

Abstract
The affinity and specificity of drugs with human serum albumin (HSA) are crucial factors influencing the bioactivity of drugs. To gain insight into the carrier function of HSA, the binding of levamlodipine with HSA has been investigated as a model system by a combined experimental and theoretical/computational approach. The fluorescence properties of HSA and the binding parameters of levamlodipine indicate that the binding is characterized by one binding site with static quenching mechanism, which is related to the energy transfer. As indicated by the thermodynamic analysis, hydrophobic interaction is the predominant force in levamlodipine-HSA complex, which is in agreement with the computational results. And the hydrogen bonds can be confirmed by computational approach between levamlodipine and HSA. Compared to predicted binding energies and binding energy spectra at seven sites on HSA, levamlodipine binding HSA at site I has a high affinity regime and the highest specificity characterized by the largest intrinsic specificity ratio (ISR). The binding characteristics at site I guarantee that drugs can be carried and released from HSA to carry out their specific bioactivity. Our concept and quantification of specificity is general and can be applied to other drug-target binding as well as molecular recognition of peptide-protein, protein-protein, and protein-DNA interactions.
Introduction
Biological function at the molecular level is realized by the interactions and recognitions among bio-molecules. There are two crucial factors determining the bio-molecular recognition and binding process. One is the affinity that measures the stability of associating two molecules together. The other is the specificity of binding of one molecule with a specific one but not with others (discriminating against others) (1). Conventionally, high affinity has been used as the criterion for the stability and virtual screening of drug targets in the pharmaceutical industry. However, high affinity fails to always guarantee high specificity (2), yet high specificity is crucial for molecular recognition and practice of drug design.
The conventional way of defining specificity is the capability of discrimination of a specific ligand against different receptors. To prove the specificity of a ligand to a receptor, one has to search all the related receptors (Fig. 1 a). This is not always practical. For a specific ligand binding with different receptors, we are probing interactions between the ligand and different receptors through the change of sequences of the receptors (1,3,4). During the process of a specific ligand binding with a specific receptor, different intermediate binding modes (states) have different structures and binding energies with different set of contact interactions between the ligand and the receptor (Fig. 1 b). By exploring different structures of various binding modes, the binding probes different interactions between the ligand and the receptor. If the receptor is large and there are sufficient number of contact interactions between the ligand and the receptor, probing interactions through different structures and sequences should be statistically equivalent (because exploring different binding structures mean exploring different spatial contacts and different contacts explore different sequences). We can therefore explore the specificity by looking at the different intermediate binding modes between a ligand and a receptor, which is much easier to carry out, rather than looking at the whole universe of the receptors to test the specificity of a ligand, which is essentially impractical.
Figure 1.
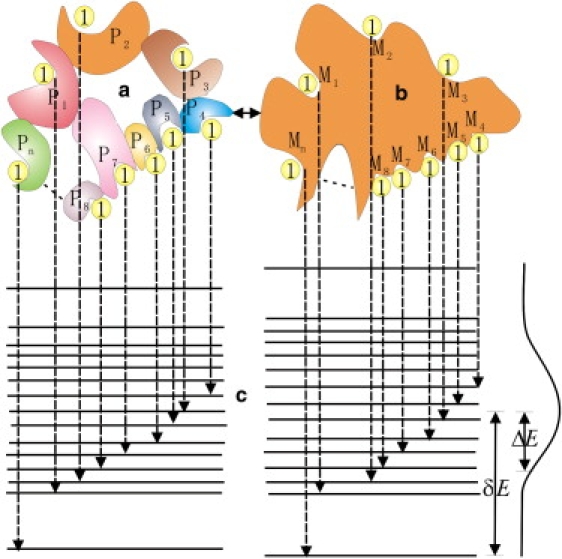
Illustration of the equivalent concept of the conventional specificity to intrinsic specificity ratio (ISR) as well as the corresponding energy spectrum. (a) A specific ligand binding to different receptors, P1–Pn represent the different proteins with different binding sites. (b) Different binding modes of a specific ligand to a specific receptor, M1–Mn represent the different modes with different set of contact interactions. (c) Similar energy spectrum and the Gaussian distribution, δE represents the energy gap between the native or lowest energy state and the average binding energy state, and ΔE represents the energy variance of the nonnative states.
The collections of the energies associated with different intermediate binding modes of a ligand with a receptor form a binding energy spectrum. The ground state with lowest energy can be represented as “native” state whereas the population of the rest of the other weakly intermediate bound states (binding modes) is expected to follow a Gaussian distribution due to the large number theorem (Fig. 1 c). The two important energy terms are δE, which represents the energy gap between the native or lowest energy state and the average binding energy state, and ΔE, which defines the energy variance of the “nonnative” states. The ratio of the two energy terms δE/ΔE is defined as the intrinsic specificity ratio (ISR) (1,4). (Intrinsic specificity here means the capability of discriminating native binding state (mode) from different binding states (modes) for a ligand binding with a receptor.) Because the population follows Boltzmann distribution P ∼ exp[−E/kT], a large ISR indicates a high level of discrimination of the minimum energy state (native binding mode) from the weaker binding states (binding modes) for a particular ligand-receptor binding complex (Fig. 1 b). From the discussion on the equivalence of Fig. 1, a and b, the ISR can serve as a quantitative measure for specificity.
Human serum albumin (HSA) as a main carrier protein is the most abundant in serum and binds many compounds with high affinity (5). Crystal structure analyses have shown that HSA has binding sites for compounds at site I and site II in subdomains IIA and IIIA, respectively. And the sole tryptophan residue (Trp214) of HSA is in subdomain II A (6). Almost all hydrophobic amino acids form hydrophobic cavities that play an important role on transportation of drugs to their targets. The previous studies on interactions between HSA and drugs have provided information of its structural features, which were based mainly on the size and polarity rather than affinity and specificity of drugs (7,8). No investigations were made to clarify why site I serves as a primary binding site for mostly small molecules and why HSA does possess the carrier function. Thus, the study on the binding characteristics of drugs to HSA is of importance in drug research field.
