

Abstract
A model alpha-helical peptide encapsulated in a reverse micelle is used to study the structure and dynamics of proteins under constrained environments that mimic the membrane-water environment in cells. Molecular dynamics simulations of the self assembly of systems composed of a peptide, sodium bis(2-ethylhexyl) sulfosuccinate (AOT), water, and isooctane show that the peptide prefers to be located at the water/AOT headgroups interface. We explore the effect of the AOT headgroup charge and the peptide charge and find that the peptides migrate to the interface in all cases. These results show that the peptides prefer the constrained hydration environment of the AOT headgroups. The driving force for this configuration is the gain in entropy by released water molecules that otherwise would solvate the protein and surfactant headgroups
Main Text
Reverse micelles (RM) formed with sodium bis(2-ethylhexyl) sulfosuccinate (AOT) surfactant, water and nonpolar solvents have been widely used to encapsulate proteins in nonpolar solvents (1,2). AOT RM can serve as a model for studying the effect of crowding and protein confinement in highly charged polar environments. The AOT surfactant does not require cosurfactants and simplifies the formation of RM. A wide variety of proteins are able to partition into RM. Recently, protein-RM systems have been proposed to study the structure and dynamics of large biomolecular systems, taking advantage of the fast tumbling time of protein-containing RM dissolved in low viscosity nonpolar solvents like CO2 or isooctane (3–5). Proof of principle studies on ubiquitin, α-chymotrypsin, and flavodoxin have shown that the protein structure does not change significantly, although there are some changes in the dynamics (3–5). Early studies of proteins in RM have shown that the proteins are active and that the enzymatic activity is largely maintained (6). The confinement of proteins in the small volumes of RM also forces the folding of proteins of marginal stability (7).
The highly electrostricted environment in the water phase of RM is known to change the thermodynamics and dynamics of water (8). The properties of AOT RM depend on the molecular ratio w0 = [water]/[AOT]. At low w0 (<8), the water inside the RM has lower activity than in bulk (9). The sequence specific hydration of peptides has been implicated in the stability of α-helices (10–12), where water competes with the α-helix hydrogen bonds. Therefore, changing the activity near the protein may affect the peptide stability. Infrared spectroscopic measurements of Ala-based peptides (13,14) and an amphipatic peptide (15) in conjunction with a local infrared marker have been used to report the hydration environment around helical peptides in AOT RM with different water contents. These experiments showed that the stability of α-helical peptides can be tuned with w0, where the helical content at w0 = 6 is higher than in bulk. Also, the peptide backbone and hydrophobic side chains were mostly dehydrated at w0 = 6 and that the backbone becomes partially hydrated at w0 = 20. The lysine side chains were shown to point toward AOT headgroups.
The behavior of helical peptides in AOT RM can be compared and contrasted with the behavior inside carbon nanotubes (CN) (16–20). The stabilization of α-helical peptides in CN depend on sequence, confinement volume, and the hydrophobicity and water activity in the interior of the CN (16–19). In contrast to CN, the RM presents a highly electrostricted environment to the biomolecule.
To study the dynamics, hydration, and motions of the helical peptides in the highly charged and crowded environment of RM, we conducted a series of extensive molecular dynamics simulations of the structure and dynamics of AOT/water RM in isooctane, with and without peptides. The simulations were started from a random distribution of the system components and have been simulated for 200 ns. Two water/surfactant ratios are studied, w0 = 6 and w0 = 11. The numbers of AOT and water molecules were chosen to be two times larger than the optimal number in a single RM (for a given w0). This way, a self-assembled system will contain multiple RM. The w0 = 11 system consists of 286 AOT, 282 sodium ions (Na), 3,249 water molecules, 16,710 isooctane, and one peptide, for a total of 162,595 atoms. We simulated the peptide AK4 = NH3+-YG(AKAAA)4AG-COO−, similar to the peptide studied by Mukherjee et al. (13,14). The w0 = 6 system contains 128 AOT, 124 Na, 768 water, 3,838 isooctane, and two octapeptides, for a total of 41,666 atoms. The peptide sequence is NH3+-AK(A)4KA-COO−. We used the AOT parameters described by Abel et al. (21) and Amber ff99SB (22) force field for the peptide, and the united atom force field of Siepman (23) for isooctane. Simulations were conducted with NAMD (24). We used particle mesh Ewald, with a grid size of 180 for all directions, and unit box size (17.2, 17.2, 17.4) nm. The systems were simulated at T = 300 K and P = 1 atm. During the simulations, we observed a rapid aggregation of AOT molecules forming a large number of small RM. The RM fuse into larger RM and in some instances fission into smaller RM. The RM fusion process is diffusion limited. One crucial step for two RM to fuse together is the formation of a water channel connecting them. We did not observe fusion of more than two RM. The number of RM starts changing slowly after 30 ns for w0 = 6 and 150 ns for w0 = 11. For w0 = 6, we conducted multiple shorter (20 ns) simulations of the initial steps to ensure reproducibility of the results. Fig. 1 B shows the time evolution of the number of RM along the simulation trajectory. The reduction in the number of RM decays exponentially, with faster equilibration time for w0 = 6 than for w0 = 11. Fig. 1 A shows final configuration of the w0 = 11 system. Eight RM were formed, with different sizes, and the AK4 peptide was encapsulated into the largest RM. The RM containing the peptide has a slightly larger w0 (w0 = 13.98 ± 0.04) than other RM in the system. The AK4 peptide stays α-helical through the 200 ns simulation. Fig. 2 shows the position of the AK4 peptide at the RM water/isooctane interface. Some nonpolar Ala side chains make contact with the isooctane solution. Two Lys side chains are well solvated, whereas two other Lys chains pointing toward the RM exterior make extensive contacts with the negatively charged AOT headgroup. Na ions are coordinated to the AOT headgroups.
Figure 1.
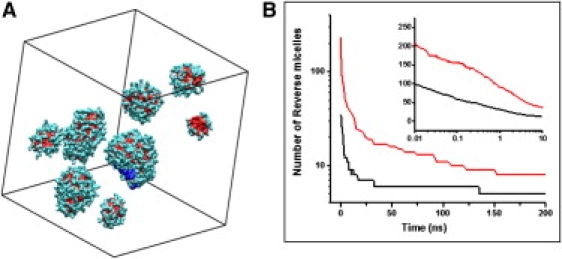
(A) Image of the final configuration showing RM of the w0 = 11 simulation. The peptide encapsulated in the largest RM is colored blue to ease the view. (B) Number of RM along the simulation trajectory. The inset shows the number of RM versus log(t) for short times, black for w0 = 6 and red for w0 = 11.
Figure 2.

Atomic representation of the position of the AK4 peptide relative to the RM. (A) View from the outside of the RM. One side of the α-helix is exposed to the hydrocarbon solvent. (B) View from the inside the RM. The charged side chains point toward the RM interior and make contact with the AOT headgroups.
To gain an understanding of which factors affect the location of the peptide within the RM, we performed simulations of the peptides NH3+-AK(A)4KA-COO− and NH3+-A8-COO− in a w0 = 12 AOT RM and in a RM formed with neutral (but polar) AOT. All four simulations showed that the peptides bind to the headgroup interface, away from the “free water” region of the RM. This suggests that the positioning of the peptides may be driven by the low entropy of the water near the headgroups. By sharing the solvation shells of the peptides with the ions and headgroups, water that would otherwise be bound to the peptide is freed into the water phase, where it will have larger translational and rotational entropy.
Fig. 3 shows the proximity distribution function of AOT headgroups and tails, waters, and ions around the α-helical peptide, and the coordination of water around the backbone carbonyl oxygen for the α-helix in the RM and in bulk water. The proximity distribution function clearly shows that the peptide interacts largely with both the AOT tails and the charged headgroups. Although water molecules in the RM interact with the backbone, the interactions are much less than in bulk. Fig. 3 B shows a drastic reduction in the number of water coordinated to the backbone. This picture is consistent with the preferential placement of the peptide at the RM interface and not at the center, where most of the free waters are. Our calculations are in agreement with the measurements by Mukherjee et al. (13,14) and provide an atomic picture of the arrangement of the peptide on the RM water/headgroup interface.
Figure 3.
