Characterization of (123)I-vascular endothelial growth factor-binding sites expressed on human tumour cells: possible implication for tumour scintigraphy. Int J Cancer. 2001;91:789–796. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1126>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 40.Myoken Y, Kayada Y, Okamoto T, Kan M, Sato GH, Sato JD. Vascular endothelial cell growth factor (VEGF) produced by A-431 human epidermoid carcinoma cells and identification of VEGF membrane binding sites. Proc Natl Acad Sci U S A. 1991;88:5819–5823. doi: 10.1073/pnas.88.13.5819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thieme H, Aiello LP, Takagi H, Ferrara N, King GL. Comparative analysis of vascular endothelial growth factor receptors on retinal and aortic vascular endothelial cells. Diabetes. 1995;44:98–103. doi: 10.2337/diab.44.1.98. [DOI] [PubMed] [Google Scholar]
- 42.Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem. 1994;269:26988–26995. [PubMed] [Google Scholar]
- 43.Wang D, Lehman RE, Donner DB, Matli MR, Warren RS, Welton ML. Expression and endocytosis of VEGF and its receptors in human colonic vascular endothelial cells. Am J Physiol Gastrointest Liver Physiol. 2002;282:G1088–1096. doi: 10.1152/ajpgi.00250.2001. [DOI] [PubMed] [Google Scholar]
- 44.Whitaker GB, Limberg BJ, Rosenbaum JS. Vascular endothelial growth factor receptor-2 and neuropilin-1 form a receptor complex that is responsible for the differential signaling potency of VEGF(165) and VEGF(121) J Biol Chem. 2001;276:25520–25531. doi: 10.1074/jbc.M102315200. [DOI] [PubMed] [Google Scholar]
- 45.Forsten KE, Lauffenburger DA. The role of low-affinity interleukin-2 receptors in autocrine ligand binding: alternative mechanisms for enhanced binding effect. Mol Immunol. 1994;31:739–751. doi: 10.1016/0161-5890(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 46.Dembo M, Kagey-Sobotka A, Lichtenstein LM, Goldstein B. Kinetic analysis of histamine release due to covalently linked IgE dimers. Mol Immunol. 1982;19:421–434. doi: 10.1016/0161-5890(82)90208-5. [DOI] [PubMed] [Google Scholar]
- 47.Shea LD, Omann GM, Linderman JJ. Calculation of diffusion-limited kinetics for the reactions in collision coupling and receptor cross-linking. Biophys J. 1997;73:2949–2959. doi: 10.1016/S0006-3495(97)78323-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dougher M, Terman BI. Autophosphorylation of KDR in the kinase domain is required for maximal VEGF-stimulated kinase activity and receptor internalization. Oncogene. 1999;18:1619–1627. doi: 10.1038/sj.onc.1202478. [DOI] [PubMed] [Google Scholar]
- 49.Berk DA, Yuan F, Leunig M, Jain RK. Fluorescence photobleaching with spatial Fourier analysis: measurement of diffusion in light-scattering media. Biophys J. 1993;65:2428–2436. doi: 10.1016/S0006-3495(93)81326-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marme D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 1996;87:3336–3343. [PubMed] [Google Scholar]
- 51.Keyt BA, Berleau LT, Nguyen HV, Chen H, Heinsohn H, Vandlen R, Ferrara N. The carboxyl-terminal domain (111–165) of vascular endothelial growth factor is critical for its mitogenic potency. J Biol Chem. 1996;271:7788–7795. doi: 10.1074/jbc.271.13.7788. [DOI] [PubMed] [Google Scholar]
- 52.Shen H, Clauss M, Ryan J, Schmidt AM, Tijburg P, Borden L, Connolly D, Stern D, Kao J. Characterization of vascular permeability factor/vascular endothelial growth factor receptors on mononuclear phagocytes. Blood. 1993;81:2767–2773. [PubMed] [Google Scholar]
- 53.Siemeister G, Schirner M, Reusch P, Barleon B, Marme D, Martiny-Baron G. An antagonistic vascular endothelial growth factor (VEGF) variant inhibits VEGF-stimulated receptor autophosphorylation and proliferation of human endothelial cells. Proc Natl Acad Sci U S A. 1998;95:4625–4629. doi: 10.1073/pnas.95.8.4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kendall RL, Wang G, DiSalvo J, Thomas KA. Specificity of vascular endothelial cell growth factor receptor ligand binding domains. Biochem Biophys Res Commun. 1994;201:326–330. doi: 10.1006/bbrc.1994.1705. [DOI] [PubMed] [Google Scholar]
- 55.Dixelius J, Makinen T, Wirzenius M, Karkkainen MJ, Wernstedt C, Alitalo K, Claesson-Welsh L. Ligand-induced vascular endothelial growth factor receptor-3 (VEGFR-3) heterodimerization with VEGFR-2 in primary lymphatic endothelial cells regulates tyrosine phosphorylation sites. J Biol Chem. 2003;278:40973–40979. doi: 10.1074/jbc.M304499200. [DOI] [PubMed] [Google Scholar]
- 56.Huang K, Andersson C, Roomans GM, Ito N, Claesson-Welsh L. Signaling properties of VEGF receptor-1 and -2 homo- and heterodimers. Int J Biochem Cell Biol. 2001;33:315–324. doi: 10.1016/s1357-2725(01)00019-x. [DOI] [PubMed] [Google Scholar]
- 57.Autiero M, Waltenberger J, Communi D, Kranz A, Moons L, Lambrechts D, Kroll J, Plaisance S, De Mol M, Bono F, Kliche S, Fellbrich G, Ballmer-Hofer K, Maglione D, Mayr-Beyrle U, Dewerchin M, Dombrowski S, Stanimirovic D, Van Hummelen P, Dehio C, Hicklin DJ, Persico G, Herbert JM, Shibuya M, Collen D, Conway EM, Carmeliet P. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat Med. 2003;9:936–943. doi: 10.1038/nm884. [DOI] [PubMed] [Google Scholar]
- 58.Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, Wu Y, Bono F, Devy L, Beck H, Scholz D, Acker T, DiPalma T, Dewerchin M, Noel A, Stalmans I, Barra A, Blacher S, Vandendriessche T, Ponten A, Eriksson U, Plate KH, Foidart JM, Schaper W, Charnock-Jones DS, Hicklin DJ, Herbert JM, Collen D, Persico MG. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med. 2001;7:575–583. doi: 10.1038/87904. [DOI] [PubMed] [Google Scholar]
- 59.Li B, Fuh G, Meng G, Xin X, Gerritsen ME, Cunningham B, de Vos AM. Receptor-selective variants of human vascular endothelial growth factor. Generation and characterization. J Biol Chem. 2000;275:29823–29828. doi: 10.1074/jbc.M002015200. [DOI] [PubMed] [Google Scholar]
- 60.Muller YA, Christinger HW, Keyt BA, de Vos AM. The crystal structure of vascular endothelial growth factor (VEGF) refined to 1.93 A resolution: multiple copy flexibility and receptor binding. Structure. 1997;5:1325–1338. doi: 10.1016/s0969-2126(97)00284-0. [DOI] [PubMed] [Google Scholar]
- 61.Pan B, Fairbrother WJ. 1H, 13C, and 15N resonance assignment of the vascular endothelial growth factor receptor-binding domain in complex with a receptor-blocking peptide. J Biomol NMR. 2002;22:189–190. doi: 10.1023/a:1014297525543. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.