Levamlodipine, S-(−)-2-[(2-aminoethoxy)methyl]-4-(2-chlorophenyl)-3-ethoxy-carbonyl-5-methoxycarbonyl-6-methyl-1,4-dihydropyridine (Fig. 2), is used widely in the treatment of hypertension and angina (9). So far, very little knowledge is available about the mode of interaction of levamlodipine with HSA at molecular level. The occurrence and nature of levamlodipine binding HSA described in this study have been investigated as a model system to identify the biological function of HSA based on two factors, affinity and specificity, by a combined experimental and computational approach. Characterizations of affinity and specificity may advance our understanding of this unique carrier protein. The expected output should ultimately help design levamlodipine derivatives with altered HSA-binding properties.
Figure 2.
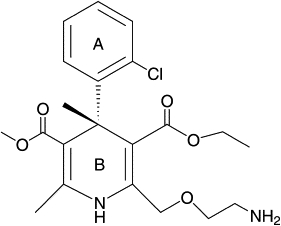
Chemical structure of levamlodipine.
Materials and Methods
Reagents
HSA was purchased from Sigma Chemical Co. (St. Louis, MO). Levamlodipine was obtained from the National Institute for Control of Pharmaceutical and Products, China. All chemicals were of analytical reagent grade and MilliQ water was used throughout.
Ultraviolet-vis measurements
Ultraviolet (UV)-vis absorption spectra were recorded on a double beam Cary 500 Scan UV-vis/NIR spectrophotometer (Varian, Japan) at 298 K in the range 250–450 nm using a quartz cell with 1.0 cm pathlength.
Fluorescence quenching measurements
Fluorescence measurements were carried out on a laser-induced fluorescence spectrofluorimeter equipped with a 150 W xenon lamp and a thermostat bath (PTI, London Ontario, Canada). The excitation wavelength was 290 nm, and the emission spectra were recorded with maximum observed at 340 nm. Each titration was prepared in a 2 mL quartz cuvette and incubated in dark for 1.0 min. The bandwidth for measuring emission was 5 nm. The temperature of sample was kept by recycled water throughout experiment.
Theoretical calculations
The possible quenching mechanism between levamlodipine and HSA at 298 K and 309 K can be analyzed by the Stern-Volmer equation (10):
| (1) |
here F and F0 are the steady state fluorescence intensities in the presence and absence of quencher, respectively; Kq, τ0, and [Q] are the quenching rate constant, the average lifetime of the molecule without quencher, and the concentration of quencher, respectively. Taking fluorescence lifetime of Trp in HSA at ∼10−8 s (11), an approximate quenching rate constant (Kq, M−1 s−1) can be obtained by the slope of Stern-Volmer curves.
The binding constant KA (M−1) and the number of binding sites n can be elicited based on the following two equations: (10):
| (2) |
| (3) |
The thermodynamic parameters were evaluated according to the equation (7):
| (4) |
Here, R is the gas constant. The free energy change (ΔG) can be estimated based on the following typical thermodynamics relationship (7):
| (5) |
The distance between Trp214 and the bound small molecule can be calculated according to the Förster theory (12). The efficiency of energy transfer, E, is calculated using the following equation:
| (6) |
Here, r is the distance between donor and acceptor; R0 is the critical distance when the transfer efficiency is 50%, which can be calculated by
| (7) |
where K2 is the spatial orientation factor of the dipole; N is the refractive index of the medium; Ф is the fluorescence quantum yield of the donor; and J is the overlap integral of the fluorescence emission spectrum of the donor and the absorption spectrum of the acceptor. In this case, K2 = 2/3, N = 1.36, and Ф = 0.15 (13). J is given by the following equation:
| (8) |
where F(λ) is the fluorescence intensity of the fluorescent donor of wavelength λ; and ɛ(λ) is the molar absorption coefficient of the acceptor at wavelength λ.
Molecular modeling and quantitative measure of specificity
Docking calculations were carried out on the system of levamlodipine-HSA with the Autodock4.0 package (14), which possesses a free-energy scoring function based on a linear regression analysis. Two parameters, the AMBER force field and a larger set of diverse protein-ligand complexes with known inhibition constants, were used in Autodock 4.0. The standard error (SE) is ∼2.5 kcal/mol, which is enough to discriminate between leads with milli-, micro-, and nanomolar inhibition constants. The structure of HSA was obtained from the protein data bank (PDB 1h9z (15)), which has been further refined and optimized using pdb2pqr1.3 software package (Molecular Networks GmbH, Germany) to add the missing side chains of some residues and to remove clashes. The 3D structure of levamlodipine was built by using the web-tools corina3D. Site I was used as the potential site for target docking simulations. Autodock tools were used to prepare the protein and the ligand. All hydrogen atoms were added; and simultaneously, gasteiger charges were assigned to the protein and the ligand. The nonpolar hydrogen atoms were merged for the protein and the ligand. The part of myristic acid retained as a “plug” in the original position was prepared by using the Chimera package (Molecular Networks GmbH), adding the hydrogen atoms and the AM1BCC charges (17).