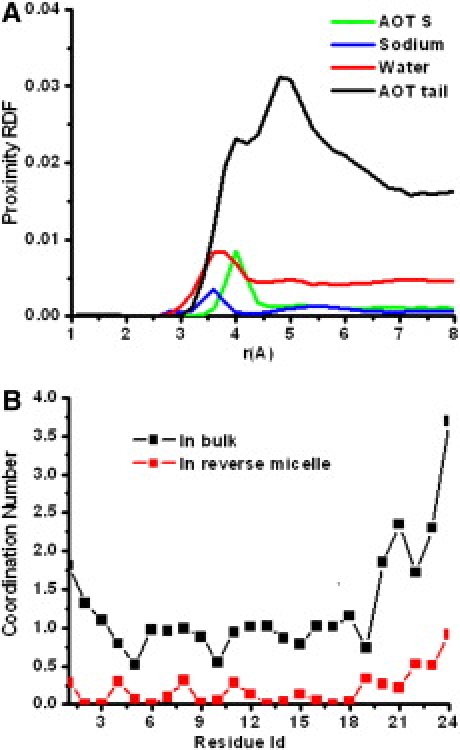
Coordination of waters, ions, AOT headgroups, and AOT tails around the α-helical peptide. (A) Unnormalized proximity radial distribution functions around the α-helix. (B) Water coordination number around each peptide carbonyl oxygen.
The overall picture gained from these simulations is that the α-helical peptide prefers to reside at the AOT RM interface such that the loss in entropy due to the coordination of water around the AOT headgroups is minimal. The peptide and the headgroups will share water molecules in their coordination shells. The water-enriched layer formed around the AOT headgroups reduces the water activity in such a way that it promotes binding of the peptide to the headgroups (19).
In conclusion, we have conducted 200 ns molecular dynamics simulations of self assembly of systems with compositions of Na AOT, water, isooctane, and Ala-based α-helical peptides. The α-helical peptides encapsulated into the RM are located at the interface of the RM, where the entropy loss of the water molecules is reduced. The peptide is more dehydrated compared to the peptide in bulk water. Charged Lys side chains interact strongly with the surfactant headgroups. Given the heterogeneous amino acid composition of water-soluble proteins, their interaction with AOT RM may be different from what we observe in simple α-helical peptides (25,26). Molecular dynamics studies of other proteins in AOT RM will provide insights into the structure and dynamics of proteins in highly polar constrained environments.
Acknowledgments
This work was supported primarily by the Nanoscale Science and Engineering Initiative of the National Science Foundation under National Science Foundation Award No. DMR-0642573. We gratefully acknowledge a SUR grant by IBM that provided us with a BlueGene/L computer used in the calculations presented here.
Reference and Footnotes
- 1.Goklen K.E., Hatton T.A. Protein extraction using reverse micelles. Biotechnol. Prog. 1985;1:69–74. doi: 10.1002/btpr.5420010113. [DOI] [PubMed] [Google Scholar]
- 2.Goklen K.E., Hatton T.A. Liquid-liquid extraction of low molecular weight proteins by selective solubilization in reverse micelles. Separ. Sci. Technol. 1987;22:831–841. [Google Scholar]
- 3.Wand A.J., Ehrhardt M.R., Flynn P.F. High-resolution NMR of encapsulated proteins dissolved in low-viscosity fluids. Proc. Natl. Acad. Sci. USA. 1998;95:15299–15302. doi: 10.1073/pnas.95.26.15299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lefebvre B.G., Liu W., Peterson R.W., Valentine K.G., Wand A.J. NMR spectroscopy of proteins encapsulated in a positively charged surfactant. J. Magn. Reson. 2005;175:158–162. doi: 10.1016/j.jmr.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 5.Pitre F., Pileni M.P. Structural study in AOT reverse micelle containing native and modified α-chymotrypsin. Prog. Colloid Polym. Sci. 1993;93:333–335. [Google Scholar]
- 6.Creagh A.L., Prausnitz J.M., Blanch H.W. Structural and catalytic properties of enzymes in reverse micelles. Enzyme Microb. Technol. 1993;15:383–392. doi: 10.1016/0141-0229(93)90124-k. [DOI] [PubMed] [Google Scholar]
- 7.Peterson R.W., Anbalagan K., Tommos C., Wand A.J. Forced folding and structural analysis of metastable proteins. J. Am. Chem. Soc. 2004;126:9498–9499. doi: 10.1021/ja047900q. [DOI] [PubMed] [Google Scholar]
- 8.Faeder J., Ladanyi B.M. Molecular dynamics simulations of the interior of aqueous reverse micelles. J. Phys. Chem. B. 2000;104:1033–1046. [Google Scholar]
- 9.Luisi P.L., Giomini M., Pileni M.P., Robinson B.H. Reverse micelles as hosts for proteins and small molecules. Biochim. Biophys. Acta. 1988;947:209–246. doi: 10.1016/0304-4157(88)90025-1. [DOI] [PubMed] [Google Scholar]