The Autogrid, 60 × 60 × 60 grid size, with a spacing of 0.375 Å centered on the special position in the potential binding site was prepared by using autodock tools. Docking was carried out by using the empirical free energy function and the Lamarckian genetic algorithm. The molecular modeling was carried out based on the following parameters: the energy evaluations of 100,000, the maximum number of 27,000 iterations for an initial population of 100 randomly placed individuals with a mutation rate of 0.02, a crossover rate of 0.80, and an elitism value of 1.0. The other parameters were defaults. The number of docking runs was 10,000. Evaluation of the results was carried out by sorting the binding energy predicted by docking conformations. A cluster of analysis based on the root mean-square deviation value that is <2.0 Å was carried out subsequently. Next, we have investigated the binding modes at the other six binding sites, considering levamlodipine as the common ligand. All preparations and parameters were consistent with the contents described as above. For each binding site, we collected different binding modes with different binding free energy. We can calculate the affinity, the energy gap between the lowest binding energy state and average binding energy state as well as the variance of the free energies of different binding modes. In this way, ISR as the ratio of gap versus square root of variances of binding energies can be obtained, and the SE is ∼0.1.
The resulting seven protein-ligand complexes were subjected to energy minimization and molecular dynamics (MD). The initial structures were obtained after levamlodipine docking into the structure of HSA as described above. These structures were prepared using the leap module of Amber 8 (18). The Amber 8 suite of programs together with AMBER FF99 force field was used to carry out all MD simulations. Each system was solvated in a truncated octahedron TIP3P water box (19). Counterions were added to maintain electroneutrality of the whole system. Minimizations were carried out in three steps, using the AMBER force field supplemented with parameters for the nonnatural amino acids. First, holding the protein and ligand fixed, the solvent molecules were relaxed and optimized, followed by the side chain atoms and the finally whole protein-ligand complex including the backbone. The MD was carried out with respect to each system first, applying harmonic restraints with force constants of 10 kcal/mol/Å2 to all solute atoms, by heating from 0 to 300 K over 20 ps in the canonical ensemble, followed by equilibrating to adjust the solvent density under 1 atm pressure over 50 ps in the isothermal-isobaric ensemble simulation with force constants of 0.5 kcal/mol/Å2 to the atoms within 6.0 Å from the ligand. The harmonic restraints were then reduced to zero with 100 ps isothermal-isobaric ensemble simulations for the stability of the whole system. The resulting structures were the starting points of the production MD simulations. A 1-ns production run was carried out with the resultant snapshots collected every 1 ps. For all simulations, 2 fs time step and 10 Å nonbonded cutoff were used. The particle mesh Ewald method (20) was used to treat long-range electrostatics, and bond lengths involving bonds to hydrogen atoms were constrained by SHAKE (21). The other parameters were defaults.
Next, we used the trajectories obtained from the above MD simulations, which was started with the seven complexes to carry out protein-ligand binding free energy (ΔG) calculations using the solvated interaction energies method (22). The binding free energies for the protein-ligand complexes were estimated using the Sietraj program. Sietraj is an alternative to the MM-PBSA software provided by the AMBER distribution. It calculates ΔG for snapshot structures from the MD simulation with a rigid infinite separation of the protein and ligand (22). ΔG is the sum of the intermolecular van der Waals and Coulomb interactions plus the change in reaction field energy (determined by solving the Poisson-Boltzmann equation) and nonpolar salvation energy (proportional to the solvent-accessible surface area) (22). ΔG is then scaled by an empirically determined factor, α, obtained by fitting to a training set of 99 protein-ligand complexes. The scaling can be considered a crude treatment of entropy-enthalpy compensation (22). The SE is ∼0.05.
Results
Characterization of interaction between levamlodipine and HSA
UV-vis absorption measurement can be used to explore the complex formation (23). The maximum absorption peak of levamlodipine seems to be 358 nm in ethyl alcohol; however, it is evident that the UV absorption spectrum of levamlodipine shows a slightly bathochromic shift to 361 nm and an appreciable enhancement of absorption intensity in levamlodipine-HSA system (Fig. 3). In this case, the UV absorption intensity of HSA at 278 nm increases with the addition of levamlodipine, and simultaneously an appreciable absorbance at 259 nm is observed too. Accordingly, the absorption intensity increasing of HSA in levamlodipine-HSA system can be interpreted as the result that the binding of levamlodipine to HSA alters the microenvironment around HSA but not secondary structure (24). In conclusion, the observation has provided several lines of evidence in support of the presence of the interaction between levamlodipine and HSA, and that levamlodipine accommodates to its binding site on HSA.
Figure 3.
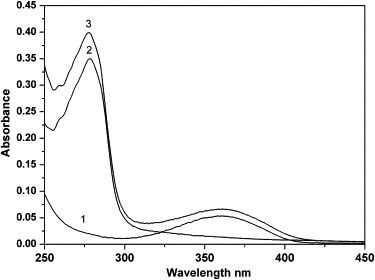
UV-vis absorption spectra of HSA in the presence of levamlodipine. Curve 1: [levamlodipine] = 1.0 × 10−5 M; curve 2: [HSA] = 1.0 × 10−5 M; and curve 3: levamlodipine-HSA complex, [levamlodipine] = [HSA] = 1.0 × 10−5 M. T = 298 K, pH = 7.4.
HSA shows a characteristic emission maximum at 340 nm when it is excited at 290 nm, and the single Trp214 residue can explain the phenomenon (6). The intrinsic fluorescence of HSA is very sensitive to its microenvironment, namely when local surrounding of HSA is slightly altered its intrinsic fluorescence weakens. Thus, the characteristics reflecting local environmental changes can attribute to HSA conformation change, bio-molecule binding and denaturation, etc. The fluorescence quenching of HSA induced by levamlodipine is displayed in Fig. 4. Obviously, with increasing of the concentration of levamlodipine the fluorescence intensity of HSA decreases. An appreciable blue shift from 340 nm to 332 nm is observed for the maximum emission wavelength of HSA with progressive titrating levamlodipine solution to HSA solution. The fluorescence emission wavelength can be used to estimate the binding mode, because the wavelength is strongly dependent on the microenvironment especially the hydrophobicity around the protein. The blue shift of maximum emission wavelength indicates the chromophore of protein, Trp214, is placed in a more hydrophobic environment after the addition of levamlodipine (25). Under the experimental temperatures and pH, the conformation of HSA is not changed (24), and so the denaturation can leave out of account. Moreover, the 50 nm of Δλ, the value of difference between excitation and emission wavelengths, is close to 60 nm that is a characteristic Δλ of the synchronous fluorescence of Trp residue, indicating that levamlodipine is closed to Trp residue (23). Thus, the strong fluorescence quenching indicates that levamlodipine binds to HSA and the binding site is adjacent to the sole tryptophan residue (25). However, it is worth mentioning that there are probably other binding modes between them but most of them are so unstable that they disappear very quickly and the fluorescence quenching of HSA carries through the dynamic equilibrium.
Figure 4.
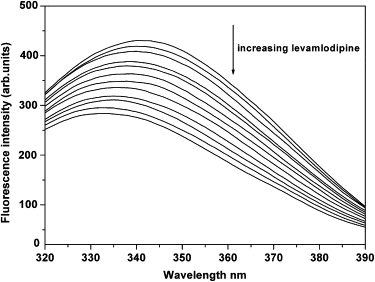
Fluorescence quenching spectrum of HSA in the presence of levamlodipine. Concentration of HSA was 2.0 × 10−5 M whereas the corresponding concentrations of levamlodipine were 0, 4, 12, 20, 24, 32, 40, 50, 60, 70, 80, and 90 × 10−6 M, respectively, as the arrow indicates. T = 298 K; λex = 290 nm; and λem = 340 nm.
Levamlodipine has no fluorescence by fixing the excitation wavelength at 290 nm, thus, the direct excitation of levamlodipine is avoided at this wavelength. With progressive increasing concentration of levamlodipine to HSA solution, the fluorescence intensity of HSA decreases with a regularly concomitant increase in the levamlodipine emission (Fig. 5 a). To exclude a false concentration factor and further to confirm the observed spectral behavior, various concentrations of levamlodipine solution added to PBS buffer without HSA were used as control samples to monitor spectral changes. It can be noted that the emission of levamlodipine in the absence of HSA differs from that in the presence of HSA (Fig. 5 b), which shows that the increase of levamlodipine emission excited at 374 nm predominantly originates from the completely HSA-bound levamlodipine. The large enhancement of the fluorescence emission can be attributed to the reduced polarity of the environment due to less polar of the hydrophobic interior of HSA. Thus, the enhancement of fluorescence intensity in presence of HSA indicates that levamlodipine partitions to a restrictive and hydrophobic binding site in HSA.
Figure 5.
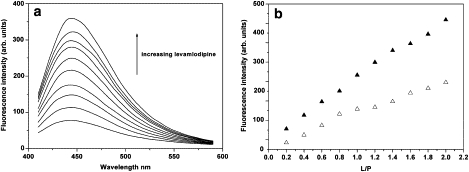
Fluorescence emission spectra of levamlodipine in presence of HSA (a, b ▴) and of alone PBS (b ▵). [HSA] = 2.0 × 10−5 M; [PBS] = 0.1 M, pH 7.4; L/P (levamlodipine/HSA molar ratio) from 0.1 to 2.0; λex = 374 nm.
Binding mechanism and binding constants
Clearly, the increase of levamlodipine emission is predominantly attributable to this quenching of Trp fluorescence by levamlodipine in levamlodipine-HSA system, which is involved in a fluorescence resonance energy transfer mechanism (12). Fluorescence resonance energy transfer occurs as long as the fluorescence emission spectrum of fluorophore (donor) overlaps with the UV absorption spectrum of small molecule (acceptor). The UV-vis absorption peak of levamlodipine is mostly in the range of fluorescence emission wavelength of HSA (Fig. 6), the fluorescence energy of HSA therefore can be transferred to levamlodipine. The calculated distance, r = 4.91 nm, is >8 nm, which agrees exactly with the nonradiative energy transfer theory (26) and facilitates the intermolecular energy transfer to the drug (27). The conclusion can be drawn that the energy transfer from HSA to levamlodipine occurs and quenches the florescence of HSA.
Figure 6.
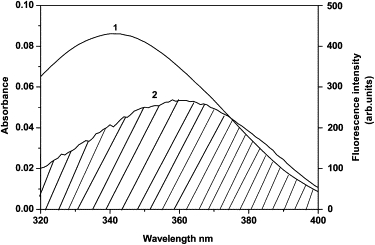
Overlapping of the fluorescence emission spectrum of HSA (λex = 290 nm) (1) with UV absorption spectrum of levamlodipine (2). [levamlodipine] = 1.0 × 10−5 M, [HSA] = 1.0 × 10−5 M, pH = 7.4; T = 298 K.
For the binding of small molecules to protein, the fluorescence measurements can provide some important information, such as binding mechanism, binding constants, binding sites, the number of binding sites, etc. As shown in Fig. 7, the plots show well linear relation at every experimental temperature, which indicates that only one kind of quenching mechanisms is predominant, either dynamic one or static one (28). The corresponding constants of Kq are found to be 4.42 × 1011 M−1 s−1 (r = 0.999) at 298 K and 1.2 × 1012 M−1 s−1 (r = 0.998) at 309 K, respectively. The maximum scatter collision quenching constant Kq of various quenchers for biopolymers is ∼2.0 × 1010 M−1 s−1 (29). In this study, Kq of binding of levamlodipine with HSA is greater than the maximum value of collision quenching Kq, which means that the quenching is not initiated by dynamic collision but by static one, i.e., the formation of levamlodipine-HSA complex.
Figure 7.