- 10.Vila J.A., Ripoll D.R., Scheraga H.A. Physical reasons for the unusual α-helix stabilization afforded by charged or neutral polar residues in alanine-rich peptides. Proc. Natl. Acad. Sci. USA. 2000;97:13075–13079. doi: 10.1073/pnas.240455797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia A.E., Sanbonmatsu K.Y. α-Helical stabilization by side chain shielding of backbone hydrogen bonds. Proc. Natl. Acad. Sci. USA. 2002;99:2782–2787. doi: 10.1073/pnas.042496899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silva R.A.G.D., Nguyen J.Y., Decatur S.M. Probing the effect of side chains on the conformation and stability of helical peptides via isotope-edited infrared spectroscopy. Biochemistry. 2002;41:15296–15303. doi: 10.1021/bi026507z. [DOI] [PubMed] [Google Scholar]
- 13.Mukherjee S., Chowdhury P., Gai F. Tuning the cooperativity of the helix-coil transition by aqueous reverse micelles. J. Phys. Chem. B. 2006;110:11615–11619. doi: 10.1021/jp062362k. [DOI] [PubMed] [Google Scholar]
- 14.Mukherjee S., Chowdhury P., Gai F. Infrared study of the effect of hydration on the amide I band and aggregation properties of helical peptides. J. Phys. Chem. B. 2007;111:4596–4602. doi: 10.1021/jp0689060. [DOI] [PubMed] [Google Scholar]
- 15.Mukherjee S., Chowdhury P., DeGrado W.F., Gai F. Site-specific hydration status of an amphipathic peptide in AOT reverse micelles. Langmuir. 2007;23:11174–11179. doi: 10.1021/la701686g. [DOI] [PubMed] [Google Scholar]
- 16.O'Brien E.P., Stan G., Thirumalai D., Brooks B.R. Factors governing helix formation in peptides confined to carbon nanotubes. Nano Lett. 2008;8:3702–3708. doi: 10.1021/nl8019328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaitheeswaran S., Thirumalai D. Interactions between amino acid side chains in cylindrical hydrophobic nanopores with applications to peptide stability. Proc. Natl. Acad. Sci. USA. 2008;105:17636–17641. doi: 10.1073/pnas.0803990105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sorin E.J., Pande V.S. Nanotube confinement denatures protein helices. J. Am. Chem. Soc. 2006;128:6316–6317. doi: 10.1021/ja060917j. [DOI] [PubMed] [Google Scholar]
- 19.Zhou H.-X. Helix formation inside a nanotube: possible influence of backbone-water hydrogen bonding by the confining surface through modulation of water activity. J. Chem. Phys. 2007;127:245101–245104. doi: 10.1063/1.2812282. [DOI] [PubMed] [Google Scholar]
- 20.Abel S., Waks M., Urbach W., Marchi M. Structure, stability, and hydration of a polypeptide in AOT reverse micelles. J. Am. Chem. Soc. 2006;128:382–383. doi: 10.1021/ja053043u. [DOI] [PubMed] [Google Scholar]
- 21.Abel S., Sterpone F., Bandyopadhyay S., Marchi M. Molecular modeling and simulations of AOT-water reverse micelles in isooctane. Structural and dynamic properties. J. Phys. Chem. B. 2004;108:19458–19466. [Google Scholar]
- 22.Hornak V., Abel R., Okur A., Strockbine B., Roitberg A. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin M.G., Siepmann J.I. Novel configurational-bias Monte Carlo method for branched molecules. Transferable potentials for phase equilibria. 2. United-atom description of branched alkanes. J. Phys. Chem. B. 1999;103:4508–4517. [Google Scholar]
- 24.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Horn W.D., Simorellis A.K., Flynn P.F. Low-temperature studies of encapsulated proteins. J. Am. Chem. Soc. 2005;127:13553–13560. doi: 10.1021/ja052805i. [DOI] [PubMed] [Google Scholar]
- 26.Pometun M.S., Peterson R.W., Babu C.R., Wand A.J. Cold denaturation of encapsulated ubiquitin. J. Am. Chem. Soc. 2006;128:10652–10653. doi: 10.1021/ja0628654. [DOI] [PMC free article] [PubMed] [Google Scholar]