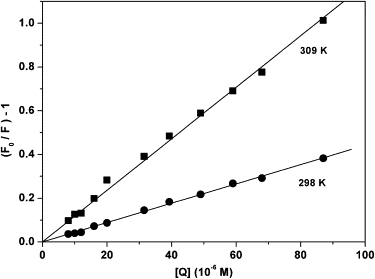
Stern-Volmer curves for quenching various concentrations of levamlodipine with HSA at 298 K and 309 K. [HSA] = 2.0 × 10−5 M; λex = 290 nm.
The binding constant KA at various temperatures can be obtained from Fig. 8: KA = 4.3 × 103 M−1 (r = 0.996) at 298 K and KA = 1.2 × 104 M−1 (r = 0.998) at 309 K. The linearity of both the curves indicates that levamlodipine binds independently to one class of sites on HSA. The values of n are noticed to be (1.02 ± 0.01) at 298 K and (0.91 ± 0.02) at 309 K, respectively. From the data of n, it can be suggested that there is one independent binding site on HSA for levamlodipine.
Figure 8.
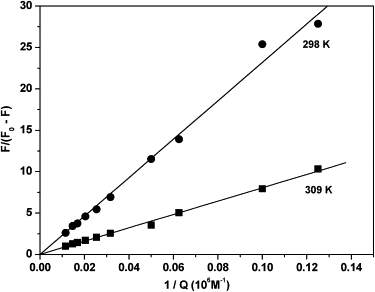
Lineweaver-Burk curves for quenching levamlodipine with HSA at 298 K and 309 K. [HSA] = 2.0 × 10−5 M; λex = 290 nm.
Interaction forces between levamlodipine and HSA
According to the dependence of binding constants on temperature, the thermodynamic parameters considered to be responsible for the formation of the levamlodipine-HSA complex were analyzed to further analyze the type of interaction forces between levamlodipine and HSA. Usually, the type of interaction forces between small molecules and macromolecules mainly include hydrogen bonds, van der Waals forces, electrostatic interactions, and hydrophobic interactions (30). To estimate the binding mode, the thermodynamic parameters, enthalpy changes (ΔH), entropy changes (ΔS), and free energy changes (ΔG), are mainly considered. On the basis of the characteristic signs of the thermodynamic parameters, positive ΔH and ΔS are frequently taken as evidences for hydrophobic interactions (31). This attraction of hydrophobic species is known as hydrophobic bonding or hydrophobic interaction, which resulted from their unwelcome reception in water. Here, both the positive values of ΔH (71.43 kJ mol−1) and ΔS (309.3 J mol−1 K−1) listed in Table 1 show that hydrophobic interaction plays an absolutely key role in the binding of levamlodipine to HSA. Furthermore, it is clear that the binding of levamlodipine to HSA is an exothermic process accompanied by negative values of ΔG and a positive value of ΔS. Both the negative values of ΔG at various temperatures imply the tendency of spontaneous binding of levamlodipine to HSA. Moreover, it is worth mentioning that contribution to ΔG arises more from the TΔS rather than from ΔH, so the binding process is entropy driven.
Table 1.
Thermodynamic parameters of binding of levamlodipine to HSA
| T (K) | ΔG (kJ mol−1) | ΔH (kJ mol−1) | ΔS (J mol−1 K−1) |
|---|---|---|---|
| 298 | −20.73 | 71.43 | 309.30 |
| 309 | −24.13 |
Molecular modeling of levamlodipine-HSA complex
In this study, we have shown the validity of the binding of levamlodipine with HSA in an experimental way. To further realize the information of levamlodipine binding HSA, molecular modeling was applied to predict the binding mode of levamlodipine in HSA. It can be seen that the entrance of site I, which is surrounded by positively charged residues such as Arg218, Arg222, Arg257, His242, and Lys199, is the binding site (Fig. 9). Of interest is the observation that the A- and B-rings of levamlodipine are practically nonplanar, ring A rotates vertically around ring B. The phenyl of Trp214 is vertically close to the hydrophobic part of A-ring in levamlodipine with 5.61 nm distance, which is close to the calculated value and means that the fluorescence energy of Trp can transfer to levamlodipine to bring the quenching (27). Within 6 Å around levamlodipine, the surrounding microenvironment of levamlodipine in site I is shown to be rich in nonpolar amino acid residues, such as Ala215, Ala261, Ala291, Ile264, Ile290, Leu219, Leu238, Phe223, and Trp214. Accounting for the hydrophobic property of levamlodipine (insoluble in water), it can be concluded that hydrophobic force is the major interaction in the binding between levamlodipine and HSA.
Figure 9.
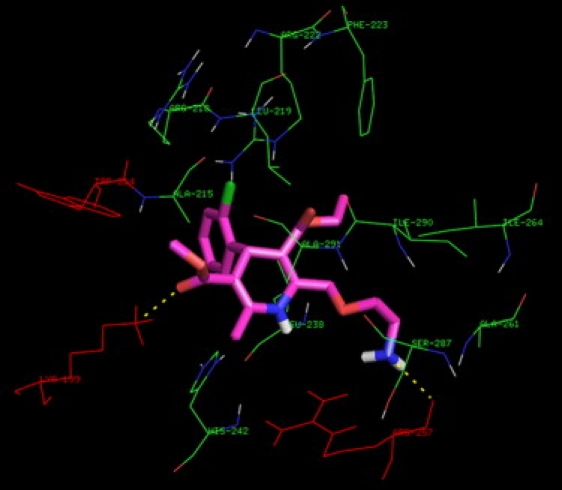
Binding modeling of levamlodipine to HSA in the entrance of site I. The displayed residues are within 6 Å around levamlodipine. The H-bonds are shown by broken line. Levamlodipine is shown as cylinder model (C, magenta; O, red; H, white; N, blue; and Cl, green).
The interaction between ligands and HSA in site I was dominated by hydrophobic interactions, but there were also specific interactions (15). The interaction between levamlodipine and HSA is not exclusively hydrophobic in nature due to several polar residues in the proximity of the bound ligand playing a role of stabilizing the ligand via H-bonds (Fig. 9). As shown, Lys199 and Arg257 are found to be important in firming the binding environment of the ligand. The side chain nitrogen atom of Lys199 is in suitable position to form intermolecular H-bond with the oxygen atom of 5-methoxycarbonyl of levamlodipine. Additionally, the intermolecular H-bond is also formed between the main chain carbonyl oxygen of Arg257 and the nitrogen atom of (2-aminoethoxy) methyl of levamlodipine. However, H-bonds fail to be found experimentally, which may be explained as the following deductions. On the one hand, microenvironment difference surrounding HSA exists in the binding system between experimental study under physiological conditions and molecular modeling under ideal conditions. On the other hand, according to the two molecular properties, the role of hydrophobic force is much greater than that of H-bonds through comparing their contributions to the binding of levamlodipine with HSA. Accordingly, this finding not only provides an optimal structural basis to explain the very efficient fluorescence quenching of HSA emission in the presence of levamlodipine, but also supplements the formation of intermolecular H-bond that can not be validated by experimental method.
Comparison of the binding energies and ISR values at seven sites
To gain insight into the carrier function of HSA in more detail, the binding energies at seven binding sites were calculated by computational method. In addition, ISR was used to represent the specificity of binding between receptors and ligands. The binding at site 5 is apparently looser than that at site 7, and the ISR value is much smaller than that at site 7 (Table 2). Thus, both the two principal findings strongly suggest that the complex formation between levamlodipine and HSA at the site is the most unstable and nonspecific. This further implies that it is impossible for HSA to bind and carry levamlodipine in the body through using site 5. For sites 2, 3, and 4, the bindings are slightly tighter, but the ISR values are apparently smaller than that at site 7. From the observation, it can be concluded that the complex formation is tight but not enough specific at site 2, 3, or 4, which suggests that levamlodipine bound at site 2, 3, or 4 is relatively less specific than at site 7 to release from HSA to plasma.
Table 2.
Comparison of the predicted binding energies and ISR values at seven sites on HSA
| Binding site | Binding energy (kcal mol−1) | ISR |
|---|---|---|
| 1 | −9.88 | 3.87 |
| 2 | −8.99 | 2.60 |
| 3 | −8.52 | 1.72 |
| 4 | −9.25 | 1.38 |
| 5 | −6.49 | 2.72 |
| 6 | −8.00 | 3.80 |
| 7 | −8.34 | 4.11 |
In the common practice of binding, high affinity is used as the screening criterion. The affinity, however, may not always guarantee the specificity. The intrinsic specificity is often strongly correlated with the structural fit (4), that is a good measure of the structure specificity. In the high affinity zone, the small molecules can still have different specificities. In current report, the binding at site 1 belongs to the high affinity regime, but the specificity is less than that at site 7. We will choose the small molecules with both high affinity and high specificity as the potential good binding. According to this opinion, the binding of levamlodipine to HSA at site1 is unfeasible to carry out its carrier and transportation functions based on the view of drug efficacy that only portion of a drug unbound to plasma is generally bioactive. Moreover, in the body, site 1 allows both electrostatic and hydrophobic interactions to contribute to the progressive increase affinity in the presence of fatty acids (32). This suggests that it is impossible to displace the fatty acid from site 1 because HSA carries ∼0.1–2 mol of fatty acid per mol protein under normal physiological conditions (33).
Simultaneously, drug-binding experiments indicated that it was possible to displace fatty acids from sites 6 and 7, suggesting that they may be of relatively lower affinity (34), which was in agreement with our molecular modeling results. Experimentally, levamlodipine binds to site 7, one high-affinity site on HSA, with a binding constant of 1.2 × 104 M−1 under physiological conditions. Although the binding constant is <3.4 × 105 M-1 of high-affinity binding of warfarin, a probe of site I, it is in the range of typical association constants of 104–106 M−1. It is possible for high-affinity levamlodipine binding to site 7 thus because the in vivo concentration of drugs is always much lower than that of HSA (35). Comparing of binding energies and ISR values at sites 6 and 7, it is clear that the affinity at site 6 is close to that at site 7, but the ISR is smaller than that at site 7. The observation implies that levamlodipine specifically binds at site 7 rather than site 6 with the high regime.
Discussion
HSA serves as carrier function for many drugs in the body. A common set of seven binding sites on HSA, site 1 in subdomain IB, site 2 in subdomain IA and IIA, sites 3 and 4 in subdomain IIIA, site 5 in subdomain IIIB, site 6 in domain II, and site 7 in subdomain IIA, have been shown (36). To characterize the protein that is responsible for carrying drugs in more detail, a total of seven binding sites that were docked by levamlodipine have been analyzed through comparing their affinity and specificity.
According to the major idea of drug efficacy, both the sites, sites 6 and 7, can be principal drug-binding sites for levamlodipine. Why, then, is site 7 the primary binding site for levamlodipine but not site 6 as indicated experimentally in this study? To our knowledge, ligands binding to site II, i.e., site 6, are often surrounded by negatively charged aromatic carboxylic acids. However, levamlodipine is a positively charged compound. Site II seems to be smaller, or less flexible, than site I, thus, it seems to be more restricted than site I (35). Furthermore, drugs binding to HSA can be modulated by simultaneous binding of endogenous compounds, such as fatty acids. From the view of circulation in the body, site 6 has a high affinity for long-chain fatty acids, whereas site 7 seems to be a primary site for short-chain, but not long-chain, fatty acids. In addition, long-chain fatty acids are normally most prevalent in the circulation, and they can enhance the affinity of the protein for certain site I ligands by induction of conformational changes in the albumin molecule (35). The medium-chain fatty acids bind with high affinity to site II, but only bind to site I with low affinity. Therefore the displacing effect on site I drugs by competition is probably very small, which further guarantees levamlodipine smoothly binds HSA at site I rather than site II. At higher fatty acid concentrations it is suggestive of a low affinity interaction between fatty acids and site 7 due to the absence of specific interactions (15). Importantly, at higher drug concentrations the affinity for fatty acids falls off, which probably attributes to the direct competition between the drug and fatty acids (36). These findings further support the opinion that levamlodipine is preferential to locate in site 7 rather than site 6 on HSA under physiological conditions, especially in presence of fatty acids.
Of particular importance is the finding in Table 2 that the affinity at site 7 is in the high regime with the highest specificity. We believe our new, to our knowledge, intrinsic specificity definition has the advantages of quickly identifying and quantifying the specificity of the ligand to the receptor without going through all the receptors as the conventional definition of specificity. This gives an absolute measure of specificity with a dimensionless quantity, ISR, which can be calculated once the binding spectrum from the collection of each intermediate binding mode of a ligand and a receptor is known. The binding spectra can be obtained through binding/docking and detailed free energy calculation/conformational search (Fig. 10). The binding, at site 7, with a highest ISR value of 4.11 displays a mostly lowest binding energy that is well set apart energetically from the nonnative binding states, which exhibits a high intrinsic specificity as is expected for a binding (4). These bindings at other sites but site 7 have a smaller energy difference between the native and average nonnative states relative to the spread of the energy spectrum of nonnative states. In our recent work, we have suggested that the origin of high intrinsic specificity seems to be the underlying hydrophobic interactions (4). It has been observed that hydrophobic interactions dominate in the levamlodipine-HSA complex experimentally. The high ISR value obtained from the physical binding spectrum thus agrees with the experimental conclusion that hydrophobic interactions are mainly responsible for the intrinsic specificity.
Figure 10.
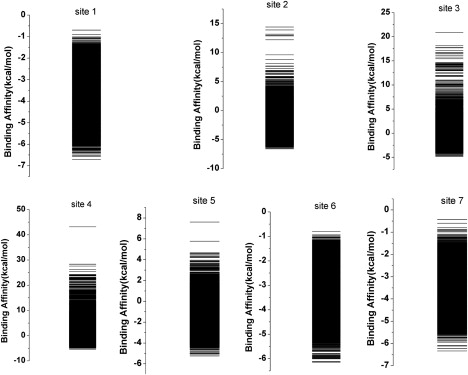
Comparison of the predicted levamlodipine energy spectra at seven sites on HSA.
In most cases, drug-HSA interactions will significantly affect the distribution volume and the elimination rate of drugs as a result of their binding to HSA (5). Only that portion of a drug-free in plasma can produce pharmacological effect through free transferring to the target organ. Contrary to this, the drug tightly bound to HSA hardly passes through the blood capillary walls to reach the action site due to its larger molecular weight. Thus, the drug specifically bound in site 7 can free release from HSA due to its relatively low affinity, which further guarantees HSA to server as the carrier protein. This rational is in agreement with the drug soaking experiments (36) and further confirms that levamlodipine may be easier to locate and to displace in site I as shown experimentally in this study.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (20575063, 90713022 and 20735003) and the Chinese Academy of Sciences (KJCX2.YW.H09). Financial support was also provided by a National Science Foundation Career Award (J.W.) and the American Chemical Society Petroleum Research Fund (J.W.). The authors have no conflicting financial interests.
Contributor Information
Xiurong Yang, Email: xryang@ciac.jl.cn.
Erkang Wang, Email: ekwang@ciac.jl.cn.
Jin Wang, Email: jin.wang.1@stonybrook.edu.
References
- 1.Wang J., Verkhivker G.M. Energy landscape theory, funnels, specificity, and optimal criterion of biomolecular binding. Phys. Rev. Lett. 2003;90:1881011–1881014. doi: 10.1103/PhysRevLett.90.188101. [DOI] [PubMed] [Google Scholar]
- 2.Wlodawer A., Erickson J.W. Structure-based inhibitors of HIV-1 protease. Annu. Rev. Biochem. 1993;62:543–585. doi: 10.1146/annurev.bi.62.070193.002551. [DOI] [PubMed] [Google Scholar]
- 3.Liu Z., Zheng X., Wang J., Wang E. Molecular analysis of thymopentin binding to HLA-DR molecules. PLoS ONE. 2007;2:e1348. doi: 10.1371/journal.pone.0001348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang J., Zheng X., Yang Y., Drueckhammer D., Yang W. Quantifying intrinsic specificity: A potential complement to affinity in drug screening. Phys. Rev. Lett. 2007;99:1981011–1981014. doi: 10.1103/PhysRevLett.99.198101. [DOI] [PubMed] [Google Scholar]
- 5.Kragh-Hansen U. Molecular aspects of ligand binding to serum albumin. Pharmacol. Rev. 1981;33:17–22. [PubMed] [Google Scholar]
- 6.He X.M., Carter D.C. Atomic structure and chemistry of human serum albumin. Nature. 1992;358:209–215. doi: 10.1038/358209a0. [DOI] [PubMed] [Google Scholar]
- 7.Il'ichev Y.V., Perry J.L., Simon J.D. Interaction of Ochratoxin A with human serum albumin. Preferential binding of the dianion and pH effects. J. Phys. Chem. B. 2002;106:452–459. [Google Scholar]
- 8.Takamura N., Haruta A., Kodama H., Tsuruoka M., Yamasaki K. Mode of interaction of loop diuretics with human serum albumin and characterization of binding site. Pharm. Res. 1996;13:1015–1019. doi: 10.1023/a:1016098305796. [DOI] [PubMed] [Google Scholar]
- 9.Kelly J.G., O'Malley D.K. Clinical pharmacokinetics of calcium antagonists. Clin. Pharmacokinet. 1992;22:416–433. doi: 10.2165/00003088-199222060-00002. [DOI] [PubMed] [Google Scholar]
- 10.Gelamo E.L., Silva C.H.T.P., Imasato H., Tbak M. Interaction of bovine (BSA) and human serum albumins (HSA) with ionic surfactants: spectroscopy and modelling. Biochim. Biophys. Acta. 2002;1594:84–99. doi: 10.1016/s0167-4838(01)00287-4. [DOI] [PubMed] [Google Scholar]
- 11.Lakowicz J.R., Weber G. Quenching of fluorescence by oxygen. Probe for structural fluctuations in macromolecules. Biochemistry. 1973;12:4161–4170. doi: 10.1021/bi00745a020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Förster T., Sinanoglu O. Academic Press; New York: 1996. Modern Quantum Chemistry. [Google Scholar]
- 13.Cyril L., Earl J.K., Sperry W.M. E. & F. N. Spon; London: 1961. Biochemists Handbook. [Google Scholar]
- 14.Hetenyi C., Spoel D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Letters. 2006;580:1447–1450. doi: 10.1016/j.febslet.2006.01.074. [DOI] [PubMed] [Google Scholar]
- 15.Petitpas A.A., Bhattacharya S., Twine M.E., Curry S. Crystal structure analysis of warfarin binding to human serum albumin. J. Biol. Chem. 2001;276:22804–22809. doi: 10.1074/jbc.M100575200. [DOI] [PubMed] [Google Scholar]
- 16.Reference deleted in proof.
- 17.Pettersen E.F., Goddard T.C., Huang C.C., Couch G.S., Greenblatt D.M. UCSF chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 18.Case D.A., Cheatham T.E., Darden T., Gohlke H., Luo R. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 20.Darden T., York D., Pedersen L. Particle mesh Ewald: an N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 21.Ryckaert J.P., Ciccotti G., Berendsen H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 22.Naim M., Bhat S., Rankin K.N., Dennis S., Chowdhury S.F. Solvated interaction energy (SIE) for scoring protein-ligand binding affinities. 1. Exploring the parameter space. J. Chem. Inf. Model. 2007;47:122–133. doi: 10.1021/ci600406v. [DOI] [PubMed] [Google Scholar]
- 23.Hu Y.J., Liu Y., Wang J.B., Xiao X.H., Qu S.S. Study of the interaction between monoammonium glycyrrhizinate and bovine serum albumin. J. Pharm. Biomed. Anal. 2004;36:915–919. doi: 10.1016/j.jpba.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 24.He W., Li Y., Si H., Dong Y., Sheng F. Molecular modeling and spectroscopic studies on the binding of guaiacol to human serum albumin. J. Photochem. Photobiol. A. 2006;182:158–167. [Google Scholar]
- 25.Yuan T., Weljie A.M., Vogel H.J. Tryptophan fluorescence quenching by methionine and selenomethionine residues of calmodulin: orientation of peptide and protein binding. Biochemistry. 1998;37:3187–3195. doi: 10.1021/bi9716579. [DOI] [PubMed] [Google Scholar]
- 26.Feng X.Z., Lin Z., Yang L.J., Wang C., Bai C.L. Investigation of the interaction between acridine orange and bovine serum albumin. Talanta. 1998;47:1223–1229. doi: 10.1016/s0039-9140(98)00198-2. [DOI] [PubMed] [Google Scholar]
- 27.Sytnik A., Litvinyuk I. Energy transfer to a proton-transfer fluorescence probe: tryptophan to a flavonol in human serum albumin. Proc. Natl. Acad. Sci. USA. 1996;93:12959–12963. doi: 10.1073/pnas.93.23.12959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lakowicz J.R. Plenum Press; New York: 1983. Principles of Fluorescence Spectroscopy. [Google Scholar]
- 29.Ware W.R. Oxygen quenching of fluorescence in solution: an experimental study of the diffusion process. J. Phys. Chem. 1962;66:455–458. [Google Scholar]
- 30.Klotz I.M. Physiochemical aspects of drug-protein interactions: a general perspective. Ann. N. Y. Acad. Sci. 1973;226:18–35. doi: 10.1111/j.1749-6632.1973.tb20465.x. [DOI] [PubMed] [Google Scholar]
- 31.Ross P.D., Subramanian S. Thermodynamics of protein association reactions: forces contributing to stability. Biochemistry. 1981;20:3096–3102. doi: 10.1021/bi00514a017. [DOI] [PubMed] [Google Scholar]
- 32.Reynolds J.A., Herbert S., Steinhardt J. Binding of some long-chain fatty acid anions and alcohols by bovine serum albumin. Biochemistry. 1968;7:1357–1361. doi: 10.1021/bi00844a016. [DOI] [PubMed] [Google Scholar]
- 33.Fredrickson S.D., Gordon R.S. The metabolism of albumin bound C14-labeled unesterified fatty acids in normal human subjects. J. Clin. Invest. 1958;37:1504–1515. doi: 10.1172/JCI103742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Curry S., Mandelkow H., Brick P., Franks N. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat. Struct. Biol. 1998;5:827–835. doi: 10.1038/1869. [DOI] [PubMed] [Google Scholar]
- 35.Kragh-Hansen U., Chuang V.T.G., Otagirl M. Practical aspects of the ligand-binding and enzymatic properties of human serum albumin. Biol. Pharm. Bull. 2002;25:695–704. doi: 10.1248/bpb.25.695. [DOI] [PubMed] [Google Scholar]
- 36.Bhattacharya A.A., Grune T., Curry S. Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin. J. Mol. Biol. 2000;303:721–732. doi: 10.1006/jmbi.2000.4158. [DOI] [PubMed] [Google Scholar]